

Summary
Anti-cancer drugs kill only a fraction of cells within a population at any given time. Here, we describe a protocol to quantify drug-induced fractional killing over time using high-throughput imaging. This protocol can be used to compare the effect of hundreds of conditions in parallel. We show how this protocol can be used to examine fractional killing in response to inhibitors of the mitogen-activated protein kinase pathway.
For complete details on the use and execution of this protocol, please refer to Inde et al. (2020).
Subject areas: Cell biology, Cell-based assays, Cancer, High-throughput screening, Microscopy
Graphical Abstract
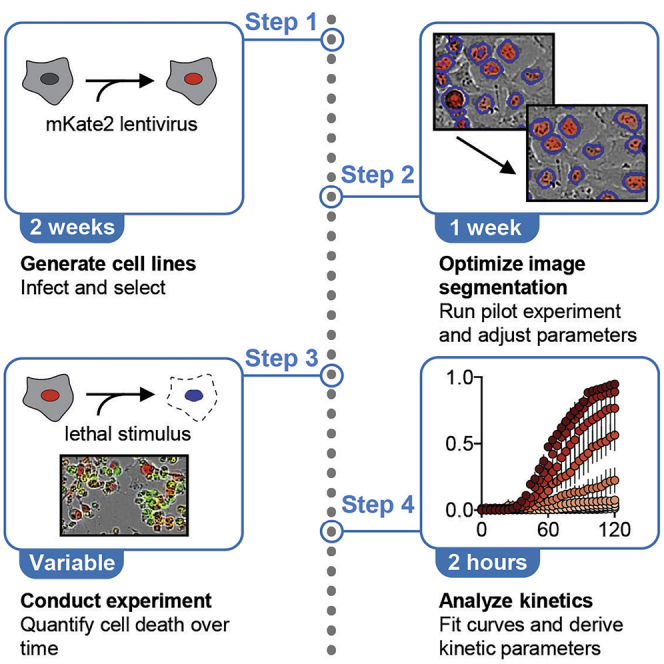
Highlights
-
•
Protocol for high-throughput analysis of drug-induced fractional killing in vitro
-
•
Detailed methods for comparing fractional killing between conditions
-
•
Description of MEK1/2 inhibitor-induced fractional killing variability
Anti-cancer drugs kill only a fraction of cells within a population at any given time. Here, we describe a protocol to quantify drug-induced fractional killing over time using high-throughput imaging. This protocol can be used to compare the effect of hundreds of conditions in parallel. We show how this protocol can be used to examine fractional killing in response to inhibitors of the mitogen activation protein kinase pathway.
Before you begin
Generate mKate2-expressing cell lines
Timing: 2 weeks
To assess drug-induced fractional killing, it is necessary to count both live and dead cells within the population (Inde et al., 2020). Live cells can be conveniently detected via expression of a nuclear-localized fluorescent protein, mKate2. As described below, this enables live cells to be counted using a microscope; these counts are necessary to compute fractional killing in the main protocol.
Note: The methods for generating cell lines and analyzing data described in this protocol should be generalizable to any imaging platform. For imaging live and dead cells, this protocol utilizes an Incucyte microscope installed in a standard tissue culture incubator maintained at 37°C with 5% CO2 (Figure 1A). Specific imaging parameters may need to be adjusted if using a different imaging platform.
Figure 1.

Generation of mKate2+ cell lines
(A) An Incucyte installed in a standard tissue culture incubator for the STACK protocol.
(B) HT-1080 cells seeded in a tissue culture plate after adhering to the plate surface. Cells are imaged using the Incucyte 10× objective in the phase-contrast channel. Cells were infected with NLR lentivirus at a MOI of ~3 (right) or vehicle control (left). Cells were then treated with 1 μg/mL puromycin for 24 h (bottom). Scale bar, 100 μm.
Note: The protocol is optimized for adherent cell lines, which remain in a single focal plane in culture. It is conceivable that this protocol could be applied to non-adherent cell lines, but additional optimization would be required, including for the centrifugation of culture plates to collect cells at the bottoms of wells prior to imaging.
CRITICAL: Cell should be thawed and cultured in appropriate culture medium, as recommended by the suppler. Choice of culture medium or additives should not affect the execution of the protocol. As with any cell-based assay, using early passage cells is preferred. However, the protocol will work with cells at any passage number. The protocol is compatible with cell culture requiring coated culture vessels (e.g., Matrigel); confirm successful Incucyte imaging of cells in coated plates before beginning.
-
1.Determine antibiotic selection dose for cell line(s) of interest.
-
a.Seed cells in 6 wells of a 12-well culture plate. Seeding density should result in approximately 75% confluence the following day when antibiotic is added.
-
b.Allow cells to adhere to plate 18–24 h.
-
c.Apply antibiotic for selection in a dose response.
-
i.Dilute antibiotic in culture media to a dose of 10 μg/mL. Dilute 1:1 in culture media four times to yield a two-fold dose response ranging from 625 ng/mL to 10 μg/mL.
-
ii.Aspirate media from cultured cells and replace with antibiotic selection media. Replace normal media in remaining well for an untreated control.
-
iii.Monitor cells daily with a standard light microscope or by imaging using an Incucyte. For most cell lines, puromycin selection will be complete within 48 h.
-
iv.Determine the minimum antibiotic dose at which selection is complete in uninfected cells (Figure 1B). If death is not apparent from morphological features of the selected cells, SYTOX Green may be used, as described below, to confirm cell death.
-
v.Note the minimum dose required to kill >99% of uninfected cells and use this dose for selection of NucLight Red-infected cell lines in step 3.
-
i.
-
a.
Note: Lentiviral NucLight Red vectors are available from Sartorius carrying either puromycin- or bleomycin-selectable markers. These vectors express mKate2 fluorescent protein fused to a nuclear localization sequence that targets this protein into the nucleus. Protein import into the nucleus is an ATP-dependent process, and therefore only active in live cells. NucLight Green vectors are also available expressing GFP, if desired. Puromycin is more rapidly lethal to mammalian cells than bleomycin. Selection for bleomycin resistance using bleomycin-family antibiotic zeocin often requires passaging cells several times in antibiotic-containing medium. Thus, if compatible with other experimental conditions, selection with puromycin is generally preferable. Alternatively, or in addition, infected cells may be selected using the optional fluorescence activated cell sorting (FACS) step below. FACS sorting may also be utilized to enrich for high-expressing mKate2 positive (i.e.’+’) cells.
-
2.Infect cell line(s) with NucLight Red lentivirus.
-
a.Seed cell line(s) in 12 well culture plate at approximately 25% confluence. Seed two wells per cell line, one for infection and one as a mock-infected control to confirm later antibiotic selection.
-
b.Allow cells to adhere to plate 18–24 h.
-
c.Consult packaging or supplier website to determine viral titer of NucLight Red lentivirus, expressed in transduction units (TU). Calculate volume of virus to be applied to cells to yield a multiplicity of infection (m.o.i.) of approximately 3.
-
i.Multiply the number of cells per well by 3 to obtain the necessary TU. Depending on the proliferation rate of the cell line in question, the cell number will be greater than the number of cells seeded; multiply the number of cells by 1.5 for an approximate cell number at time of infection.
-
ii.Based on the titer of the lentivirus, calculate the volume of virus required for each well (desired TU divided by TU/mL)
-
i.
-
d.On the day of infection, prepare polybrene and lentivirus solutions in culture media.
-
i.Polybrene stocks can be made at 1,000× in water at a concentration of 8 mg/mL (store at −20°C for up to 1 year). Dilute to 2× in a volume of 500 μL media per infection or control well.
-
ii.Dilute calculated volume of virus in 500 μL media per infection well.
-
i.
-
e.Aspirate media from culture plate.
-
f.Add 500 μL media containing diluted 2× polybrene per well.
-
g.To infection well(s), add 500 μL diluted virus media. To uninfected control well(s), add 500 μL culture media without virus.
-
h.Monitor expression of mKate2 over the following days by imaging using an Incucyte or another imaging platform. mKate2 expression should be apparent after 24–48 h.
-
i.The day following infection, remove and properly dispose of virus- and polybrene-containing media from all wells. Replace with normal culture media.
-
a.
-
3.Select infected cells.
-
a.Two days after infection, prepare 1 mL antibiotic media per well at the concentration determined in step 1.
-
b.Remove and properly dispose of media from wells of infection plate.
-
c.Add antibiotic selection media to all wells.Optional: Infected cells can be expanded before selection, and a portion of infected, unselected cells can be separately maintained in culture until successful selection is confirmed.
-
d.Monitor selection and confluence of infected cells by imaging using the Incucyte.
-
e.If cells reach 100% confluence during selection, passage cells to a 6 well culture plate while maintaining selection. Trypsinizing and reseeding cells in antibiotic-containing media will increase the sensitivity of cells to antibiotic; if necessary, reseed cells in antibiotic-free media and resume selection once cells have adhered to new plate.
-
f.Maintain identical passaging and selection procedures for infected cells and uninfected controls. Selection is complete when no viable cells are present in uninfected control wells (Figure 1B, bottom left).
-
g.Expand selected mKate2+ cells in normal culture media. If desired, cryopreserve vials of mKate2+ cells for later use.
 Pause point: Frozen cells can be maintained in this state indefinitely until investigators are prepared to undertake the next steps in this protocol.Alternatives: If preferred, or if antibiotic selection is not possible, select NucLight Red-infected cells by FACS. mKate2 fluorescence is detectable in the PE-Texas Red channel found in most FACS instruments; its excitation/emission maxima are 588/633. Before sorting, infected cells should be expanded so that at least 10,000 mKate2+ cells can be isolated for further expansion. An uninfected control population can be used to gate all mKate2+ cells, or the desired population can be selected based simply on the distribution of mKate2 signal in the infected cells.Note: Either of these approaches, bulk sorting of mKate2+ cells or antibiotic selection, will yield a polyclonal mKate2+ population for downstream applications. In other words, the precise number of transgene insertions and the location of these insertions in the genome will vary among the infected cells. If polyclonality is not desirable for the downstream applications of interest, mKate2-expressing cell lines may be produced by single-cell sorting of infected cells from most cell lines (not all cell lines are suitable for single-cell cloning). After sorting and expansion of clonal cell lines from the infected population, these clonal cell lines may be examined for mKate2 expression and other phenotypes of interest, then further utilized as described below.Note: Investigators may wish to validate that mKate2 expression does not affect cell death phenotypes. In our experience, mKate2 expression does not alter sensitivity to lethal stimuli. However, this can be confirmed by applying a given lethal stimulus to both the parental and mKate2-expressing cell lines in parallel and looking for similar responses using an independent readout of cell death (e.g., Annexin-V positivity).
Pause point: Frozen cells can be maintained in this state indefinitely until investigators are prepared to undertake the next steps in this protocol.Alternatives: If preferred, or if antibiotic selection is not possible, select NucLight Red-infected cells by FACS. mKate2 fluorescence is detectable in the PE-Texas Red channel found in most FACS instruments; its excitation/emission maxima are 588/633. Before sorting, infected cells should be expanded so that at least 10,000 mKate2+ cells can be isolated for further expansion. An uninfected control population can be used to gate all mKate2+ cells, or the desired population can be selected based simply on the distribution of mKate2 signal in the infected cells.Note: Either of these approaches, bulk sorting of mKate2+ cells or antibiotic selection, will yield a polyclonal mKate2+ population for downstream applications. In other words, the precise number of transgene insertions and the location of these insertions in the genome will vary among the infected cells. If polyclonality is not desirable for the downstream applications of interest, mKate2-expressing cell lines may be produced by single-cell sorting of infected cells from most cell lines (not all cell lines are suitable for single-cell cloning). After sorting and expansion of clonal cell lines from the infected population, these clonal cell lines may be examined for mKate2 expression and other phenotypes of interest, then further utilized as described below.Note: Investigators may wish to validate that mKate2 expression does not affect cell death phenotypes. In our experience, mKate2 expression does not alter sensitivity to lethal stimuli. However, this can be confirmed by applying a given lethal stimulus to both the parental and mKate2-expressing cell lines in parallel and looking for similar responses using an independent readout of cell death (e.g., Annexin-V positivity).
-
a.
Optimize the detection of live and dead cells
-
4.Determine appropriate seeding density for plate type and selected experimental design.
-
a.For a 96 well plate experiment, seed a range of cell densities in a test plate.
-
i.Trypsinize mKate2+ cells, then quench the trypsinization reaction with medium and count the cells suspended in medium. Resuspend in culture medium at 50,000 cells/mL. Dilute 1:1 in media 3× to yield cell concentrations of 25,000, 12,500, and 6,250 cells/mL. Prepare sufficient cell suspension to test a range of doses for lethal compound(s) of interest at each seeding density.
-
ii.Dispense cells into tissue culture plate in a volume of 200 μL/well. To prevent uneven distribution of cells when they adhere to the plate, centrifuge plate briefly (place in a tabletop swing bucket centrifuge, start spin, and stop when speed reaches approximately 55 × g) before placing in tissue culture incubator 18–24 h.
-
i.
-
b.Replace seeding media with treatment media.
-
i.Add SYTOX Green dye to appropriate culture media at a concentration of 20 nM. SYTOX Green is a live cell impermeable nucleic acid dye first characterized in (Langsrud and Sundheim, 1996). Uptake of SYTOX Green following plasma membrane permeabilization marks dead cells.
-
ii.Prepare treatment media by diluting compounds of interest in SYTOX Green-containing media. To test multiple doses, perform serial dilutions of compounds in SYTOX Green-containing media. Include vehicle control treatment(s) as appropriate.
-
iii.Invert seeded plate and flick sharply into a basin to remove seeding media. Adherent cells should not detach and will remain adhered to the plate. Working quickly to prevent wells from drying completely, replace with 200 μL of treatment media.
-
i.
-
c.Begin Incucyte imaging of plate.
-
i.Place plate in open slot of Incucyte. Ensure that plate is seated securely in slot and level along all plate edges.
-
ii.In Incucyte Zoom software, select appropriate culture plate definition and scanning pattern. Add plate to repeating scan schedule to scan once every 4 h.
-
i.
-
d.Monitor death and proliferation of cells over time (Figure 2A). Stop experiment when untreated wells have reached full confluence and started to die or when death induced by compound(s) of interest is complete.
-
e.If necessary, repeat optimization experiment, adjusting plate format, seeding density, compound treatments, or scan timing to adequately capture phenomena of interest.
-
a.
Note: Many lethal compounds can be used as robust controls for cell death induction and to optimize the Incucyte analyzer. A selection of suitable lethal compounds is listed in the Key resources table: bortezomib is a proteasome inhibitor that induces apoptotic cell death; staurosporine is a natural product that induces apoptosis through inhibition of multiple kinases; erastin2 is an inhibitor of the cystine/glutamate antiporter system xc- and induces ferroptotic (i.e., non-apoptotic) cell death.
-
5.Optimize analyzer for mKate2+ cell line
-
a.Select images from seeding optimization experiment(s) and add to a new image collection. Include sufficient variety of images to ensure robust object recognition across conditions: varying confluence, varying cell viability, and both drug-treated and vehicle control-treated cells. Note that drug treatment may alter cell morphology (and therefore object recognition) even if viability is unaffected.
-
b.Generate new analyzer based on image collection. Include quantification of mKate2+, SYTOX Green+, and overlap+ objects. (Overlap+ objects are identified as objects in both the red and green channels by the analyzer. Typically, these are dead cells that have not yet fully lost mKate2 fluorescence.) Recommended settings include adaptive color processing and setting min and max area filters to determine counting of red and green objects. General guidelines for adjusting analyzer parameters are discussed in the following note.
-
c.Set edge sensitivity such that red nuclei and green puncta are counted only once. Setting sensitivity too high may result in objects being segmented and counted multiple times (Figure 2B).
-
d.Examine analyzer masks and determine quality of object recognition. Adjust parameters as necessary to reduce false negative and false positive objects (Figure 2C).
-
a.
Note: The Incucyte image analyzer software has two main components for each color channel: parameters and cleanup. We use the no background subtraction, adaptive parameter, with a threshold adjustment of 10 green calibrated units (GCU) in the green channel and 1 red calibrated unit (RCU) in the red channel. This means that while the background brightness in each image is identified, it is not explicitly subtracted from the intensities of the identified objects. Instead, an object is called as long as it is 10 GCUs above the background threshold in the green channel or 1 RCU above the threshold in the red channel. The green channel GCU threshold is set higher because we have observed a green “haze” in many images (as in Figure 2A, left). True SYTOX green positive objects are very bright in comparison, and so this strict threshold cutoff correctly identifies green puncta while excluding the haze.
Note: We also use the edge split setting, which is recommended for separating closely spaced objects. The edge sensitivity is set to −7 in the green channel and −3 in the red channel. A lower edge sensitivity means that the analyzer is less likely to segment pixels into multiple objects (less likely to “call” multiple red or green objects when there is only one true object) whereas a higher edge sensitivity means that the analyzer is more likely to segment objects. For example, in Figure 2B an edge sensitivity of −3 allows for proper segmentation of red nuclei, whereas an edge sensitivity of +10 over-segments the nuclei and counts multiple red objects erroneously. Our analyzer has a lower edge sensitivity in the green channel than the red channel (−7 vs −3) because it is often necessary to separate closely packed red objects. For example, a very confluent well may have cells whose nuclei are pushed closely together. The red nuclei in this scenario should be properly counted as independent objects.
Note: For cleanup settings, the user can set min and max thresholds for area, eccentricity, mean intensity, and integrated intensity. In the green channel, we set an area filter for a minimum of 40 μm2 and a maximum of 750 μm2. In the red channel, we restrict the area from 40 μm2 to 8,100 μm2. We have found that the stricter area cutoff in the green channel also prevents erroneous counting of the green “haze” mentioned above. The eccentricity setting will define a range of roundness for objects. We do not recommend setting eccentricity threshold cutoffs. Mean and integrated intensity settings will allow for cutoffs based on the intensity levels of called objects. It can be valuable to set minimum thresholds to prevent the analyzer calling very faint objects. Particularly, this can be useful in the red channel, to exclude dead cells that still retain very faint mKate2+ signal. We set a minimum mean intensity of 5.00 A.U. in the red channel, where objects less intense are not counted by the analyzer.
Note: Cells may behave differently if they are from different tissues of origin, or if they are exposed to different drugs or seeding conditions. Therefore, there is no true “one size fits all” setting for the image analyzer. Overall, we recommend that the user alter each of these parameters as appropriate such that red and green objects are appropriately counted by the analyzer for a given lethal treatment of interest.
Figure 2.

Optimization of Incucyte analyzer
(A) HT-1080N cells following initial seeding and after treatment with vehicle control (DMSO) or the lethal proteasome inhibitor bortezomib for 48 h. Scale bar, 100 μm. Cells are imaged using the 10× objective; SYTOX green marks dead cells in the green channel.
(B) Screenshots of Incucyte software showing results of low (top) and high (bottom) edge sensitivity setting on object counting. Scale bars, 20 μm.
(C) Screenshots of Incucyte software showing optimized masking for red, green, and overlap objects. Scale bars, 100 μm.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Incucyte NucLight Red Lentivirus (EF1α, Puro) | Sartorius | Cat#4625 |
| Incucyte NucLight Red Lentivirus (EF1α, Bleo) | Sartorius | Cat#4627 |
| Chemicals, peptides, and recombinant proteins | ||
| Puromycin dihydrochloride | Thermo Fisher Scientific | Cat#A1113803 |
| Zeocin | Thermo Fisher Scientific | Cat#R25001 |
| Hexadimethrine bromide (polybrene) | Millipore Sigma | Cat#H9268 |
| SYTOX green | Thermo Fisher Scientific | Cat#S7020 |
| Bortezomib | Fisher Scientific | Cat#NC0587961 |
| Staurosporine | ApexBio | Cat#A8192 |
| Erastin2 | Cayman Chemical | Cat#27087 |
| Pimasertib | Selleck Chemicals | Cat#S1475 |
| Trametinib | Selleck Chemicals | Cat#S2673 |
| AZD8055 | Selleck Chemicals | Cat#S1555 |
| S63845 | Selleck Chemicals | Cat#21131 |
| Experimental models: cell lines | ||
| A549N | Forcina et al., 2017 | N/A |
| A549N Control | Inde et al., 2020 | N/A |
| A549N MCL1LOF | Inde et al., 2020 | N/A |
| U-2 OSN | Forcina et al., 2017 | N/A |
| Calu-6N | Inde et al., 2020 | N/A |
| HT-29N | This paper | N/A |
| H23Cas9; N | This paper | N/A |
| SW48N | This paper | N/A |
| T98GN | Forcina et al., 2017 | N/A |
| SW620N | This paper | N/A |
| NCI-H1299N | Inde et al., 2020 | N/A |
| NCI-H2291N | Inde et al., 2020 | N/A |
| Software and algorithms | ||
| Prism | GraphPad Software | N/A |
| Excel | Microsoft | N/A |
| IncuCyte Base Software | Sartorius | N/A |
| Other | ||
| Incucyte microscope | Sartorius | N/A |
| 96-well plate | Corning | Cat# 3904 |
Step-by-step method details
Conduct Incucyte experiment
In this step, cell seeding, treatment, and imaging are conducted to generate raw counts of live and dead cells at each timepoint over the course of an experiment. These counts provide the basis for further analysis of cell death kinetics in subsequent steps.
-
1.Seed mKate2+ cells for treatment.
-
a.Trypsinize, quench, and count mKate2+ cells. Resuspend in culture media for seeding at the density determined in pilot experiments.
-
b.Seed cells in appropriate tissue culture vessel, centrifuge 55 × g for 5 s, and place in tissue culture incubator. Allow to adhere 18–24 h.
-
a.
-
2.Apply drug treatment.
-
a.Add SYTOX Green dye to appropriate culture medium at a concentration of 20 nM.
-
b.Prepare treatment media by diluting compounds of interest in SYTOX Green-containing medium. If desired, perform serial dilutions of compounds to test multiple doses. Include vehicle control treatment(s) as appropriate.
-
c.Invert seeded plate and flick sharply into a basin to remove seeding medium. Working quickly to prevent wells from drying completely, replace with 200 μL of treatment medium.
-
a.
-
3.Begin Incuycte imaging of plate.
-
a.Place plate in open slot of Incucyte. Ensure that plate is seated securely in slot and level along all plate edges.
-
b.In Incucyte Zoom software, select the plate definition for the specific vessel (e.g., Corning 96 well, Cat# 3904) being employed in the experiment from the list of preset option. Add the plate to the repeating scan schedule to scan once every 4 h.
-
a.
-
4.Complete Incucyte experiment and obtain raw counts for analysis.
-
a.Stop experiment at endpoint determined in pilot experiments.
-
b.Run analysis of collected images using cell line-optimized analyzer.
-
c.Export time course data for each treatment condition for three object count parameters: SYTOX Green+ objects per mm2, mKate2+ objects per mm2, and overlap objects per mm2.
-
a.
Analyze fractional killing kinetics
This step describes the processing of raw object counts to calculate cell death kinetic parameters. Specifically, raw counts are converted to a lethal fraction (LF) metric as described in (Forcina et al., 2017). Curve fitting of LF values over the course of an experiment allows for calculation of the death onset (DO) and death rate (DR) parameters. DR values are a useful means to quantify the rate of drug-induced fractional killing (FK), as described in (Inde et al., 2020). These parameter values can then be compared between diverse conditions acquired in parallel.
-
5.Calculate and plot LF for each time point in each experimental condition. Refer to “Quantification and statistical analysis” for a sample dataset.Optional: Examine the overlap+ counts for the tested conditions. For some cell line/drug combinations, or individual timepoints, these may be a substantial fraction of all mKate2+ object. If overlap+ counts are high (> 5% of all mKate2+ counts), generate corrected mKate2+ object counts by subtracting the overlap object count from the corresponding mKate2 object count at each timepoint.
-
a.Using the raw or corrected mKate2+ counts, calculate LF according to the formula below. MaxSG is the maximum value for SYTOX Green+ counts observed up to the current timepoint.
-
b.Using GraphPad Prism software, plot the LF data over time in an XY scatter plot
-
a.
-
6.Fit one phase association curve to data. This curve, also referred to as a lag-exponential death (LED) model, includes a plateau phase (up until DO) where little or no death occurs and an exponential phase (after DO) where death begins at a given rate (DR) and then slows over time.
-
a.In the Prism software, create a new analysis from the data table containing the LF data.
-
b.In the “Create New Analysis” window, select “Nonlinear regression (curve fit)” under the “XY analyses” category.
-
c.In the “Parameters: Nonlinear Regression” window, in the “Model” tab, select “Plateau followed by one phase association” under the “Exponential” category.
-
d.Also, in the “Parameters: Nonlinear Regression” window, in the “Constrain” tab, change the Constraint Type for X0 (corresponding to DR) to “No constraint.”
-
e.Click “OK” to perform the curve-fitting analysis. A new “Results” sheet will appear when the analysis is complete.
-
a.
-
7.Extract kinetic parameters from fitted curve.
-
a.In the Results sheet corresponding to the LF data table, confirm a successful curve fit. If the nonlinear regression analysis is unable to determine parameters for the curve, the column will be labeled “Ambiguous” and will not provide 95% CI values. In these cases, often where little death is observed in the treated cells, the exponential curve fit is not appropriate for the data, and DR and DO values should not be used for comparisons with other conditions.
-
b.For each time course, kinetic parameters correspond to calculated “Best-fit values” for the fitted curves. X0 corresponds to DO in hours; Y0 corresponds to the baseline LF preceding DO; Plateau corresponds to LF at the conclusion of the experiment; and K corresponds to DR.
-
a.
Expected outcomes
Kinetic parameters extracted from the fitted curves should reasonably match qualitative observations about the observed onset of death in the population and the rate of death after onset. STACK analysis and kinetic parameter extraction allows comparison of drug-induced death kinetics across different drugs (alone or in combination), doses, and cell lines. Example data adapted in part from (Inde et al., 2020) are shown in Figure 3.
Figure 3.

Utilizing kinetic parameters to compare FK across genotypes and drug combinations
(A) Cell death over time in A549N WT and MCL1 CRISPR-mutated loss of function (LOF) cells. Cells are treated with lethal (5 μM) or sublethal (500 nM) dose of the MEK inhibitor pimasertib to induce cell death, and the effect of combination with an mTOR inhibitor (AZD8055, 500 nM) or MCL1 inhibitor (S63845, 5 μM) is tested in each genetic background. Data represent mean ± SD from three independent biological replicates.
(B) Extracted cell death kinetic parameter values and their interpretations for the curves presented in A.
Quantification and statistical analysis
Here we present a sample dataset generated from the treatment of ten cell lines treated for 120 h with dose responses for three drugs: two MEK inhibitors (pimasertib and trametinib) and a proteasome inhibitor (bortezomib) (Figure 4). To illustrate data processing steps, raw and processed data are shown for a 5 μM dose of pimasertib in H2291 lung carcinoma cells (Table 1).
Figure 4.

Utilizing kinetic parameters to compare FK for lethal compounds across cell lines
(A) Example lethal fraction (LF) values for ten cancer cell lines treated with lethal compounds over 120 h. Data represent mean ± SD from three independent biological replicates. Raw data for a representative replicate of pimasertib-treated H2291N cells are shown in Table 1.
(B) Kinetic parameters calculated from selected curves depicted in (A).
Table 1.
Raw object counts and calculated lethal fraction values
| Time | Raw data |
Calculated values |
|
|---|---|---|---|
| SYTOX Green+ counts/mm2 | mKate2+ counts/mm2 | Lethal fraction (LF) | |
| 0 | 1.856391 | 118.809 | 0.015 |
| 4 | 2.320489 | 132.2679 | 0.017 |
| 8 | 1.392293 | 154.0805 | 0.015 |
| 12 | 1.856391 | 147.5831 | 0.015 |
| 16 | 1.392293 | 161.0419 | 0.014 |
| 20 | 1.392293 | 168.0034 | 0.014 |
| 24 | 4.17688 | 175.429 | 0.023 |
| 28 | 4.17688 | 180.0699 | 0.023 |
| 32 | 5.105075 | 185.6391 | 0.027 |
| 36 | 7.425564 | 186.5673 | 0.038 |
| 40 | 14.38703 | 189.816 | 0.070 |
| 44 | 13.92293 | 187.9596 | 0.071 |
| 48 | 18.56391 | 195.8493 | 0.087 |
| 52 | 20.4203 | 190.2801 | 0.097 |
| 56 | 23.66899 | 184.2468 | 0.114 |
| 60 | 27.84587 | 184.7109 | 0.131 |
| 64 | 40.8406 | 189.816 | 0.177 |
| 68 | 38.05602 | 191.6724 | 0.176 |
| 72 | 48.26617 | 192.6006 | 0.200 |
| 76 | 52.90715 | 185.175 | 0.222 |
| 80 | 58.94042 | 183.7827 | 0.243 |
| 84 | 64.97369 | 175.8931 | 0.270 |
| 88 | 84.00169 | 181.4622 | 0.316 |
| 92 | 83.5376 | 175.429 | 0.324 |
| 96 | 95.60414 | 176.8212 | 0.351 |
| 100 | 95.14005 | 173.1085 | 0.356 |
| 104 | 100.2451 | 168.0034 | 0.374 |
| 108 | 106.7425 | 160.1137 | 0.400 |
| 112 | 129.9474 | 164.7547 | 0.441 |
| 116 | 138.7652 | 168.0034 | 0.452 |
| 120 | 136.9088 | 159.6496 | 0.465 |
Data are shown for H2291N cells treated with 5 μM pimasertib at the indicated timepoints.
Limitations
As demonstrated in the example dataset above, robust calculation of DO and DR parameter values can only be performed for conditions where sufficient lethality is observed during the course of the experiment, typically with a final lethal fraction of greater than 0.2 (Forcina et al., 2017). Thus, a sufficiently lethal dose of drug must be applied, and the experiment must run long enough for lethality to be observed. Empirically, we have observed that when these conditions are met, DO and DR can be calculated robustly even if the time course does not run until the lethal fraction values have reached a final plateau. Similarly, these parameters can typically be calculated accurately using a scanning interval of 4 h or longer; however, in some cases, such as very rapid lethal treatments, a shorter measurement interval (e.g., 1 h) may better capture fast kinetics.
Note: In addition to extracted kinetic parameter values, other information derived from these curves, including the maximum lethal fraction values and the area under the lethal fraction curve could potentially be of use in summarizing and comparing fractional killing under different lethal conditions. These alternative values, which are easily extracted from lethal fraction data over time, may provide a useful surrogate measure of fractional killing for cases where cell death exhibits low or unusual kinetics over time that cannot be effectively captured by the described curve-fitting approach.
Troubleshooting
Problem 1
Heterogeneous expression of mKate2 in viable cells (Before you begin, step 3)
Potential solution
Reapply antibiotic selection until more uniform marker expression is observed or employ the alternative FACS-based selection strategy described after step 3g under “Before you begin.”
Problem 2
False positive or false negative objects recognized by Incucyte image analyzer (Before you begin, step 5)
Potential solution
Repeated adjustments to analyzer parameters may be required until a final version is established for a given cell line. Accurate quantification of mKate2+ objects is particularly difficult at 100% confluence or greater; cell seeding densities or experiment time courses may also be modified to prevent overcrowding of cells and subsequent quantification errors.
Problem 3
Changes in cell morphology occur during or after cell death (Before you begin, step 5)
Potential solution
If necessary, analyzer should be optimized for each combination of cell line and lethal stimulus, with parameters set to count mKate2+ objects even if their shape is affected by drug treatment. After death, some dead cells may lose SYTOX Green fluorescence over time. This should not affect LF calculations, since they are based on the maximum SYTOX Green values observed up to a given timepoint, as described (Forcina et al., 2017).
Problem 4
Non-specific SYTOX Green signal (Step-by-step method details, step 2)
Potential solution
20 nM is an appropriate SYTOX Green concentration for almost all cell lines. In some rare cases, this concentration may yield dim green signal in the cytoplasm of viable cells; in these cases, lower doses of SYTOX Green may be tested. Non-specific SYTOX Green signal may also indicate the presence of contamination such as mycoplasma.
Problem 5
Other non-specific fluorescence (Step-by-step method details, step 2)
Potential solution
Some tested compounds may be autofluorescent and therefore interfere with imaging, typically producing false positives for SYTOX Green fluorescent objects. Autofluorescent compounds can be preliminarily identified by identifying conditions where SYTOX Green counts are high at early timepoints, without obvious evidence for cell death. Subsequent visual inspection can confirm that this fluorescence in the green channel is not due to true positive dead cells. In some cases, autofluorescent material is present outside cells. Conditions with autofluorescent compounds may either be removed from consideration or analyzed based on the live cell counts alone.
Resource availability
Lead contact
Requests for further information should be directed to and will be fulfilled by the Technical Contact, Zintis Inde (zinde@hsph.harvard.edu). Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Scott J. Dixon (sjdixon@stanford.edu).
Materials availability
Cell lines expressing nuclear mKate2 are available from the Lead Contact.
Data and code availability
There is no additional data associated with this protocol.
Acknowledgments
We thank Leslie Magtanong and other members of the Dixon lab for comments on this work. S.J.D. is supported by the NIH (1R01GM122923).
Author contributions
Conceptualization, Z.I. and S.J.D.; Methodology, Z.I. and J.R.; Writing – Original Draft, Z.I.; Writing – Review & Editing, Z.I., J.R., and S.J.D.; Funding Acquisition and Supervision, S.J.D.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Zintis Inde, Email: zinde@hsph.harvard.edu.
Scott J. Dixon, Email: sjdixon@stanford.edu.
References
- Forcina G.C., Conlon M., Wells A., Cao J.Y., Dixon S.J. Systematic quantification of population cell death kinetics in mammalian cells. Cell Syst. 2017;4:600–610.e6. doi: 10.1016/j.cels.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inde Z., Forcina G.C., Denton K., Dixon S.J. Kinetic heterogeneity of cancer cell fractional killing. Cell Reports. 2020;32:107845. doi: 10.1016/j.celrep.2020.107845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langsrud S., Sundheim G. Flow cytometry for rapid assessment of viability after exposure to a quaternary ammonium compound. J. Appl. Bacteriol. 1996;81:411–418. doi: 10.1111/j.1365-2672.1996.tb03527.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
There is no additional data associated with this protocol.