

Abstract
Organophosphate (OP) compounds are widely used as pesticides and herbicides and exposure to these compounds has been associated with both chronic and acute forms of neurological dysfunction including cognitive impairment, neurophysiological problems and cerebral ataxia with evidence of mitochondrial impairment being associated with this toxicity. In view of the potential mitochondrial impairment, the present study aimed to investigate the effect of exposure to commonly used OPs, dichlorvos, methyl-parathion (parathion) and chloropyrifos (CPF) on the cellular level of the mitochondrial electron transport chain (ETC) electron carrier, coenzyme Q10 (CoQ10) in human neuroblastoma SH-SY5Y cells. The effect of a perturbation in CoQ10 status was also evaluated on mitochondrial function and cell viability. A significant decreased (P < 0.0001) in neuronal cell viability was observed following treatment with all three OPs (100 µM), with dichlorvos appearing to be the most toxic to cells and causing an 80% loss of viability. OP treatment also resulted in a significant diminution in cellular CoQ10 status, with levels of this isoprenoid being decreased by 72% (P < 0.0001), 62% (P < 0.0005) and 43% (P < 0.005) of control levels following treatment with dichlorvos, parathion and CPF (50 µM), respectively. OP exposure was also found to affect the activities of the mitochondrial enzymes, citrate synthase (CS) and mitochondrial electron transport chain (ETC) complex II+III. Dichlorvos and CPF (50 µM) treatment significantly decreased CS activity by 38% (P < 0.0001) and 35% (P < 0.0005), respectively compared to control levels in addition to causing a 54% and 57% (P < 0.0001) reduction in complex II+III activity, respectively. Interestingly, although CoQ10 supplementation (5 μM) was able to restore cellular CoQ10 status and CS activity to control levels following OP treatment, complex II+III activity was only restored to control levels in neuronal cells exposed to dichlorvos (50 µM). However, post supplementation with CoQ10, complex II+III activity significantly increased by 33% (P < 0.0005), 25% (P < 0.005) and 35% (P < 0.0001) in dichlorvos, parathion and CPF (100 µM) treated cells respectively compared to non-CoQ10 supplemented cells. In conclusion, the results of this study have indicated evidence of neuronal cell CoQ10 deficiency with associated mitochondrial dysfunction following OP exposure. Although CoQ10 supplementation was able to ameliorate OP induced deficiencies in CS activity, ETC complex II+III activity appeared partially refractory to this treatment. Accordingly, these results indicate the therapeutic potential of CoQ10 supplementation in the treatment of OP poisoning. However, higher doses may be required to engender therapeutic efficacy.
Keywords: Organophosphate, Methyl-parathion, Dichlorvos, Chloropyrifos, Mitochondria, Coenzyme Q10
Introduction
Organophosphate (OP) compounds are used as pesticides and herbicides [1]. They are generally derived from esters of phosphoric acid [2]. These toxic synthetic compounds including dichlorvos, methyl-parathion (parathion) and chloropyrifos (CPF) can cause a neurological syndrome called OP poisoning, which can display itself in both acute and chronic forms [3]. OP poisoning appears to be less prevalent in the UK, compared to developing countries [4]. However, it is becoming increasingly apparent that sheep farmers exposed to OP-based sheep dip are at a high risk of developing OP poisoning [5, 6]. As well as agricultural farmers, air craft cabin crew [7] and military personnel (Gulf war syndrome) are susceptible [8]. In farmers, OP poisoning as a result of exposure to OP based-sheep dip, via both inhalation and direct skin contact, can result in chronic health defects including neurological problems, muscle pain and fatigue [9]. Interestingly similar clinical presentations have been observed in Gulf war syndrome, an illness which developed not long after deployment of war veterans to the 1991 Gulf war [10, 11]. However, although the origin of Gulf war syndrome remains to be uncertain, it has been attributed to exposure of pesticides [12].
The mechanism of action of these OPs involves the inhibition of the nerve transmission enzyme acetylcholinesterase (AChE), which is required for the degradation of acetylcholine [13]. Persistence of acetylcholine at the cholinergic synapses due to AChE inhibition results in insect death due to hyper-excitation [14], and indeed, this also remains the most common adverse side effect of OP exposure in humans [15, 16]. In addition to this, OP poisoning has been associated with mitochondria dysfunction [17]. Clinical presentations have also been observed in OP patients which indicate evidence of mitochondrial dysfunction including fatigue [18], peripheral neuropathy [19] and cerebral ataxia [20]. Interestingly, cerebral ataxia is also a prominent feature of severe Coenzyme Q10 (CoQ10) deficiency [21, 22].
Support for the potential mitochondrial toxicity of OPs has been provided by identified mitochondrial dysfunction as a result of exposure to OPs. This includes reduction in the activity of mitochondrial electron transport chain (ETC) enzymes, complex I (NADH: ubiquinone reductase), complex II (succinate dehydrogenase: ubiquinone reductase) and complex IV (cytochrome c oxidase) enzymes in rat studies [23], reduction in mitochondrial complex I-III (NADH: cytochrome c reductase) activity, which can in turn affect the production of ATP [24]. Mitochondrial swelling and elevated cytochrome c levels have also been observed in rabbit livers treated with the OP, chloropyrifos [17]. Interestingly, these alterations also appeared to correlate with elevated oxidative stress levels in rabbit’s liver. Furthermore, evidence of oxidative stress as a result of OP exposure has also been observed in vivo as elevations of oxidative stress marker malondialdehyde has been identified in mice blood levels post OP administration [25]. In view of the association between mitochondria dysfunction and cellular oxidative stress [17, 26], the ability of OPs to induce an increase in reactive oxidant species (ROS) generation may further support the potential mitochondrial toxicity of these group of compounds.
The increased oxidative stress and mitochondria dysfunction associated with OP treatment [17], may result from diminution in cellular CoQ10 status as the result of exposure to these compounds. CoQ10 functions as an electron carrier in the ETC as well as serving as a potent lipid soluble antioxidant [27]. Administration of CoQ10 has previously been reported to alleviate mitochondrial oxidative stress [28], as well as enhancing the activities of ETC complexes I-III and complex IV in rats treated with the OP, dichlorvos [24]. CoQ10 administration for 12 weeks prior to dichlorvos treatment was also found to improve cognitive performance, mitochondrial physiological features and attenuate neuronal damage [29]. These findings suggest a potential role for CoQ10 supplementation in the treatment of OP toxicity. Although, at present there is a paucity of information available on the therapeutic efficacy of CoQ10 in the treatment of the clinical symptoms associated with OP poisoning in human subjects. However, veterans suffering with Gulf War syndrome, a condition associated with OP poisoning, have been reported to show some clinical improvement following CoQ10 treatment [30]. The factors responsible for biochemical and clinical efficacy of CoQ10 in the treatment of OP toxicity have yet to be elucidated. However, the possibility arises that the exogenous CoQ10 may be replenishing an endogenous CoQ10 deficiency induced by these synthetic compounds.
The present study evaluated the effect of OPs dichlorovos, chloropyrifos and methyl-parathion on human neuronal cellular CoQ10 status and mitochondrial function. In addition, the effect of CoQ10 supplementation on mitochondrial function following OP exposure was also investigated.
Materials and Methods
Materials
All reagents were of analytical grade and obtained from Sigma-Aldrich Chemical (Poole, UK).
Cell Culture
SH-SY5Y human neuroblastoma cells (passage 17–24) were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 1% penicillin and streptomycin and 10% foetal bovine serum (FBS). Cultures were maintained in an incubator with 5% CO2 at 37 °C. At around 70–90% confluence, cells were harvested, washing twice with phosphate-buffered saline (PBS) followed by the addition of 3 ml of trypsin. Cells were seeded in plastic 96-well plates at a density of 20,000 cells/well or T75 flasks at 150,000 cells/ml prior to treatment with OPs. OP (dichlorvos, chloropyrifos and methyl-parathion) treatment was carried out in 10% DMEM at concentrations of 10 µM, 50 µM and 100 µM and SH-SY5Y cells were treated with the OPs for 5 days. These concentrations were selected in the present study as these OP concentrations have been reported in the blood of individuals who are regularly exposed to OPs [31, 32], as well as in patients who have developed OP poisoning [33]. These concentrations also parallel those used in previous studies [34–36]. Because the OPs were dissolved in ethanol, control cells were treated with the vehicle ethanol. Cells were further supplemented with CoQ10 (5 µM) for 2 days with a further addition of the OPs (50 µM and 100 µM) according to the methods by Duberley et al. [37].
Cell Viability Assay
SH-SY5Y human neuroblastoma cell viability was obtained using the tetrazolium dye MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay. This colorimetric assay is based on the ability of the mitochondrial enzyme NADPH-dependant cellular oxidoreductase enzymes to reduce MTT to its purple coloured insoluble formazan. Trypan blue dye solution was added to the cells and dye excluding cells were counted using a haemocytometer. Cells were plated and treated with OPs as previously described. MTT solution (20 µl) was added to each well of the 96 well plate post a 5 day incubation with the OPs. After a 3–4 h incubation with MTT at 37 °C, the formazan product was solubilized using 100 µl of DMSO and the absorbance of each well was read at 570 nm on a CLARIOstar® Plus.
ETC Enzyme Activities
The activities of the ETC complex II/III (succinate: cytochrome C reductase) and citrate synthase were determined spectrophotometrically at wavelengths of 412 nm and 550 nm respectively, according to methods described by Duberley et al. [38]. Complex II/III activities were expressed as a ratio to citrate synthase activity to account for mitochondrial enrichment [39].
Total Protein Determination
Cellular protein concentration was determined by the method of Lowry et al. [40]. Protein standards were prepared by diluting BSA (0.2 mg/ml) in H2O (0, 25, 50, 75, 100, 200 µg/ml). Samples were diluted 1/100 in H2O in Eppendorf tubes. 100 µl of reagent A and 800 µl of reagent B were added to the Eppendorf tubes and standards. Absorbance was measured spectrophotometrically at 750 nm. Protein concentrations can be determined using relative absorbance of protein standards expressed as (mg/ml).
Lactate Measurement
Following a 5-day incubation with the OPs, 1 ml of culture media from control and OP treated cells was transferred to an Eppendorf tube and frozen at − 80 °C. Samples were thawed and immediately transferred to hospital grade grey topped Fluoride (K EDTA) tubes. The samples were analysed on the Roche Cobas c analyser using Roche 2nd generation LACT2 colorimetric lactate assay. This assay utilises lactate oxidase to oxidise lactate to pyruvate with the generation of hydrogen peroxide. The hydrogen peroxide formed then reacts under a reaction catalysed by a peroxidase with phenol and 4-aminophenazone to form a red-violet quinoneimine dye which absorbs at 500 nm. The resulting increase in absorption is proportional to the amount of lactate present in the sample.
Quantification of CoQ10 Status
The CoQ10 status of SH-SY5Y cells was determined using high pressure liquid chromatography with UV detection at 275 nm according to the method of Duncan et al. [41]. The results were expressed pmol/mg of protein.
Statistical Analysis
All results are expressed as a mean ± standard deviation. One-way ANOVA with Tukey’s multiple comparison post hoc test was used for comparison of groups > 2 using statistical software GraphPad Prism. P < 0.05 was considered significant.
Results
Organophosphates Induced a Significant Reduction in Cell Viability
The MTT cytotoxicity assay was used to assess cellular metabolic activity post treatment with OPs. The percentage (%) of viable cells following a 5 day incubation with a concentration of 10 µM of the OPs showed very little effect on cell viability (data not shown). However, at 50 µM MTT conversion was reduced significantly by 20% (P < 0.05) for parathion treated cells compared to control levels, which directly corresponds to a significant reduction in cell viability (Fig. 1). Following a 5 day incubation with OPs (100 µM) dichlorvos, parathion and CPF there was an 80%, 55% and 25% (P < 0.0001) significant reduction in cell viability respectively compared to control levels (Fig. 1).
Fig. 1.
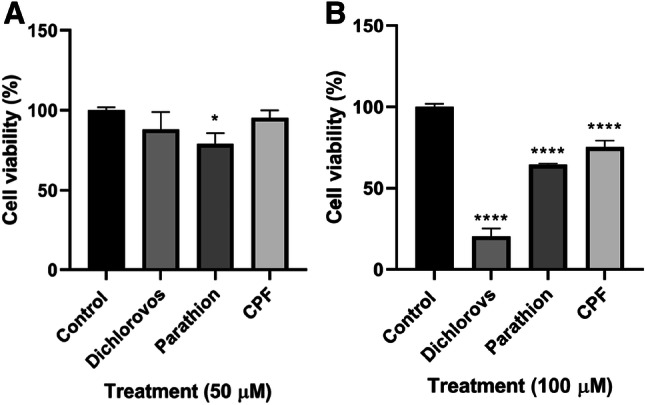
Cell viability (%) of SH-SY5Y cells following a 5 day incubation with OPs dichlorvos, parathion and CPF for concentrations of a 50 µM (n = 3) b 100 µM (n = 3). Error bars represent standard deviation; statistical analysis was carried out using one-way ANOVA with Tukey’s multiple comparison post hoc test; levels of significance: *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001 compared to control levels
Organophosphate Treatment had no Effect on Lactate Concentration
Incubation of SH-SY5Y cells with OPs dichlorvos, parathion and CPF (100 µM) resulted in no significant difference in lactate medium concentrations (mmol/l) compared to control levels (Fig. 2).
Fig. 2.
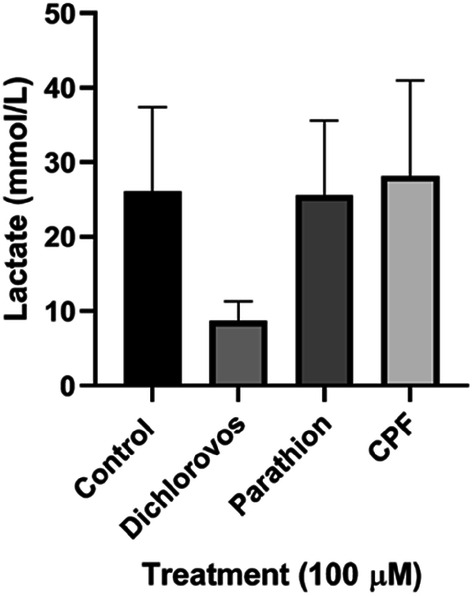
Effect of 5 day treatments of OPs dichlorvos, parathion and CPF (100 µM) on SH-SY5Y cellular medium lactate concentration (mmol/L) (n = 3). Error bars represent standard deviation; statistical analysis was carried out using one-way ANOVA with Tukey’s multiple comparison post hoc test; levels of significance: *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001 compared to control levels
Organophosphates Induced Neuronal Coenzyme Q10 Deficiency
The CoQ10 status of SH-SY5Y cells decreased following a 5 day incubation with the OPs. Cellular CoQ10 status significantly decreased 72% (P < 0.0001), 62% (P < 0.0005) and 43% (P < 0.005) post treatment with dichlorvos, parathion and CPF (50 µM) respectively compared to control levels (Fig. 3). Cellular CoQ10 status significantly decreased (P < 0.0001) by 52%, 64% and 40% post treatment with dichlorvos, parathion and CPF (100 µM) respectively compared to control levels (Fig. 3).
Fig. 3.
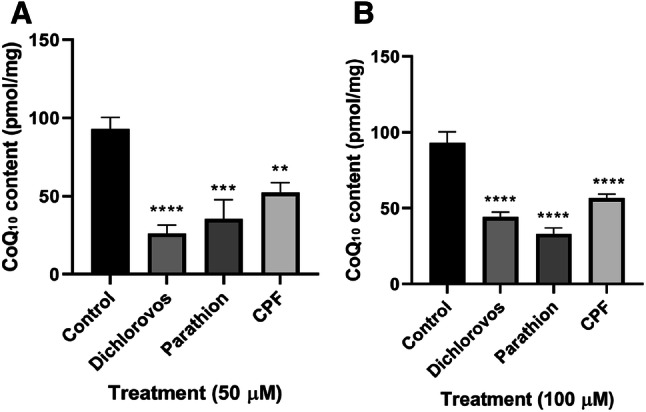
Effect of 5 day treatments of OPs dichlorvos, parathion and CPF on CoQ10 content (pmol/mg) in SH-SY5Y cells. a 50 µM concentration of OPs (n = 3), b 100 µM concentration of OPs (n = 3). Error bars represent standard deviation; statistical analysis was carried out using one-way ANOVA with Tukey’s multiple comparison post hoc test; levels of significance: *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001 compared to control levels
CoQ10 Supplementation Demonstrated Protective Properties Against OP-Induced Cellular Death
MTT cytotoxicity assay was used to measure cell viability (%) following 5 day incubations with OPs dichlorvos, parathion and CPF (100 µM), and then a further 2 day incubation with CoQ10 (5 µM). Post treatment with CoQ10 parathion and CPF treated cells resulted in a significant increase in cell viability, increasing 30% (P < 0.0005) and 18% (P < 0.0001) respectively compared to non-CoQ10 supplemented cells (Fig. 4).
Fig. 4.
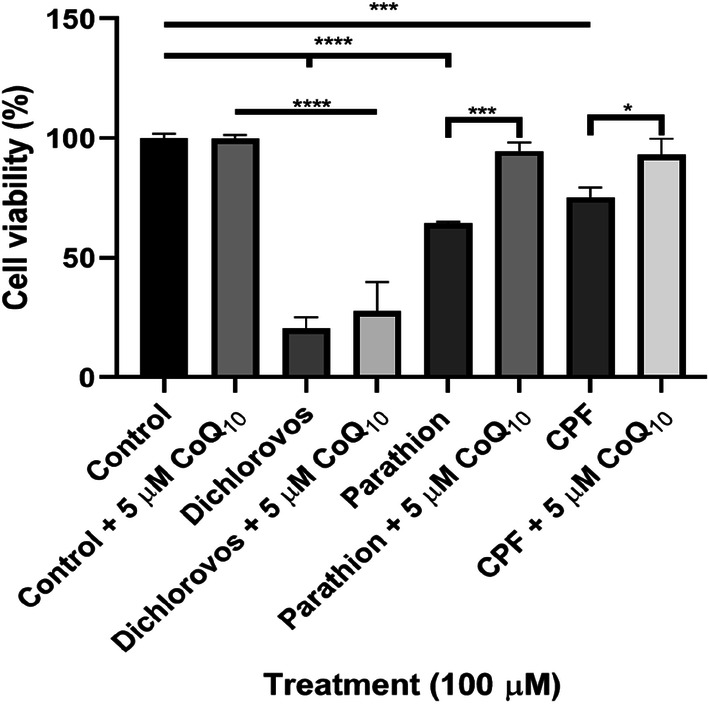
Effect of a 2 day supplementation with CoQ10 following a 5 day incubation with OPs dichlorvos, parathion and CPF (100 µM) in SH-SY5Y cells on cell viability (%). Error bars represent standard deviation; statistical analysis was carried out using one-way ANOVA with Tukey’s multiple comparison post hoc test; levels of significance: *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001
CoQ10 Supplementation Improved OP-Induced Alterations on CS Activity
Incubation of SH-SY5Y cells with OPs dichlorvos, parathion and CPF at 50 µM and 100 µM resulted in alterations in citrate synthase (CS) (mitochondrial matrix marker enzyme) activity. At 50 µM concentrations, dichlorvos and CPF showed a significant reduction in CS activity, decreasing 38% (P < 0.0001) and 35% (P < 0.0005) respectively compared to control levels (Fig. 5). At 100 µM concentrations, dichlorvos and CPF showed a significant reduction (P < 0.0001) in CS activity, reducing 86% and 42% respectively compared to control levels. However, parathion showed a significant increase (P < 0.0001) in CS activity, increasing 39% compared to control levels (Fig. 5).
Fig. 5.
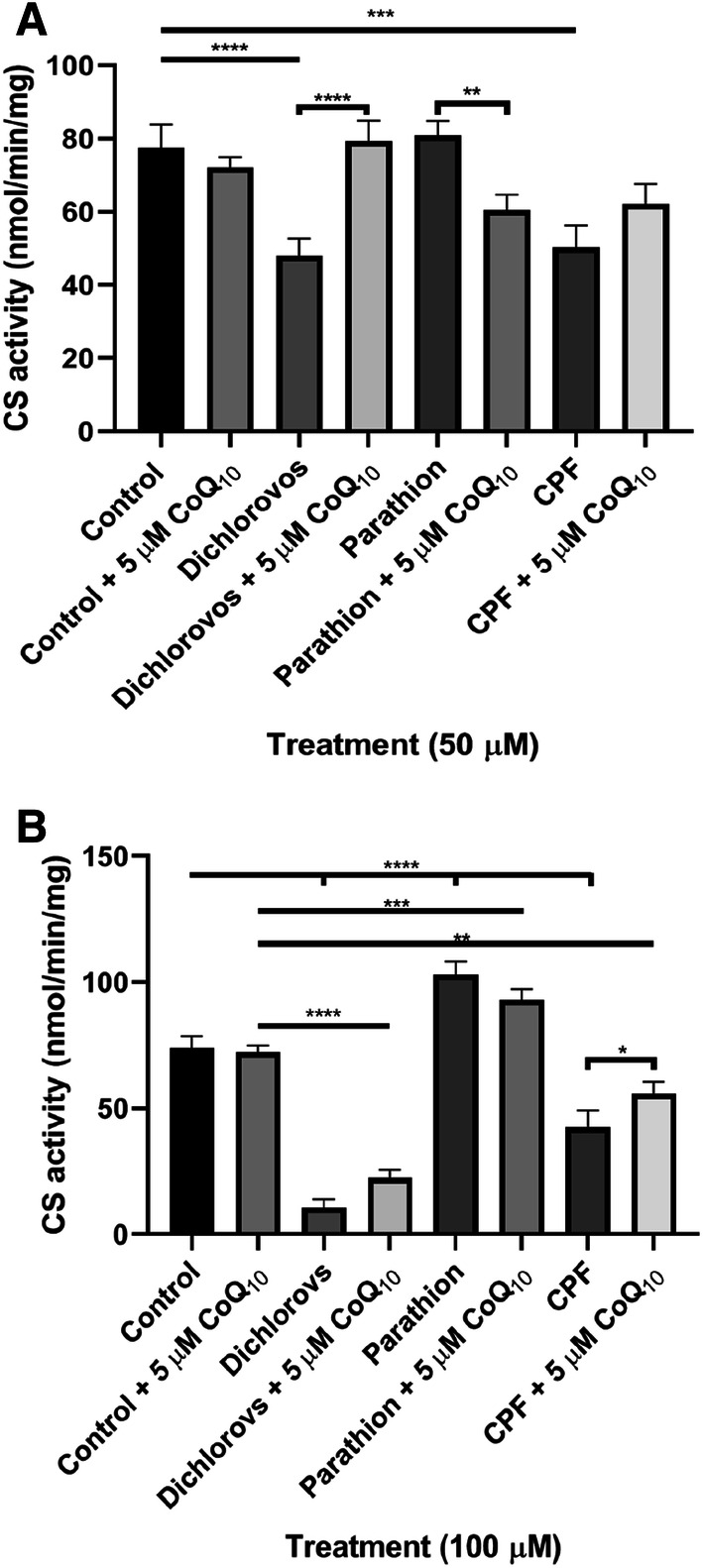
Effect of a 5 day incubation with OPs dichlorvos, parathion and CPF on citrate synthase activity in SH-SY5Y cells, followed by a further 2 day supplementation with CoQ10 (5 µM) compared to controls. a Citrate synthase activity post treatment with 50 µM of OPs, with and without a further 2 day supplementation with CoQ10 (5 µM) (n = 3), b citrate synthase activity post treatment with 100 µM of OPs, with and without a further 2 day supplementation with CoQ10 (5 µM) (n = 3). Error bars represent standard deviation; statistical analysis was carried out using one-way ANOVA with Tukey’s multiple comparison post hoc test; levels of significance: *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001
Post supplementation with CoQ10, CS activity significantly increased by 47% (P < 0.0001) in dichlorvos (50 µM) treated cells compared to non-CoQ10 supplemented cells thus showing no significant difference from controls post treatment with CoQ10. Post supplementation with CoQ10, CS activity significantly increased (P < 0.05) by 20% in CPF (100 µM) treated cells compared to non-CoQ10 supplemented cells (Fig. 5).
CoQ10 Supplementation Improved OP-Induced Alterations in Complex II+III Activity
Incubation of SH-SY5Y cells with OPs dichlorvos, parathion and CPF at 50 µM and 100 µM resulted in alterations in complex II+III activity. At 50 µM OP concentrations, dichlorvos and CPF showed a significant reduction (P < 0.0001) in complex II+III activity, reducing 54% and 57% respectively compared to control levels. Treatment with 100 µM of dichlorvos showed no complex II+III activity. Parathion and CPF (100 µM) treated cells showed a significant reduction (P < 0.0001) in complex II+III activity, reducing 66% and 67% respectively compared to control levels (Fig. 6).
Fig. 6.
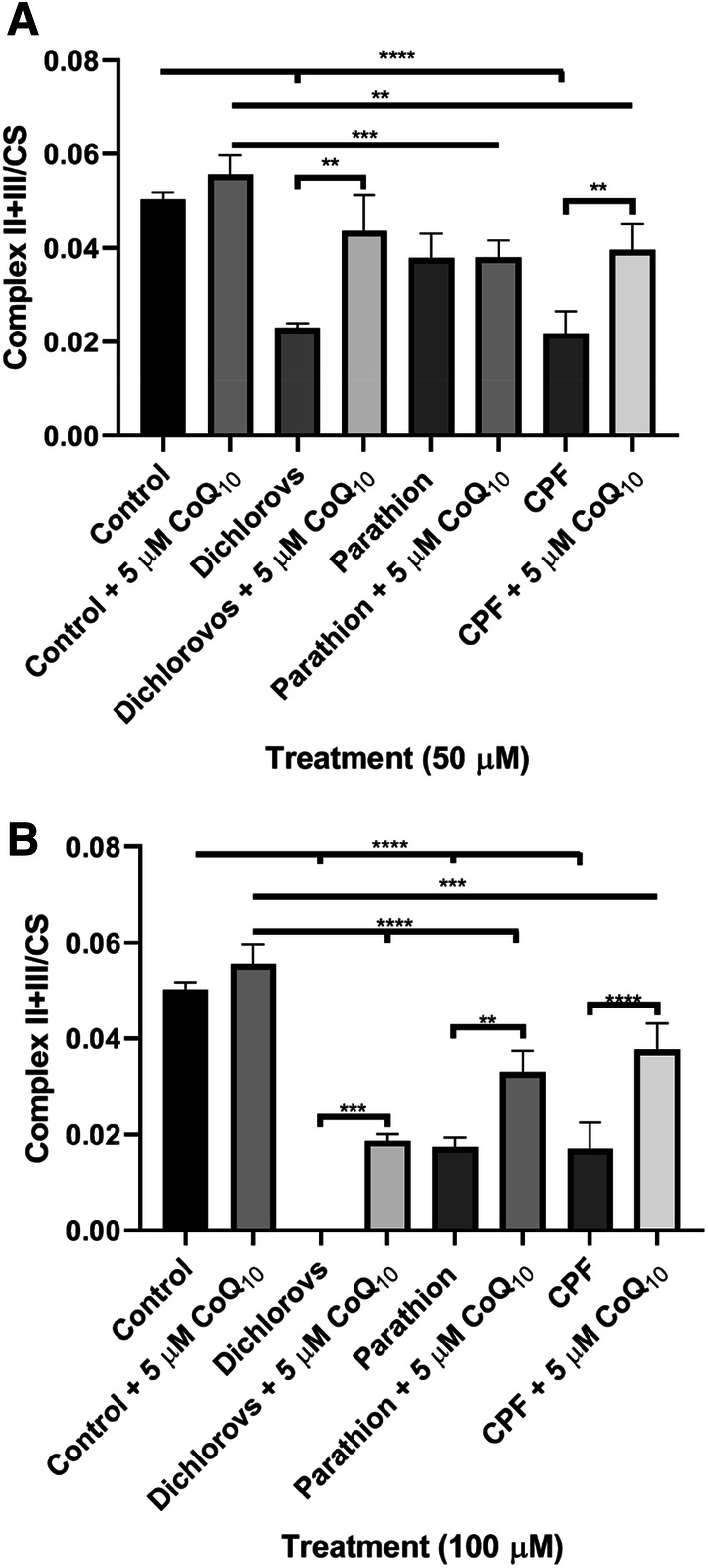
Effect of a 5 day incubation with OPs dichlorvos, parathion and CPF on complex II + III activity in SH-SY5Y cells, followed by a further 2 day supplementation with CoQ10 (5 µM) compared to controls. a Complex II + III activity post incubation with 50 µM of OPs, with and without a further 2 day supplementation with CoQ10 (5 µM) (n = 3), b Complex II + III activity post incubation with 100 µM of OPs, with and without a further 2 day supplementation with CoQ10 (5 µM) (n = 3). Error bars represent standard deviation; statistical analysis was carried out using one-way ANOVA with Tukey’s multiple comparison post hoc test; levels of significance: *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001
Post supplementation with CoQ10, complex II+III activity significantly increased (P < 0.005), increasing 32% and 28% in dichlorvos and CPF (50 µM) treated cells compared to non-CoQ10 supplemented cells. Post supplementation with CoQ10, complex II+III activity significantly increased, increasing 33% (P < 0.0005), 25% (P < 0.005) and 35% (P < 0.0001) in dichlorvos, parathion and CPF (100 µM) treated cells compared to non-CoQ10 supplemented cells (Fig. 6).
Discussion
The results of the present study have provided evidence of OP induced neuronal cell CoQ10 deficiency which was associated with an impairment of ETC complex II+III activity. As far as the authors are aware, this is the first study to report an association between OP exposure and a deficit in cellular CoQ10 status and this may offer an explanation for the ability of CoQ10 supplementation to ameliorate some of the neurological symptoms associated with OP poisoning [42]. Furthermore, cerebral ataxia has been associated with OP poisoning which is one of the main clinical presentations of CoQ10 deficiency [20].
A deficit in cellular CoQ10 status was detected following treatment with OPs dichlorvos, parathion and CPF (50 and 100 µM) indicating the ability of OPs to induce neuronal CoQ10 deficiency with dichlorvos, inducing the most pronounced deficit in the status of this isoprenoid. The cause of the cellular CoQ10 deficiency associated with OP exposure has yet to be elucidated, and may be due to the ability of these synthetic compounds to directly inhibit the CoQ10 biosynthetic pathway or as the result of some other secondary mechanism. OPs associated with increased ROS generation may result in an increase in oxidative catabolism as well as a possible inhibition of enzymes in the CoQ10 biosynthetic pathway [17].
In view of the dependence of ETC complex II+III activity upon endogenous CoQ10 status [43], we also assessed the effect of OP treatment on complex II+III activity. Interestingly, the decrease in cellular CoQ10 status was found to accompany a significant loss in complex II+III activity, supporting the requirement for CoQ10 for optimal complex II+III activity. This therefore indicates the potential for a deficit in CoQ10 status to impair ETC activity. The activity of complex II+III was expressed as a ratio to CS to account for mitochondria enrichment. Therefore, we can assume that the reduction observed to complex II+III activity reflects impairment to oxidative phosphorylation rather than a loss in mitochondrial number [39]. ETC inhibition generates free radicals, leading to increased oxidative stress [44]. Since the ETC is highly susceptible to oxidative damage and ROS can directly damage mitochondrial enzymes [45], the possibility arises that the loss in complex II+III activity may also have resulted from OP-induced ROS generation [17, 25].
Importantly, the 65% reduction in complex II+III activity observed in chloropyrifos and parathion (100 µM) treated cells may not be sufficient to impair oxidative phosphorylation, as it has been previously reported that complex III activity has to be inhibited by 70–80% before oxidative phosphorylation is compromised [46]. In support of this, the assessed lactate medium levels post treatment with the OPs (100 µM) wasn’t found to be elevated compared to control levels, suggesting an upregulation of glycolytic metabolism was not required to compensate for impairment of oxidative phosphorylation [47]. Alternatively, this may be due to a significant reduction in cell viability due to OP-induced toxicity.
The significant increase in CS activity following treatment of the neuronal cells with parathion (100 µM) would indicate that exposure to this synthetic compound induced an increase in mitochondrial biogenesis in the SH-SY5Y cells, which has been reported to occur as an adaptive response to impaired oxidative phosphorylation [48]. In contrast to the increase in CS activity, the level of neuronal CoQ10 was found to decrease following treatment with parathion and since approximately 45% of cellular CoQ10 is found within the mitochondria [49], this would suggest that the diminution in cellular CoQ10 status does not reflect the loss of mitochondrial enrichment. Contrary to parathion, CPF and dichlorvos induced a loss of neuronal cell CS activity which may reflect a loss of mitochondrial enrichment. A decrease in CS activity has previously been associated with a decrease in cellular ATP production, an increase in ROS production and accelerated cell death [50]. This may explain to some degree the evidence of cellular toxicity and loss of cell viability indicated by the MTT assay following exposure to OPs at concentrations of 50 µM and 100 µM. However, no evidence of toxicity was apparent when OPs were used at a concentration of 10 µM (data not shown).
There is a paucity of information available on the ability of CoQ10 to ameliorate some of the adverse clinical effects associated with OP toxicity. However, a study by Bayir et al. [51] reported the ability of CoQ10 to attenuate OP induced cardiac toxicity. Furthermore, a recent study by Belousova et al. [42] demonstrated that CoQ10 improves neurological function in transient focal brain ischemia in rats [42]. Similarly, the present study showed that CoQ10 supplementation (5 µM) was able to restore parathion and CPF (100 µM) treated cell viability to control levels. OPs may induce toxicity to cells by their ability to upregulate ROS production and downregulate antioxidant enzymes [52, 53]. Thus, CoQ10 may play roles in ameliorating OP induced toxicity by acting as a free radical scavenger to mitigate DNA damage and lipid peroxidation from OP-induced oxidative stress, as well as elevating activities of antioxidant enzymes including OP detoxifying enzyme, paraxonase [29, 54].
CoQ10 supplementation was able to restore CS activity to control levels following treatment with the OPs dichlorvos and CPF (50 µM), but not in cells treated with 100 µM of OPs. Although the precise mechanism by which exogenous CoQ10 induces this effect is uncertain, in its ability to act as an antioxidant CoQ10 may protect the cell against OP induced oxidative stress and therefore prevent an impairment of oxidative phosphorylation or increased loss of mitochondrial enrichment [55]. However, further work will be required to evaluate the effect of CoQ10 supplementation on OP-induced cellular oxidative stress. Alternatively, CoQ10 may activate mitochondria biogenesis as indicated by the study of Jing et al. [56] which reported an increase in the ratio of COX-1 mitochondrial DNA to that of SDH-A nuclear DNA following treatment of cells with CoQ10. This would therefore explain the restoration of CS activity (marker for mitochondrial enrichment) following treatment with CoQ10 in cells exposed to OPs, dichlorvos and CPF (50 µM).
In addition, CoQ10 treatment was found to increase ETC complex II+III activity, however the activity of this enzyme only reached control levels in cells treated with 50 µM of dichlorovos. Treatment with 100 µM of this OP resulted in an apparent complete inhibition of complex II+III activity, possibly reflecting the significant loss of cell viability indicated by the MTT assay. Subsequent CoQ10 supplementation was found to increase complex II+III activity to 34% of control levels in neuronal cells treated with 100 µM dichlorovos. The failure of CoQ10 supplementation to fully restore neuronal cell ETC complex II+III activity to control levels following exposure to OPs (CPF, parathion and dichlorovos at 100 µM) may indicate that the loss of enzyme activity was not solely the result of a diminution in cellular CoQ10 status and possibly the result of some other inhibitory mechanism. However, some studies have suggested that only 11% of an exogenous quinone is able to reach the mitochondrion [57] and therefore the failure of CoQ10 supplementation to restore ETC complex II+III activity may be due to the dosage of CoQ10 used in the study which was 5 µM. In agreement with this, a study by Duberley et al. [37] reported that a dosage of CoQ10 above 10 µM may be required to fully restore ETC enzyme activity following a diminution of CoQ10 status. complex II+III was not fully recovered post treatment with OPs (100 µM). However, in view of the MTT results, perbutation of complex II+III may not be sufficient to impair oxidative phosphorylation.
In conclusion, this study for the first time has indicated the possibility that OP exposure may result in a loss of cellular CoQ10 status which therefore may be a contributory factor to some of the clinical adverse side effects associated with OP exposure. Supplementation with CoQ10 was able to restore OP-induced CS activity and cell viability to control levels in some cases. However, future work to evaluate the effect of OPs on oxidative stress levels and the ability for CoQ10 to ameliorate OP-induced oxidative stress is required to fully identify the molecular mechanism by which CoQ10 improves mitochondria functioning in the present study. CoQ10 supplementation was unable to fully restore ETC complex II+III activity to control levels in this study. This may reflect the dosage of the quinone used and higher doses (> 10 µM) may be required to elicit biochemical efficacy. Thus, targeted strategies aimed at restoring cellular CoQ10 status following OP exposure should be considered, although further studies are first required to assess evidence of cellular CoQ10 deficiency in patients following OP exposure.
Acknowledgements
We are grateful to Pharma Nord UK for providing funding for this study and to Dr. David Mantle for his help and advice with the study.
Abbreviations
- OP
Organophosphate
- ETC
Electron transport chain
- CoQ10
Coenzyme Q10
- CS
Citrate synthase
- CPF
Chloropyrifos
Compliance with Ethical Standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Special Issue: In Honor of Professor Juan Bolanos.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Aktar MW, Sengupta D, Chowdhury A. Impact of pesticides use in agriculture: their benefits and hazards. Interdisc Toxicol. 2009;2(1):1–12. doi: 10.2478/v10102-009-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rathnayake LK, Northrup SH. Structure and mode of action of organophosphate pesticides: a computational study. Comput Theor Chem. 2016;1088:9–23. [Google Scholar]
- 3.Hung D-Z, et al. The long-term effects of organophosphates poisoning as a risk factor of CVDs: a nationwide population-based cohort study. PLoS ONE. 2015;10(9):e0137632–e0137632. doi: 10.1371/journal.pone.0137632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parmod KS, Ashok S. Organophosphate poisoning : A review. Med J Indonesia. 2003;12(2):120–126. [Google Scholar]
- 5.Solomon C, et al. Neuropsychiatric symptoms in past users of sheep dip and other pesticides. Occup Environ Med. 2007;64(4):259–266. doi: 10.1136/oem.2005.023879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pilkington A, et al. An epidemiological study of the relations between exposure to organophosphate pesticides and indices of chronic peripheral neuropathy and neuropsychological abnormalities in sheep farmers and dippers. Occup Environ Med. 2001;58(11):702–710. doi: 10.1136/oem.58.11.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schindler BK, et al. Occupational exposure of air crews to tricresyl phosphate isomers and organophosphate flame retardants after fume events. Arch Toxicol. 2013;87(4):645–648. doi: 10.1007/s00204-012-0978-0. [DOI] [PubMed] [Google Scholar]
- 8.Terry AV., Jr Functional consequences of repeated organophosphate exposure: potential non-cholinergic mechanisms. Pharmacol Ther. 2012;134(3):355–365. doi: 10.1016/j.pharmthera.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamel F, Hoppin JA. Association of pesticide exposure with neurologic dysfunction and disease. Environ Health Perspect. 2004;112(9):950–958. doi: 10.1289/ehp.7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White RF, et al. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: effects of toxicant exposures during deployment. Cortex. 2016;74:449–475. doi: 10.1016/j.cortex.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hom J, Haley RW, Kurt TL. Neuropsychological correlates of Gulf War syndrome. Archives of Clinical Neuropsychology. 1997;12(6):531–544. [PubMed] [Google Scholar]
- 12.McCauley LA. Chapter 6—organophosphates and the Gulf War syndrome. In: Gupta RC, editor. Toxicology of organophosphate & carbamate compounds. Burlington: Academic Press; 2006. pp. 69–78. [Google Scholar]
- 13.Fukuto TR. Mechanism of action of organophosphorus and carbamate insecticides. Environ Health Perspect. 1990;87:245–254. doi: 10.1289/ehp.9087245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y. Organophosphate-induced brain damage: mechanisms, neuropsychiatric and neurological consequences, and potential therapeutic strategies. NeuroToxicology. 2012;33(3):391–400. doi: 10.1016/j.neuro.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Shenouda J, Green P, Sultatos L. An evaluation of the inhibition of human butyrylcholinesterase and acetylcholinesterase by the organophosphate chlorpyrifos oxon. Toxicol Appl Pharmacol. 2009;241(2):135–142. doi: 10.1016/j.taap.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lionetto MG, et al. Acetylcholinesterase as a biomarker in environmental and occupational medicine: new insights and future perspectives. Biomed Res Int. 2013;2013:321213–321213. doi: 10.1155/2013/321213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toualbia N, Rouabhi R, Salmi A. Evaluation of cytochrome c level and mitochondrial dysfunction biomarkers of Oryctolagus cuniculus liver exposed to Chlorpyrifos. Toxicol Environ Health Sci. 2017;9(5):325–331. [Google Scholar]
- 18.Perry J, et al. Organophosphate exposure and the chronic effects on farmers: a narrative review. Rural Remote Health. 2020;20(1):4508. doi: 10.22605/RRH4508. [DOI] [PubMed] [Google Scholar]
- 19.Ergün SS, et al. Delayed neuropathy due to organophosphate insecticide injection in an attempt to commit suicide. Hand (New York, NY) 2009;4(1):84–87. doi: 10.1007/s11552-008-9126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fonseka MM, et al. Self-limiting cerebellar ataxia following organophosphate poisoning. Hum Exp Toxicol. 2003;22(2):107–109. doi: 10.1191/0960327103ht341cr. [DOI] [PubMed] [Google Scholar]
- 21.Quinzii CM, Hirano M, Naini A. Cerebellar Ataxia and CoQ10 Deficiency. J Neurol Disord Stroke. 2013;1(1):1004–1004. [PMC free article] [PubMed] [Google Scholar]
- 22.Musumeci O, et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56(7):849–855. doi: 10.1212/wnl.56.7.849. [DOI] [PubMed] [Google Scholar]
- 23.Masoud A, Kiran R, Sandhir R. Impaired mitochondrial functions in organophosphate induced delayed neuropathy in rats. Cell Mol Neurobiol. 2009;29(8):1245–1255. doi: 10.1007/s10571-009-9420-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binukumar BK, et al. Mitochondrial energy metabolism impairment and liver dysfunction following chronic exposure to dichlorvos. Toxicology. 2010;270(2–3):77–84. doi: 10.1016/j.tox.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 25.Yurumez Y, et al. Beneficial effect of N-Acetylcysteine against organophosphate toxicity in mice. Biol Pharm Bull. 2007;30(3):490–494. doi: 10.1248/bpb.30.490. [DOI] [PubMed] [Google Scholar]
- 26.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 27.Hargreaves IP. Ubiquinone: cholesterol's reclusive cousin. Ann Clin Biochem. 2003;40(Pt 3):207–218. doi: 10.1258/000456303321610493. [DOI] [PubMed] [Google Scholar]
- 28.Nakazawa H, et al. Coenzyme Q10 protects against burn-induced mitochondrial dysfunction and impaired insulin signaling in mouse skeletal muscle. FEBS Open Bio. 2019;9(2):348–363. doi: 10.1002/2211-5463.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Binukumar BK, et al. Protective efficacy of coenzyme Q10 against DDVP-induced cognitive impairments and neurodegeneration in rats. Neurotox Res. 2012;21(4):345–357. doi: 10.1007/s12640-011-9289-0. [DOI] [PubMed] [Google Scholar]
- 30.Golomb BA, et al. Coenzyme Q10 benefits symptoms in Gulf War veterans: results of a randomized double-blind study. Neural Comput. 2014;26(11):2594–2651. doi: 10.1162/NECO_a_00659. [DOI] [PubMed] [Google Scholar]
- 31.Pluth JM, et al. Increased frequency of specific genomic deletions resulting from in vitro malathion exposure. Can Res. 1996;56(10):2393. [PubMed] [Google Scholar]
- 32.Huen K, et al. Organophosphate pesticide levels in blood and urine of women and newborns living in an agricultural community. Environ Res. 2012;117:8–16. doi: 10.1016/j.envres.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eyer F, et al. Extreme variability in the formation of chlorpyrifos oxon (CPO) in patients poisoned by chlorpyrifos (CPF) Biochem Pharmacol. 2009;78(5):531–537. doi: 10.1016/j.bcp.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu C-Y, Chang P-A, Wu Y-J. Trichlorfon induces apoptosis in SH-SY5Y neuroblastoma cells via the endoplasmic reticulum? Chem Biol Interact. 2009;181(1):37–44. doi: 10.1016/j.cbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Bharate SB, et al. Thionate versus oxon: comparison of stability, uptake, and cell toxicity of (14CH3O)2-labeled methyl parathion and methyl paraoxon with SH-SY5Y cells. J Agric Food Chem. 2010;58(14):8460–8466. doi: 10.1021/jf100976v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amani N, et al. Chlorpyrifos toxicity in mouse cultured cerebellar granule neurons at different stages of development: additive effect on glutamate-induced excitotoxicity. Cell J. 2016;18(3):464–472. doi: 10.22074/cellj.2016.4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duberley KE, et al. Effect of Coenzyme Q10 supplementation on mitochondrial electron transport chain activity and mitochondrial oxidative stress in Coenzyme Q10 deficient human neuronal cells. Int J Biochem Cell Biol. 2014;50:60–63. doi: 10.1016/j.biocel.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 38.Duberley KE, et al. Human neuronal coenzyme Q10 deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: implications for pathogenesis and treatment. J Inherit Metab Dis. 2013;36(1):63–73. doi: 10.1007/s10545-012-9511-0. [DOI] [PubMed] [Google Scholar]
- 39.Selak MA, et al. Mitochondrial activity in Pompe's disease. Pediatr Neurol. 2000;23(1):54–57. doi: 10.1016/s0887-8994(00)00145-4. [DOI] [PubMed] [Google Scholar]
- 40.Lowry OH, et al. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 41.Duncan AJ, et al. Determination of coenzyme Q10 status in blood mononuclear cells, skeletal muscle, and plasma by HPLC with di-propoxy-coenzyme Q10 as an internal standard. Clin Chem. 2005;51(12):2380–2382. doi: 10.1373/clinchem.2005.054643. [DOI] [PubMed] [Google Scholar]
- 42.Belousova M, et al. Intravenous treatment with coenzyme Q10 improves neurological outcome and reduces infarct volume after transient focal brain ischemia in rats. J Cardiovasc Pharmacol. 2016;67(2):103–109. doi: 10.1097/FJC.0000000000000320. [DOI] [PubMed] [Google Scholar]
- 43.Rahman S, et al. Neonatal presentation of coenzyme Q10 deficiency. J Pediatr. 2001;139(3):456–458. doi: 10.1067/mpd.2001.117575. [DOI] [PubMed] [Google Scholar]
- 44.Guo C, et al. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regener Res. 2013;8(21):2003–2014. doi: 10.3969/j.issn.1673-5374.2013.21.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jo S-H, et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem. 2001;276(19):16168–16176. doi: 10.1074/jbc.M010120200. [DOI] [PubMed] [Google Scholar]
- 46.Davey GP, Peuchen S, Clark JB. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem. 1998;273(21):12753–12757. doi: 10.1074/jbc.273.21.12753. [DOI] [PubMed] [Google Scholar]
- 47.Solaini G, et al. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta (BBA) Bioenerg. 2010;1797(6):1171–1177. doi: 10.1016/j.bbabio.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 48.Poole OV, Hanna MG, Pitceathly RDS. Mitochondrial disorders: disease mechanisms and therapeutic approaches. Discov Med. 2015;20(111):325–331. [PubMed] [Google Scholar]
- 49.Ericsson J, Dallner G. Distribution, biosynthesis, and function of mevalonate pathway lipids. In: Borgese N, Harris JR, editors. Endoplasmic reticulum. Boston, MA: Springer US; 1993. pp. 229–272. [DOI] [PubMed] [Google Scholar]
- 50.Cai Q, et al. Reduced expression of citrate synthase leads to excessive superoxide formation and cell apoptosis. Biochem Biophys Res Commun. 2017;485(2):388–394. doi: 10.1016/j.bbrc.2017.02.067. [DOI] [PubMed] [Google Scholar]
- 51.Bayir A, et al. The effects of ubiquinone (CoQ10) on heart tissue in cardiac toxicity related to organophosphate poisoning. Hum Exp Toxicol. 2013;32(1):45–52. doi: 10.1177/0960327112455070. [DOI] [PubMed] [Google Scholar]
- 52.Gultekin F, Ozturk M, Akdogan M. The effect of organophosphate insecticide chlorpyrifos-ethyl on lipid peroxidation and antioxidant enzymes (in vitro) Arch Toxicol. 2000;74(9):533–538. doi: 10.1007/s002040000167. [DOI] [PubMed] [Google Scholar]
- 53.Wang L, et al. Chlorpyrifos exposure in farmers and urban adults: Metabolic characteristic, exposure estimation, and potential effect of oxidative damage. Environ Res. 2016;149:164–170. doi: 10.1016/j.envres.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 54.Nadjarzadeh A, et al. Effect of Coenzyme Q10 supplementation on antioxidant enzymes activity and oxidative stress of seminal plasma: a double-blind randomised clinical trial. Andrologia. 2014;46(2):177–183. doi: 10.1111/and.12062. [DOI] [PubMed] [Google Scholar]
- 55.Noh YH, et al. Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis. 2013;4(10):e820–e820. doi: 10.1038/cddis.2013.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jing L, et al. Coenzyme Q10 protects astrocytes from ROS-induced damage through inhibition of mitochondria-mediated cell death pathway. Int J Biol Sci. 2015;11(1):59–66. doi: 10.7150/ijbs.10174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bentinger M, Brismar K, Dallner G. The antioxidant role of coenzyme Q. Mitochondrion. 2007;7(Suppl):S41–50. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]