

Abstract
The enumeration of circulating tumor cells (CTCs) in the peripheral bloodstream of metastatic cancer patients has contributed to improvements in prognosis and therapeutics. There have been numerous approaches to capture and counting of CTCs. However, CTCs have potential information beyond simple enumeration and hold promise as a liquid biopsy for cancer and a pathway for personalized cancer therapy by detecting the subset of CTCs having the highest metastatic potential. There is evidence that epithelial cell adhesion molecule (EpCAM) expression level distinguishes these highly metastatic CTCs. The few previous approaches to selective CTC capture according to EpCAM expression level are reviewed. A new two-stage microfluidic device for separation, enrichment and release of CTCs into subpopulations sorted by EpCAM expression level is presented here. It relies upon immunospecific magnetic nanoparticle labeling of CTCs followed by their field- and flow-based separation in the first stage and capture as discrete subpopulations in the second stage. To fine tune the separation, the magnetic field profile across the first stage microfluidic channel may be modified by bonding small Vanadium Permendur strips to its outer walls. Mathematical modeling of magnetic fields and fluid flows supports the soundness of the design.
1. Introduction
Mortality in patients suffering from carcinomas (cancers derived from epithelial cells) is, in the majority of cases, due to the metastasis of the disease. Metastasis involves the release of cancer cells from the primary tumor into the blood circulation, resulting in their transport to other sites in the body where they give rise to the growth of secondary tumors. Although cancer cells may also be found in the lymph nodes draining the primary tumor, metastasis is believed to occur principally via the blood circulation [1]. Metastasis is a complicated, multistep process that is still not fully understood [2–9]. For metastasis to succeed, a cancer cell must survive a series of steps involving invasion of surrounding tissue, intravasation to the blood microvasculature, transport in the blood circulation to a capillary bed at some distant site, extravasation, survival in the local environment, and subsequent proliferation to form a secondary tumor. More than a million cells may be shed per gram of tumor in a day [10]. This would be equivalent to over 200 circulating tumor cells (CTCs) being introduced to each mL of the bloodstream each day (assuming a 5 L blood volume). Fortunately, very few of these cells remain in the bloodstream for very long let alone survive to initiate new tumor growth [11]. In fact, CTCs in the bloodstream are extremely rare For example, for the U.S. Food and Drug Administration (FDA)-approved CellSearch System (Menarini Silicon Biosystems Inc., Bologna, Italy) the critical threshold for CTC captured from a 7.5 mL blood sample is set at just 5 for a positive diagnosis of metastatic breast or prostate cancer and just 3 for metastatic colorectal cancer [12–15]. This compares with approximately 5 × 107 leukocytes typically found in a 7.5 mL blood sample. It is a challenge to isolate small numbers of CTC from this background of leukocytes, and it may be that the low thresholds may reflect a less than prefect capture efficiency.
Cancer cells derived from epithelial cells tend to be much larger (most are 20 to 30 μm in diameter) than normal blood cells (leukocytes are between 8 and 14 μm, and erythrocytes are 6 to 9 μm) and are not able to pass through the capillary beds of the lungs, for example, where capillaries may be only 3 to 8 μm in diameter. Erythrocytes can pass through the capillaries due to their extreme deformability, while leukocytes are able to extravasate. In fact, leukocytes spend most of their time in the tissues and enter the bloodstream for transport to different parts of the body. One might therefore expect all CTCs to be trapped within minutes of release. Most of those that become trapped in a capillary bed or in the bone marrow undergo apoptosis while some may become dormant [16–20]. Apoptosis may be induced in many of the cells when they detach from the extracellular matrix and neighboring cells in the primary tumor [18]. Of course, some do survive, remain active, and go on to cause the spread of disease.
The fact that dissemination of cancer cells occurs primarily through the bloodstream opened the possibility of monitoring the progression of disease, or its response to treatment, by enumeration of the CTCs in the blood. This has become a very active area of research in recent years for good reason. Compared to the taking of a biopsy, the collection of a blood sample is easy, minimally intrusive, and may be repeated as necessary [21–23]. In addition to providing a CTC count, the characterization and culturing of captured CTCs may provide information concerning the mechanisms of metastasis as well as mutational responses to chemotherapy [24, 25].
2. Heterogeneity of CTCs
The cells in the primary tumor, as well as in secondary tumors, are heterogeneous, as are the cells that are shed into the bloodstream. It is generally accepted that CTCs undergo an epithelial to mesenchymal transition (EMT) where epithelial cell adhesion molecule (EpCAM) surface expression becomes downregulated. The cells become more mesenchymal in nature with increased ability to migrate and invade tissues [26–29]. Mesenchymal to epithelial transition (MET) is thought to occur with the formation of metastatic tumors which, like the primary tumor, tend to exhibit a mixed epithelial/mesenchymal phenotype [30, 31].
It is not known if all CTCs are capable of initiating secondary tumor growth. It has been suggested that the growth of a metastatic tumor requires the presence of cancer stem cells (CSCs), a subset of CTCs possessing some of the characteristics of normal stem cells, such as self-renewal and ability to differentiate into various phenotypes, in this case the phenotypes present in the primary tumor [32]. CSCs also appear to exhibit other traits relevant to metastasis such as increased motility and invasiveness, and heightened resistance to apoptosis. It has been speculated that CSCs must be present in the primary tumor (intrinsic CSCs), and are necessary for its growth. In both primary and secondary tumors, the CSCs are then assumed to give rise to rapidly expanding progenitor cells that eventually differentiate into other phenotypes [33–35]. It may be assumed that CSCs would have reduced EpCAM expression, but the degree of downregulation is not yet established, and some EpCAM expression may actually be necessary to maintain cancer cell and CSC attributes [36, 37]. This is consistent with findings of Jolly et al. that CTCs showing partial EMT, and therefore reduced EpCAM expression, have much greater tumor-initiating and metastatic potential than cells that have undergone complete EMT, and that partial EMT may be associated with stemness [38–40].
3. CTC capture technologies
3.1. Capture of CTC
The vast majority of CTC capture techniques are capable of achieving only binary selection where cells are either captured or discarded depending on some intrinsic or induced property. Capture of CTCs must be highly efficient and specific. There may be some flexibility in setting the capture threshold for some approaches, but they do not generally separate cells into multiple fractions that differ in expression level of EpCAM, for example.
There have been numerous reviews of the different technologies proposed for CTC isolation, reflecting current interest and the potential benefits that may arise from their study (see, for example, refs. [41–45]). The isolation of CTCs from a blood sample requires the exploitation of differences in either their physical or biological properties from those of the other cells in the sample. Physical properties that can be exploited include size, density, deformability, electrical polarizability, and intrinsic magnetic susceptibility. For isolation of intact cells, the biological property that can be exploited is limited to the immunochemistry of their surface membranes. Selection according to surface antigens can simply be via interaction with antibodies attached to surfaces, or by enhancing the physical difference of the targeted cells from non-targeted cells by, most commonly, immunomagnetic labeling or by attachment of size-density amplification beads [46].
Immunomagnetic labeling of EpCAM imparts relatively high magnetic susceptibility to EpCAM+ CTCs, sufficient for their capture by application of a magnetic field gradient to the bulk sample, as used by the CellSearch System. Density gradient centrifugation may also be applied to bulk samples. In both cases, the transport distances for the targeted cells are relatively large and CTC migration is hindered by the other cells present. These factors contribute to poor capture efficiency. There has been an attempt to avoid these drawbacks for magnetic selection by movement of a plastic-sheathed magnetic stirring rod through the cell suspension; the MagSweeper device [47].
Microfluidic systems offer more options for cell selection, and the recent emphasis has been overwhelmingly in their favor (see recent reviews [48–58]). In microfluidic systems, cells are carried by laminar fluid flow into regions where they can be influenced by well controlled fields or where interaction with active surfaces is enhanced. Cells may be selected for by size or deformability using fluid inertial effects in specially shaped microchannels. Dielectrical selection according to differences in electrical polarizability can influence cells across only the small thicknesses of microfluidic channels [59–61]. Resonant acoustic frequencies between 1 and 10 MHz are suited to manipulation of particles and cells of 1 to 20 μm in diameter [62]. Acoustofluidic separations require use of microfluidic channels where a single focusing node at the channel midpoint is obtained when channel thickness corresponds to half the resonant wavelength [63]. The separation or capture of magnetically labeled CTCs is enhanced by the short transport distances in microfluidic devices and the high magnetic fields and field gradients that can be generated in localized regions.
Transport distances under the influence of an applied field in microfluidic channels are typically less than 0.5 mm. The dipole magnet described in this work is predicted to generate a field greater than 1 T and a field gradient of more than 0.5 T/mm over a range of 0.5 mm. (A gradient of 0.5 T/mm over a distance of 0.5 mm would correspond to a decrease of 0.25 T, which is consistent with Fig. S3 of the Supplementary Material.) Cell tracking velocimetry studies of immunomagnetically labeled Jurkat cells (immortalized T lymphocyte cell line) in field gradients of 0.131 T/mm showed mean magnetophoretic velocities of 30.9 μm/s[64] so that velocities greater than 118 μm/s would be expected at gradients > 0.5 T/mm, giving a transport time of < 4.2 seconds over a distance of 0.5 mm. (It should be noted that velocities up to about 73 μm/s could be induced by increasing the amount of immunomagnetic reagent used. Magnetic selection from bulk samples may involve transport across a vessel of 1 cm in diameter or more. A dipole magnet placed against the wall of such a vessel may generate a field of over 1 T at the inner wall close to the magnet, but the field will decay approximately exponentially with distance from the magnet as will the field gradient. The Supplemetary Matrial shows the results of two-dimensional modeling a simple dipole magnet using Finite Element Method Magnetics (FEMM 4.2) [65]. Each magnet has a cross section of 1 cm × 1 cm, and they are separated by 1 mm. Fig. S1 shows magnetic flux lines and a flux density plot around the magnets, and Fig. S2 the fall in magnetic field B and field gradient with distance from the dipole. It is clear that even if the field gradient exceeds 0.5 T/mm close to the near wall, such field gradients cannot extend far into the vessel. Field induced transport velocities would initially fall in proportion to the field gradient when the nanoparticle labels are magnetically saturated, but fall more rapidly as the label magnetization falls below saturation with reduction of B. This, combined with the increased transport distance, would lead to a greatly increased time for cell transport. In terms of sample volume depleted per unit time, this will be offset only to some extent by the larger volume under the influence of the magnetic field.
3.2. Separation and isolation of cells according to antigen expression level
Few attempts have been made to isolate CTCs into different fractions according to some surface expression level. Nagrath et al. [66] proposed that two successive binary separations be carried out, the first to select for high EpCAM expressing cells (having greater than 2/3 coverage of 1-μm immunomagnetic beads), and the second to select for moderately expressing cells (with 1/3 to 2/3 bead coverage). For cultured PANC-1 cells (a human pancreatic carcinoma, epithelial-like cell line), they were thereby able to select for high, moderate and low (less than 1/3 label coverage) expression of EpCAM. FACS analysis of the fractions showed only moderate differences between the populations, but a scratch assay of the cultured fractions appeared to show a more significant difference. They were also able to show differences in the relative abundance of the different fractions for blood samples taken from six patients suffering from pancreatic ductal adenocarcinoma.
Kwak et al. [67] modified an earlier approach of Kang et al. [68] in which magnetically labeled CTCs were captured in a series of small chambers on the side of a microfluidic channel by application of a magnetic field gradient across the channel breadth. The new design, called the Mag-Gradient Chip, used a simple block permanent magnet, as in the original work, but it used a serpentine channel with five straight channel segments parallel to the magnet surface, each segment successively closer to the magnet. Cells that are highly expressing in EpCAM and therefore carry the most magnetic labels were expected to be captured in the side chambers of the furthest channel segment, while those that are lower EpCAM expressing should be captured in the segments closer to the magnet. The device was tested using cultured MCF-7 (high EpCAM) and MDA-NB-231 (low EpCAM) cells, and it showed some degree of selective capture.
Kelley and co-workers have developed the Velocity Valley Chip (or VV Chip) [69–72] and the related magnetic ranking cytometry (MagRC) device [73–75]. The VV Chip uses a series of thin channels containing arrays of small X-shaped obstacles. A non-uniform magnetic field is applied across the thickness of the channels using arrays of 1.5 mm diameter NdFeB magnets placed against their major faces. The mean fluid velocity is arranged to decrease in successive channels either by branching from one into two, two into four, and four into eight channels or by altering their relative thicknesses or breadths. The purpose of the obstacles is to impede the cell motion thereby improving capture efficiency. If a cell is carried into a region close to an obstacle where the fluid drag force is exceeded by the magnetic force then it is captured. The highest EpCAM-expressing cells are expected to be captured in the first channel with lower expressing cells being captured in successive channels where fluid drag force is lower. The device captured cells spiked into whole blood more efficiently than CellSearch, and it was confirmed that different cell types exhibiting different levels of EpCAM expression were captured in different channels [69, 70]. Some preliminary work was also reported for blood samples taken from metastatic prostate cancer patients. Different patients showed different distributions of CTCs among the channels, possibly indicating different prevalence of EMT [69]. Preliminary work involving an animal model was also reported [71].
The initial MagRC device [73, 74] has many features in common with the VV Chip. It uses a single, 50-μm thick, microfluidic channel with arrays of small NdFeB permanent magnets applied to the exterior walls of the channel to generate transverse magnetic field gradients, and an internal array of 700 X-shaped obstacles, 500 μm in diameter. Below each of the X-shaped elements is a thin (1.5 μm) circular nickel element on the lower wall. The diameters of these nickel elements gradually increase along the length of the channel from 272 μm at the inlet to 470 μm at the outlet. The nickel elements are magnetized by the external permanent magnets, creating circular cell-capture zones that increase in size with the size of the elements and with the level of magnetic labeling. Since the nickel elements are located only on the lower channel wall, it is not clear why arrays of NdFeB magnets are applied to both walls. Nevertheless, strongly labeled cells were expected to be quickly captured near the channel inlet while less strongly labeled cells were expected to be captured further along the channel. To be captured by a nickel element, a cell must be driven close to the lower wall under the influence of the transverse magnetic field gradient and aided by gravity. It must also be carried into the capture zone by the fluid flow. There is an element of chance in cell capture, as in the VV Chip, but the probability of capture must increase with the size of the zones along the channel length. They were able to capture different cultured cell lines in different regions of the channel according to EpCAM expression level. Clinical samples were also studied, and significant differences in apparent expression levels correlated with prostate cancer aggression (as rated by Gleason score).
A new generation of MagRC [75] has just 10 capture zones characterized by different size nickel elements (290 μm to 470 μm in diameter at 20 μm increments) beneath the X-shaped obstacles. The channel breadth increases in stepwise fashion along its length resulting in stepwise reduction of mean channel flow velocity and therefore stepwise reduction in fluid drag force close to the nickel elements. Arrays of small NdFeB magnets were again arranged on both sides of the channel. The device successfully retained labeled cancer cells spiked into whole blood (100 cells into 50 μL blood), with different EpCAM-expressing cell types being retained in different regions of the channel. They also created orthotopic xenografts of human prostate cancer cells in immunodeficient mice and followed the progression of disease by examining the distribution profile of labeled cells captured from blood samples in the device as a function of time for the different type cancer cells. They also studied the effect of docetaxel treatment on the cell profiles.
A macro-scale device, the dipole magnetic fractionator (DMF) developed in our laboratory, also has the capacity for immunomagnetically separating cells into different subpopulations [76–79]. The DMF separates immunomagnetically labeled cells across the breadth of a thin parallel-walled channel on a continuous basis. It has many features in common with multi-outlet split-flow thin channel (SPLITT) fractionation [80], another member of the family of field- and flow-based separation techniques to which field-flow fractionation (FFF) also belongs. The cell suspension is introduced as a narrow stream, using hydrodynamic focusing, close to one of the side walls of the channel, with cell-free media introduced to the rest of the channel breadth. As they are carried along the length of the channel, the cells are driven across the channel breadth by their interaction with an applied magnetic field gradient. Those cells that strongly express the targeted antigen, and therefore carry more of the magnetic nanoparticle labels, are driven further across the channel breadth than cells that have lower expression levels. The cells are carried to a series of outlets arranged across the channel breadth and are thereby separated on a continuous basis into different fractions depending on the level of targeted antigen expression. In the macro system the magnetic field was generated using specially shaped, soft iron pole-pieces to obtain the required variation of field across the channel breadth. The channels were about10 and 15 mm broad and 0.75 to 1.8 mm thick. The use of magnetic nanoparticles for cell labeling results in a strong correlation between expression level and magnetophoretic mobility that reflects the number of bound nanoparticles [78, 79]. Table 1 lists the mean EpCAM expression level for a number of cell lines, as reported by Ozkumur et al. [81], estimated using flow cytometry measurements. Also listed are the mean magnetophoretic mobilities for the magnetically labeled cells as measured by cell tracking velocimetry [82–84] in our laboratory. The correlation is remarkably good. The magnetophoretic velocity v (in units of μm/s) of a labeled cell at high field (> 0.5 T) may be approximated via the relationship given by Leigh et al. [64] that may be written
| (1) |
where m is the magnetophoretic mobility in units of mm3/TAs and dB/dx is the magnetic field gradient in units of T/mm.
Table 1.
Mean EpCAM expression level obtained from Ozkumur et al. [81] and mean magnetophoretic mobilities for a set of immunomagnetically labeled cell lines.
| EpCAM | Magnetophoretic | |
|---|---|---|
| Cell line | expression | mobility (mm3/TAs) |
| LBX1 | 900 | 3.70 × 10−5 |
| MDA-MB-231 | 2000 | 8.23 × 10−5 |
| PC3–9 | 4000 | 1.65 × 10−4 |
| SKBR3 | 20000 | 7.41 × 10−4 |
| MCF 10A | 40000 | 1.65 × 10−3 |
We present here the design for a microfluidic DMF coupled to a magnetic capture module. The system is designed to efficiently isolate immunomagnetically labeled CTCs from a patient blood sample, separate them according to their targeted antigen expression level, and capture them for microscopic examination on a single optical plane in an area of about 30 mm2. Due to the gentle treatment of the cells during separation, they should remain viable and the separated subpopulations may be subsequently collected for further analysis or culture.
4. Microfluidic chip design
As mentioned above, the microfluidic system has two stages. The first is a microfluidic version of the DMF while the second is based on another instrument developed in our laboratory – magnetic deposition microscopy (MDM) [85–88]. In their microfluidic forms, they take advantage of reduced transport distances and reduced dispersion. There are additional important advantages associated with the microfluidic system as discussed below.
4.1. The microfluidic DMF stage
4.1.1. Separation channel
The separation channel is constructed as a simple sandwich of a spacer of 250 μm thickness, with the outline of the channel and the inlet and outlet ports excised, between two plane glass wafers of 400 μm thickness. The excised spacer is shown in Fig. 1. There are three inlet ports (on the left of the figure) and six outlet ports (on the right). The inlet and outlet ports are 250 μm in breadth. The main body of the separation channel is 60 mm long and has a breadth of 1.74 mm. The suspension of magnetically tagged cells is introduced to the center inlet port, and cell-free buffer solution to the other two ports. The relative volumetric flow rates are set so that the sample is initially confined to a narrow stream close to a side wall of the channel (corresponding to the lower edge wall in the figure). The ratio of the sample flow rate to that of the total flow rate determines the cross-sectional area of the sample stream within the overall flow following the merging of the fluids. Computational fluid dynamics has been used to predict the initial confines of the sample stream for a macro-scale DMF (of similar aspect ratio to the current micro channel) at a sample to overall flow rate ratio of 0.04:1 [78]. The sample may be confined to a smaller stream at the center of the channel thickness by reducing this ratio, but there must be a compromise between sample stream cross section and sample throughput. A ratio between 0.03 and 0.05 to 1 can be assumed to be acceptable. An advantage of the microfluidic system is that cells, particularly the larger cells, will be continually driven away from the channel walls by hydrodynamic effects and will tend to occupy the fluid streams in the central region of the channel thickness (between about 20% and 80% of the distance across the channel thickness) [89, 90]. The sample stream then flows without deviation or significant dispersion until it encounters the magnetic field gradient. From this point the magnetically labeled cells are driven across the breadth of the channel (corresponding to the upward direction in the figure) as they are carried along the channel length (from left to right). The channel flow is distributed to the six outlets, and under the laminar flow conditions there will be no remixing of the separated cell subpopulations downstream of the region under the magnetic field gradient. The flow rates at the inlets and outlets are set such that the cell subpopulation with highest expression of the targeted antigen (tagged with the highest number of magnetic nanoparticles) will be driven to the uppermost channel outlet. The untagged cells will not migrate across the channel breadth and will exit at the bottom outlet. Those with intermediate levels of antigen expression will be distributed to the intermediate outlets.
Fig. 1.
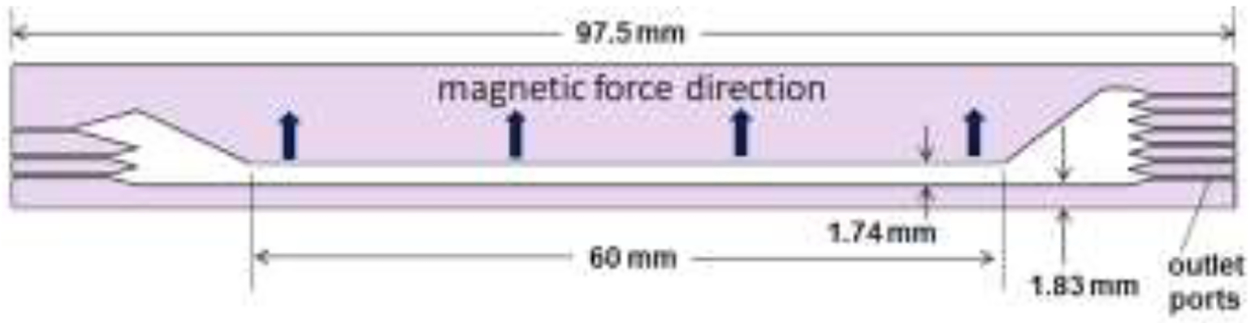
The spacer that is sandwiched between wafers to form the DMF channel. The figure shows the three inlets on the left and six outlets on the right.
4.1.2. Magnetic field
As mentioned above, the channel must be subjected to a magnetic field gradient that acts across its breadth. This is accomplished by placing it between the pole pieces of a dipole magnet. The placement of the channel relative to the pole pieces and the shape of the pole pieces influence the field gradient across the channel. In previous approaches for the macro DMF, the field gradient or the gradient in the square of the field was arranged to be fairly constant across the breadth [76, 77]. This was implemented using soft iron pole pieces machined to precise cross-sectional profiles determined using field modeling software (Magneto, Integrated Engineering Software, Winnipeg, Manitoba, Canada). Constant gradient in the square of the field is predicted to induce constant transverse migration velocity for cells labeled with paramagnetic particles. Constant field gradient is predicted to drive cells labeled with superparamagnetic nanoparticles at constant transverse velocity. This is because superparamagnetic nanoparticles tend to be magnetically saturated at the magnetic fields used.
Two different design objectives were set for the Micro DMF magnetic field: a field gradient that decreases across the channel breadth from a maximum at the side corresponding to sample introduction (Micro DMF I), and a constant field gradient across the channel breadth (Micro DMF II). The first design provides lower selectivity for separation of a wide range of magnetophoretic mobility while the second provides high selectivity over a limited range of cell mobility, as shown below.
4.1.2.1. Decreasing field gradient across channel breadth (Micro DMF I)
The magnet assembly is very similar to that used for constant field gradient, that is shown in Fig. 2, but differs in the shape and depth of the pole pieces. The magnet assembly is 35 mm along the length of the dipole. The custom NdFeB magnet block is prism-shaped with an isosceles trapezoidal cross section. It has a height of 20.5 mm, base breadth of 18 mm and upper breadth of 6 mm. It has a nominal magnetic energy product of 40 MGOe. The overall assembly has a breadth of 20 mm. The pole pieces have a depth of 3 mm and are separated by a constant gap of 1.20 mm, and the overall height of the assembly is 26 mm. The magnet and yoke assembly were constructed by Dexter Magnetic Technologies (Elk Grove Village, IL) according to our design. The channel is placed so that the channel edge is 2.10 mm from the surface of the magnet block. This requires a thin spacer of thickness 0.27 mm placed between the magnet block and the edge of the glass wafer-channel spacer assembly. (The channel, in the orientation shown in Figure 1, is lowered into the gap between the pole pieces of the magnet shown in Figure 2.)
Fig. 2.
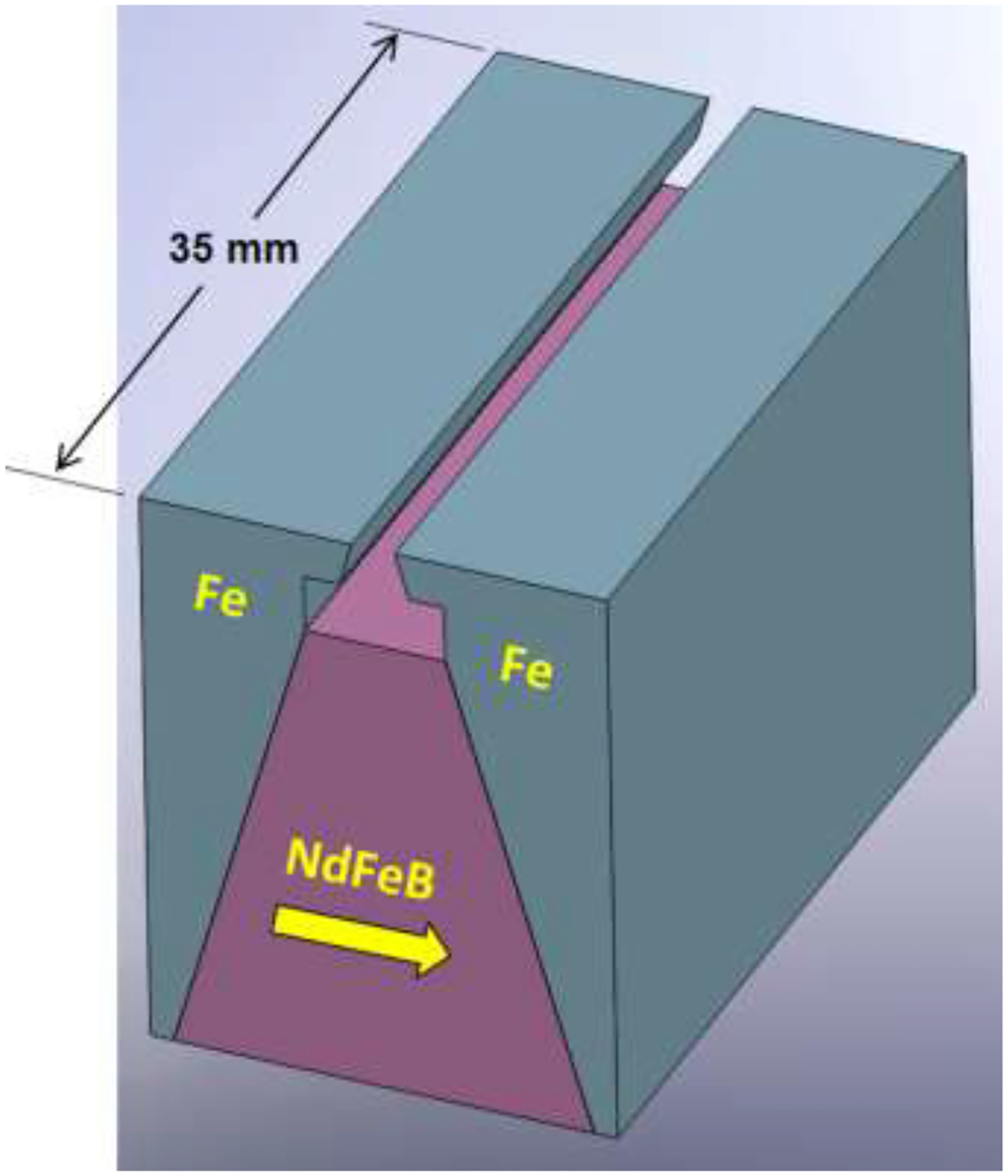
The DMF magnet block (NdFeB) and pole pieces (Fe) used in combination with Vanadium Permendur attachments for the Micro DMF II. The direction of magnetization of the magnet block is indicated by the arrow.
4.1.2.2. Constant field gradient (Micro DMF II)
On the scale of the Micro DMF, it is not feasible to accurately machine iron pole pieces to obtain particular field gradients. To obtain an approximately constant field gradient across the Micro DMF channel, the pole pieces were machined as simple wedges so that the dipole gap varied in a linear fashion across the breadth of the channel. The dipole gap has a depth of 2 mm and its width varied linearly from 3.00 mm to 1.85 mm. The overall height of the assembly is 25 mm because of the smaller pole piece depth. The total depth available for the channel placement is 4.5 mm measured from the top face of the poles to the surface of the trapezoidal magnet block. To enhance the field and to modify the field gradient across the channel, a set of small, parallel, soft magnetic strips are bonded to the two outer walls of the channel. The effect of such magnetizable strips on the magnetic field has been described previously [91]. In the current system, the field-modifying strips are laser-cut into 150 μm thick Vanadium Permendur (49% Co, 49% Fe, 2% V). The gradient in polar gap and the spacing of the strips were optimized by field modeling (Magneto). Figure 3 shows a schematic of a small section of the Vanadium Permendur strips. Each of the 10 strips is 75 μm wide, and the spacing between successive strips varies as follows: 185, 144, 123, 109, 99, 109, 123, 144, and 185 μm. Support bridges of 40 μm breadth were left in place between strips at intervals of 1.68 mm to maintain uniform spacing. The strip attachments are 48.0 mm long and 1.97 mm wide. The channel is mounted between the poles as shown in Fig. 4 which shows a cross section of the channel. The spacer and wafers are slid into contact with the magnet and the exact position of the channel may be deduced from the dimensions given in the text and figures. The Vanadium Permendur strips are also not exactly centered on the channel breadth. The inner strip is placed at 2.11 mm from the edge of the spacer, and 0.28 mm from the inner edge of the channel.
Fig. 3.
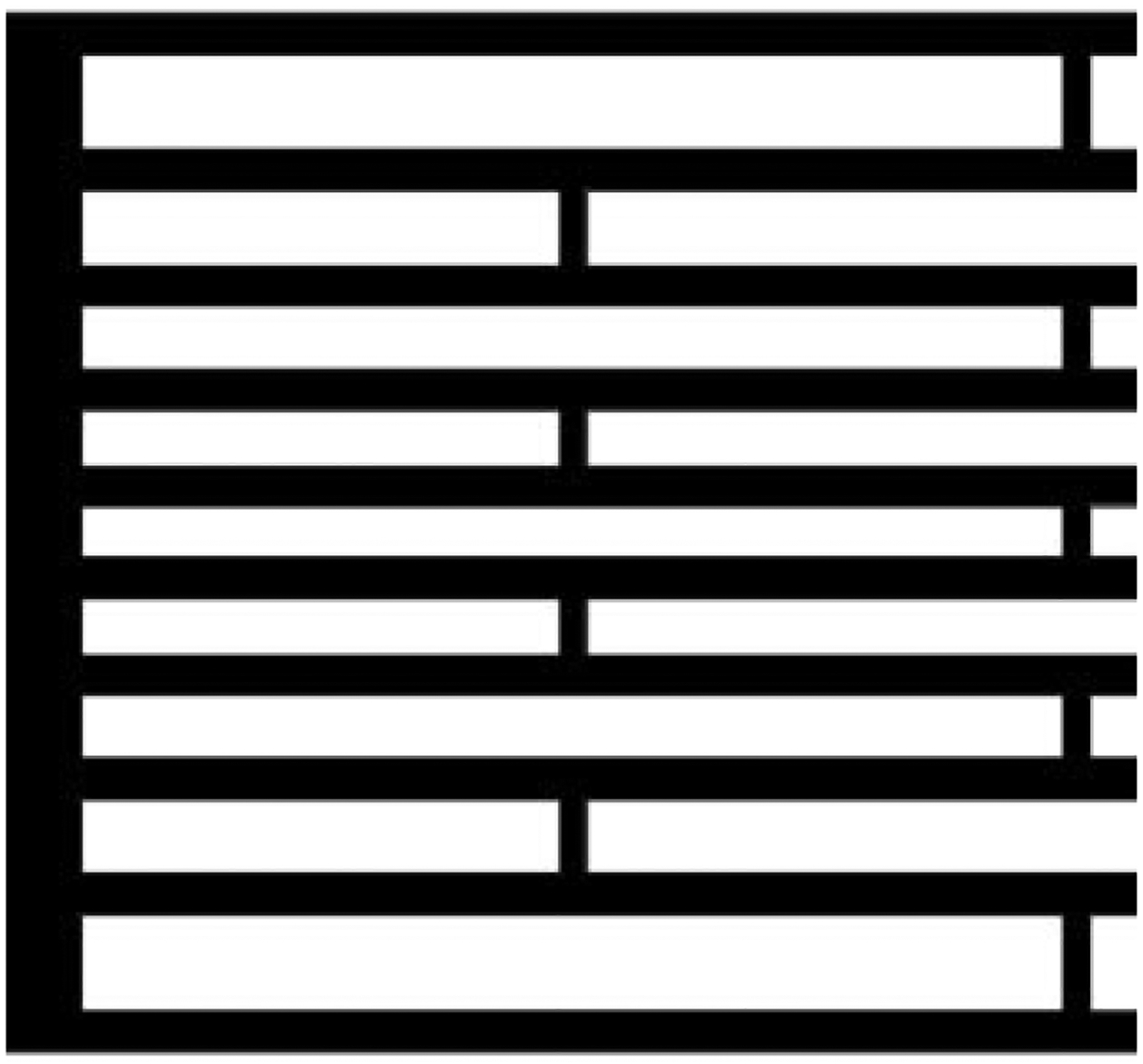
An end section of one of the Vanadium Permendur attachments. The dimensions are given in the text.
Fig. 4.
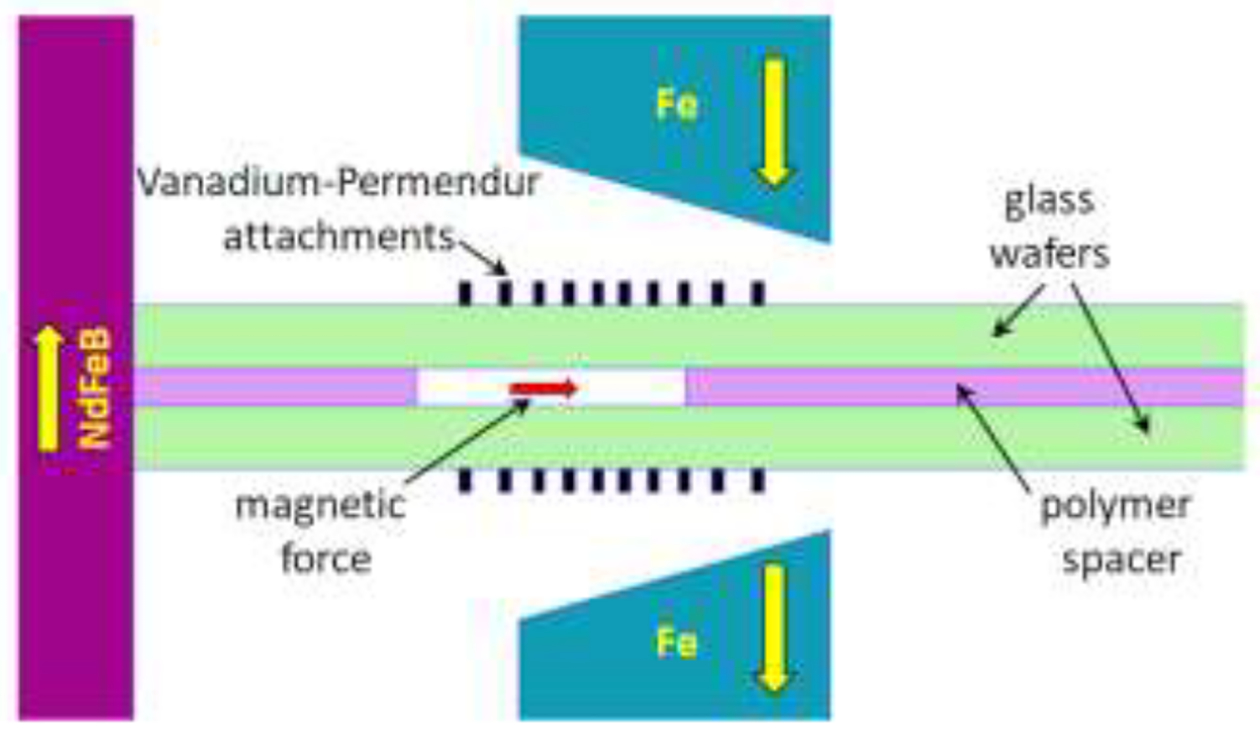
Schematic of the cross section of the Micro DMF II channel placed between the magnet pole pieces, pushed into contact with the surface of the magnet block on the left. The Vanadium Permendur attachments are seen on each side of the channel. The direction of magnetization of the magnet block (NdFeB) and pole pieces (Fe) are indicated by the yellow arrows.
4.1.2.3. Selectivity and dynamic range of the two magnetic fields
Figure 5 shows the variation of field gradient dB/dx with x, the axis across the separation channel breadth at the center line of the channel thickness for the two magnetic fields (red curve for the decreasing gradient of Micro DMF I and the blue curve for the approximately constant field gradient of Micro DMF II). The origin for the x-axis in Figure 5 is located at the surface of the magnet block inside the soft iron yoke, and the horizontal bars show the extent of the channel breadths with colors corresponding to each of the respective cases. The field modeling was carried out using Magneto software, assuming NdFeB magnet properties of remanence Br = 1.29 T, coercivity Hc = 979 kA/m, and relative permeability μ = 1.049, corresponding to a typical 40 MGOe magnet, the included library magnetic properties of Vanadium Permendur, and 1010 low-carbon steel. Figure S3 of the Supplementary Marterial shows the variation of field B across the channel breadth for the two cases. In each case the field exceeds 1 T at distance x of about 1.8 mm. Superparamagnetic nanoparticles tend to be magnetically saturated at about 0.1 to 0.2 T, and it can be safely assumed that they are magnetically saturated in the DMF channels. The force FM on a magnetically labeled biological cell is then given by
| (2) |
where Ms is the saturation magnetization of the iron oxide component of the nanoparticles attached to the cell, VM is the volume of iron oxide contained in the nanoparticles attached to the cell, and dB/dx is the local gradient in magnetic field. The local transverse velocity of the cell is then given by
| (3) |
where f is the friction coefficient for the cell. In the region of transverse magnetic field gradient, the final transverse position x of a cell following its residence time tDMF in the DMF field is obtained by integration of Eq. (3):
| (4) |
where x0 is the initial position. By evaluating the integral in Eq. (4) over the range of available x we can predict the final transverse position as a function of tDMFVMMs / f. The range of VMMs / f captured by the different channel outlets is easily adjusted by changing the total channel flow rate which sets the residence time in the region of magnetic field gradient.
Fig. 5.
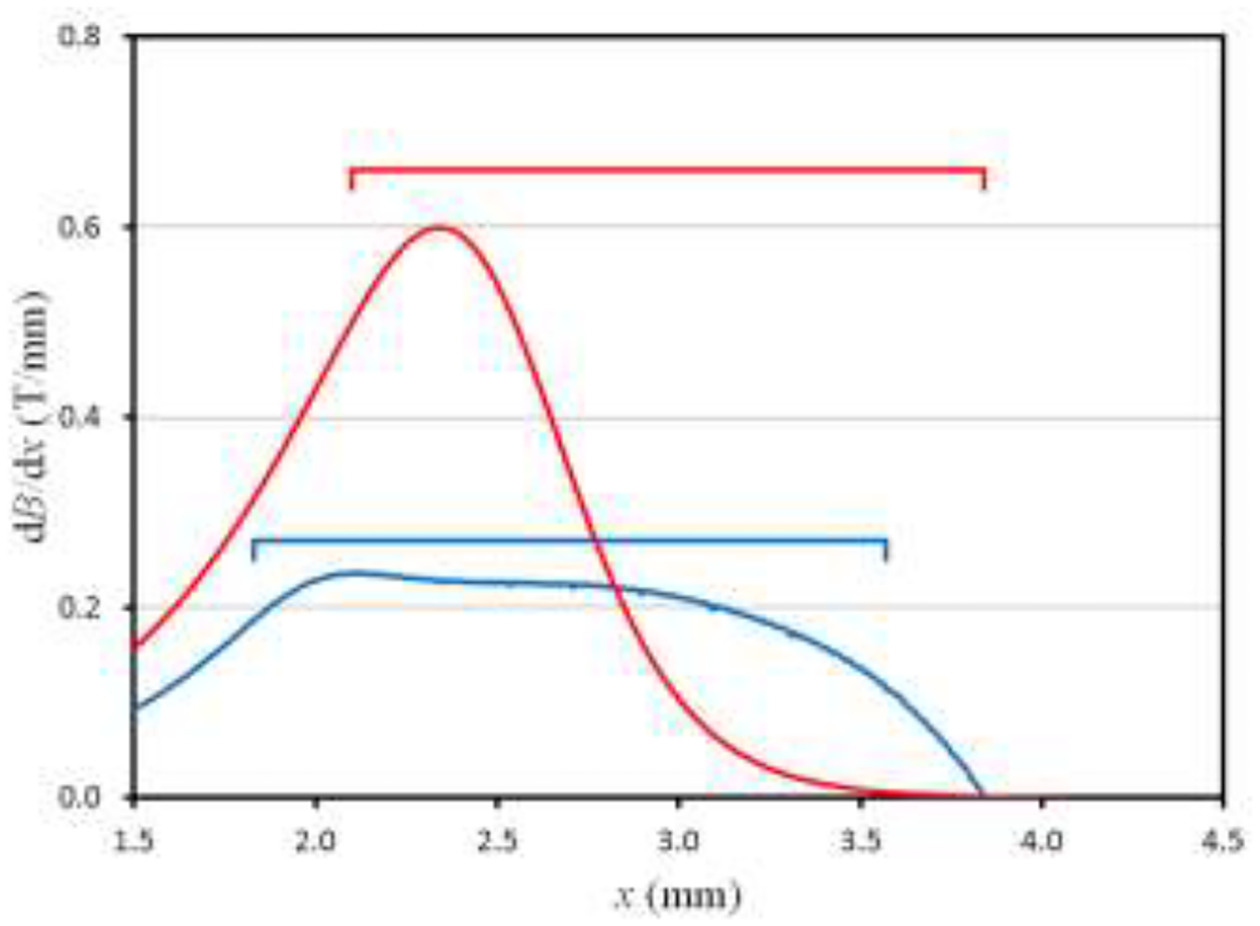
Plots of the gradient in magnetic field B across the breadth of the channel for Micro DMF I (decreasing field gradient, red curve) and Micro DMF II (approximately constant field gradient, blue curve). The horizontal bars with corresponding colors indicate the positions of the channel in each case.
Figure 6 shows the plots of log(tDMFVMMs / f) versus the final position across the breadth of the DMF channel for the two magnetic fields described above. For this figure, the origin for the x-axis is assumed to coincide with the side of the channel that is close to the sample input stream. An initial position x0 of 0.05 mm from this channel side wall was assumed. For the case of decreasing field gradient (Micro DMF I), the range of VMMs / f distributed across the channel breadth from about 0.2 mm to the far wall at 1.74 mm is almost three orders of magnitude. As mentioned above, the absolute range of VMMs / f may be selected by adjusting the flow rate and thereby the residence time tDMF of the cells. For the approximately constant field gradient (Micro DMF II) the range of VMMs / f that is distributed across the channel outlets corresponds to about a single order of magnitude. The decreasing field gradient is therefore suitable for selecting between widely differing expression levels of labeled antigens, while the constant field gradient is suited to a finer selection between populations of cells that differ more subtly in expression level. In either case, the flow rate can be adjusted so as to distribute cells of interest across the central channel outlets, with the highest and lowest expressing cells of interest being carried to the outlets close to the channel edge walls. The system is therefore extremely flexible in application by the use of optimized magnetic field and flow rates.
Fig. 6.
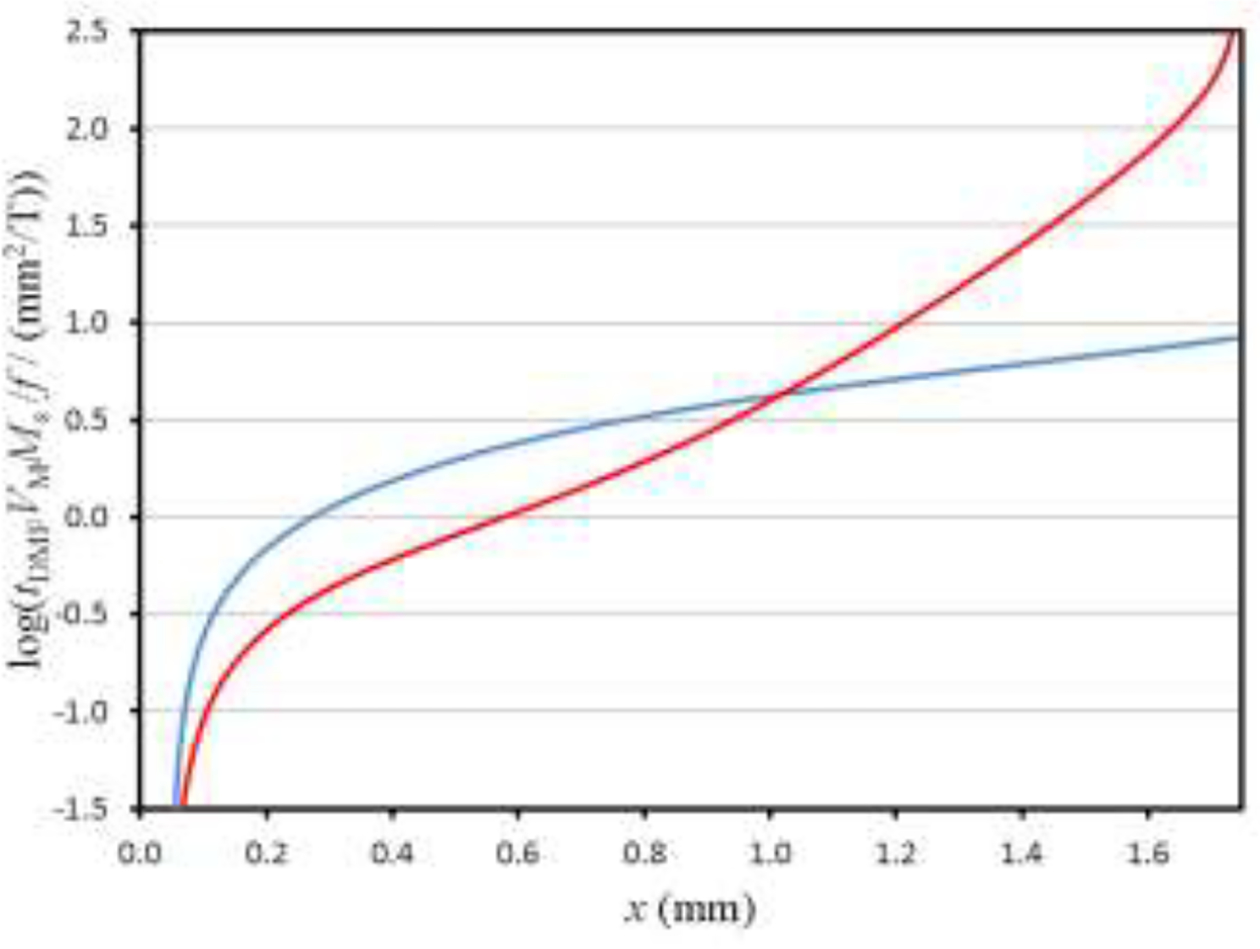
Plots of log(tDMFVMMs/f) versus the final migration distance across the breadth of the DMF channel for the two assumed magnetic fields: decreasing gradient in B (Micro DMF I, red curve) and approximately constant gradient in B (Micro DMF II, blue curve). Initial position x0 of 0.05 mm assumed in each case and the horizontal axis corresponds to the breadth of the DMF channel.
4.2. The MDM stage
Magnetic deposition microscopy is based upon the earlier technology of ferrography [92] used for capture and study of mostly ferromagnetic wear particles found in lubricating oils. The particles were captured from a continuous fluid flow through a channel by application of a magnetic field gradient across the channel thickness. Since the particles were relatively strongly magnetized by the applied field, a relatively weak magnetic field gradient was sufficient for their capture. A gradually increasing field gradient along the direction of flow captured particles of gradually decreasing magnetic dipole moment. The system was modified to capture weakly magnetized biological materials by using a thinner channel, thereby reducing the migration distance for capture, and using a high gradient magnetic dipole field with the dipole gap arranged across the breadth of the channel [85, 86]. Later designs used two or four successive dipole fields to improve the capture efficiency [87, 88].
The MDM module is suitably dimensioned to accept the streams of separated immunomagnetically labeled cells produced by the DMF. These streams flow into six parallel channels placed in contact with a system of magnets that give rise to a series of four regions of high magnetic field gradient which attract the labeled cells to the channel wall where they are captured. The parallel channels are constructed in the same manner as the DMF channel. The channel outlines are cut into a 250 μm thick spacer that is sandwiched between two plane glass wafers of 400 μm thickness.
The MDM magnet assembly was constructed in the laboratory at the Cleveland Clinic. It includes four 1×1/2×1/2 inch, four 1×1/4×1/4 inch, sixteen 1/4×1/8×1/8 inch, and sixteen 1×1/4×1/16 inch NdFeB magnet blocks, all transversely magnetized and nominally 42 MGOe energy product, except for the 1/16 inch thick blocks which were 45 MGOe. The blocks were assembled in an aluminum housing along with specially shaped soft iron pieces in the arrangement shown in Fig. S4 of the Supplementary Material, and held in place using non-magnetic stainless steel nuts and bolts. The upper, capture surface of the magnet and soft iron assembly is 3.55 inch long and 1.0 inch broad, and it had a height of 1.0 inch. All magnet blocks were placed with horizontal magnetization, the direction of magnetization for each collection opposing that of its neighbors. The predicted contours in magnetic field B (calculated using Magneto) are shown in Fig. S5 of the Supplementary Material where the four regions of high field gradient correspond to the edges of the 1/16 inch blocks. Figure S6 of the Supplementary Material shows the variation in the gradient in magnetic field B in the direction across the channel thickness along the length of the assembly at distances of 0.400, 0.525, and 0.650 mm from the surface, corresponding to the two inner walls of the MDM channel and the midpoint of the channel, assuming the channel is placed in contact with the MDM assembly. Figure S7 of the Supplementary Material shows the variation in the gradient in B for the same positions, but in the direction of flow along the channel.
It is important to confirm that cells that are deflected significantly in the DMF will be captured in the MDM channel. Transverse local cell velocity u in the MDM channel is given by
| (5) |
where dB / dy is the local field gradient across the channel thickness. This field gradient depends on both the position across the channel thickness and the distance along the channel relative to the MDM magnet assembly. The local cell velocity along the channel v is given by
| (6) |
where again the field gradient measured in the direction of flow depends on cell position across the channel thickness and the position relative to the magnet assembly, 〈v〉 is the mean fluid velocity, and ξ is the ratio of transverse position y to channel thickness w. Trajectories of cells may be calculated by dividing Eq. (5) by Eq. (6) and rearranging to the form
| (7) |
Consideration of small intervals δz in z allows calculation of cumulative corresponding intervals δy in y from any arbitrary initial position. Note that the gradient dB / dz has negligible influence on cell trajectories at typical fluid flow velocities. From Eq. (7) it is apparent that there will be a lower limit to VMMs / f 〈v〉 for capture of cells initially located at the far channel wall where y = w, or ξ = 1. For the particular 4-dipole MDM magnet assembly, assumed channel thickness of 250 μm, and wafer thickness of 400 μm placed in contact with the magnet, the limit was found to be 0.00213 mm/T. Figure S8 of the Supplementary Material shows a set of cell trajectories for this limiting condition, with initial cell positions corresponding to ξ0 of 0.992, 0.8, 0.6, 0.4, and 0.2. (It is not possible to obtain a solution using Eq. (7) for ξ0 of 1.0.)
5. Simulation of cell separation
It is relatively simple to compare the limiting capture capability of the MDM to the smallest value of tDMFVMMs / f that is deflected significantly by the magnetic field in the DMF. Suppose the mean fluid velocity is the same for the DMF and MDM channels. This can be achieved by fabricating the six parallel MDM channels with identical breadths, and with their collective breadth approximately equal to that of the DMF channel. The residence time in the DMF field can be calculated with the assumption that the cells proceed at a velocity approaching the maximum fluid velocity vmax at the channel midpoint. The DMF residence time tDMF is therefore equal to LDMF / vmax where LDMF is the 35 mm length of the DMF magnet assembly. The limiting capture capability of the MDM is equivalent to VMMs / f〈v〉 0.00213 mm/T. This is equivalent to tDMFVMMs / f = 0.050 mm2/T, or −1.30 on the logarithmic scale of Fig. 6. This indicates that the MDM module is indeed capable of capturing all cells that are deflected to any significant degree in the DMF module. If this had not been the case, it would be a simple matter to modify the breadths of the individual parallel channels of the MDM module. It is not necessary that the channels be of equal breadth, and those carrying the less deflected cells from the DMF could be made broader than the others to compensate for the reduced capture rate. We can be certain, however, that the current design using equal breadth MDM channels can capture all cells deflected in the DMF module.
A simulation of the separation of a mixture of immunomagnetically labeled cell populations differing in antigen expression level, and consequently differing in magnetophoretic mobility, is shown in Fig. 7. The constant magnetic field gradient for the Micro DMF II was assumed. Ansys CFX was used to model the fluid flow with Lagrangian particle tracking for the suspended cells. For this preliminary simulation, the fluid properties of phosphate buffered saline were assumed, with total flow rate of 0.250 mL/min and sample flow rate of 0.010 mL/min, or 0.60 mL/hour. It must be emphasized that these flow rates are not optimized. Assumed cell mobilities correspond to the colors: magenta 1.6 × 10−3, purple 1.4 × 10−3, cyan 1.1 × 10−3, green 7.1 × 10−4, yellow 3.5 × 10−4, red 3.7 × 10−5, and black 0.0 mm3/TAs. The black cell tracks correspond to unlabeled cells and the red to very low expressing cells. The figure shows that the black and red cell tracks exit the channel at outlet 1 in line with the inlet while the higher mobility cell populations are predicted to be distributed across the other five outlets with very little cross contamination between populations. The simulation is in excellent agreement with the predictions of Section 4 which suggest that the lateral displacement of cells is linearly proportional to their magnetophoretic mobility when the magnetic field gradient is constant.
Fig. 7.
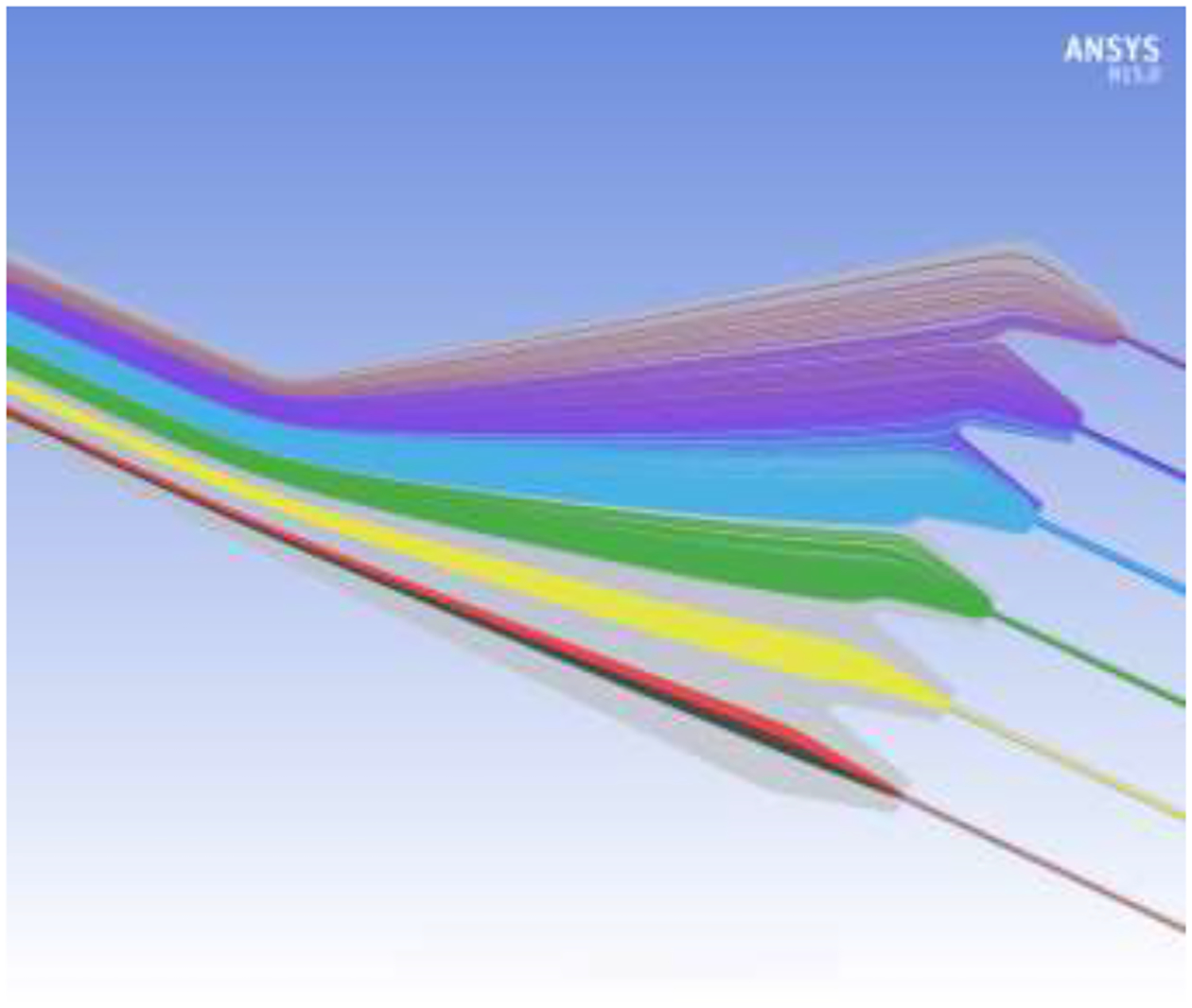
Simulation of separation of different immunomagnetically labeled cell populations having different antigen expression levels and, consequently, different magnetophoretic mobilities. The different color cell tracks correspond to different magnetophoretic mobilities as described in the text.
6. Conclusions
A new microfluidic device for capturing CTCs from a patient blood sample has been described. It will be capable of not only selecting for CTCs using a specific surface marker, such as EpCAM, but of separating the cells into several subpopulations according to marker expression level and placing these into localized regions for counting and optical examination. The subpopulations may be subsequently collected into small fluid volumes for culture and further examination simply by removing the magnetic field from the collection chip. The design is based upon the combination of two thoroughly tested macro devices constructed and used in our laboratory. The CTCs in the blood sample are immunomagnetically labeled for the specific surface marker using nanoparticle labels so that the response to an applied magnetic field gradient is well correlated with marker expression level. The importance of separating CTCs into subgroups according to EpCAM expression level was discussed. The property may be related to EMT and invasiveness of the CTCs, and may indicate progression of disease or response to treatment. It will be possible to examine these aspects in greater detail using the described device. The convenience of operation of this device is predicted to be superior to that of other approaches described in the literature.
Supplementary Material
Highlights.
Circulating tumor cells differ in their surface antigen expression
Magnetically labeled tumor cells isolated by their interaction with magnetic field
Microfluidic device designed to capture subpopulations of circulating tumor cells
The characterization of subpopulations will better inform response to treatment
Acknowledgements
This work was supported, in part by grants from the U.S. National Institutes of Health (HL131720 and CA62349) and the U.S. National Science Foundation (IIP-1926986).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.chroma.2020.461823.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Joyce JA, Pollard JW, Microenvironmental regulation of metastasis, Nat. Rev. Cancer 9(4) (2009) 239–252. 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chambers AF, Groom AC, MacDonald IC, Dissemination and growth of cancer cells in metastatic sites, Nat. Rev. Cancer 2(8) (2002) 563–572. 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- [3].Fidler IJ, The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited, Nat. Rev. Cancer 3(6) (2003) 453–458. 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- [4].Talmadge JE, Fidler IJ, AACR Centennial Series: The biology of cancer metastasis: Historical perspective, Cancer Res. 70(14) (2010) 5649–5669. 10.1158/0008-5472.CAN-10-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chaffer CL, Weinberg RA, A perspective on cancer cell metastasis, Science 331(6024) (2011) 1559–1564. 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- [6].Plaks V, Koopman CD, Werb Z, Circulating tumor cells, Science 341(6151) (2013) 1186–1188. 10.1126/science.1235226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lambert AW, Pattabiraman DR, Weinberg RA, Emerging biological principles of metastasis, Cell 168(4) (2017) 670–691. 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pandya P, Orgaz JL, Sanz-Moreno V, Modes of invasion during tumour dissemination, Mol. Oncol 11(1) (2017) 5–27. 10.1002/1878-0261.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Riggi N, Aguet M, Stamenkovic I, Cancer metastasis: A reappraisal of its underlying mechanisms and their relevance to treatment, Annu. Rev. Pathol.: Mech. Dis 13(1) (2018) 117–140. 10.1146/annurev-pathol-020117-044127. [DOI] [PubMed] [Google Scholar]
- [10].Butler TP, Gullino PM, Quantitation of cell shedding into efferent blood of mammary adenocarcinoma, Cancer Res. 35(3) (1975) 512–516. [PubMed] [Google Scholar]
- [11].Price JE, Aukerman SL, Fidler IJ, Evidence that the process of murine melanoma metastasis is sequential and selective and contains stochastic elements, Cancer Res. 46(10) (1986) 5172–5178. [PubMed] [Google Scholar]
- [12].Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LWMM, Hayes DF, Circulating tumor cells, disease progression, and survival in metastatic breast cancer, New Engl. J. Med 351(8) (2004) 781–791. 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- [13].Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AGJ, Uhr JW, Terstappen LWMM, Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases, Clin. Cancer Res 10(20) (2004) 6897–6904. 10.1158/1078-0432.CCR-04-0378 [DOI] [PubMed] [Google Scholar]
- [14].Hayes DF, Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Miller MC, Matera J, Allard WJ, Doyle GV, Terstappen LWWM, Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival, Clin. Cancer Res 12(14) (2006) 4218–4224. 10.1158/1078-0432.CCR-05-2821. [DOI] [PubMed] [Google Scholar]
- [15].Riethdorf S, O’Flaherty L, Hille C, Pantel K, Clinical applications of the CellSearch platform in cancer patients, Adv. Drug Deliv. Rev 125 (2018) 102–121. 10.1016/j.addr.2018.01.011. [DOI] [PubMed] [Google Scholar]
- [16].Fidler IJ, Metastasis: Quantitative analysis of distribution and fate of tumor emboli labeled with 125I-5-Iodo-2′ -deoxyuridine, J. Natl. Cancer Inst 45(4) (1970) 773–782. 10.1093/jnci/45.4.773. [DOI] [PubMed] [Google Scholar]
- [17].Weiss L, Metastatic Inefficiency, in: Van de Woude GF, Klein G (Eds.), Advances in Cancer Research, Academic Press, San Diego, 1990, pp. 159–211. 10.1016/S0065-230X(08)60811-8. [DOI] [PubMed] [Google Scholar]
- [18].Frisch SM, Francis H, Disruption of epithelial cell-matrix interactions induces apoptosis, J. Cell Biol 124(4) (1994) 619–626. 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC, Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases, Am. J. Pathol 153(3) (1998) 865–873. 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gomis RR, Gawrzak S, Tumor cell dormancy, Mol. Oncol 11(1) (2017) 62–78. 10.1016/j.molonc.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pantel K, Alix-Panabières C, The potential of circulating tumor cells as a liquid biopsy to guide therapy in prostate cancer, Cancer Discov. 2(11) (2012) 974–975. 10.1158/2159-8290.cd-12-0432. [DOI] [PubMed] [Google Scholar]
- [22].Alix-Panabières C, Pantel K, Circulating tumor cells: liquid biopsy of cancer, Clin. Chem 59(1) (2013) 110–118. 10.1373/clinchem.2012.194258. [DOI] [PubMed] [Google Scholar]
- [23].Pantel K, Alix-Panabières C, Liquid biopsy: Potential and challenges, Mol. Oncol 10(3) (2016) 371–373. 10.1016/j.molonc.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Poudineh M, Sargent EH, Pantel K, Kelley SO, Profiling circulating tumour cells and other biomarkers of invasive cancers, Nat. Biomed. Eng 2(2) (2018) 72–84. 10.1038/s41551-018-0190-5. [DOI] [PubMed] [Google Scholar]
- [25].Millner LM, Linder MW, Valdes R, Circulating tumor cells: A review of present methods and the need to identify heterogeneous phenotypes, Ann. Clin. Lab. Sci 43(3) (2013) 295–304. [PMC free article] [PubMed] [Google Scholar]
- [26].Thompson EW, Newgreen DF, Carcinoma invasion and metastasis: A role for epithelial mesenchymal transition?, Cancer Res. 65(14) (2005) 5991–5995. 10.1158/0008-5472.can-05-0616. [DOI] [PubMed] [Google Scholar]
- [27].Książkiewicz M, Markiewicz A, Żaczek AJ, Epithelial-mesenchymal transition: A hallmark in metastasis formation linking circulating tumor cells and cancer stem cells, Pathobiology 79(4) (2012) 195–208. 10.1159/000337106. [DOI] [PubMed] [Google Scholar]
- [28].Yeung KT, Yang J, Epithelial–mesenchymal transition in tumor metastasis, Mol. Oncol 11(1) (2017) 28–39. 10.1002/1878-0261.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Micalizzi DS, Haber DA, Maheswaran S, Cancer metastasis through the prism of epithelial-to-mesenchymal transition in circulating tumor cells, Mol. Oncol 11(7) (2017) 770–780. 10.1002/1878-0261.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chaffer CL, Thompson EW, Williams ED, Mesenchymal to epithelial transition in development and disease, Cells Tiss. Org 185(1–3) (2007) 7–19. 10.1159/000101298. [DOI] [PubMed] [Google Scholar]
- [31].Gunasinghe NPAD, Wells A, Thompson EW, Hugo HJ, Mesenchymal–epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer, Cancer Metastasis Rev. 31(3) (2012) 469–478. 10.1007/s10555-012-9377-5. [DOI] [PubMed] [Google Scholar]
- [32].Li F, Tiede B, Massague J, Kang Y, Beyond tumorigenesis: cancer stem cells in metastasis, Cell Res. 17(1) (2007) 3–14. 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- [33].Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur M-H, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS, Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature, Cancer Res. 69(4) (2009) 1302–1313. 10.1158/0008-5472.can-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pang R, Law WL, Chu ACY, Poon JT, Lam CSC, Chow AKM, Ng L, Cheung LWH, Lan XR, Lan HY, Tan VPY, Yau TC, Poon RT, Wong BCY, A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer, Cell Stem Cell 6(6) (2010) 603–615. 10.1016/j.stem.2010.04.001. [DOI] [PubMed] [Google Scholar]
- [35].Clevers H, The cancer stem cell: premises, promises and challenges, Nat. Med 17(3) (2011) 313–319. 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- [36].Munz M, Baeuerle PA, Gires O, The emerging role of EpCAM in cancer and stem cell signaling, Cancer Res. 69(14) (2009) 5627–5629. 10.1158/0008-5472.can-09-0654. [DOI] [PubMed] [Google Scholar]
- [37].Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, Klein C, Saini M, Bauerle T, Wallwiener M, Holland-Letz T, Hofner T, Sprick M, Scharpff M, Marme F, Sinn HP, Pantel K, Weichert W, Trumpp A, Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay, Nat. Biotechnol 31(6) (2013) 539–544. 10.1038/nbt.2576. [DOI] [PubMed] [Google Scholar]
- [38].Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben-Jacob E, Onuchic JN, Levine H, Implications of the hybrid epithelial/mesenchymal phenotype in metastasis, Front. Oncol 5 (2015) 155 10.3389/fonc.2015.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jolly MK, Huang B, Lu M, Mani SA, Levine H, Ben-Jacob E, Towards elucidating the connection between epithelial–mesenchymal transitions and stemness, J. R. Soc. Interface 11(101) (2014) 20140962 10.1098/rsif.2014.0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jolly MK, Tripathy SC, Somarelli JA, Hanash SM, Levine H, Epithelial/mesenchymal plasticity: how have quantitative mathematical models helped improve our understanding?, Mol. Oncol 11(7) (2017) 739–754. 10.1002/1878-0261.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bankó P, Lee SY, Nagygyörgy V, Zrínyi M, Chae CH, Cho DH, Telekes A, Technologies for circulating tumor cell separation from whole blood, J. Hematol. Oncol 12(1) (2019) 48 10.1186/s13045-019-0735-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Huang Q-Q, Chen X-X, Jiang W, Jin S-L, Wang X-Y, Liu W, Guo S-S, Guo J-C, Zhao X-Z, Sensitive and specific detection of circulating tumor cells promotes precision medicine for cancer, J. Cancer Metastasis Treat 5 (2019) 34 10.20517/2394-4722.2018.94. [DOI] [Google Scholar]
- [43].Akpe V, Kim TH, Brown CL, Cock IE, Circulating tumour cells: a broad perspective, J. R. Soc. Interface 17(168) (2020) 20200065 10.1098/rsif.2020.0065. [DOI] [Google Scholar]
- [44].Boya M, Chu C-H, Liu R, Ozkaya-Ahmadov T, Sarioglu AF, Circulating tumor cell enrichment technologies, in: Schaffner F, Merlin J-L, von Bubnoff N (Eds.), Tumor Liquid Biopsies, Springer International Publishing, Cham, 2020, pp. 25–55. 10.1007/978-3-030-26439-0_2. [DOI] [PubMed] [Google Scholar]
- [45].Li X, Li Y, Shao W, Li Z, Zhao R, Ye Z, Strategies for enrichment of circulating tumor cells, Transl. Cancer Res 9(3) (2020) 2012–2025. 10.21037/tcr.2020.01.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Park J-M, Lee J-Y, Lee J-G, Jeong H, Oh J-M, Kim YJ, Park D, Kim MS, Lee HJ, Oh JH, Lee SS, Lee W-Y, Huh N, Highly efficient assay of circulating tumor cells by selective sedimentation with a density gradient medium and microfiltration from whole blood, Anal. Chem 84(17) (2012) 7400–7407. 10.1021/ac3011704. [DOI] [PubMed] [Google Scholar]
- [47].Talasaz AH, Powell AA, Huber DE, Berbee JG, Roh K-H, Yu W, Xiao W, Davis MM, Pease RF, Mindrinos MN, Jeffrey SS, Davis RW, Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device, Proc. Natl. Acad. Sci. U.S.A 106(10) (2009) 3970–3975. 10.1073/pnas.0813188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zou D, Cui D, Advances in isolation and detection of circulating tumor cells based on microfluidics, Cancer Biol. Med 15(4) (2018) 335–353. 10.20892/j.issn.2095-3941.2018.0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Iliescu FS, Poenar DP, Yu F, Ni M, Chan KH, Cima I, Taylor HK, Cima I, Iliescu C, Recent advances in microfluidic methods in cancer liquid biopsy, Biomicrofluidics 13(4) (2019) 041503 10.1063/1.5087690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shen Y, Yalikun Y, Tanaka Y, Recent advances in microfluidic cell sorting systems, Sensors and Actuators B: Chemical 282 (2019) 268–281. 10.1016/j.snb.2018.11.025. [DOI] [Google Scholar]
- [51].Tian F, Liu C, Lin L, Chen Q, Sun J, Microfluidic analysis of circulating tumor cells and tumor-derived extracellular vesicles, TrAC, Trends Anal. Chem 117 (2019) 128–145. 10.1016/j.trac.2019.05.013. [DOI] [Google Scholar]
- [52].Yin J, Deng J, Du C, Zhang W, Jiang X, Microfluidics-based approaches for separation and analysis of circulating tumor cells, TrAC, Trends Anal. Chem 117 (2019) 84–100. 10.1016/j.trac.2019.07.018. [DOI] [Google Scholar]
- [53].Ajanth P, Sudeepthi A, Sen AK, Microfluidics technology for label-free isolation of circulating tumor cells, J. Inst. Eng. India Ser. C 101 (2020) 1051–1071. 10.1007/s40032-020-00617-z. [DOI] [Google Scholar]
- [54].Hu D, Liu H, Tian Y, Li Z, Cui X, Sorting technology for circulating tumor cells based on microfluidics, ACS Comb. Sci 10.1021/acscombsci.0c00157 (2020). 10.1021/acscombsci.0c00157. [DOI] [PubMed] [Google Scholar]
- [55].Lei KF, A review on microdevices for isolating circulating tumor cells, Micromachines 11(5) (2020) 531 10.3390/mi11050531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liang W, Liu J, Yang X, Zhang Q, Yang W, Zhang H, Liu L, Microfluidic-based cancer cell separation using active and passive mechanisms, Microfluid. Nanofluid 24(4) (2020) 26 10.1007/s10404-020-2331-x. [DOI] [Google Scholar]
- [57].Lin Z, Luo G, Du W, Kong T, Liu C, Liu Z, Recent advances in microfluidic platforms applied in cancer metastasis: circulating tumor cells’ (CTCs) isolation and tumor-on-a-chip, Small 16(9) (2020) 1903899 10.1002/smll.201903899. [DOI] [PubMed] [Google Scholar]
- [58].Pei H, Li L, Han Z, Wang Y, Tang B, Recent advances in microfluidic technologies for circulating tumor cells: enrichment, single-cell analysis, and liquid biopsy for clinical applications, Lab Chip 20(21) (2020) 3854–3875. 10.1039/D0LC00577K. [DOI] [PubMed] [Google Scholar]
- [59].Huang Y, Wang X-B, Becker FF, Gascoyne PRC, Introducing dielectrophoresis as a new force field for field-flow fractionation, Biophys. J 72(2) (1997) 1118–1129. 10.1016/S0006-3495(97)78144-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shim S, Stemke-Hale K, Tsimberidou AM, Noshari J, Anderson TE, Gascoyne PRC, Antibody-independent isolation of circulating tumor cells by continuous-flow dielectrophoresis, Biomicrofluidics 7(1) (2013) 011807 10.1063/1.4774304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li M, Li WH, Zhang J, Alici G, Wen W, A review of microfabrication techniques and dielectrophoretic microdevices for particle manipulation and separation, J. Phys. D: Appl. Phys 47(6) (2014) 063001 10.1088/0022-3727/47/6/063001. [DOI] [Google Scholar]
- [62].Lenshof A, Evander M, Laurell T, Nilsson J, Acoustofluidics 5: Building microfluidic acoustic resonators, Lab Chip 12(4) (2012) 684–695. 10.1039/c1lc20996e. [DOI] [PubMed] [Google Scholar]
- [63].Williams PS, Martin M, Hoyos M, Acoustophoretic mobility and its role in optimizing acoustofluidic separations, Anal. Chem 89(12) (2017) 6543–6550. 10.1021/acs.analchem.7b00685. [DOI] [PubMed] [Google Scholar]
- [64].Leigh DR, Steinert S, Moore LR, Chalmers JJ, Zborowski M, Cell tracking velocimetry as a tool for defining saturation binding of magnetically conjugated antibodies, Cytometry A 66A(2) (2005) 103–108. 10.1002/cyto.a.20155. [DOI] [PubMed] [Google Scholar]
- [65].Meeker DC, Finite Element Method Magnetics, Version 4.2 (15 November 2013. Mathematica Build; ). https://www.femm.info/wiki/HomePage. [Google Scholar]
- [66].Jack R, Hussain K, Rodrigues D, Zeinali M, Azizi E, Wicha M, Simeone DM, Nagrath S, Microfluidic continuum sorting of sub-populations of tumor cells via surface antibody expression levels, Lab Chip 17(7) (2017) 1349–1358. 10.1039/C6LC01496H. [DOI] [PubMed] [Google Scholar]
- [67].Kwak B, Lee J, Lee D, Lee K, Kwon O, Kang S, Kim Y, Selective isolation of magnetic nanoparticle-mediated heterogeneity subpopulation of circulating tumor cells using magnetic gradient based microfluidic system, Biosens. Bioelectron 88 (2017) 153–158. 10.1016/j.bios.2016.08.002. [DOI] [PubMed] [Google Scholar]
- [68].Kang JH, Krause S, Tobin H, Mammoto A, Kanapathipillai M, Ingber DE, A combined micromagnetic-microfluidic device for rapid capture and culture of rare circulating tumor cells, Lab Chip 12(12) (2012) 2175–2181. 10.1039/c2lc40072c. [DOI] [PubMed] [Google Scholar]
- [69].Mohamadi RM, Besant JD, Mepham A, Green B, Mahmoudian L, Gibbs T, Ivanov I, Malvea A, Stojcic J, Allan AL, Lowes LE, Sargent EH, Nam RK, Kelley SO, Nanoparticle-mediated binning and profiling of heterogeneous circulating tumor cell subpopulations, Angew. Chem. Int. Edit 54(1) (2015) 139–143. 10.1002/anie.201409376. [DOI] [PubMed] [Google Scholar]
- [70].Besant JD, Mohamadi RM, Aldridge PM, Li Y, Sargent EH, Kelley SO, Velocity valleys enable efficient capture and spatial sorting of nanoparticle-bound cancer cells, Nanoscale 7(14) (2015) 6278–6285. 10.1039/C5NR00797F. [DOI] [PubMed] [Google Scholar]
- [71].Muhanna N, Mepham A, Mohamadi RM, Chan H, Khan T, Akens M, Besant JD, Irish J, Kelley SO, Nanoparticle-based sorting of circulating tumor cells by epithelial antigen expression during disease progression in an animal model, Nanomed. Nanotechnol. Biol. Med 11(7) (2015) 1613–1620. 10.1016/j.nano.2015.04.017. [DOI] [PubMed] [Google Scholar]
- [72].Green BJ, Kermanshah L, Labib M, Ahmed SU, Silva PN, Mahmoudian L, Chang IH, Mohamadi RM, Rocheleau JV, Kelley SO, Isolation of phenotypically distinct cancer cells using nanoparticle-mediated sorting, ACS Appl. Mater. Interfaces 9(24) (2017) 20435–20443. 10.1021/acsami.7b05253. [DOI] [PubMed] [Google Scholar]
- [73].Poudineh M, Aldridge PM, Ahmed S, Green BJ, Kermanshah L, Nguyen V, Tu C, Mohamadi RM, Nam RK, Hansen A, Sridhar SS, Finelli A, Fleshner NE, Joshua AM, Sargent EH, Kelley SO, Tracking the dynamics of circulating tumour cell phenotypes using nanoparticle-mediated magnetic ranking, Nat. Nanotechnol 12(3) (2017) 274–281. 10.1038/nnano.2016.239. [DOI] [PubMed] [Google Scholar]
- [74].Poudineh M, Sargent EH, Kelley SO, Amplified micromagnetic field gradients enable high-resolution profiling of rare cell subpopulations, ACS Appl. Mater. Interfaces 9(31) (2017) 25683–25690. 10.1021/acsami.7b04677. [DOI] [PubMed] [Google Scholar]
- [75].Kermanshah L, Poudineh M, Ahmed S, Nguyen LNM, Srikant S, Makonnen R, Pena Cantu F, Corrigan M, Kelley SO, Dynamic CTC phenotypes in metastatic prostate cancer models visualized using magnetic ranking cytometry, Lab Chip 18(14) (2018) 2055–2064. 10.1039/C8LC00310F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Chalmers JJ, Zborowski M, Sun L, Moore L, Flow through, immunomagnetic cell separation, Biotechnol. Progr 14(1) (1998) 141–148. 10.1021/bp970140l. [DOI] [PubMed] [Google Scholar]
- [77].Moore LR, Zborowski M, Sun L, Chalmers JJ, Lymphocyte fractionation using immunomagnetic colloid and a dipole magnet flow cell sorter, J. Biochem. Biophys. Methods 37(1–2) (1998) 11–33. 10.1016/S0165-022X(98)00010-4. [DOI] [PubMed] [Google Scholar]
- [78].Schneider T, Moore LR, Jing Y, Haam S, Williams PS, Fleischman AJ, Roy S, Chalmers JJ, Zborowski M, Continuous flow magnetic cell fractionation based on antigen expression level, J. Biochem. Biophys. Methods 68(1) (2006) 1–21. 10.1016/j.jbbm.2006.02.011. [DOI] [PubMed] [Google Scholar]
- [79].Schneider T, Karl S, Moore LR, Chalmers JJ, Williams PS, Zborowski M, Sequential CD34 cell fractionation by magnetophoresis in a magnetic dipole flow sorter, Analyst 135(1) (2010) 62–70. 10.1039/b908210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Giddings JC, A system based on split-flow lateral-transport thin (SPLITT) separation cells for rapid and continuous particle fractionation, Sep. Sci. Technol 20(9–10) (1985) 749–768. 10.1080/01496398508060702 [DOI] [Google Scholar]
- [81].Ozkumur E, Shah AM, Ciciliano JC, Emmink BL, Miyamoto DT, Brachtel E, Yu M, Chen P.-i., Morgan B, Trautwein J, Kimura A, Sengupta S, Stott SL, Karabacak NM, Barber TA, Walsh JR, Smith K, Spuhler PS, Sullivan JP, Lee RJ, Ting DT, Luo X, Shaw AT, Bardia A, Sequist LV, Louis DN, Maheswaran S, Kapur R, Haber DA, Toner M, Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells, Sci. Transl. Med 5(179) (2013) 179ra47 10.1126/scitranslmed.3005616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chalmers JJ, Zborowski M, Moore L, Mandal S, Fang B, Sun L, Theoretical analysis of cell separation based on cell surface marker density, Biotechnol. Bioeng 59(1) (1998) 10–20. 10.1002/(SICI)1097-0290(19980705)59:1<10::AID-BIT3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- [83].Chalmers JJ, Haam S, Zhao Y, McCloskey K, Moore L, Zborowski M, Williams PS, Quantification of cellular properties from external fields and resulting induced velocity: Magnetic susceptibility, Biotechnol. Bioeng 64(5) (1999) 519–526. 10.1002/(SICI)1097-0290(19990905)64:5<519::AID-BIT2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- [84].McCloskey KE, Chalmers JJ, Zborowski M, Magnetophoretic mobilities correlate to antibody binding capacities, Cytometry 40(4) (2000) 307–315. 10.1002/1097-0320(20000801)40:4<307::AID-CYTO6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- [85].Zimmerman PA, Thompson JM, Fujioka H, Collins WE, Zborowski M, Diagnosis of malaria by magnetic deposition microscopy, Am. J. Trop. Med. Hygiene 74(4) (2006) 568–572. [PMC free article] [PubMed] [Google Scholar]
- [86].Melnik K, Sun J, Fleischman A, Roy S, Zborowski M, Chalmers JJ, Quantification of magnetic susceptibility in several strains of Bacillus spores: Implications for separation and detection, Biotechnol. Bioeng 98(1) (2007) 186–192. 10.1002/bit.21400. [DOI] [PubMed] [Google Scholar]
- [87].Karl S, David M, Moore L, Grimberg BT, Michon P, Mueller I, Zborowski M, Zimmerman PA, Enhanced detection of gametocytes by magnetic deposition microscopy predicts higher potential for Plasmodium falciparum transmission, Malaria J 7 (2008) 66 10.1186/1475-2875-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Nath P, Strelnik J, Vasanji A, Moore LR, Williams PS, Zborowski M, Roy S, Fleischman AJ, Development of multistage magnetic deposition microscopy, Anal. Chem 81(1) (2009) 43–49. 10.1021/ac8010186. [DOI] [PubMed] [Google Scholar]
- [89].Williams PS, Particle trajectories in field-flow fractionation and SPLITT fractionation channels, Sep. Sci. Technol 29(1) (1994) 11–45. 10.1080/01496399408002468. [DOI] [Google Scholar]
- [90].Williams PS, Lee S, Giddings JC, Characterization of hydrodynamic lift forces by field-flow fractionation. Inertial and near-wall lift forces, Chem. Eng. Commun 130(1) (1994) 143–166. 10.1080/00986449408936272. [DOI] [Google Scholar]
- [91].Nath P, Moore LR, Zborowski M, Roy S, Fleischman A, A method to obtain uniform magnetic-field energy density gradient distribution using discrete pole pieces for a microelectromechanical-system-based magnetic cell separator, J. Appl. Phys 99(8) (2006) 08R905 10.1063/1.2171936. [DOI] [Google Scholar]
- [92].Seifert WW, Westcott VC, A method for the study of wear particles in lubricating oil, Wear 21(1) (1972) 27–42. 10.1016/0043-1648(72)90247-5. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.