

Abstract
The clinical presentation of liver disease is highly variable in homozygous ZZ alpha-1 antitrypsin (AAT) deficiency, and not all patients with the homozygous ZZ genotype develop liver disease. Although not fully identified, there is likely a strong influence of genetic and environmental modifiers of the intracellular injury cascade and fibrotic response. Most ZZ children are well and remain undiagnosed. Of those who come to medical attention, the most common pediatric presentation is neonatal cholestatic hepatitis, sometimes referred to as “neonatal hepatitis syndrome”. The gold standard for diagnosis of AAT deficiency is analysis of the AAT protein phenotype in the patient serum or the genotype of their DNA genome. Careful follow up of all diagnosed children is important. Heterozygotes for S and Z alleles of AAT (SZ) may develop progressive liver disease similar to ZZ patients and also require close monitoring. There is no specific treatment for AAT deficiency induced liver disease and current therapy remains supportive with management of complications. Rarely, patients require liver transplant and typically the patient outcomes are excellent. With improved understanding of liver injury mechanisms, new strategies for treatment are now being explored, including siRNA technology, molecules to modulate secretion, and enhancers of proteolysis.
Keywords: Autophagy, proteolysis, ER-associated degradation (ERAD), protein polymer, siRNA
Introduction
Alpha-1 antitrypsin (AAT) is an abundant serum protein synthesized in the liver (1) and secreted into the blood. The classic form of AAT deficiency is defined as individuals who are homozygous for the mutant Z allele AAT variant. This classic form of AAT deficiency is referred to as the ZZ, or “PiZZ” in World Health Organization nomenclature, and accounts for the majority of AAT-associated liver disease. However, in certain circumstances, compound heterozygotes with only one Z allele are implicated in liver disease. The SZ genotype is by far the most common compound heterozygote associated with increased risk of liver disease. The wild type AAT protein referred to as M type is normally synthesized in the hepatocyte and rapidly excreted by the endoplasmic reticulum in large quantities. In contrast, the Z variant inefficiently folds into its final conformation thereby resulting in 85% retention as large protein polymers in the hepatocytes through endogenous protein quality control pathways (1,2). Accumulation of the Z variant of AAT protein in liver cells triggers an intracellular injury cascade that may result in chronic liver disease, cirrhosis, and hepatocellular carcinoma (HCC) (1,2). There is likely an important role for genetic and environmental modifiers because the clinical course among this group of patients is highly variable.
It is particularly important to consider genetic and environmental modifiers when examining the clinical course of children with AAT deficiency. Multiple European and North American birth screening studies indicate the majority of ZZ children are undiagnosed and remain well. However, a small number of ZZ neonates develop cholestatic hepatitis, which is commonly referred to as the “neonatal hepatitis syndrome”. This is characterized by elevated conjugated bilirubin in the serum with variable elevations of serum alanine transaminase (ALT) and aspartate transaminase (AST). In European and North American medical systems, these infants are typically diagnosed efficiently and the majority will spontaneously improve. However, some exhibit severe complications of liver disease with a variable progression to cirrhosis, portal hypertension, and liver failure from pre-school through early adulthood (3). Some ZZ infants who did not exhibit neonatal cholestasis will also develop liver failure later in childhood. The factors that influence this highly variable disease progression in this population are still unknown; however, several new studies are producing data that may contribute to our understanding of this progression. There is no specific treatment for AAT deficiency; however, if a child develops life-threatening liver failure, liver transplantation has been highly successful with excellent results.
Clinical presentation
AAT deficiency is associated with a variety of clinical presentations in the neonatal period (1,4-7). The most common presentation is the “neonatal hepatitis syndrome”, sometimes referred to as “neonatal cholestasis” or “cholestatic hepatitis”. The clinical spectrum varies from a well-appearing, but irritable, neonate to ill-appearing and tachycardic. The neonatal liver responds to a variety of insults with a stereotypical pattern of hepatitis that is detected on physical exam as jaundice, scleral icterus, and variable degrees of hepatosplenomegaly. The laboratory workup is also non-specific. Liver tests may demonstrate ALT and AST elevations ranging from slightly above normal to more than a ten-fold elevation, while the gamma-glutamyl transpeptidase (GGT) may be normal or high. Typically, the total serum bilirubin is elevated, most notably with a high conjugated (“direct”) fraction. The CBC may be normal or with have non-specific and variable changes. The differential diagnosis of this syndrome is very broad with more than a 100 potential conditions to consider, including congenital anomalies, acquired congenital infections and inborn errors of metabolism. Extrahepatic biliary atresia is relatively rare but extremely important etiology of neonatal hepatitis syndrome to identify, and thus is commonly investigated. Of note, in Caucasian populations particularly with northern European heritage, AAT deficiency is one of the more common causes of neonatal hepatitis syndrome.
After a diagnosis of neonatal hepatitis (“neonatal cholestasis”) is established the neonate is commonly referred to a pediatric gastroenterologist or hepatologist for further evaluation. In the United States and Europe, if initial screens for perinatal infection and congenital anomalies are unremarkable, specific testing for AAT deficiency is generally performed (7,8). After a diagnosis of AAT deficiency is established, the patient’s family is given extensive education, provided genetic counseling and offered genetic testing of family members. A liver biopsy is not required for the diagnosis of AAT deficiency, but may be indicated depending on the clinical scenario. However, liver biopsy alone may not reliably differentiate the various causes of cholestatic liver diseases of infancy. Findings of liver biopsy in ZZ infants include giant cell transformation, significant steatosis, lobular hepatitis, fibrosis, hepatocellular necrosis, bile duct paucity, or bile duct proliferation (Figure 1A) (4,5,9). Under conventional hematoxylin and eosin (HE) stains, eosinophilic inclusions are seen in some but not all hepatocytes, and referred to as “globules”. These structures represent dilated ER membranes engorged with polymerized AAT mutant Z protein (Figure 1B) (10). Globules are more easily identified after using the histological stain periodic acid-Schiff (PAS) followed by digestion with diastase as this combination results in red glycoproteins on a neutral background. Normal liver cells do not usually contain accumulations of PAS-positive material. It is important to note that globules are also not present in all hepatocytes and can be absent in the first few weeks of life. Moreover, similar eosinophilic inclusions can be seen in other diseases and therefore the diagnosis should not depend on the liver biopsy alone.
Figure 1.

Human ZZ liver stained with HE (A) and PAS followed by digestion with diastase (B). (A) demonstrates significant steatosis seen in the ZZ liver; (B) depicts eosinophilic inclusions referred to as globules, indicated by black arrows, which appear red on a neutral background after applying. Normal liver cells do not contain accumulations of PAS-positive material. HE, hematoxylin and eosin; PAS, periodic acid-Schiff.
AAT deficiency is most common in individuals with European heritage. A landmark newborn screening study undertaken in Sweden in the early 1970s produced important information about this disease. In the study 127 infants with the ZZ variant were identified following an unbiased birth screening of over 200,000 Swedish neonates (Figure 2) (7). Among the many important observations, the authors noted that >80% of neonates with cholestasis had spontaneous resolution of their liver disease and remained healthy at follow up at age 18 years. Only about 3% had progressed to severe, life-threatening liver disease (13). The majority of these infants had normal physical exams and some had mild liver function test enzyme elevations indicating that these infants would have likely gone undiagnosed without unbiased newborn screening. Similar, but less extensive studies in the US echoed these findings. However, it is unclear if the same results would hold true in a more heterogeneous population associated with a different set of modifier genes, such as the United Kingdom or North America (14). This is because many retrospective reports of ZZ children from the US and other parts of Europe, such as the French cohort of Ruiz and colleagues, have described a small number of children who progressed to severe disease later in childhood, including as teenagers, and some who developed severe disease who did not exhibit neonatal cholestasis (15). Both of these group observations are absent from the Swedish cohort. It is possible that there are genetic differences in these populations, or that these events in later childhood are less common than 1 in 127 (2). Other rare symptomatic presentations in infants, excluding neonatal cholestasis, include vitamin K deficiency coagulopathy, poor feeding, failure to thrive, GI bleeding, and acute liver failure. These rarely reported presentations were also absent in the Swedish study cohort.
Figure 2.
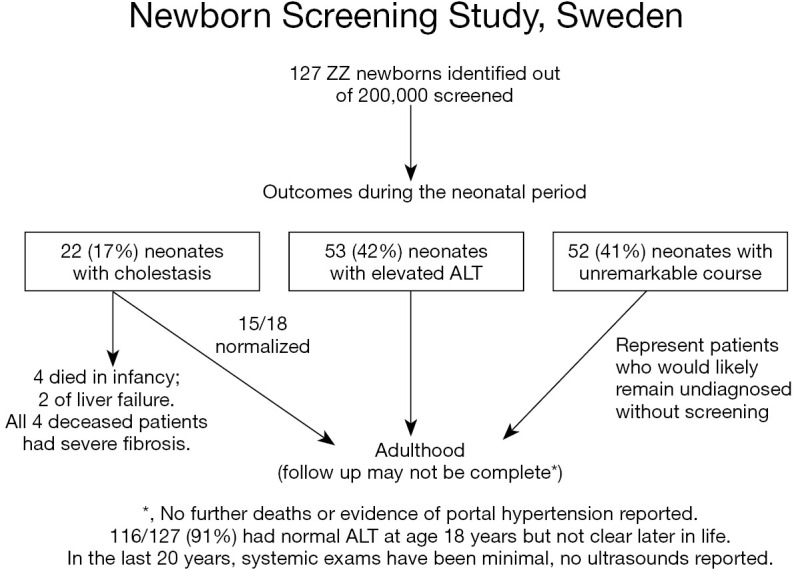
Outcomes of the Swedish Newborn Screening Program (7,11,12). Figure 2 demonstrates the condensed outcomes of 127 ZZ neonates that were identified after an unbiased newborn screen of over 200,000 Swedish neonates. Of those identified, the majority of the ZZ infants were healthy in the neonatal period and without the unbiased newborn screen, this group would have likely remained undiagnosed. Moreover, only 16% of neonates exhibited neonatal cholestasis. Of those, approximately 3% developed severe disease including severe fibrosis, liver failure, and death. The majority of the cohort at 18 years of age, including most of the neonatal cholestasis group, demonstrated unremarkable laboratory exams and pulmonary function tests. Follow up since that time has been published but has not been as stringent or as complete as the original report. ALT, alanine transaminase.
The presentation of older children with AAT deficiency is also highly variable, and may be incidentally identified on screening bloodwork or with signs or symptoms of liver disease, including feeding difficulties, failure to thrive, or asymptomatic hepatosplenomegaly identified on routine exams. Rarely, some children may even acutely develop symptoms or signs of portal hypertension with GI bleeding, thrombocytopenia, or other signs of acute liver failure. Commonly, well-appearing ZZ children have isolated mild, but persistent, elevations of ALT over many years with no other evidence of liver damage. While not commonly required, liver biopsy in older children is also usually remarkable for large globules in periportal hepatocytes, with mild microvesicular steatosis, and mild lobular inflammation. Children with previous neonatal hepatitis, who have spontaneously recovered, will sometimes develop severe liver disease later in the first or second decade of life with compensated, but stable cirrhosis with portal hypertension (5,16-18). If these children are closely monitored and avoid GI bleeding and other complications, their chronic disease may not progress to liver failure for years to decades. New onset, severe liver disease in young and middle aged ZZ adults is uncommon (6,13,19,20). However, the risk of severe liver disease and cirrhosis increases with age in later adulthood. The lifetime risk of cirrhosis maybe as high as 40% (19). Adults may also have chronic hepatitis or more rarely, develop HCC (19).
Pathophysiology of AAT ZZ liver disease
The understanding of liver injury in AAT deficiency is ongoing as the consequences of ZZ protein accumulation are wide-ranging, not entirely understood, and a full discussion is out of the scope of this article. Early studies conducted by Perlmutter and colleagues examined the clearance of mutant Z protein in patient tissue and found that reduced intracellular clearance of mutant Z protein correlated with life-threatening liver disease (21). These findings supported the hypothesis that accumulation of the mutant Z protein in the liver was the key trigger of liver injury.
The hepatocyte uses a variety of proteolytic processes to reduce Z protein burden, including ubiquitin-dependent and ubiquitin-independent proteasomal pathways as well as ER-associated degradation (ERAD) (22). Additionally, autophagy is an intracellular degradation pathway that was also identified as playing an important role in compensating for the accumulation of misfolded mutant Z proteins. ERAD is thought to be the primary route for degradation of AAT mutant Z molecules. Studies of human fibroblast cells derived from ZZ homozygous patients suggest that patients susceptible to liver disease have less efficient ERAD of AAT mutant Z protein than ZZ patients not affected by liver disease (23,24).
Liver biopsy of ZZ patients show varied accumulation of AAT mutant Z protein among individual hepatocytes. Recent studies suggest cellular injury cascade resulting in increased susceptibility to apoptosis is triggered within a small subset of hepatocytes with the highest accumulation of mutant Z protein (1,25). The result is low but slightly increased rate of hepatocyte death in ZZ liver tissue compared to normal tissue. In order to maintain functional liver mass, the cells with lower polymer accumulation proliferate. With time the continued stress, death, and repair of hepatocytes results in liver fibrosis, cirrhosis, and HCC.
Diagnosis
The gold standard for diagnosis of AAT deficiency is analysis of the “AAT protein phenotype” in patient serum (an isoelectric focusing protein gel), or genotype (PCR of genomic DNA, usually leukocytes, to detect mutant alleles) (1). If the protein phenotype test is performed, then the genotype is inferred from the result. Liver biopsy is not required for the diagnosis. Phenotype gel analysis requires sophisticated technical attention and is best performed in an experienced reference laboratory. Some centers use serum AAT level as a screening test and pursue the gold standard test if results are outside the normal range. It is important to note that AAT is an acute phase reactant and thus ZZ patients may have increases in serum AAT during active inflammatory episodes. Therefore, clinicians should be cautious when interpreting an isolated AAT level. Theoretically, ZZ patients cannot produce AAT levels in normal to near normal range; however, the author’s anecdotal experience suggests that SZ patients with active liver disease and inflammation occasionally have AAT levels in normal range. Neonates appear to have higher serum AAT levels that will then rapidly decrease over the first few months of life. However, this fact may not be reflected in the reference range of many laboratories. Moreover, clinicians should avoid obtaining serum AAT levels or phenotype tests if the patient recently received any plasma transfusion because the results may reflect the donor plasma rather than the patient’s status. Similarly, this concept may be applied to patients with emphysema who regularly receive protein replacement for ATT. In these scenarios, liver biopsy may be a very helpful tool to assess the degree and extent of liver injury as well as fibrosis. Liver biopsy still remains the gold standard test for that determination.
Prognosis and management
Many authorities propose frequent close monitoring of ZZ patients, or at least annually, for possible progression of liver disease (4). Clinicians familiar with liver disease signs and management of potential complications should perform these evaluations. The rationale for annual monitoring is the extremely unpredictable progression of AAT liver disease. Routine evaluation should consist of a history and physical exam with particular attention paid to presence of hepatosplenomegaly, laboratory studies including white blood cell (WBC) count, platelet count, ALT, AST, alkaline phosphatase, bilirubin, total protein, albumin, and international normalized ratio (INR). Worse outcomes are loosely associated with elevated AST and ALT; however, progression of disease is so variable that it is not predictive in individual patients. Some data indicates GGT elevation may be an especially sensitive indicator of liver disease, and may be particularly ominous if it is very highly elevated in the first few years of life. Yet, despite the fact that patients with neonatal cholestasis as their initial presentation of AAT deficiency (as described above) are statistically associated with a higher risk of severe disease, most will do well. Of note, the absence of a history of neonatal cholestasis does not rule out the possibility of progression to cirrhosis and liver failure (15).
In all patients with chronic liver disease, including AAT deficiency, the presence of thrombocytopenia, worsening granulocytopenia, rising liver enzymes and bilirubin, and coagulopathy are very concerning as these findings suggest disease progression. However, life-threatening cirrhosis and portal hypertension can present with relatively unremarkable liver function studies. Therefore, clinicians should not be overly reassured by normal liver laboratory results and always maintain a high index of suspicion for the possibility of silent cirrhosis. In adults, male sex is associated with more severe liver disease, but this is less of a predictor for children. The degradation of the mutant Z protein in cells from children with severe liver disease is less efficient in those with no liver disease (21,23). Additionally, new data suggests specialized analyses of Z protein in serum may assist predicting liver disease in children. However, these investigational reports have yet to be incorporated into routine clinical laboratory testing.
As an adjunct to physical examination, clinicians should also obtain a baseline liver and hepatobiliary ultra-sonogram to evaluate general hepatobiliary health as well as liver and spleen sizes. According to American Association for the Study of Liver Diseases (AASLD) guidelines, liver ultrasound should be performed every 6 months for detection of HCC for patients at >2% per year risk of HCC (26). However, there is a lack of good predictive data to evaluate the risk of developing HCC in patients with AAT deficiency. Therefore, guidelines for performing routine ultrasound assessments for HCC applies only to patients with AAT deficiency and evidence of portal hypertension, cirrhosis and abnormal laboratory findings with persistently elevated liver enzymes and other liver function tests mentioned above.
At present, there is no specific treatment for AAT deficiency induced liver disease. Treatment for progressive liver injury is supportive, including attention to adequate nutrition, prevention of rickets, and early recognition and treatment of coagulopathy. Clinicians should recognize and manage complications such as portal gastropathy, variceal bleeding and ascites. Commonly, children and adults with AAT-related liver cirrhosis remain clinically stable and live relatively unaffected lives for many years. Moreover, available data indicates that children with cirrhosis usually have normal growth, development, and anthropometric measurements. With this in mind, it is still critical to identify patients with portal hypertension and cirrhosis early so they can be appropriately counseled and monitored. Such patients should be cautioned against splenic injury from intense contact sports, advised to abstain from alcohol consumption, and should be nutritionally supplemented with fat-soluble vitamins. Additionally, patients should be surveyed for esophageal or gastric variceal bleeding and advised to avoid non-steroidal anti-inflammatory drugs (NSAIDs) as these medications can result in precipitation of severe, life-threatening variceal bleeding, even in well compensated patients with otherwise minimal symptoms of liver injury.
Patients with cirrhosis of any cause are routinely cautioned against the use of NSAIDs. However, animal model studies of AAT deficiency suggest that, even in the absence of liver cirrhosis, NSAIDs could be specifically more toxic to the ZZ liver. NSAIDs, which block prostaglandin inhibition, are predicted to increase AAT synthesis, and potentially increase misfolded AAT protein accumulation (1). Similarly, as an acute phase reactant protein, the increased synthesis of AAT in the presence of active inflammation is also believed to increase the quantity of misfolded AAT proteins and subsequently increase risk of liver damage. Therefore, NSAID use contributes to a cycle that potentially augments the progression of liver injury. The pattern of increased liver injury associated with NSAIDs in the ZZ liver has thus far only been reported in animal models, and has not been tested in humans. However, even normal doses of NSAIDs are still believed to be more toxic to ZZ liver patients than standard doses of acetaminophen. Although high doses of acetaminophen may also injure any liver, including ZZ phenotypes, many authorities favor moderate dosing of acetaminophen for treatment of pain or fever and suggest avoiding NSAIDs entirely. There is minimal data that supports recommendations to avoid alcohol consumption in patients with ZZ phenotype AAT deficiency without any evidence of liver damage. According to AASLD guidelines for adults with hepatitis C virus and without any evidence of liver damage, up to three alcoholic drinks per week may be considered safe. It is not known whether this fact is directly applicable to AAT patients.
All AAT deficiency patients are strongly advised to avoid cigarette smoke and other irritant inhalation exposures, regardless of presence of liver and/or lung disease. Studies suggest that exposure to even secondhand smoke and environmental air pollutants in childhood are considered as a very important risk factor for development of adult emphysema associated with AAT deficiency (6,27,28). For these reasons, even in patients who seek medical attention primarily for liver disease, ZZ patients and their household contacts are strongly cautioned against smoking. Studies have concluded that AAT deficiency-associated ZZ children do not usually develop clinically significant or detectable emphysema but they may be at risk of developing childhood asthma and may encounter a variety of respiratory symptoms (29,30). Commonly, ZZ patients are referred to an adult pulmonologist for baseline evaluation at 18 years of age. If a patient develops any other respiratory symptoms or asthma prior to 18 years, an early referral for pulmonary evaluation may be warranted.
There is ongoing development of novel approaches to management of ZZ patients that focus on mitigating the inflammatory cascade triggered by ZZ accumulation. For example, the development of RNA inhibition technology that interrupts Z protein production in PiZZ mouse model resulted in elimination of liver injury and the return of the liver to wild type health (11). Currently, two phase II trials of siRNA-based drugs, to eliminate Z protein production and thereby eliminate liver damage, are also enrolling. Moreover, the development of chemical chaperones that improve proper protein folding or augment secretion of AAT mutant Z have been a longstanding focus of therapeutic intervention. The primary barrier identified to chaperone therapy is the sheer mass of AAT protein synthesized daily because, assuming there is 1-to-1 binding stoichiometry between chaperone and protein, a huge mass of chaperone would have to be delivered to the ER of hepatocytes. Nevertheless, studies are promising and have identified compounds that promote secretion of AAT mutant Z. Currently, there is a human phase II trials of new small molecule chaperones led by Vertex Corporation have now begun, which could theoretically benefit both the lungs and the liver. Finally, there have been many studies that examined methods to accelerate the degradation of mutant Z protein. Studies have demonstrated augmentation of autophagy in hepatocytes results in decreased misfolded mutant Z protein in both cell culture and mouse models (1,2,12). In model systems, a regimen of carbamazepine, sirolimus, and a bile acid or ursodeoxycholic acid, combined with a genetic approach to augment expression of key autophagy regulators, resulted in enhanced autophagy. Thus, reduced accumulation of the mutant Z protein and decreased liver cell injury. A human trial of low-dose carbamazepine in ZZ patients with cirrhosis was performed but results are yet to be reported.
Liver transplant and outcome in AAT deficiency
Liver transplantation is not a common outcome in children with AAT deficiency, but it is unavoidable in some patients. Data from the French pediatric cohort suggests that only about 10% of subjects received a liver transplant with a mean follow up of 4.7 years. However, data from a recently reported US cohort had a greater proportion of pediatric patients listed for liver transplantation, but that group was more focused on tertiary care and the experience of centers with active liver transplant programs (2).
Liver transplantation is considered when progressive liver failure begins to limit the child’s growth, development, activities of daily life, or when complications such as portal hypertension with variceal bleeding or hepatic encephalopathy become more frequent, and in cases of fulminant liver failure. Regardless of their age at diagnosis or initial presentation, any child with AAT deficiency is at risk, albeit low, for progression to liver failure or cirrhosis. Therefore, regular follow up is indicated for all children with AAT deficiency to monitor for progressive of liver disease or decompensation. The child should be referred to a liver transplantation center if there is evidence of progressive liver injury or fibrosis, including under-nutrition, progressive splenomegaly, hard hepatomegaly, ascites, encephalopathy, GI bleeding, coagulopathy, thrombocytopenia, and elevated bilirubin. Due to the slowly progressive nature that characterizes this disease, it is common for many children with AAT deficiency to be referred for transplantation consideration, but many experience a significant delay in listing or progression to actual transplantation.
The specifics of listing for transplantation and allocation mechanisms for organs vary by country. United Network for Organ Sharing, a not-for-profit, non-governmental organization, manages the organ allocation system in the US. This organization utilizes the Pediatric End-Stage Liver Disease (PELD) scoring system to prioritize children for organs who are at risk for death in the next 3–6 months. This is within the constraints of body size and blood type compatibility. The PELD score consists of an empiric formula of patient characteristics that was originally derived from previous studies of the liver transplant waiting list database. Since most of the patient data in the original database came from patients with biliary atresia, the PELD system has generally been successful for allocating livers to children with this disease. However, this has led to criticism of this system for potentially contributing to delays in transplantation for more rare conditions, such as AAT deficiency. Some centers also increase the flexibility of organ assignment by offering living donor transplants, in which the left lateral segment can be resected from an adult to an infant, or larger portions resected for larger recipients (27). Another transplantation option includes splitting a cadaveric donor liver assigned to an adult, with the left lateral segment allocated to a child recipient. The specifics for transplantation and allocation mechanisms are highly variable among European countries. Some countries participate in a multi-national cooperative that share organs and expertise while others use a modification of the PELD scoring system. Another model seen in Europe focuses all transplants on one or two highly experienced and supported centers, which are often given control over recipient allocation at the local level. In reports from the past 25 years, pediatric liver transplant outcomes in children with AAT deficiency have repeatedly been reported as excellent, and often above average when compared to other diseases (3,28). One-year survival rates are approximately 90%, or greater, with more than an 83% 5-year survival (29).
Heterozygotes and other genotypes
It is reasonably well accepted that compound heterozygotes for S and Z alleles of AAT (SZ) may develop progressive liver disease similar to ZZ patients (e.g., PAS-positive globules resistant to diastase digestion). However, due to the lack of specific evidence-based studies the risk of disease in SZ patients is not well defined. The S allele is commonly found in North America and Western Europe, particularly among the populations of Portugal and Spain. Studies have demonstrated that mutant S protein can only heteropolymerize within the cell if it is co-expressed with another mutant Z protein. Thus, this fact may explain why progressive liver injury occurs in SZ patients while liver disease is absent in SS individuals (6,23,25). Therefore, it is reasonable for clinicians to evaluate and monitor SZ children in a pattern identical to ZZ patients.
Individuals carrying one normal M allele and one mutant Z allele (AAT heterozygotes ‘PiMZ’ or ‘MZ’ carriers) represent 2% of Caucasian populations, and are generally considered healthy in regards to lung and liver disease (6). However, several retrospective studies with data from referral centers show a 3- to 5-fold increased over-representation of MZ cohort of patients in groups with chronic liver conditions (e.g., cryptogenic cirrhosis or in association with concurrent viral hepatitis) (6,16). A reasonable and widely accepted explanation for this finding is that the MZ heterozygote state likely represents a genetic modifier of other liver diseases. Overall, MZ children appear to be healthy and the MZ phenotype in adults is not accepted as the etiology of otherwise unexplained liver disease without a detailed and extensive liver evaluation. No special health care or screening assessments is indicated for MZ individuals, above that for the general population.
Summary
At present, no country includes Homozygous ZZ AAT deficiency on the newborn screen and most such children are well and remain undiagnosed. However, a minority of ZZ neonates present with neonatal cholestasis and are subsequently diagnosed with AAT deficiency. Still others are diagnosed later in childhood with evidence of liver disease or as a result of family testing. Some ZZ children may have systemic symptoms and signs, such as poor growth, and may exhibit mild-to-moderate evidence of liver dysfunction. Infrequently, ZZ children develop progressive liver fibrosis, cirrhosis, and liver failure. At present, there is no specific treatment for AAT deficiency and patients are typically managed by addressing liver disease complications. Rarely, disease progression necessitates liver transplantation with excellent results in comparison to other diagnoses. All children diagnosed with AAT deficiency should be followed carefully. Both patients and their families should be cautioned to avoid smoking, counseled in other good health practices, and offered genetic counseling as well as family testing.
Acknowledgments
Funding: None
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Footnotes
Provenance and Peer Review: This article was commissioned by the Guest Editors (Luca Fabris and Mario Strazzabosco) for the series “Recent Advances in Rare Liver Diseases” published in Translational Gastroenterology and Hepatology. The article was sent for external peer review organized by the Guest Editors and the editorial office.
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tgh.2020.02.23). The series “Recent Advances in Rare Liver Diseases” was commissioned by the editorial office without any funding or sponsorship. JHT reports grants and personal fees from Arrowhead Pharma, grants and personal fees from Dicerna, grants and personal fees from Vertex, grants from NIH, grants from Alpha-1 Foundation, personal fees from Proteostasis, personal fees from Retrophin, personal fees from RestoreBio, grants and personal fees from KorroBio, personal fees from Ironwood Pharmaceuticals, grants and personal fees from Casma, grants and personal fees from Camp4 Therapeutics, grants and personal fees from Intellia, grants and personal fees from Editas, grants and personal fees from Alnylam, personal fees from EcoR1, personal fees from BioMarin, outside the submitted work. The other authors have no conflicts of interest to declare.
References
- 1.Teckman JH, Mangalat N. Alpha-1 antitrypsin and liver disease: mechanisms of injury and novel interventions. Expert Rev Gastroenterol Hepatol 2015;9:261-8. 10.1586/17474124.2014.943187 [DOI] [PubMed] [Google Scholar]
- 2.Teckman JH, Rosenthal P, Abel R, et al. Baseline analysis of a young α-1-antitrypsin deficiency liver disease cohort reveals frequent portal hypertension. J Pediatr Gastroenterol Nutr 2015;61:94-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Németh A. Liver transplantation in alpha(1)-antitrypsin deficiency. Eur J Pediatr 1999;158 Suppl 2:S85-8. 10.1007/PL00014329 [DOI] [PubMed] [Google Scholar]
- 4.Nelson DR, Teckman J, Di Bisceglie AM, et al. Diagnosis and management of patients with α1-antitrypsin (A1AT) deficiency. Clin Gastroenterol Hepatol 2012;10:575-80. 10.1016/j.cgh.2011.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlmutter DH. Alpha-1-antitrypsin deficiency: diagnosis and treatment. Clin Liver Dis 2004;8:839-59, viii-ix. 10.1016/j.cld.2004.06.001 [DOI] [PubMed] [Google Scholar]
- 6.American Thoracic Society, European Respiratory Society American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003;168:818-900. 10.1164/rccm.168.7.818 [DOI] [PubMed] [Google Scholar]
- 7.Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976;294:1316-21. 10.1056/NEJM197606102942404 [DOI] [PubMed] [Google Scholar]
- 8.Sveger T. alpha 1-antitrypsin deficiency in early childhood. Pediatrics 1978;62:22-5. [PubMed] [Google Scholar]
- 9.Eriksson S. Alpha 1-antitrypsin deficiency. J Hepatol 1999;30 Suppl 1:34-9. [PubMed] [Google Scholar]
- 10.Alboni P, Gianfranchi L, Pacchioni F, et al. Antiarrhythmic drugs in patients with recurrent atrial fibrillation: where are we? Ital Heart J 2005;6:169-74. [PubMed] [Google Scholar]
- 11.Piitulainen E, Sveger T. Respiratory symptoms and lung function in young adults with severe alpha(1)-antitrypsin deficiency (PiZZ). Thorax 2002;57:705-8. 10.1136/thorax.57.8.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo S, Booten SL, Aghajan M, et al. Antisense oligonucleotide treatment ameliorates alpha-1 antitrypsin-related liver disease in mice. J Clin Invest 2014;124:251-61. 10.1172/JCI67968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sveger T, Eriksson S. The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology 1995;22:514-7. [DOI] [PubMed] [Google Scholar]
- 14.Cruz PE, Mueller C, Cossette TL, et al. In vivo post-transcriptional gene silencing of alpha-1 antitrypsin by adeno-associated virus vectors expressing siRNA. Lab Invest 2007;87:893-902. 10.1038/labinvest.3700629 [DOI] [PubMed] [Google Scholar]
- 15.Ruiz M, Joly P, Lachaux A. Severe liver disease in children with α-1 antitrypsin deficiency in France (DEFI-ALPHA Cohort). J Pediatr Gastroenterol Nutr 2015;61:e25. 10.1097/MPG.0000000000000932 [DOI] [PubMed] [Google Scholar]
- 16.Sveger T. The natural history of liver disease in alpha 1-antitrypsin deficient children. Acta Paediatr Scand 1988;77:847-51. 10.1111/j.1651-2227.1988.tb10767.x [DOI] [PubMed] [Google Scholar]
- 17.Mowat AP. Alpha 1-antitrypsin deficiency (PiZZ): features of liver involvement in childhood. Acta Paediatr Suppl 1994;393:13-7. 10.1111/j.1651-2227.1994.tb13200.x [DOI] [PubMed] [Google Scholar]
- 18.Pittschieler K, Massi G. Alpha 1 antitrypsin deficiency in two population groups in north Italy. Padiatr Padol 1988;23:307-11. [PubMed] [Google Scholar]
- 19.Eriksson S. Alpha-1-antitrypsin deficiency: natural course and therapeutic strategies. In: Boyer JL, Blum HE, Maier KP, et al. editors. Falk Symposium 115: liver cirrhosis and its development. Dordrecht: Kluwer Academic Publishers, 2001:307-15. [Google Scholar]
- 20.Eriksson S. A 30-year perspective on alpha 1-antitrypsin deficiency. Chest 1996;110:237S-42S. 10.1378/chest.110.6_Supplement.237S [DOI] [PubMed] [Google Scholar]
- 21.Wu Y, Whitman I, Molmenti E, et al. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc Natl Acad Sci U S A 1994;91:9014-8. 10.1073/pnas.91.19.9014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sifers RN. Medicine. Clearing conformational disease. Science 2010;329:154-5. 10.1126/science.1192681 [DOI] [PubMed] [Google Scholar]
- 23.Teckman JH, Perlmutter DH. The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J Biol Chem 1996;271:13215-20. 10.1074/jbc.271.22.13215 [DOI] [PubMed] [Google Scholar]
- 24.Perlmutter DH. Alpha-1-antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates. Annu Rev Med 2011;62:333-45. 10.1146/annurev-med-042409-151920 [DOI] [PubMed] [Google Scholar]
- 25.Lindblad D, Blomenkamp K, Teckman J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology 2007;46:1228-35. 10.1002/hep.21822 [DOI] [PubMed] [Google Scholar]
- 26.He XX, Li Y, Ren HP, et al. 2010 guideline for the management of hepatocellular carcinoma recommended by the American Association for the Study of Liver Diseases. Zhonghua Gan Zang Bing Za Zhi 2011;19:249-50. [PubMed] [Google Scholar]
- 27.Tannuri AC, Gibelli NE, Ricardi LR, et al. Living related donor liver transplantation in children. Transplant Proc 2011;43:161-4. 10.1016/j.transproceed.2010.11.013 [DOI] [PubMed] [Google Scholar]
- 28.Hughes MG, Jr, Khan KM, Gruessner AC, et al. Long-term outcome in 42 pediatric liver transplant patients with alpha 1-antitrypsin deficiency: a single-center experience. Clin Transplant 2011;25:731-6. 10.1111/j.1399-0012.2010.01371.x [DOI] [PubMed] [Google Scholar]
- 29.Kemmer N, Kaiser T, Zacharias V, et al. Alpha-1-antitrypsin deficiency: outcomes after liver transplantation. Transplant Proc 2008;40:1492-4. 10.1016/j.transproceed.2008.02.075 [DOI] [PubMed] [Google Scholar]
- 30.Sveger T, Piitulainen E, Arborelius M., Jr Clinical features and lung function in 18-year-old adolescents with alpha 1-antitrypsin deficiency. Acta Paediatr 1995;84:815-6. 10.1111/j.1651-2227.1995.tb13765.x [DOI] [PubMed] [Google Scholar]