

Abstract
Amyloid A nephropathy is a possible complication of chronic inflammatory disease. Proteinuria and kidney failure are the main features of the disease. Tocilizumab (TCZ), an IL6-R antibody approved for rheumatoid arthritis, is a promising choice for histologically demonstrated nephropathy. We describe a case of kidney amyloid associated with Sweet syndrome treated with TCZ. The patient was affected by Sweet syndrome associated with proteinuria. Kidney biopsy showed amyloid deposits. During the follow-up, cutaneous and renal findings were refractory to many immunosuppressive regimen (cyclophosphamide, leflunomide, interferon and steroid). After few years, the patient developed rapidly progressive nephropathy associated with nephrotic syndrome (proteinuria up to 6 g/die). A second kidney biopsy was performed and it showed worsening of amyloid nephropathy. Thus, TCZ was administrated (8 mg/kg once a month) and it stabilized kidney function and induced partial remission of the nephrotic syndrome in the following 2 years.
Keywords: Amyloidosis A, Sweet syndrome, Interleukin 6, Tocilizumab, Nephrotic syndrome, Kidney biopsy
Introduction
Systemic amyloidosis due to the deposit of serum amyloid A (SAA) is a possible complication of chronic inflammatory diseases. Kidney involvement is very prevalent being present in up to 97% of patients at the diagnosis. The signs of kidney damage are proteinuria, often in the nephrotic range, and reduced glomerular filtration rate (GFR), end-stage renal disease (ESRD) can occur in a large number of patients, either at the diagnosis (11%) or after few years (23%). The relative risk of ESRD is 1.24 for each twofold increase of SAA concentration [1]. Tocilizumab (TCZ) is an IL6-R antibody that has been approved for rheumatoid arthritis (RA) and more recently for large vessel vasculitis. It has the potential to be used in patients with renal amyloidosis too. Emerging literature demonstrates that renal involvement due to several inflammatory diseases can benefit from the use of TCZ. In fact, it has already been administrated not only in patients affected by RA, but also in systemic inflammatory diseases complicated by histologically proven SAA amyloidosis nephropathy. The present paper describes the case of a patient with renal inflammatory amyloidosis successfully treated with TCZ and the literature revision that reports the use of tocilizumab in SAA amyloid nephropathy.
Case report
A 63-year-old man was admitted in March 2016 for progressive worsening of proteinuria and renal function.
The patient’s past medical history showed anaphylaxis to penicillin and tetracyclines and hypertension. Since 10 years, he was affected by multiple skin lesions on his back and face, elevated and inflamed, like wheals associated with axillary lymphadenopathy. For this reason, he received corticosteroid for several years and then interferon (for 5 months), but both therapies had only a sub-optimal control of skin lesions.
Monoclonal gammopathy of undetermined significance (MGUS) was diagnosed 3 years before admission to the hospital. The histology of a lymph node biopsy was compatible with diagnosis of Kikuchi–Fujimoto disease. Serum creatinine value at the time was 1.08 mg/dlL and proteinuria was 0.2 g/24 h, but serum level of SAA was found to be twice as high as the maximum value range. During the follow-up, proteinuria remained 0.2–0.5 g/24 h and serum creatinine was stable. In the weeks preceding hospitalization, an increase in proteinuria (up to 2.4 g/24 h) associated with worsening of skin lesions occurred (Fig. 1).
Fig. 1.
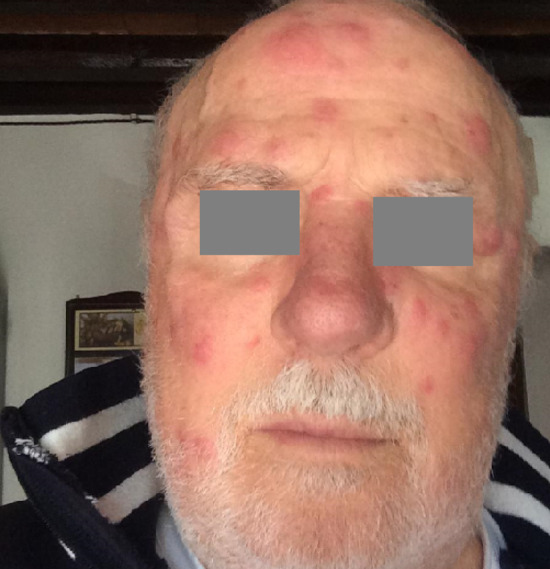
Skin lesions at the time of the diagnosis
At admission to the hospital, the patient referred mild asthenia, but not dyspnea. Physical examination showed skin lesions on the face, back and chest; they were in keeping with the diagnosis of Sweet syndrome. There were no signs of edema, or pulmonary hyperhydration. Arterial blood pressure resulted to be 140/85 mmHg. Biochemistry showed serum creatinine to be 1.1 mg/dl, hemoglobin 11.3 g/dl, complement C3 85 mg/dl, C4 26.1 mg/dl, C-reactive protein 5.9 mg/dl and SAA 40 µg/ml. Urinalysis confirmed overt proteinuria and hyaline cast proteinuria in the 24-h urine collection resulted to be 2–2.6 g.
The patient underwent biopsy of the periumbilical fat that resulted negative for amyloidosis deposits. Kidney biopsy was performed and histology findings reported: 22 glomeruli, six of them with global sclerosis, two of them with focal deposition of amorphous material, Jones silver stain negative, Congo red positive, tubular atrophy up to 5% and vessels free from deposits. The conclusive diagnosis was kidney amyloidosis.
Diagnosis of amyloidosis nephropathy due to systemic inflammation was made so that the patient was initially treated with an oral steroid course. Because of only partial benefit, prednisone was associated with oral cyclophosphamide from May 2017 to September 2017 and then to leflunomide. All these therapeutic regimes resulted in no benefit to proteinuria (1.8–2.5 g/24 h) and kidney function (creatinine serum levels 1.2–1.4 mg/dl).
In December 2017, there was a rapid decline of the kidney function (serum creatinine 2.5 mg/dl) coupled with increased proteinuria (10 g/24 h), worsening of skin lesions and SAA increment up to 530 µg/ml. In December 2017, a second kidney biopsy was performed, which showed the following findings: 14 glomeruli, 3 of them sclerotic, 7 of them with diffuse hyaline deposits, Congo red-positive staining with apple green birefringence, mainly localized on the vascular pole; tubular atrophy up to 10%. Hyaline deposits appear in some tubular sections and in small and medium vessels. Electron microscopy examination demonstrated SAA deposits (as detected by immunogold). Conclusion: worsening features of kidney amyloidosis (Figs. 2 and 3).
Fig. 2.

a Periodic acid–Schiff staining: deposit of SAA localized at the vascular pole (blue arrow) and in the glomerular arterioles (yellow arrow). b Congo red staining: deposits in a vessel (yellow arrow) and at the vascular pole of the glomerulus (blue arrow). c Masson trichrome staining: deposit of SAA at the vascular pole of the glomerulus (yellow arrow) and concomitant expansion of mesangial matrix. d Deposit of amyloid at the vascular pole negative for Jones silver staining (yellow arrow)
Fig. 3.
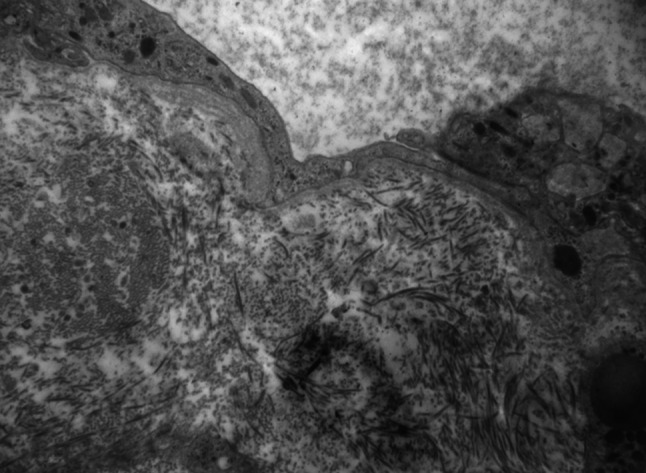
Electron microscopy shows amorphous aggregates in the urinary space of the glomerulus and complete effacement of podocytes
In January 2018, serum creatinine rose up to 3.5 mg/dl, consistent with a rapidly progressive nephropathy. Hence, leflunomide was stopped and high dose of intravenous steroid (methylprednisolone 250 mg days 1, 2 and 3) was administered, followed by oral prednisolone 25 mg daily. In the following weeks, only partial benefits on kidney function and urine protein loss (serum creatinine 3 mg/dl and proteinuria 6 g/24 h) were observed.
Hence, in February 2018, TCZ was started with the aim of controlling the inflammatory status and the steroid sparing. It was administrated at the dose of 8 mg/Kg once a month. Since that time, a slow but consistent reduction of proteinuria was observed leading to incomplete remission of the nephrotic syndrome (proteinuria 1.5 g/24 h, at January 2020). Progressive loss of GFR was halted and residual kidney function remained stable over time (Fig. 4). During the follow-up from March 2018 to January 2020, SAA levels were in the normal range and Sweet syndrome symptoms were well controlled with the same dosage of TCZ for all of time and in absence of side effects. Only low doses of oral steroid were required for short time for mild skin exacerbations.
Fig. 4.

Proteinuria trend and eGFR with time, before and after TCZ. O represents pulse steroid therapy (Solu-Medrol 500 mg three times)
Discussion
Amyloidosis AA is a systemic disease characterized by extracellular deposition of serum amyloid A, due to chronic inflammatory disorders. SAA is a small acute-phase protein (104 amino acids) produced by the liver. It was the first non-immunoglobulin protein identified in amyloid depositions; this is the reason why it is called serum amyloid A. The circulating level of SAA can increase over 1000-fold the normal values within 24 h after an acute event and it falls down rapidly after the resolution of the acute-phase trigger. IL-1 and IL-6 are the cytokines that induce SAA synthesis. SAA mediates its effect through the activation of NLRP3 inflammasome. This is a multimeric protein complex that initiates an inflammatory type of cell death and triggers the release of proinflammatory cytokines. There are two ways of activation of NLRP3 inflammasome. The first is directly mediated by SAA that activates Toll-like receptors (TLRs) inducing intracellular pathway of nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB). The second is an indirect activation by phagocytosis of fibrils of SAA that induce NIMA-related kinase 7 (NEK7). These mechanisms of NLRP3 inflammasome activation are both mediated by caspase-1, that induces inflammatory cascade and production of cytokines such as IL-1β, IL-18, IL-6, TNFα and monocyte chemoattractant protein-1 (MPC-1). Cytokines and TNF induce themselves the production of SAA from the liver, worsening the inflammatory condition [2, 3].
The biological role of SAA is connected to the recovery and recycle of cholesterol from damaged tissue for cell repair, redirecting them to macrophages. SAA is bound to high-density lipoproteins (HDL) in the blood and replaces APOA-I. Moreover, high value of SAA induces endothelial dysfunction, and both these mechanisms promote lipid metabolism abnormalities and atherogenesis [4].
The up-regulated inflammatory pathway and the lipid abnormalities also induce liver injury and promote non-alcoholic steato-hepatitis (NASH). In addition, this pathway is mediated by NLRP3; the inflammasome activates TLRs by damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). TLRs activate the cell to release of proinflammatory cytokines. Furthermore, there is the oligomerization of NLRP3 with procaspase-1 and apoptosis-associated speck-like protein to CARD (ASC), and the complex promotes production and secretion of proinflammatory cytokines (IL-1β and IL-18) [5].
A persistent or recurrent increase in the circulating SAA is the hallmark for inflammatory diseases, but it also facilitates the aggregation and extracellular deposition of SAA in several tissues. SAA is poorly soluble in aqueous solutions. Amino-terminal fragment is the site that allows formation of organized insoluble fibrils. The C-terminal regions are removed by proteolysis that causes fibril formation, mostly composed by 76 amino acids, but also SAA fragments from 45 to 94 amino acids can be found in the tissues. These deposits shared a characteristic birefringence when stained with planar dyes such as Congo red. The kidney is a major target for systemic AA, and the deposits involve the whole structures: glomeruli (especially in the mesangial area), vessels (arteries, peritubular vessels and veins), interstitial and peritubular space. The possible different sites that are affected explain the various presentation of kidney involvement. Proteinuria, ranging from up to nephrotic values, reflects a glomerular involvement, whereas worsening loss of function, acute/rapidly progressive or/and chronic kidney injury may also reflect a vascular and tubular-interstitial damage. Often, the simultaneous presentation of these findings is suggestive for the presence of fibrils in the whole kidney structures. Delayed diagnosis has a negative impact on patients’ prognosis. If the inflammatory cascade and the deposition process are not halted, organ failure is certain. In fact, control of the inflammation and, therefore, the production of SAA in patients with AA amyloidosis may halt amyloid deposition and enable the regression and/or stabilization of kidney function and proteinuria [6–8].
However, AA remains a systemic disease. Bowel, gastric, liver, thyroid, testicles, heart and nervous system can be involved and the diagnosis requires histological demonstration of amyloid deposits in the tissue by immunohistochemistry or by proteomics-based techniques.
IL-6 is a cytokine involved in the pro-inflammatory pathway. It is produced and secreted by T cells and macrophages in the inflammatory site and then it moves to the liver. Its secretion induces inflammatory cascade and fever mediated by an extensive range of acute phase proteins, such as C-reactive protein (CRP), SAA, fibrinogen, hepcidin and others. IL-6 is also produced by myocyte, osteoblasts and other human tissues, and modulates cellular expression.
IL-6 overproduction is involved in autoimmune disease, both T and B cell pathways; it can promote T cell differentiation in cytotoxic T cell and Th17 by modifying the balance of immunological tolerance and developing T cell-mediated autoimmunity and chronic inflammation.
IL-6 stimulates B cells and immunoglobulin production and promotes the secretion of several pro-osteoclast mediators, such as RANKL, IL1 and prostaglandin. This bone environment changes cause reduced bone mass and osteoporosis [9–11].
IL-6 also induces production of VEGF that in turn increases angiogenesis and vascular permeability. This leads to the typical inflammatory features of rheumatoid arthritis or also to the edema of remitting seronegative synovitis. In the synovia, IL-6 elicits fibroblast-like synoviocytes to produce tissue damage.
Sweet syndrome is associated with IL6 increase. Since 1993, some authors had shown that in patients affected by Sweet syndrome, IL6 was steadily high in spite of TNF, G-CSF, IL1-beta and gamma interferon [12–14]. Therefore, high level of IL6 at the time of the diagnosis and its reduction after therapy and clinical remission demonstrate the pathological link between this disease and this chemokine [15].
Not only acute inflammatory disease but also chronic illnesses, such as diabetes, obesity, atherosclerosis and cancer can stimulate IL-6 production, but to a lower extent l than in the acute phase. High level of IL-6 is linked to higher risk of morbidity (such as atherosclerosis, stroke and ictus) and mortality [16–18].
Hagihara et al. found that an antibody with affinity for IL-6 receptor blocked the acute phase reaction, much stronger than IL-1 receptor antagonist or anti-tumor necrosis factor (TNF) antibody. This antibody was called tocilizumab (TCZ).
TCZ is a humanized monoclonal antibody that competitively inhibits the binding of IL-6 to its receptor. After site saturation occurs, TCZ blocks the production of endothelial chemokines and consequent recruitment of immune cells (B cell stimulation and T cell differentiation). The use of TCZ was approved for the treatment of rheumatoid arthritis in 2009 [19]. Renal amyloidosis due to SAA deposition may be a potential novel indication of TCZ. In fact, several diseases with inflammatory kidney involvement can benefit from the use of this humanized antibody (Fig. 4). It had already been administrated in patients with demonstrated nephropathy by inflammatory amyloidosis due to Behcet’s disease [20], latent tuberculosis [21], Castleman disease [22], rheumatoid arthritis [23] and others [24–27]. Blocking the inflammatory cascade, TCZ allows the removal of SAA depositions and improves or stabilizes renal function and proteinuria. In patients with Sweet syndrome, the use of TCZ is mostly supported, not only by the aim of inhibition of the inflammatory cascade, but also by the evidence of the pathophysiology correlation between IL6 and the disease [12–15].
All of reports describing autoimmune disease with inflammatory kidney involvement share the same findings; amyloidosis AA histologically proved, clinical features of nephrotic syndrome, worsening of renal function and scarce control of the inflammatory disease with the standard therapy. Uda H et al. [28] used TCZ and demonstrated the superiority compared to the standard oral therapy in a comparative prospective study; in fact, IL-6 receptor-blocking agent postponed the start of hemodialysis by the suppression of the inflammatory response more than the other drugs. The main limit of this study was the absence of histological definition of kidney involvement. However, TCZ treatment may be a chance also for other types of glomerulopathies due to inflammatory systemic disease. Different authors reported the use of the IL-6 receptor blocker for IgA nephropathy and for necrotizing crescentic glomerulonephritis in systemic Castleman disease [29, 30]. These papers suggested that TZC could work not only on inflammatory cascade but could also modulate immune system and resolve kidney evolvement in autoimmune diseases.
Our findings confirm that TCZ is able to reduce proteinuria and to halt progressive loss of kidney function in nephropathy due to amyloidosis AA. The novelty of this case report consists in the description of a SAA-associated nephropathy associated with Sweet syndrome, which to our knowledge is the first report. The beneficial effect of TCZ induced an incomplete remission of nephrotic syndrome and stabilization of residual kidney function. The current literature on TCZ-treated SAA nephropathy is reported in Table 1.
Table 1.
The literature of use of TCZ for kidney amyloid involvement
| Author | Clinical findings | Kidney histological findings | Disease | Dosage of TCZ | Adverse reaction | Outcome |
|---|---|---|---|---|---|---|
| Magro-Checa C. 2011 [21] | Nephrotic syndrome | Diffuse deposition of amyloid in glomeruli, vessels and interstitium | Tuberculosis | 8 mg/kg once a month | None | Complete remission of nephrotic syndrome |
| Redondo-Pachón M.D. 2013 [20] | Nephrotic syndrome | Diffuse deposition of amyloid in glomeruli, vessels, tubular basement membrane and interstitium | Behcet's disease | 8 mg/kg once a month | None | Complete remission of nephrotic syndrome |
| Hasegawa S. 2013 [27] | Nephrotic syndrome and worsening of renal function | Deposition of SAA in glomeruli | Rheumatoid arthritis | 8 mg/kg once a month | Not reported | Complete remission of nephrotic syndrome and improvement of renal function |
| Iijima T. 2013 [24] | Nephrotic syndrome and worsening of renal function | Diffuse deposition of SAA in glomeruli, vessels and tubules | Rheumatoid vasculitis/necrotizing crescentic glomerulonephritis | 8 mg/kg once a month | Not reported | Partial remission of nephrotic syndrome and improvement of renal function |
| Yamada S 2014 [26] | Nephrotic syndrome and worsening of renal function | Deposition of SAA in glomeruli associated with fibrocellular crescent. Mesangial expansion and fibrocellular crescents | Rheumatoid arthritis | 8 mg/kg every 4 weeks | Not reported | Complete remission of nephrotic syndrome and improvement of renal function |
| Serelis J. 2015 [25] | Nephrotic syndrome | Diffuse deposition of amyloid in glomeruli e vessels | Familiar Mediterranean fever | 8 mg/kg once a month | Not reported | Partial remission of nephrotic syndrome |
| Yamagata A. 2017 [23] | Nephrotic syndrome | Deposition of SAA in glomeruli | Rheumatoid arthritis | Unknown | Not reported | Complete remission of nephrotic syndrome |
| Yamada Y. 2018 [22] | Decline of renal function and proteinuria | Diffuse deposition of amyloid in glomeruli, vessels and interstitium | Castleman disease | 8 mg/kg once a month | Not reported | Demonstration of clearance of amyloid in the kidney |
In conclusion, tocilizumab has been usefully used for the treatment of a case of histologically proven amyloidosis A nephropathy. TCZ therapy was well tolerated and safe with beneficial effects on the residual kidney function and proteinuria. On the basis of our case and of the existing literature, it is conceivable that TCZ may a possible option for the treatment of nephropathies linked to inflammatory systemic diseases.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lackmann HJ, Goodman HJ, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Eng J Med. 2007;356(23):2361–2371. doi: 10.1056/NEJMoa070265. [DOI] [PubMed] [Google Scholar]
- 2.Sack GH., Jr Serum amyloid—a review. Mol Med. 2018;24:46. doi: 10.1186/s10020-018-0047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scapioni R, et al. Renal involvement in autoinflammatory diseases and inflammasome-mediated chronic kidney damage. Clin and Exp Rheum. 2018;110(1):54–60. [PubMed] [Google Scholar]
- 4.Benditt EP, et al. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc Natl Acad Sci USA. 1977;74:4025–4028. doi: 10.1073/pnas.74.9.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Del Campo JA, et al. Role of inflammatory response in liver diseases: therapeutic strategies. World J Hepatol. 2018;10(1):1–7. doi: 10.4254/wjh.v10.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillmore JD, et al. Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet. 2001;358(9275):24–29. doi: 10.1016/S0140-6736(00)05252-1. [DOI] [PubMed] [Google Scholar]
- 7.Kuroda T, et al. Significant association between renal function and area of amyloid deposition in kidney biopsy specimens in reactive amyloidosis associated with rheumatoid arthritis. Amyloid. 2012;32:3155–3162. doi: 10.1007/s00296-011-2148-8. [DOI] [PubMed] [Google Scholar]
- 8.Scarpioni R, et al. Renal involvement in autoinflammatory diseases and inflammasome-mediated chronic kidney damage. Clin Exp Rheumatol. 2018;36(Suppl. 110):S54–S60. [PubMed] [Google Scholar]
- 9.Di Munno O, et al. The effect of biologic agents on bone homeostasis in chronic inflammatory rheumatic diseases. Clin Exp Rheumatol. 2019;37(3):502–507. [PubMed] [Google Scholar]
- 10.Toshio T, et al. IL-6 in Inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6(10):a016295. doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagihara K, et al. IL-6 plays a critical role in the synergistic induction of human serum amyloid A (SAA) gene when stimulated with proinflammatory cytokines as analyzed with an SAA isoform real-time quantitative RT-PCR assay system. Biochem Biophys Res Commun. 2004;314:363–369. doi: 10.1016/j.bbrc.2003.12.096. [DOI] [PubMed] [Google Scholar]
- 12.Reuss-Borst MA, et al. Sweet's syndrome associated with myelodysplasia: possible role of cytokines in the pathogenesis of the disease. Br J Haematol. 1993;84(2):356–358. doi: 10.1111/j.1365-141.1993.tb03083.x. [DOI] [PubMed] [Google Scholar]
- 13.Loraas A, et al. Cytokine response pattern in Sweets syndrome associated with myelodysplasia. Br J Haematol. 1994;87(3):669. doi: 10.1111/j.1365-2141.1994.tb08340.x. [DOI] [PubMed] [Google Scholar]
- 14.Hattori H, et al. Sweets syndrome associated with recurrent fever in a patient with trisomy 8 myelodysplastic syndrome. Int J Hematol. 2003;77(4):383–386. doi: 10.1007/BF02982648. [DOI] [PubMed] [Google Scholar]
- 15.Takano Y, et al. Serum cytokine profile in pediatric Sweets Syndrome: a case report. J Med Case Rep. 2017;11(1):178. doi: 10.1186/s13256-017-1317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarla NS, et al. Effects of olive oil on TNF-α and IL-6 in humans: implication in obesity and frailty. Endocr Metab Immune Disord Drug Targets. 2018;18(1):63–74. doi: 10.2174/1871530317666171120150329. [DOI] [PubMed] [Google Scholar]
- 17.Bastard JP, et al. Evidence for a link between adipose tissue interleukin-6 content and serum C-reactive protein concentrations in obese subjects. Circulation. 1999;99(16):2219–2222. doi: 10.1161/01.CIR.99.16.2219.c. [DOI] [PubMed] [Google Scholar]
- 18.Qu D, et al. IL-6 in diabetes and cardiovascular complications. Br J Pharmacol. 2014;171(15):3595–3603. doi: 10.1111/bph.12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biggioggero M, et al. Tocilizumab in the treatment of rheumatoid arthritis: an evidence-based review and patient selection. Drug Des Devel Ther. 2019;13:57–70. doi: 10.2147/DDDT.S150580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redondo-Pachón MD, et al. Tocilizumab treatment for nephrotic syndrome due to amyloidosis in Behcet’s disease. Ren Fail. 2013;35(4):547–550. doi: 10.3109/0886022X.2013.773913. [DOI] [PubMed] [Google Scholar]
- 21.Magro-Checa C, et al. Successful use of tocilizumab in a patient with nephrotic syndrome due to a rapidly progressing AA amyloidosis secondary to latent tuberculosis. Amyloid. 2011;18(4):235–239. doi: 10.3109/13506129.2011.613962. [DOI] [PubMed] [Google Scholar]
- 22.Yamada Y, et al. Tocilizumab histologically improved AA renal amyloidosis in a patient with multicentric Castleman disease: a case report . Clin Nephrol 2018. PMID 29701172 [DOI] [PubMed]
- 23.Yamagata A, et al. Rapid clinical improvement of amyloid A amyloidosis following treatment with tocilizumab despite persisting amyloid deposition: a case report. BMC Nephrol. 2017;18:377. doi: 10.1186/s12882-017-0799-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iijima T, et al. Tocilizumab improves systemic rheumatoid vasculitis with necrotizing crescentic glomerulonephritis. Mod Rheumatol 2013 [DOI] [PubMed]
- 25.Serelis J, et al. Remission of nephrotic syndrome due to AA-amyloidosis, complicating familiar Mediterranean fever, with tocilizumab. Clin Exp Rheumatol 2015. [PubMed]
- 26.Yamada S, et al. Tocilizumab-induced remission of nephrotic syndrome accompanied by secondary amyloidosis and glomerulonephritis in a patient with rheumatoid arthritis. CEN Case Rep 2014 [DOI] [PMC free article] [PubMed]
- 27.Hasegawa S, et al. [Case report; a case of rheumatoid arthritis with renal amyloidosis and nephrotic syndrome effectively treated with tocilizumab]. Nihon Naika Gakkai Zasshi 2011 [DOI] [PubMed]
- 28.Uda H, et al. Tocilizumab postpones the start of hemodialysis compared to conventional or al treatment in amyloid A amyloidosis patients with advanced renal insufficiency by suppressing serum SAA levels. Amyloid. 2017;24(1):62–63. doi: 10.1080/13506129.2017.1301420. [DOI] [PubMed] [Google Scholar]
- 29.Iijima T, et al. Tocilizumab for AA Amyloidosis after treatment of multicentric Castleman disease with steroids, chemotherapy and rituximab for Over 20 Years. Intern Med. 2015;54:3215–3219. doi: 10.2169/internalmedicine.54.4183. [DOI] [PubMed] [Google Scholar]
- 30.Matsunami M, et al. The efficacy and safety of anti-interleukin-6 receptor monoclonal blockade in a renal transplant patient with Castleman disease: early post-transplant outcome. BMC Nephrol. 2018;19(1):263. doi: 10.1186/s12882-018-1065-4. [DOI] [PMC free article] [PubMed] [Google Scholar]