

Abstract
Motivation
When metabolites are analyzed by electrospray ionization (ESI)-mass spectrometry, they are usually detected as multiple ion species due to the presence of isotopes, adducts and in-source fragments. The signals generated by these degenerate features (along with contaminants and other chemical noise) obscure meaningful patterns in MS data, complicating both compound identification and downstream statistical analysis. To address this problem, we developed Binner, a new tool for the discovery and elimination of many degenerate feature signals typically present in untargeted ESI-LC-MS metabolomics data.
Results
Binner generates feature annotations and provides tools to help users visualize informative feature relationships that can further elucidate the underlying structure of the data. To demonstrate the utility of Binner and to evaluate its performance, we analyzed data from reversed phase LC-MS and hydrophilic interaction chromatography (HILIC) platforms and demonstrated the accuracy of selected annotations using MS/MS. When we compared Binner annotations of 75 compounds previously identified in human plasma samples with annotations generated by three similar tools, we found that Binner achieves superior performance in the number and accuracy of annotations while simultaneously minimizing the number of incorrectly annotated principal ions. Data reduction and pattern exploration with Binner have allowed us to catalog a number of previously unrecognized complex adducts and neutral losses generated during the ionization of molecules in LC-MS. In summary, Binner allows users to explore patterns in their data and to efficiently and accurately eliminate a significant number of the degenerate features typically found in various LC-MS modalities.
Availability and implementation
Binner is written in Java and is freely available from http://binner.med.umich.edu.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Untargeted LC-MS metabolomics studies can detect thousands of signals, commonly called features. Only a small portion of these can be readily identified. There is a growing recognition in the field that a significant proportion of the detected metabolome is highly redundant due to the presence of isotopes, adducts and in-source fragments. The abundance of these redundant or ‘degenerate features’ (Mahieu and Patti, 2017; Mahieu et al., 2016) complicates efforts to identify true metabote ions, and often leads to inflated false discovery rates in downstream statistical analysis.
A number of computational approaches for the identification of degenerate features has been developed (Alonso et al., 2011; Broeckling et al., 2014; Brown et al., 2011; Bueschl et al., 2014; Daly et al., 2014; DeFelice et al., 2017; Kuhl et al., 2012; Silva et al., 2014; Tikunov et al., 2012; Uppal et al., 2017). Most of them take advantage of retention time (RT) similarity and strong intensity correlations among features originating from the same metabolite. One early approach was to annotate pairs of features meeting fixed thresholds for correlation, RT and mass tolerance (Alonso et al., 2011; Brown et al., 2011). Although this can help eliminate many redundancies in the data, there is no objective way to select optimal thresholds across different experiments. Furthermore, pairwise approaches are limited in their ability to assess the full range of relationships within groups of features, which can impede identification of a correct neutral mass.
More recent tools employ a variety of computational techniques to group-related features by RT and correlation. These include graph-based clustering of co-eluting features by peak shape (Senan et al., 2019; Kuhl et al., 2012), unsupervised clustering (Broeckling et al., 2014; Tikunov et al., 2012), and Bayesian probabilistic sampling (Daly et al., 2014; Silva et al., 2014).
Some tools supplement feature grouping with a mechanism for specifying common adducts and neutral loss events to provide a set of putative annotations (DeFelice et al., 2017; Kuhl et al., 2012; Uppal et al., 2017). A recently published tool CliqueMS uses empirical adduct and fragment frequencies to generate the most plausible set of annotations (Senan et al., 2019). The accuracy of the annotations can be markedly improved by customizing the list of reference annotations used by each tool to reflect the knowledge of the underlying analytical platform. However, to the best of our knowledge, none of the existing tools supports data exploration to assist users in developing a comprehensive list of custom reference annotations, or assessing the plausibility of the annotated features.
Despite multiple attempts to identify degenerate features in untargeted LC-MS data via computational methods, the true extent of the problem was not fully appreciated until recently. Mahieu et al. (2016) developed mz.unity to enumerate all possible isotopic, adduct and neutral loss relationships as well as complex adducts such as heterodimers and situational adducts with background ions. mz.unity analysis coupled with a C13 isotope-labeled ‘credentialing’ allowed a nearly 30-fold data reduction in Escherichia coli culture data analyzed by UPLC-MS. Similarly, Wang et al. (2019) developed PAVE to reduce feature lists using information from calculated carbon and nitrogen atom counts, commonly observed adduct relationships and weak fragmentation, accomplishing a similarly significant feature reduction in metabolomics analyses of cellular extracts. Although these efforts highlight the extent of underexplored degeneracy in untargeted metabolomics data, their application is limited to the types of samples conducive to isotope labeling. Further, since most metabolomics studies include multiple samples, computational methods designed to analyze individual runs (Senan et al., 2019; Mahieu et al., 2016) do not take advantage of aligned feature tables that can be generated by most data processing software.
To address the shortcomings of existing computational approaches and enable deeper feature annotation we developed Binner, a program for annotating degenerate features in untargeted LC-MS data. In addition to identifying commonly occurring adducts and fragments, Binner provides visualization tools useful for identifying complex adducts in the data. The application of Binner has allowed us to detect and catalog a number of previously unrecognized complex adducts containing two or more charge carriers, salts or solvent adducts and neutral losses generated during the ionization of molecules in LC-MS.
To evaluate Binner’s performance we compared the annotation results to those generated by three previously published programs (DeFelice et al., 2017; Kuhl et al., 2012; Uppal et al., 2017). We found that Binner outperformed these programs by having the highest number of correctly annotated principal ions (PIs) and the lowest number of misannotated ones.
2 Materials and methods
2.1 Binner workflow
Binner is a standalone Java application. It is platform-independent and can be used to analyze data generated by any data processing software. Supplementary Figure S1 shows a screenshot of the Binner user interface. A detailed description of the Binner input and output file formats are provided in the user manual (http://binner.med.umich.edu).
The main steps of the data analysis workflow are shown in Figure 1. The input to the program is an aligned feature table that includes mass to charge ratios (m/z), RTs and intensities for all features.
Fig. 1.

Binner analysis workflow
Data pre-processing includes removing features with a high proportion of missing intensity values to ensure meaningful correlation analysis results. Next, median feature intensity value imputation is performed for remaining missing values, followed by log-scale transformation.
The first step of the algorithm is RT-based binning. The feature list is arranged in RT order and then segmented into bins such that a new bin is initiated whenever the RT difference between consecutive features exceeds a user-specified gap. The choice of RT gap depends on the chromatographic conditions and should exceed the expected RT difference between features originating from the same metabolite thus preventing the assignment of related features to separate bins. Pearson’s or Spearman’s correlation coefficients of intensity values are then calculated across all pairs of features in a bin.
The next step of the workflow is C13 isotope detection. Within each bin, Binner locates pairs of features meeting thresholds for RT similarity, correlation and mass difference (∼1, ∼0.5 and ∼0.33 for singly, doubly and triply charged ions, respectively). For simplicity, based on the lower abundance of natural C13 isotopologs relative to C12 in small molecules, Binner also requires that corresponding isotopic features decrease in intensity as their mass increases. Isotopes not detected at this stage may be detected in subsequent annotation stages.
Following isotope detection, features in each bin are clustered by the correlation coefficients of their intensities using hierarchical clustering with average linkage where feature-to-feature distances are represented by the Euclidian distance between correlation vectors. The number of clusters is calculated by finding the cluster configuration that maximizes the average per-feature silhouette value (Rousseeuw, 1987; see Supplementary Material for further details).
The annotation process starts by identifying the most abundant feature in a cluster (determined by median intensity across samples). This feature is iteratively assigned an adduct hypothesis corresponding to the most frequent ions (e.g. M+H, M+Na etc.) for a given charge-state. For each hypothesis, an underlying neutral mass is calculated from the m/z value for the feature along with the mass and charge of the hypothesized adduct. Binner then uses each neutral mass to search for annotations that account for the reported m/z values of other features in the cluster. Employing a user-supplied annotation file, putative annotations are assigned using additive combinations of charge carriers, adducts and neutral losses. The hypothesis that maximizes the number of putatively annotated features determines PI assignment; related features are annotated accordingly. We find this method of generating annotations centered around the PI to be both more efficient and accurate than exhaustive searches for all possible adduct combinations within a cluster. If any features within the cluster remain unannotated, the full set of steps is repeated beginning with the most abundant unannotated feature, and this process continues until annotation is attempted for every cluster element.
2.2 Metabolomics data generation and feature finding
Reversed phase LC-MS (RPLC-MS) data were obtained from human plasma samples in positive and negative ionization mode as described in the Supplementary Material. Spectral data files were converted to .mzXML format using ProteoWizard MSConvert 3.0 and processed using XCMS. Raw data from this study have been deposited at the Metabolomics Workbench: (Project ID: PR000673). Hydrophilic interaction chromatography (HILIC) positive data were obtained from rat muscle tissue (courtesy of Dr Clary Clish of the Broad Institute). Sample preparation and data processing steps are detailed in the Supplementary Material.
2.3 Comparison of Binner to other annotation tools
We compared the annotations generated by Binner to those generated by CAMERA (Kuhl et al., 2012), Mass Spectral Feature List Optimizer (MS-FLO) (DeFelice et al., 2017) and xMSannotator (Uppal et al., 2017). Comparable sets of adducts and neutral losses were used for each tool (see Supplementary S4) and wherever applicable, common thresholds were used: RT tolerance 0.05 min, mass tolerance 0.005 Da (10 ppm for xMSannotator), and correlation threshold 0.7.
To perform the evaluation we selected 75 compounds that are well distributed throughout the chromatogram and relatively abundant in human plasma analyzed by the RPLC-Quadrupole Time of Flight platform. These compounds were previously identified by accurate mass, RT and/or MS/MS matching to reference spectra. For each compound, the PI (https://goldbook.iupac.org/html/P/P04847.html) has been previously validated. The union of adducts, fragments, and isotopes detected by the four programs was used as a benchmark for enumerating the number of correctly assigned isotope, adduct and fragment annotations and the number of feature groups containing these annotations. More details about how the comparison was performed are provided in the Supplementary Material.
3 Results
3.1 Using Binner to generate feature annotations
To demonstrate the functionality of Binner we analyzed RPLC-MS untargeted metabolomics data from 79 human plasma samples. After XCMS processing, the dataset contained 5953 and 4170 features in positive and negative ionization mode, respectively (see Supplementary S1 and S2 for the complete Binner reports). In addition to generating automated annotations, an important feature of Binner is the ability to visualize data to help users explore their results and generate new annotations. Figure 2 shows an example of Binner output for two clusters derived from the RP-positive mode data. The first three columns contain the user-provided feature name, mass-to-charge ratio and RT for every feature as given in the input file. The remaining columns show other Binner-generated information as detailed in the figure caption, as well as a pairwise correlation heatmap that is useful for validating feature relationships. Each of the clusters shown in Figure 2 contains seven features. In the first cluster, Binner annotated C908 and C909 as [M+Na] and [M+H], respectively. The correlation coefficient of the intensities of these two features is 0.45. In our experience it is not unusual for commonly occurring adducts such as these to have relatively low correlations that could easily be missed by a method that imposes a rigid correlation threshold. Binner was able to annotate these features as likely representing a single metabolite based on close RTs and the [M+H]/[M+Na] mass difference.
Fig. 2.

Binner output. Two clusters are shown. The first three columns come from the input file. Each row corresponds to a single feature. Binner calculates median intensity of each feature across all samples. The subsequent columns provide Binner annotations for isotopes and adducts/neutral losses. To help the user understand the origin of the annotations, Binner provides the respective calculations in the Derivations column. Features highlighted in green represent the most abundant feature in the feature group, and all other members of the feature group are highlighted in yellow. The heatmap on the right helps visualize the correlations that were used to cluster the features
In contrast, the second cluster contains more highly correlated features. C930 and C934 were identified as isotopes of C931 and C935, respectively. The subsequent annotation process identified C931 and C925 as the [M+H] and [M+Na] adducts, respectively, and C935 as a loss of an NH3 group. These five features and isotopes belong to the feature group for kynurenine, which was identified by accurate mass, RT and MS/MS compared against an authentic standard in our compound library. Two remaining unannotated features, C926 and C929, are highly correlated with the rest of the features within the cluster and elute at an identical RT. Referring to an in-house MS/MS library along with a search in METLIN (kynurenine, positive mode, 20 V, [M+H]) shows that these features matched fragment ions observed in low-energy CID fragmentation of kynurenine. This demonstrates the utility of correlation matrices in manually uncovering feature relationships that are not pre-specified or easily described, thus facilitating the discovery of additional degenerate features.
In addition to correlation heatmaps, Binner generates mass difference matrices that can provide further guidance for uncovering feature relationships and new annotations. To illustrate this process, we consider a cluster from the negative mode data of the RPLC-MS dataset containing LysoPC 16: 0 (see Supplementary S2 for the complete Binner report). Figure 3A shows Binner annotations for 12 features found in this cluster. The matrix of mass differences, shown in Figure 3C, helped generate a hypothesis that could explain the observed mass differences, e.g. the mass difference of 67.987 equates to the mass of NaCOOH, the mass difference of 57.958 corresponds to the addition of NaCl, and multiples of those values (e.g. 135.974 = 2X NaCOOH) are also clearly present in the mass matrix. When each of these mass differences is considered with respect to the PI of LPC 16:0, a pattern of complex adducts comprised of multiple additions of NaCOOH, NaCl and HCOO- emerges.
Fig. 3.

Identification of additional annotations with the matrix of mass differences generated by Binner. (A) Binner annotations for twelve features found in the cluster containing LysoPC 16: 0. (B) MS/MS on feature 812.279 showing sodium formate clusters in the fragmentation spectrum. (C) The Binner mass matrix tab of a portion of the LysoPC 16: 0 annotations
To assess the validity of complex annotations, we performed MS/MS analysis of selected features identified by Binner. For example, the MS/MS spectrum for the mass 812.2791 shown in Figure 3B supports the Binner hypothesis. The base mass of LPC 16:0 is not present in the MS/MS spectrum, indicating that the charge stays with the adduct during fragmentation. The spectrum masses match the following formulas (± 0.004 Da): 112.9855: (NaCOOH)HCOO-, 180.9727: (NaCOOH)2HCOO-, 248.9573: (NaCOOH)3HCOO-, 316.9440: (NaCOOH)4HCOO-. Similar results were obtained for the MS/MS spectra for 6 of the 12 LPC 16: 0 Binner annotations, shown in Supplementary Figure S2.
Although other programs have used correlations and mass differences to flag potential redundancies in the data, Binner attempts to make the annotation process transparent to the user, enabling deeper exploration of the data.
3.2 Using Binner to derive new annotations for complex adducts
Despite our best efforts to compile a comprehensive list of potential annotations derived from observations of our data and from the published literature (Broeckling et al., 2014; Bueschl et al., 2014; DeFelice et al., 2017; Mahieu et al., 2016), many correlated features with similar RTs remained unannotated. To understand potential relationships among such features we designed the Binner output to enable in-depth data exploration that could help discover additional annotations.
We hypothesized that a histogram highlighting the most frequent mass differences observed throughout the dataset could help reveal potential additions to the pool of neutral mass gains and losses under consideration and facilitate further annotations, similar to the strategy employed by Brown et al. (2009). We illustrate this concept applied to a Binner cluster containing the identified metabolite leucine from a HILIC positive untargeted plasma metabolomics experiment run on a ThermoFinnigan Q-Exactive instrument. Figure 4A and B shows parts of the matrix of mass differences and the Mass Difference Distribution tab. The latter is produced by tabulating all observed binwise mass difference values. Refer to Supplementary S3 for the complete Binner report.
Fig.4.

(A) Binner mass matrix showing common mass differences that can fully explain a cluster containing Leucine from a HILIC positive dataset. (B) A portion of the Binner output (mass distribution tab) shows that 111.952 is a frequent mass difference in this dataset. (C) Flow chart of all the ions in this Binner cluster shows the complex pattern of Leucine adducts
It has been noted previously that considering all possible combinations of common mass differences can inflate the number of false positive annotations, even when stringent mass accuracy limitations are used (Mahieu et al., 2016). Binner instead allows the user to explicitly define series of combinations of smaller neutral gain/loss groups (complex adducts) that are actually observed in the data. We illustrate the application of this strategy below.
The mass matrix in Figure 4A shows several common mass differences highlighted in different colors. Note that Column 3 in Figure 4A shows a mass difference between feature 176.066 and Leucine M+H (132.1021) of 43.9639 that corresponds to +2Na-2H, and Column 4 shows a mass difference of 67.9877 between features 244.0537 and 176.066 that can be annotated as +NaCOOH. We concluded that the frequently observed mass difference 111.952 shown in Figure 4B (also present in Figure 4A shown in red), results from the combination of two masses: 43.9636 and 67.987. Based on these findings, mass 111.952 was added to the annotation library as +2Na-2H+NaCOOH.
The flowchart shown in Figure 4C illustrates the series of transformations required to relate all of the ions in the cluster back to the neutral mass of the metabolite Leucine. By adding adduct definitions in an iterative fashion from real datasets to our annotation file, we were able to fully annotate all features in several clusters found in this dataset (Supplementary Fig. S3). The mass differences found in these data were similar to those found in the RPLC-MS data (see Supplementary S1 and S2), suggesting that complex adducts are both more common than previously recognized and are not specific to one instrument or chromatography type.
3.3 Comparative evaluation of annotation software
To evaluate Binner’s performance we selected three tools: CAMERA (Kuhl et al., 2012), which has been widely used in the community; the more recently published MS-FLO (DeFelice et al., 2017) and xMSannotator (Uppal et al., 2017). We chose these tools because of the similar scope of operations that they perform, i.e. they all accept aligned spectral features as input and generate feature annotations as part of their automated workflow. We analyzed the RPLC-MS dataset described earlier using Binner and the three other tools, acquiring four sets of annotations that we evaluated without further manual curation. Table 1 summarizes the results of our comparison and Supplementary S6 contains the detailed per compound results. Binner correctly identified the PIs for 64 out of 75 compounds, whereas xMSannotator, MS-FLO and CAMERA identified 51, 49 and 47 PIs, respectively. Surprisingly, the correct PI was identified by all four tools in only 32 out of 75 cases (Fig. 5A). To get a more complete assessment of the results generated by each tool we also compared the number of incorrectly annotated PIs and the number left unannotated. Notably, Binner did not misannotate any PIs. xMSannotator produced the largest number of incorrect annotations.
Table 1.
Summary of the analysis results for 75 known compounds
| Binner | CAMERA | MS-FLO | xMS annotator | |
|---|---|---|---|---|
| No. of correctly annotated PIs | 64 | 47 | 49 | 51 |
| No. of incorrectly annotated PIs | 0 | 15 | 6 | 23 |
| No. of unannotated PIs | 11 | 13 | 20 | 1 |
| Total no. of adduct/NL annotationsa | 225 | 184 | 152 | 155 |
| Total no. of isotope annotationsa | 201 | 220 | 157 | 86 |
| Total no. of feature groups | 87 | 86 | N/Ab | 133 |
Isotopes and adducts were counted if consistent with the correct PI interpretation.
MS-FLO was not included in this comparison because it does not explicitly define feature groups.
Fig. 5.
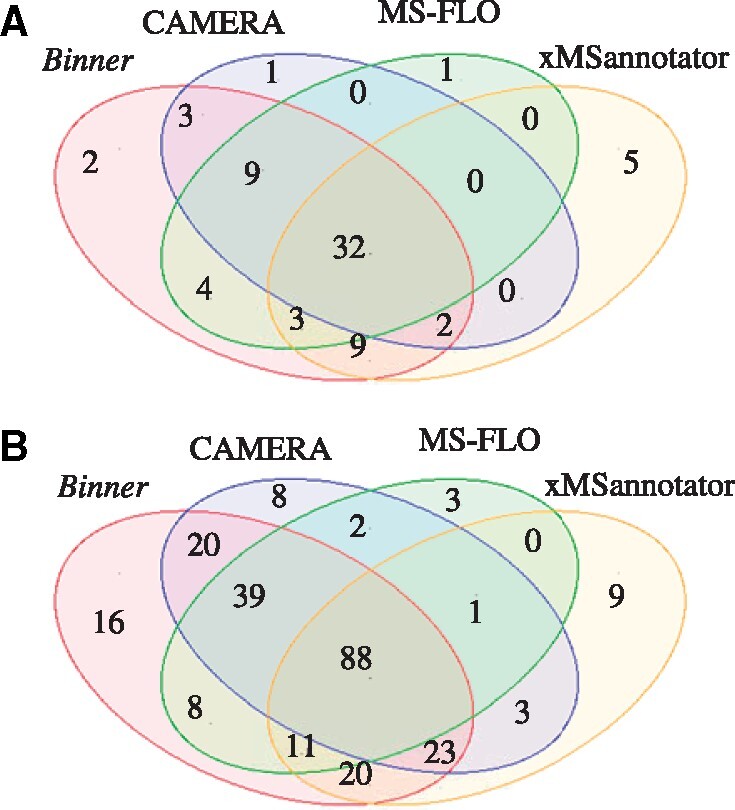
Venn diagram illustrating the overlap between annotations generated by different programs. (A) PI annotations. (B) Adducts and neutral loss annotations
Next, we compared the number of adducts and neutral loss fragments annotated by each program. Annotations were considered correct if they were consistent with the parent PI. Binner was able to correctly annotate 225 adducts and neutral losses, whereas CAMERA, MS-FLO and xMSannotator produced 184, 152 and 155 annotations, respectively. Differences in the observed number of correct PI assignments and annotations can be partially attributed to the different approaches used by the four tools. Binner and CAMERA results were most similar both in terms of group detection and accuracy of adduct and fragment annotations. However, CAMERA relies on a set of weights to generate annotations and does not consider relative feature abundance when predicting the PI. Binner’s consideration of relative feature abundance provides an advantage when fragments and adducts are produced in lower quantities than the corresponding metabolite with a single charge carrier (e.g. M+H, M+Na), which is often the case when using soft ionization techniques such as electrospray ionization (ESI).
The fixed mass difference approach used by MS-FLO failed to annotate any dimers, trimers or multiply charged adducts, which make up a large proportion of all potential annotations. We also note that while the number of annotations identified by MS-FLO could have been increased by explicitly defining certain fragment/parent ion pairs (e.g. M+H-H2O/M+H, mass diff = ∼18.01), some neutral groups (such as NH3) can be either added or lost (e.g. M+H/M+NH3 and M+H/M+NH4) and these cannot be defined simultaneously within MS-FLO.
The formula-based matching strategy employed by xMSannotator also had a number of limitations. The program assigned incorrect formulas to a significant number of features, mostly due to incidental mass matches. Further, although isotopes (including non-carbon isotopes) can be detected and used to enhance confidence in the assigned formulas, isotope detection in xMSannotator is largely limited to M+H as the charge carrier, resulting in fewer annotations. One unique feature of xMSannotator is the use of database searching coupled with pathway information, which might provide an advantage in situations where feature annotations cannot be identified solely based on pairwise correlation and mass relationships. Despite this advantage, the number of correct annotations and the overall number of confident (Level 2 or 3) assignments generated by xMSannotator were relatively small compared with the number of low confidence (Level 0 or 1) assignments.
Overall, we find Binner’s clustering and isotope detection performance to be comparable to existing metabolomics feature annotation tools and superior in terms of accurate adduct/neutral loss assignment, largely due to its consideration of relative abundances in ascribing feature labels.
4 Conclusions
In this article we introduce Binner, a new software package that provides a user-friendly solution for annotation of large untargeted datasets. Binner’s distinct data reduction strategy relies on RT-based binning followed by hierarchical clustering of intensity correlations. This approach enables the intuitive data visualization that is an important distinguishing feature of Binner. It also accounts for Binner’s capacity to generate more accurate annotations than similar tools.
We also demonstrate that visualization of correlation values and common mass differences can facilitate the detection of previously unspecified mass relationships that would likely be missed by a completely automated annotation process. Thus, compared with other tools, Binner provides a wealth of additional information that complements its automatically generated annotations. These features make Binner particularly suitable for deep data exploration and identification of complex data degeneracies.
It is rapidly becoming standard to rely on MS/MS for identifying unknown compounds in untargeted studies. Using Binner to reduce data complexity can lead to a more thorough understanding of the nature of multiple unmatched MS/MS spectra typically produced in a comprehensive data dependent MS/MS analysis.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Charles Evans for reading the article and providing valuable comments, Ms Marci Brandenburg for developing the Binner User Manual, and Dr Clary Clish (Broad Institute) and Dr Stella Aslibekyan (University of Alabama-Birmingham) for allowing us to use their unpublished data. We would also like to acknowledge the support of the CHEAR consortium and especially Dr David Balshaw for the enthusiastic support of this work. We are also grateful to the staff of the Michigan Regional Comprehensive Metabolomics Resource Core for their ongoing help and support.
Funding
This work was supported by National Institutes of Health [DK089503, DK097153, ES026553, R03CA211817, T32 CA140044 and U2COD026490].
Conflict of Interest: none declared.
Contributor Information
Maureen Kachman, Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Hani Habra, Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
William Duren, Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan Medical School, Ann Arbor, MI 48109, USA; Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Janis Wigginton, Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Peter Sajjakulnukit, Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
George Michailidis, Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan Medical School, Ann Arbor, MI 48109, USA; Department of Statistics, University of Florida, Gainesville, FL 32611, USA.
Charles Burant, Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan Medical School, Ann Arbor, MI 48109, USA; Department of Internal Medicine, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Alla Karnovsky, Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan Medical School, Ann Arbor, MI 48109, USA; Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
References
- Alonso A. et al. (2011) AStream: an R package for annotating LC/MS metabolomic data. Bioinformatics, 27, 1339–1340. [DOI] [PubMed] [Google Scholar]
- Broeckling C.D. et al. (2014) RAMClust: a novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal. Chem., 86, 6812–6817. [DOI] [PubMed] [Google Scholar]
- Brown M. et al. (2009) Mass spectrometry tools and metabolite-specific databases for molecular identification in metabolomics. Analyst, 134, 1322–1332. [DOI] [PubMed] [Google Scholar]
- Brown M. et al. (2011) Automated workflows for accurate mass-based putative metabolite identification in LC/MS-derived metabolomic datasets. Bioinformatics, 27, 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueschl C. et al. (2014) A novel stable isotope labelling assisted workflow for improved untargeted LC-HRMS based metabolomics research. Metabolomics, 10, 754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly R. et al. (2014) MetAssign: probabilistic annotation of metabolites from LC-MS data using a Bayesian clustering approach. Bioinformatics, 30, 2764–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelice B.C. et al. (2017) Mass spectral feature list optimizer (MS-FLO): a tool to minimize false positive peak reports in untargeted liquid chromatography-mass spectroscopy (LC-MS) data processing. Anal. Chem., 89, 3250–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl C. et al. (2012) CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem., 84, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahieu N.G., Patti G.J. (2017) Systems-level annotation of a metabolomics data set reduces 25000 features to fewer than 1000 unique metabolites. Anal. Chem., 89, 10397–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahieu N.G. et al. (2016) Defining and detecting complex peak relationships in mass spectral data: the mz.unity algorithm. Anal. Chem., 88, 9037–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw P.J. (1987) Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math., 20, 53–65. [Google Scholar]
- Senan O. et al. (2019) CliqueMS: a computational tool for annotating in-source metabolite ions from LC-MS untargeted metabolomics data based on a coelution similarity network. Bioinformatics, 35, 4089–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R.R. et al. (2014) ProbMetab: an R package for Bayesian probabilistic annotation of LC-MS-based metabolomics. Bioinformatics, 30, 1336–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikunov Y.M. et al. (2012) MSClust: a tool for unsupervised mass spectra extraction of chromatography-mass spectrometry ion-wise aligned data. Metabolomics, 8, 714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppal K. et al. (2017) xMSannotator: an R package for network-based annotation of high-resolution metabolomics data. Anal. Chem., 89, 1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. et al. (2019) Peak annotation and verification engine for untargeted LC-MS metabolomics. Anal. Chem., 91, 1838–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.