

Abstract
Leukemic relapse remains a major barrier to successful allogeneic hematopoietic stem cell transplantation (allo-HSCT) for aggressive hematologic malignancies. The basis for relapse of advanced lymphoid malignancies remains incompletely understood and may involve escape from the graft-versus-leukemia (GvL) effect. We hypothesized that for patients with chronic lymphocytic leukemia (CLL) treated with allo-HSCT, leukemic cell-intrinsic features influence transplant outcomes by directing the evolutionary trajectories of CLL cells. Integrated genetic, transcriptomic and epigenetic analyses of CLL cells from 10 patients revealed that the clinical kinetics of post-HSCT relapse are shaped by distinct molecular dynamics. Early relapses after allo-HSCT exhibited remarkable genetic stability; indeed single CLL cell transcriptional analysis demonstrated a cellular heterogeneity that was static over time. In contrast, CLL cells relapsing late after allo-HSCT displayed striking genetic evolution and evidence of neoantigen depletion, consistent with marked single cell transcriptional shifts that were unique to each patient. We observed a greater rate of epigenetic change for late relapses not seen in early relapses or relapses after chemotherapy alone, suggesting that the selection pressures of the GvL bottleneck are unlike those imposed by chemotherapy. No selective advantage for HLA loss was observed, even when present in pre-transplant subpopulations. Gain of stem cell modules was a common signature associated with leukemia relapse regardless of post-transplant relapse kinetics. These data elucidate the biological pathways that underlie GvL resistance and post-transplant relapse.
One Sentence Summary:
The clinical kinetics of CLL relapse after stem cell transplant are underwritten by distinct genetic and epigenetic evolutionary trajectories.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is one of the earliest forms of successful cancer immunotherapy whose study has elucidated critical insights into tumor-immune interactions (1, 2). As the only curative option for aggressive and advanced hematologic malignancies, allo-HSCT derives its potency from the underlying, donor-derived graft-versus-leukemia (GvL) effect wherein donor immune cells recognize and eradicate recipient leukemic cells. Reduced-intensity conditioning (RIC) regimens for allo-HSCT are increasingly used to reduce treatment-related morbidity and preserve the curative GvL effect (3, 4), but post-transplant disease recurrence limits their efficacy. Therefore, elucidating the mechanisms underpinning GvL resistance is vital to improve transplant outcomes.
Multiple studies have identified various pathways of GvL resistance associated with relapse of myeloid malignancies, including loss of HLA class I and II genes, upregulation of inhibitory immune checkpoint molecules, and oncogene-driven immune evasion (5–9). For lymphoid malignancies however, less is known about potential mechanisms of GvL evasion. In particular, how underlying genetic and epigenetic features inform the clinical kinetics of post-transplant relapse or whether dysregulation of HLA class I and II genes influences GvL resistance remains unclear.
To address these questions, we assembled a cohort of 10 patients with chronic lymphocytic leukemia (CLL), the majority of whom were treated with RIC allo-HSCT. We hypothesized that leukemic-intrinsic features primarily shape the evolutionary dynamics of CLL cells during GvL resistance. We therefore focused on the evaluation of longitudinal changes in genomic, epigenomic and bulk and single cell transcriptomic features of CLL cells following transplant. By defining the genetic, epigenetic and transcriptomic changes that shape CLL relapse after allo-HSCT, we demonstrate that these evolutionary paths are unique to the GvL immunologic bottleneck and identify the underlying mechanisms that influence the clinical kinetics of post-HSCT relapse.
Results
Clinical kinetics of CLL relapse after allo-HSCT correspond to distinct evolutionary paths.
We assembled a cohort of 10 CLL patients with varying time to progression after allo-HSCT (range: 83-1825 days), for whom paired pre- and post-transplant relapse specimens were available. As expected for patients with CLL undergoing allo-HSCT, patients had received multiple lines of therapy prior to transplant (median: 3, range: 1-6) and 6 of 9 assessable patients had an unmutated variable region of the immunoglobulin heavy chain (IGHV) (Fig. 1A, table S1).
Fig. 1. Timing of relapse after allo-HSCT is defined by distinct evolutionary trajectories and stem cell expression programs.
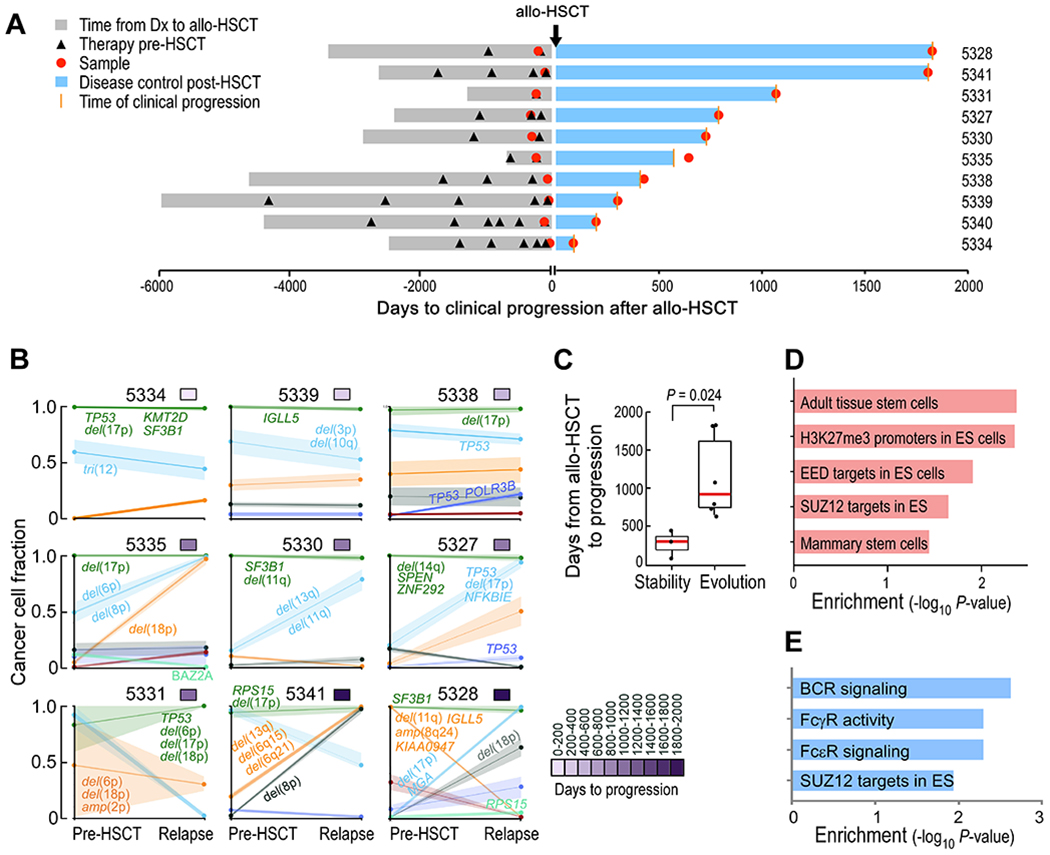
(A) Time to clinical progression after allo-HSCT for 10 patients with CLL indicated along with relevant clinical histories and times of sample procurement. (B) Evolutionary patterns of mutation clusters shown by their cancer cell fractions (CCFs), inferred using PhylogicNDT. Time to progression is shown by the purple color bar. Putative CLL drivers detected for each cluster are shown. (C) Box plot of time to relapse after allo-HSCT in patients with (n=6) and without (n=3) clonal evolution. The P value was calculated by a two-sided Wilcoxon ranked sum test. “Evolution” was defined as having any cluster with absolute difference ≥0.2 between pre-HSCT and relapse timepoints. (D) Unadjusted P values of enriched stem cell gene sets in pretreatment allo-HSCT samples per GSEA, comparing samples collected from early versus late relapses. (E) Unadjusted P values of enriched signaling pathways per GSEA, comparing post- versus pre-HSCT samples of late relapses.
We performed whole-exome sequencing (WES) of DNA isolated from purified CLL cells from paired pre- and post-transplant relapse samples and matched donor and recipient germline DNA from 9 of 10 patients (median coverage of 160x; table S2 in data file S1). Compared to HSCT-naïve CLL (10), we detected a greater number of non-silent single nucleotide variants (sSNVs) and somatic insertions and deletions (sIndels) per pre- and post-transplant exome (a median of 39 and 41), respectively (p<0.01; fig. S1A, table S3 in data file S1), consistent with the extensive chemotherapeutic exposure in patients undergoing allo-HSCT. Nevertheless, the spectrum of CLL cancer drivers in sSNVs, sIndels, and copy number alterations (CNAs) was similar to that previously observed (10) (fig. S1B, table S4 in data file S1). Consistent with the aggressive nature of these leukemias, we observed multiple patients to have mutations and CNAs involving TP53 and SF3B1, but found no somatic alterations uniquely shared among post-transplant samples.
To examine patterns of clonal dynamics associated with relapse, we used the tool PhylogicNDT (11, 12), which reconstructs phylogenetic and evolutionary trajectories of cells based on integration of somatic alterations detected from multiple samples. We clustered mutations together based on their cancer cell fractions (CCFs) and examined their change over time. Although all patients displayed multiple and diverse CLL drivers at baseline (fig. S1C, table S5 in data file S1), their evolutionary dynamics during relapse segregated into two distinct patterns: clonal stability (defined by changes in all cluster CCFs ≥0.2) and clonal evolution (changes in any cluster CCF ≥0.2) (Fig. 1B). We found that those samples with clonal stability originated from the 3 patients with the shortest times to relapse (median of 304 days; ‘early’ relapse), while clonal evolution was evident in the samples from the 6 patients with longer times to relapse (median of 798 days; ‘late’ relapse, p=0.024) (Fig. 1C).
We hypothesized that late relapses, occurring after immune reconstitution, were more likely subjected to a GvL selection pressure than early relapses. Indeed, patients with late relapse were more likely to have experienced clinical graft-versus-host disease (GvHD) (p=0.03, Fisher’s exact test; fig. S1D), often associated with GvL activity(13, 14); and all late relapsing patients with available donor chimerism data had at least a 96% peripheral blood donor chimerism before relapse, indicating immune reconstitution capable to potentially mediate GvL (table S1). Finally, we also found that late relapse was associated with in silico depletion of neoantigens. Predicted neoantigens with strong binding affinity (defined as binding affinity ≤50 nM (15, 16)) were enriched in CLL clones that contracted post-transplant (present in 25% of contracting, 4% in stable and 7% in expanding mutations; p<0.001, fig. S1E–S1F, table S6 in data file S1). Additionally, contracting neoantigens overall had stronger predicted binding affinities than those that either expanded or remained unchanged (p=0.04; p=0.00014, respectively; fig. S1G) and were observed only in late relapses, including patients 5328, 5331, and 5335 (fig. S1H). These results are consistent with the presence of GvL immune activity among late relapses.
To investigate the pre-transplant molecular profiles that may herald clonal stability or evolution, we performed differential gene expression analysis between pre-transplant CLL cells from early versus late relapses and found 853 upregulated and 793 downregulated genes (FDR<0.25). Gene set enrichment analysis (GSEA) highlighted stem cell modules in the early relapses, suggesting that the presence of stem cell-like states in the pre-transplant setting is important for subsequent rapid relapse, particularly components of the polycomb-repressive complex 2 (PRC2) such as the EED and SUZ12 gene pathways (17) (Fig. 1D, table S7 in data file S1). Consistent with the lack of genetic evolution during early relapse, we found only 66 differentially expressed genes between pre- and post-HSCT samples in early relapses suggesting little transcriptional change. However, paired differential expression analysis between pre- and post-HSCT samples in late relapses revealed 1002 differentially expressed genes (FDR<0.25) and upregulation of similar stem cell pathways in addition to Fc and B cell receptor (BCR) signaling (Fig. 1E, table S8 in data file S1). Altogether, these data support the notion that early CLL relapse after transplant is characterized by a pre-existing transcriptional state conferring resistance and harboring stem cell properties. This state therefore does not require evolution of the clonal architecture, and subsequent relapse manifests as genetic stability. In contrast, late CLL relapse, occurring after immune reconstitution, is likely subjected to a GvL selection pressure, manifested by neoantigen depletion. This immunologic bottleneck leads to acquired resistance, genetically, via clonal replacement and, transcriptionally, via upregulation of stem cell and FcR/BCR signaling pathways.
Genetic evolution of CLL cells carries phenotypic consequences.
To evaluate the functional consequences of this genetic evolution, we sought to measure changes in associated gene expression of these heterogeneous clonal populations. We adapted a droplet microfluidic-based platform (“inDrops”)(18) to obtain single cell transcriptome (scRNA-seq) data from PBMC samples collected from pre- and post-HSCT samples from 2 patients with early relapse and clonal stability (patients 5339 and 5338, relapsing 304 and 443 days after allo-HSCT) and 2 patients with late relapse and clonal evolution (patients 5341 and 5328, relapsing 1801 and 1825 days after allo-HSCT) (Fig. 2A, table S9 and S10 in data file S1). We profiled a median of 1,261 CLL cells (range: 1,035-3,751) per sample as assessed by CD19 and CD5 gene co-expression. The samples were integrated computationally using Conos (19) to overcome inter-patient variability and identify major cell populations (Fig. 2B, fig. S2A).
Fig. 2. Phenotypic changes in relation to the kinetics of genetic evolution.
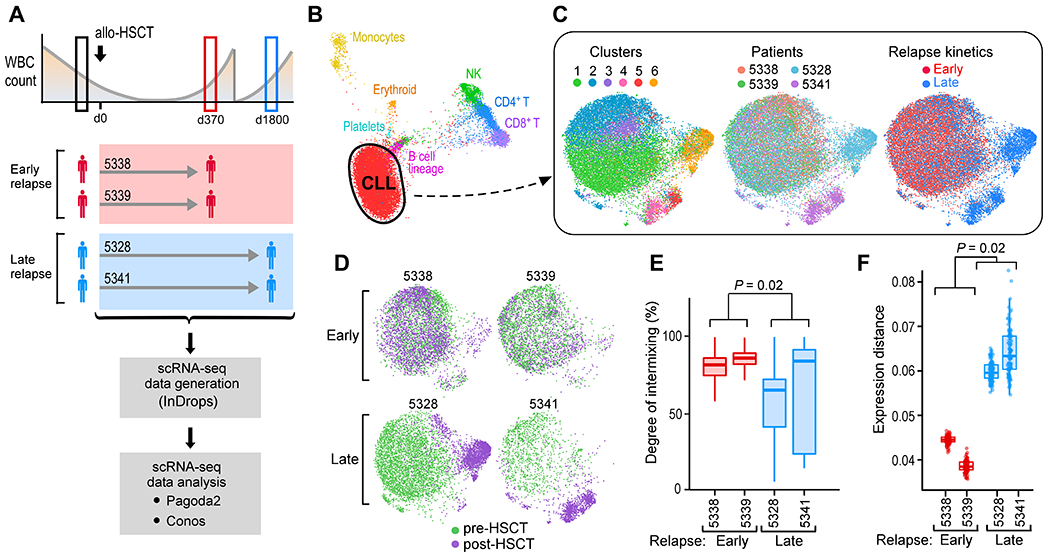
(A) Sample schematic and experimental workflow for capturing single CLL cell transcriptomes through the inDrops system from paired, pre- and post-HSCT PBMCs from two early and two late relapses. (B) Joint graph visualized using largeVis embedding showing clustering and annotation of cell subsets from PBMCs from all four patients. (C) Joint graphs using UMAP embedding of computationally identified CD19+CD5+ CLL cells, colored by cluster (left), patient (center) and relapse kinetics (right). (D) Individual joint graphs for both early and late relapse patients colored by timing. (E) kNN-based quantification of timepoint intermixing across clusters for each patient. The P value was calculated from a two-sided Welch t-test comparing means of individual cell values per patient, grouped into early (n=2) versus late (n=2) relapses. (F) Extent of gene expression change between pre- and post-transplant CLL cells for each patient. The P value was calculated from a one-sided Welch t-test.
The CLL cells consisted of 6 transcriptionally distinct clusters that segregated by patient and timing (Fig. 2C). For the patients who relapsed early, pre- and post-transplant CLL cells exhibited an indistinct population structure, evident by a high degree of intermixing of cells within clusters and consistent with their genetic and transcriptional stability seen on the bulk sample level (Fig. 2D). In contrast, analysis of patients who relapsed late revealed marked spatial segregation by timepoint with a defined population substructure evident in distinct post-transplant clusters from both late relapses. Late relapse CLL cells from pre- and post-transplant timepoints were less intermixed within clusters (p=0.02; Fig. 2E) and displayed a greater magnitude of gene expression change between the timepoints (p=0.02; Fig. 2F). Similar findings were also obtained with an alternative analysis pipeline (Seurat), demonstrating that these transcriptional shifts are not dependent on the clustering algorithm (20) (fig. S2B, C). Thus, both genetic and scRNA-seq analyses reveal early relapses as a heterogeneous population that is static over time, evidenced by a lack of genetic or transcriptional evolution during relapse after allo-HSCT.
The distinct post-transplant clusters seen in the late relapse patients also suggest the presence of multiple pathways of acquired resistance (notably cluster 6 in patient 5328 and clusters 4 and 5 in patient 5341). To delineate these pathways, we used pathway and gene set overdispersion analysis (PAGODA) to test annotated gene sets for coordinated variability across all CLL cells (21). Significantly (p<0.05) overdispersed gene sets with similar cell-separation patterns were then combined to form a single ‘aspect’ of transcriptional heterogeneity. PAGODA revealed 5 major aspects of heterogeneity that distinguished the 6 CLL clusters, corresponding to ribosomal biogenesis, antigen presentation, apoptosis regulation, proliferation, and calcium/cAMP signaling (Fig. 3A). In this manner, post-transplant pathways unique to late relapses could be identified. For example, relapse in patient 5328 was best characterized by overdispersion of apoptosis regulation, manifesting as downregulation of pro-apoptotic genes (such as TP53, DFFA, BAX) and upregulation of anti-apoptotic and cytoprotective genes (such as MTRNRL2, MTRNRL8) (22, 23). We also found loss of BACH2, a known tumor suppressor in B cell malignancies (24) in relapsed cells along with gain of known B cell leukemia-related pro-survival and anti-apoptotic genes (PIM2(25), MCL1 [pre vs. post, p<2.2 x 10−16]) (Fig. 3A, B; fig. S3A). In contrast, relapse in patient 5341 was associated with a distinct overdispersed gene set (cluster 4; calcium/cAMP signaling evidenced by TXNIP, DUSP1, ADD1, and FOS). Moreover, intra-leukemic heterogeneity between clusters 4 and 5 was evidenced by differential up- and down-regulation of oncogenic signaling and tumor suppressor genes (Fig. 3B). These data highlight the inter- and even intra-leukemic diversity of pathways leading to late relapse.
Fig. 3. Transcriptional programs define inter- and intra-leukemic heterogeneity during late relapse.
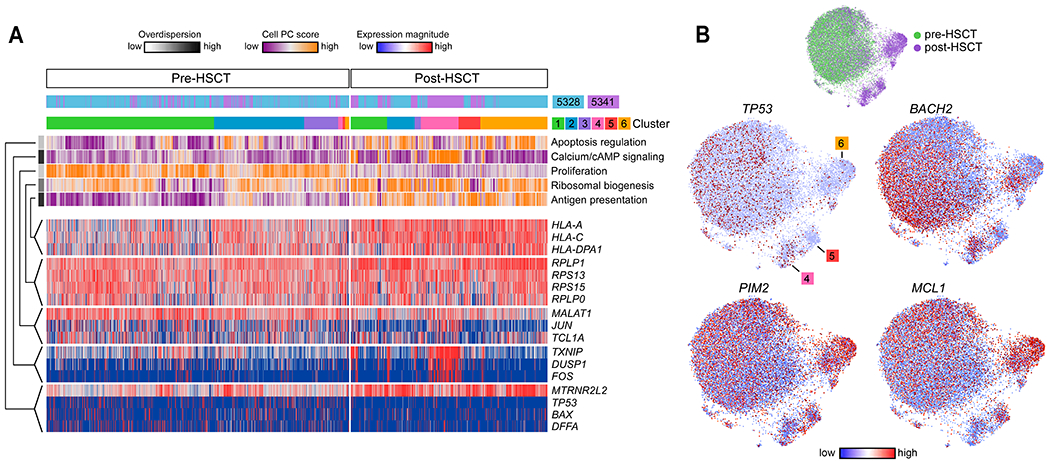
(A) Cells (columns) from patients 5328 and 5341 are organized by cluster assignment. For each cell, relapse kinetics, timing and principal component (PC)/aspect score are displayed. For each aspect, overdispersion score is shown by white/black color bar. Row labels summarize key functional annotations of gene sets for each aspect. For each aspect, gene expression patterns of top-loading genes are shown. (B) Joint graphs of CLL cells are visualized to demonstrate differential downregulation of tumor suppressor genes (TP53 and BACH2) and differential upregulation of oncogenic signalling (PIM2, MCL1) after allo-HSCT among late relapse clusters 4, 5 and 6.
HLA dysregulation provides no selective advantage for post-HSCT relapse of CLL.
We also identified shared pathways among post-transplant populations in both late relapses (clusters 4, 5 and 6). Specifically, an aspect defined by genes involved in antigen presentation was shared among all three post-transplant, late relapse clusters (Fig. 3A, 4A). Because loss of HLA expression has been found to contribute to relapse of myeloid leukemia after transplant (5, 6), we investigated further the dynamics of HLA expression variability during relapse. From our single cell data, we unexpectedly detected expression of HLA class I and II genes on post-transplant CLL cells despite relapse (Fig. 4A–B), Moreover, we observed a wide range of HLA expression by CLL cells at both pre- and post-transplant timepoints in the late relapses. Analysis of the probability distribution of HLA class I or II genes demonstrated increased HLA expression post-transplant for these two patients that was confirmed by analysis of cell surface expression for these HLA proteins (Fig. 4C, left, p<10−14 for all comparisons; fig. S3B). To extend our analysis beyond the population-level, we observed that the lowest 2.5% of HLA-expressing CLL cells consisted of mostly pre-transplant cells, suggesting that while CLL cells with very low HLA expression are detectable pre-transplant, they fail to expand at late relapse (Fig. 4C, right). At the bulk level, except for one sample with loss of HLA-A, we did not detect any somatic alterations in HLA class I, B2M or other genes involved in the antigen presentation pathway. Moreover, while baseline pre-HSCT bulk HLA class I gene expression in CLL was lower than observed in transplant-naïve samples, baseline bulk HLA class II expression was similar (Fig. 4D). Notably, transcriptional downregulation of HLA class I or II genes during relapse was not observed. These data suggest that further HLA dysregulation provides little selective advantage during CLL relapse after transplant, even when present in pre-transplant subpopulations. In addition to HLA expression, we also investigated the fold change in expression of key molecules expressed on leukemia cells known to inhibit and/or activate T cells (depending on the cognate receptor expressed) (fig. S3C). For none of these molecules did we identify a relationship between their differential regulation upon relapse and time to relapse. Thus, unlike relapses from myeloid disease after transplant, genetic and/or transcriptional alterations in HLA (or T cell ligand) genes are not common in the setting of CLL relapse.
Fig. 4. scRNA-seq analysis of late relapse clusters highlights distinctive features of post-transplant CLL cells.
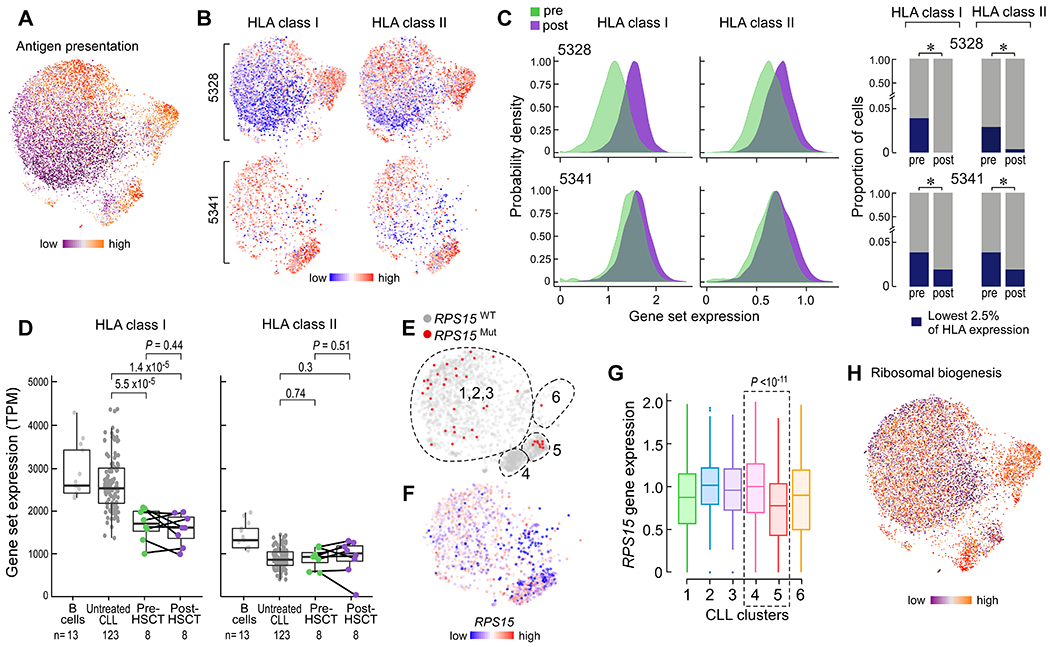
(A) CLL joint graph colored by individual cell score for the aspect defined as antigen presentation. (B) CLL joint graph displaying only cells from the indicated late relapse patient and their associated expression of HLA class I or II genes. (C) Left, probability densities of the range of HLA class I or II gene expression values. P <10−14 for all four comparisons, by two-sample Kolmogorov-Smirnov test. Right, stacked barplots indicating the lack of expansion of HLA class I or II ‘low’ expressing cells after allo-HSCT. * = P <0.01 for contraction determined from Fisher’s exact test. (D) Bulk gene set expression values for HLA class I (left) and II (right) genes from purified normal B cells, untreated CLL, and paired pre- and post-HSCT CLL cells. P value determined from Student’s t test (paired t test for pre- vs post-HSCT). (E, F) Joint graph of CLL cells from patient 5341 colored by RPS15 mutation status (E) or gene expression (F). Dotted lines indicated approximate cluster boundaries. (G) Box plots showing RPS15 gene expression by cluster for cells from patient 5341. Cluster 4 vs 5 (dotted box), P value calculated from two-sided Wilcoxon rank sum test. (H) Joint graph of all CLL cells colored by the aspect defined as ribosomal biogenesis.
Alterations in genotype can define transcriptional changes in late relapses.
The marked transcriptional change at the single cell level demonstrated by late, rather than early, relapses prompted the question of whether these transcriptional phenotypes may be driven by genetic changes. We previously reported a lack of concordance between transcriptional and genetic changes in transplant-naïve CLL cells, suggesting the presence of convergent evolution (26). For patient 5341, we could identify the presence of a putative CLL driver mutation in the ribosomal gene RPS15 (p.G139C) through WES analysis; we were also able to detect this mutation within the corresponding samples’ scRNAseq data. Consistent with the bulk WES analysis, we detected a subset of cells expressing this RPS15 mutation at the pre-transplant timepoint (clusters 1, 2, and 3) (Fig. 4E). However, within post-transplant cells, we identified two distinct expression states corresponding to absence (cluster 4) or presence (cluster 5) of the mutation (fig. S3D, P<0.01), with associated differential expression of the calcium/cAMP signaling aspect (fig. S3E) as well as TP53 and BACH2 tumor suppressors (P<0.01, cluster 4 versus 5).
Upon closer examination of the post-transplant clusters, we observed higher relative expression of the RPS15 gene (Fig. 4F, G), along with evidence of increased ribosomal activity (Fig. 4H), within cluster 4 relative to cluster 5. These data link the presence of the mutation with lower RPS15 gene expression and diminished ribosomal biogenesis; conversely, absence of the mutation is associated with higher RPS15 expression and increased ribosomal activity. This index case demonstrates that genetic variants can segregate distinct expression states in the post-HSCT relapsed setting; and that selection pressures favoring divergent, not convergent, evolutionary paths may be observed in late CLL relapse.
Methylome instability is characteristic of late relapses after allo-HSCT.
We previously demonstrated locally disordered methylation (measured by the percentage of discordant reads (PDR)) as an epigenetic mechanism of genetic variability within CLL, with increased PDR associated with a more aggressive clinical course in transplant-naïve CLL (27). We therefore investigated whether epigenetic dysregulation could underlie the observed phenotypic changes by comparing the change in PDR over time between early and late relapses (tables S11–S13 in data file S1). The change in PDR for early relapses was minimal, while late relapses exhibited greater changes in PDR across multiple genomic elements (Fig. 5A). By contrast, PDR did not change within CLL samples relapsing after chemotherapy alone, matched for timing with late relapses after transplant (Fig. 5A, fig. S4A). When we modeled the quantitative changes in PDR as a function of time, we confirmed a moderate increase in the rate of change of PDR for the late relapsers compared to the chemotherapy-treated subjects, suggesting time between samples was not a contributing factor to our PDR measurements (Fig. 5B, fig. S4B; Bayes factor=1.6) (28). Altogether, these findings are consistent with the notion of post-HSCT immune pressure selecting for increased intra-leukemic methylation variability as an underlying mechanism of phenotypic changes. Indeed, genes with increased promoter PDR in late relapses were enriched for multiple stem cell pathways, implicating a common stem-like state that is acquired during late relapses through increased methylome instability (Fig. 5C–F) and is consistent with the transcriptional upregulation of similar modules in this group (Fig. 1E).
Fig. 5. Methylome instability characterizes late relapse after allo-HSCT.
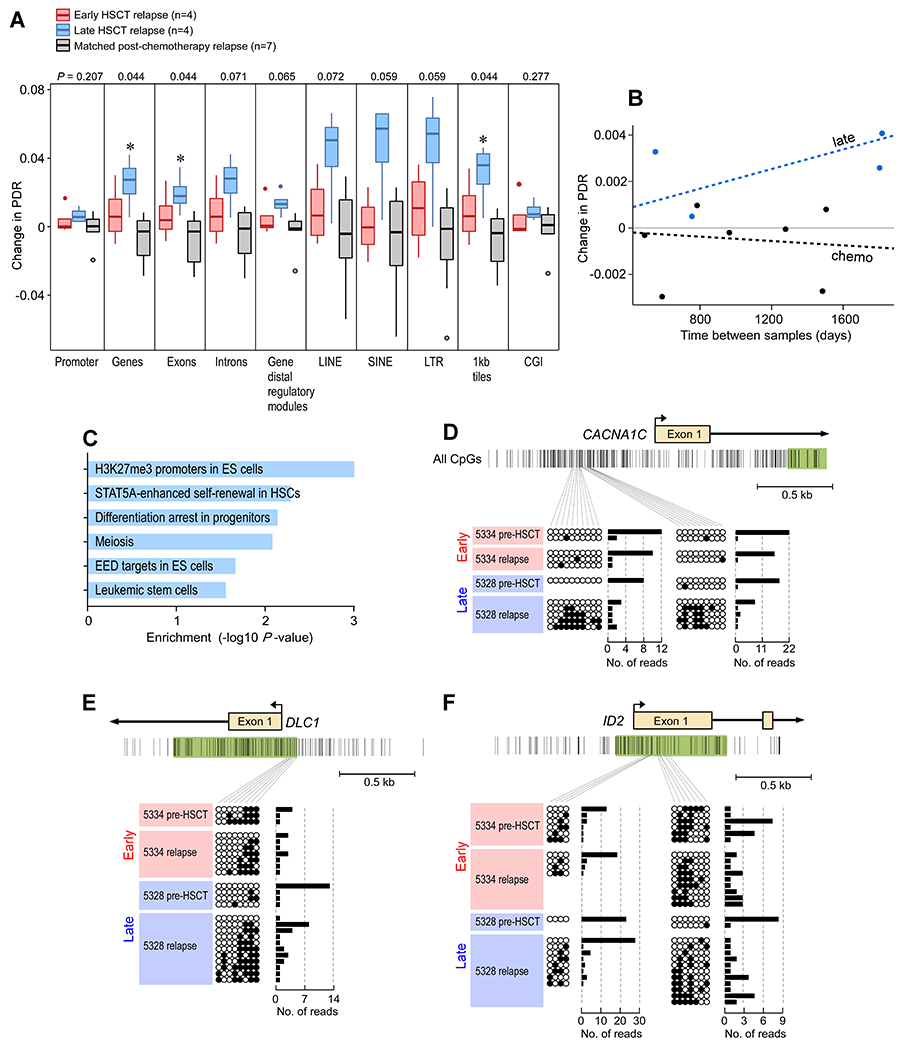
(A) Boxplots of the change in locally disordered methylation as measured by the percentage of discordant methylated reads (PDR). Shown are values for early relapse (n=4), late relapse (n=4) and matched post-chemotherapy treated relapse patients (n=7). * = p<0.05 determined from Kruskal Wallis test. (B) Graph depicts change in PDR as function of change in time to progression. Dotted line represents Bayesian linear model fitted to each group. Points represent weighted averages of all genomic regions per patient. Bayes factor=1.6. (C) Stem cell gene sets are enriched from sites of methylation differences (absolute change >10%) between pre-HSCT and relapse samples in late relapse patients. P values were calculated from Fisher’s exact test. (D-F) Ordered and disordered methylated reads for patients 5334 (early relapse) and 5328 (late relapse), respectively, for promoters corresponding to (D) CACNA1C, (E) DLC1, and (F) ID2 stem cell genes.
Discussion
We have investigated leukemic cell-intrinsic factors that contribute to GvL resistance by studying CLL relapse after transplant. We find that underlying genetic and epigenetic trajectories shape the kinetics of leukemic relapse. Indeed, early relapses after allo-HSCT were marked by a molecular stability that was remarkably consistent across genetic, transcriptional and epigenetic measurements. In contrast, late relapses exhibited striking genetic and epigenetic change along with evidence of neoantigen depletion, consistent with manifest single cell transcriptional shifts that were unique to each patient. Our analysis of genetic clonal dynamics suggest that the mechanism for these widespread phenotypic changes were likely driven by shifts in clonal composition and subpopulation selection as opposed to mutational changes within individual CLL cells.
In addition, these evolutionary paths appeared associated with the transplant setting. For example, unlike in transplant-naïve CLL samples, we found a post-HSCT relapse to exhibit concordant genotype-phenotype relationships that defined transcriptional changes and suggested divergent evolutionary paths. Furthermore, in contrast to post-chemotherapy relapses exhibiting markedly stable methylomes, recurrences late after allo-HSCT were characterized by widespread methylome instability across the epigenome. These data suggest that the evolutionary consequences of the GvL bottleneck may in fact be distinct from those imposed by chemotherapeutic ones and that therapeutic strategies to prevent or treat cancer recurrence after such immunologic bottlenecks should account for this potential diversification.
Our transcriptional and epigenetic data implicate stem cell pathways as contributory for CLL relapse after allo-HSCT given their enrichment in early relapsed CLL cells pre-HSCT and their acquisition in late relapsed CLL cells post-HSCT. Indeed, immune evasion and cell mobility are well-studied properties of stem cells (29–31). While the upregulation of pathways such as BCR and FcR signaling during late relapse suggest additional drivers of post-HSCT recurrence, our results highlight stem cell properties for future investigation as putative predictors of relapse and a mechanistic basis for GvL escape.
These studies demonstrate the applicability of single cell genomics to delineate genetic selection pressures for leukemic subpopulations within individual patients. For example, in contrast to previous reports in myeloid malignancies (5, 6), we could see no role for HLA gene downregulation during leukemic evolution. In fact, scRNA-seq enabled a high-resolution portrait of the static and even contracting dynamics of such alterations, demonstrating that even when they are present in pre-transplant subsets, no subsequent evolutionary advantage exists. These data suggest that the rules of GvL resistance differ depending on the leukemic subtype.
Looking forward, our findings should be investigated in larger patient cohorts to evaluate potential clinical confounding factors in our analysis of biological ones, such as pre-HSCT treatment history and varying times from sample collection to HSCT. Although our study focused on CLL, these results should motivate similar efforts within other lymphoid lineages where additional factors modulating GvL resistance may be discovered. Moreover, beyond leukemic cells, understanding how immune cells, particularly T cells (32), also co-evolve during post-HSCT relapse is critical for reversing GvL resistance. Finally, future studies should examine how the site of disease and the spatial microenvironment can impact these co-evolutionary interactions given their known influence on CLL behavior (33). Nevertheless, our results in studying CLL in this context highlight the roles of stem cell genes and evolutionary dynamics in promoting relapse as well as suggest biological pathways for future investigation as putative predictors of GvL escape.
Materials and Methods
Study Design
The overall objective of this study was to identify leukemia-intrinsic features that shape CLL relapse after allo-HSCT. The cohort size was determined by sample availability, and we identified 10 patients treated for CLL at the DFCI that had paired, pre- and post-allo-HSCT samples available. Patients were consented to an institutional review board (IRB)-approved research protocol (01-206). The specimens were collected after informed consent under a study protocol that was approved by the DFCI Human Subjects Protection Committee and conducted in accordance with the Declaration of Helsinki. We undertook a comprehensive assessment of genetic, epigenetic and single cell transcriptomic features from purified CLL cells and investigated longitudinal changes in a pair-wise fashion. All samples and data were included in our analyses. Availability of patient samples for each high-dimensional analysis is provided in Table S1. Briefly, paired data for the following numbers of patients were available: whole exome sequencing analysis (n=9), whole transcriptome analysis (n=8), methylome analysis (n=8), and single cell RNA sequencing analysis (n=4).
Patients and samples
As described above, a total of twenty paired CLL samples were obtained pre- and post-HSCT from patients treated at the Dana-Farber Cancer Institute (DFCI) between 2006-2016. All patients received a T-cell replete transplant. Assessments of acute and chronic GvHD were extracted from medical record as noted by treating clinician; only diagnoses requiring treatment were included. 18 of 20 samples came from cryopreserved peripheral blood mononuclear cells (PBMCs), 1 from cryopreserved bone marrow mononuclear cells (BMMCs), and 1 from a formalin-fixed (FFPE) bone marrow biopsy (see Table S1). Seven paired CLL blood samples were obtained pre- and post-chemotherapy from patients enrolled on clinical research protocols approved by the Human Subjects Protection Committee at UCSD (CLL Research Consortium). PBMCs and BMMCs from all patients were isolated by Ficoll-Hypaque density gradient centrifugation, cryopreserved with 10% dimethyl sulfoxide, and stored in vapor-phase liquid nitrogen until time of use. For all samples with at least 2×106 suspension cells (n=18 of 20 DFCI samples; n=14 of 14 UCSD samples), tumor cells were purified by flow cytometric isolation of CD19+/CD5+ cells for downstream nucleic acid extraction.
IGHV and prognostic marker analysis
As per convention, IGHV unmutated status was defined as greater than or equal to 98% homology to the closest germline match and IGHV mutated if homology was below this cut-off. Assessments for the cohort were done at the CLL Research Consortium Tissue Core (UCSD, San Diego) or at Integrated Oncology (New York, NY) as described in detail elsewhere (34). When available, routine pathological assessment of common CLL chromosomal abnormalities was included based on interphase FISH with Vysis probes (Abbott Molecular).
Whole-exome sequencing data generation and preprocessing
For whole-exome sequencing (WES), we used standard Broad Institute protocols as previously described (12). DNA of all samples was isolated using the All-prep DNA/RNA Mini kit (Qiagen Hilden, Germany). Tumor DNA was derived from CLL cells, and matched germline line DNA came from both pre-transplant CD4+ T cells as well as donor DNA derived from the transplant product. Tumour and normal DNA concentration were measured using PicoGreen® dsDNA Quantitation Reagent (Invitrogen, Carlsbad, CA). A minimum DNA concentration of 5 ng/μL was required for sequencing. All Illumina sequencing libraries were created with the native DNA. The identities of all tumour and normal DNA samples were confirmed by mass spectrometric fingerprint genotyping of 95 common SNPs by Fluidigm Genotyping (Fluidigm, San Francisco, CA). Sequencing libraries were constructed with the Illumina Rapid Capture Enrichment kit. Pooled libraries were normalized to 2nM and denatured using 0.2 N NaOH prior to sequencing. Flowcell cluster amplification and sequencing were performed according to the manufacturer’s protocols using either the HiSeq 2000 v3 or HiSeq 2500. Each run was a 76 bp paired-end with a dual eight-base index barcode read. Standard quality control metrics, including error rates, percentage-passing filter reads, and total Gb produced, were used to characterize process performance before downstream analysis. Data analysis included de-multiplexing and data aggregation.
Mutation calling.
We used the Getz Lab CGA WES Characterization pipeline [https://docs.google.com/document/d/1VO2kX_fgfUd0x3mBS9NjLUWGZu794WbTepBel3cBg08/edit] developed at the Broad Institute to call, filter and annotate point mutations, insertions and deletions. Paired-end Illumina reads were aligned to the hg19 human genome reference using the Picard pipeline [https://software.broadinstitute.org/gatk/documentation/tooldocs/4.0.1.0/picard_fingerprint_CrosscheckFingerprints.php https://software.broadinstitute.org/gatk/documentation/tooldocs/4.0.0.0/picard_analysis_CollectMultipleMetrics.php]. Cross-sample contamination was assessed with the ContEst (35) tool (5% threshold). Point mutations and indels were identified using MuTect(36) and Strelka(37), followed by annotation using Oncotator (38). Possible artifacts due to orientation bias, germline variants, sequencing and poor mapping were filtered using a variety of tools including Orientation Bias Filter (39), MAFPoNFilter (40), and RealignmentFilter. All somatic alterations thus identified were further manually inspected in IGV (41) and apparent false positives were removed.
Somatic copy number alteration identification.
Copy number events were called and filtered using GATK4 ModelSegments (42) [https://gatkforums.broadinstitute.org/dsde/discussion/11682/; https://gatkforums.broadinstitute.org/dsde/discussion/11683]. In order to minimize false positives, we utilized a copy number panel-of-normals created based on germline samples processed using the same platform. We applied a custom conversion script to format the outputs of ModelSegments (both copy ratio and allelic fraction) to be compatible with ABSOLUTE (42, 43), the tool used to estimate sample purity and ploidy as well as cancer cell fractions (CCFs). ABSOLUTE solutions were picked by manual inspection. The final chosen purity and ploidy solutions were used to estimate CCFs for detected somatic alterations in each sample.
Evaluation of clonal evolution.
Inference of clonal structure, phylogenetic relationship between clones and evolution between pre- and post-treatment time points within a sample was performed using the PhylogicNDT tool (11). For each PhylogicNDT assigned cluster within an individual, pairs of CCF values were drawn at the two points using their respective marginal posterior pdfs (N=10,000). The posterior probability that a cluster was evolving was estimated as the proportion of draws where the difference in CCF values was < 0.2. The Benjamini-Hochberg FDR correction method was used to account for multiple hypothesis testing for all clusters within an individual. An individual was considered to be evolving if there was at least one cluster with adjusted P value <0.1. Clusters were defined as ‘contracting’ or ‘expanding’ depending on the corresponding sign of the change in CCF.
HLA and neoantigen prediction.
We used Polysolver (44) for computational allele inference of major MHC class I genes from normal exome sequencing data (HLA-A, HLA-B and HLA-C). We predicted binding affinities for all possible 9 and 10-mers arising from coding mutations, insertions or deletions against all HLA alleles for a given patient using NetMHCpan-4.02 (45). Strong and weak affinity neoantigen binders were identified based on binding scores (≤ 50 nM for strong, ≤ 500 nM for weak), and those with undetectable gene expression were removed. To classify a neoantigen as contracting, expanding or stable, we evaluated whether the corresponding somatic alteration was located within a cluster that was calculated to be contracting, expanding or stable (respectively) as defined by the PhylogicNDT tool (above).
RNA sequencing data generation and analysis
A cDNA library was prepared from poly-A selected RNA and sequenced on an Illumina platform. Paired-end transcriptome reads were mapped to the UCSC hg19 reference genome using STAR. Differential expression analyses were conducted using DESeq2 R package v.1.26.0. Differentially expressed genes were identified using a cutoff of FDR p ≤0.25 and subsequently used for gene set enrichment analysis (GSEA) of the C2 subset of the Molecular Signature Database. Differentially enriched gene sets were identified using a cutoff of FDR p ≤0.25.
Single cell transcriptome generation and analysis
Sample Preparation and Processing
Cryopreserved samples were thawed the day of experiments. Dead cells were removed via an Optiprep selection protocol such that cells collected just below the PBS layer were >95% viable on trypan blue staining (18). Viable cells were subjected to immunomagnetic selection (MACS CD19 MicroBeads; Miltenyi Biotec, cat. No. 130-050-301), using MS columns, to isolate CD19+ and CD19− cells. Cells were prepared at a concentration of 1.15 x 105 cells/mL in a 15% OptiPrep solution in PBS and kept on ice until time of encapsulation.
Cell encapsulation was performed in a light-protected environment using the Olympus CKX53 microscope, Point Grey Chameleon3 1.3 MP Mono USB3 Vision camera, and OEM Syringe Pump Modules. RT/lysis mix and barcoded hydrogel beads (BHBs; from 1CellBio) were prepared as previously described (18) and directly before time of encapsulation. During encapsulation, cells and RT/lysis mix were kept at 4°C in their respective syringes using refrigerated copper coiling, and the collection tube was kept cool on an ice rack. All reagents were simultaneously loaded onto microfluidic devices (microfluidic chip; 1CellBio). The Harvard Apparatus Pump Software and the Point Grey camera software were used to control the parameters of encapsulation with the following working flow rates: 50-70 μL/hr for BHBs, 340-380 μL/hr for carrier oil, 250 μL/hr for cell suspension, and 250 μL/hr for RT/lysis mix, which produced a bead occupancy of 70-85% and droplets sizes ranging from 2.5 to 3.5 nL. Encapsulation time for 3,000 cells was approximately 9 to 12 minutes with a calculated cell doublet percentage of 5.45% to 7.22%.
Library Preparation and Sequencing
Library preparation and sequencing proceeded as previously published (18). Briefly, after the target number of cells was encapsulated, the emulsion of droplets was exposed to UV light (365 nm at ~10 mW/cm2, UVP B-100 lamp) for 8 minutes on ice. The emulsion was incubated at 50°C for 2 hr, 70°C for 15 min, and 4°C for 1 min before splitting into 3,000 cell aliquots. The emulsion was broken using 10 μL of 20% (v/v) 1H,1H,2H,2H-Perfluorooctanolin in Novec-7500 and stored in −80°C until libraries were ready to be processed. In vitro transcription was performed overnight for 16 hours at 37°C. Fragmentation of amplified RNA libraries was performed for 1.5 minutes at 70°C. The final sequence-ready libraries were amplified by 7-9 PCR cycles on average as determined by a diagnostic qPCR. Libraries were quantified by Agilent BioAnalyzer and multiplexed with unique PCR indices in sequencing batches of up to 12,000 cells. The NextSeq Illumina Sequencer was used to sequence libraries using custom sequencing primers.
scRNA-Sequencing Data Analysis
Raw count data were analyzed using the dropEst software suite(46). The dropTag tool was used to annotate reads with cellular barcodes and collapse the different read files. Reads were aligned to the hg38 genome using tophat2 (47) provided with gene annotations from UCSC. Read quantification was performed using dropEst using the simple correction model (−m) and correctional correction of UMI errors (−u).
The panel of samples was jointly analysed using the Conos suite(19) and pagoda2 packages (https://github.com/hms-dbmi/pagoda2). Briefly, normalization of individual datasets was performed using pagoda2, admitting cells with at least 500 molecules. Conos integration was performed using PCA rotation space, calculating top 30 PCs on a union of 2000 overdispersed genes for each pair of datasets. LargeVis embedding was used to visualize the joint subpopulations. Tumor cells were manually annotated as the CD19+/CD5+ cluster. The tumor cells were then re-integrated separately using Conos using CPCA rotation with 30 PCs. The resulting joint graph of CLL cells was visualized using UMAP embedding. Clusters within the CLL cells were then determined using the Leiden algorithm, with the resolution of r=0.7. Higher resolution clustering (r=1.9) was then selected to annotate distinct subpopulations appearing in late relapse samples (clusters 4-6).
An unsupervised clustering analysis was also performed using the Seurat R package v3.1.1(20). In particular, an integrated approach was taken in order to minimize individual patient effect yet maintain differences in timing. Principal component analysis (PCA) dimensionality reduction was performed with the highly variable genes as inputs. The PCs were then used to calculate a uniform manifold approximation and projection (UMAP) for the integrated data. Clusters were called at a resolution of 1 using the first 30 principal components. Tumor cell clusters were manually selected via CD5+/CD19+ and integrated again without input from additional immune cells from patients. Clusters on this tumor-specific data were called at a resolution of 0.5 using the first 30 principal components.
Methylome sequencing and analysis
RRBS libraries were generated from 25-100 ng of input DNA using the Ovation Methyl-Seq System (NuGen) following the manufacturer’s recommendation. These reads were aligned to the human hg19 genome using BSmap with flags -v 0.05 -s 16 -w 100 -S 1 -p 8 –u. An average of 23.1M reads per sample were aligned correctly. An average of 5.4M CpGs were covered per sample. The methylation state of each CpG was determined by comparing bisulfite-treated reads aligning to that CpG with the genomic reference sequence. The extent of methylation was computed by dividing the number of observed methylated cytosines (which did not undergo bisulfite conversion) by the total number of reads aligned to that CpG. The proportion of discordant reads (PDR) was calculated for each CpG as previously described(27). Gene set enrichment analysis was limited to the C2 MSigDB gene set collection, available at: https://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp?collection=C2. Gene set enrichment analysis was performed for genes that exhibit consistently elevated PDR (greater than mean promoter PDR of 0.1 in >50% of samples), and a Fisher’s exact test was used to measure the enrichment of these gene sets in late relapses (compared to early relapses). Statistical analysis for methylome data was performed with R version 3.4.0.
Flow cytometry
The following fluorochrome-conjugated antibodies were used for flow cytometric analysis and all purchased from Biolegend: CD19-BV711 (HIB19, #302246); CD5-PE-Cy7 (UCHT2, #300622); HLA-A,B,C-APC (W6/32, 311410); HLA-DR,DP,DQ (Tü39, 361716). To prevent nonspecific binding of antibodies via their constant Fc domain, human Fc block (Trustain FcX, Biolegend) was used per manufacturer’s instructions. To examine cell viability and exclude dead cells, we used a DAPI stain (BD Pharmingen). Data were acquired on a 5-laser Special Order LSR Fortessa X-20 flow cytometer (BD Bioscience) and analyzed using FlowJo software (TreeStar).
Statistical analysis
Statistical analysis was performed with R version 3.5.3, unless otherwise specified. Categorical variables were compared using the Fisher Exact test, and continuous variables were compared using the Student’s t-test, Wilcoxon rank sum test, or Kruskal-Wallis test as appropriate. Probability densities were estimated using kernel density estimations, implemented by the geom_density function within the ggplot2 package, and compared using the Kolmogorov-Smirnov test. A Bayesian linear regression model was calculated via the MCMCregress function within the MCMCpack package on weighted averages of all PDR values for all genomic regions on a per patient basis. The Bayes factors of these were calculated using the ecdf function native to R.
Analysis of cell intermixing
To quantify the degree of intermixing between pre- and post-transplant cells for each patient, we calculated the proportion of neighbors belonging to the same time point as the cell of interest. The nearest neighbors of each cell were determined as 500 closest cells on the existing embedding. The mean proportions were calculated for each patient and compared using a two-sided Welch t-test. A similar calculation was done within Seurat using the FindNeighbors function and the integrated nearest neighbors matrix along with a neighborhood size of 100 cells.
Quantification of gene expression distance
To compare the extent of expression difference in tumor cells between pre- and post-transplant timepoints for the patients with early and late relapse, for each patient we sampled a random subset of a 100 cells from each timepoint, and then calculated the expression distance as a Jensen-Shannon divergence between the vectors for the two timepoints. The values in each vector give the probability of observing a molecule of a given gene in a given time point. The statistical significance of the mean inter-timepoint distance between early and late relapse patients was assessed using a one-sided Welch t test.
Supplementary Material
Fig. S1. Frequencies of mutations, CLL cancer drivers and neoantigens.
Fig. S2. Single cell transcriptome analyses.
Fig. S3. Quantification of single cell gene and HLA protein expression values.
Fig. S4. Comparison of post-HSCT and post-chemotherapy relapses.
Table S1. Clinical characteristics.
Tables S2–S13 are in data file S1:
Table S2. Whole exome sequencing metrics.
Table S3. Somatic single nucleotide variants and insertions/deletions.
Table S4. Somatic copy number alterations (sCNAs) and ABSOLUTE-calculated cancer cell fraction estimates.
Table S5. Clustering- and phylogeny-adjusted subclone-CCFs and driver annotations.
Table S6. Neoantigens predicted per patient.
Table S7. Differentially expressed genes in pre-transplant CLL cells (early versus late).
Table S8. Differentially expressed genes in late relapses (pre- versus post-transplant).
Table S9. Single cell RNA sequencing metrics.
Table S10. Single cell transcriptomes captured per cell subset.
Table S11. Promoter methylation values.
Table S12. Promoter PDR values.
Table S13. Changes in PDR per patient per region.
Acknowledgments:
We are grateful to Rapolas Zilionis, Alex Ratner, and other members of Allon Klein’s laboratory as well as Colin Brenan, Mike Brenan, and Ilke Akartuna of 1CellBio for discussions and advice regarding setup of the inDrops platform. We appreciate the technical assistance and expertise of the Division of Hematologic Neoplasia Flow Cytometry Core Facility. We also thank the study nurses and clinical staff that obtained samples, the Pasquarello Tissue Bank in Hematologic Malignancies for processing and banking samples and the patients who generously consented for the research use of these samples. Finally, we appreciate members of the Wu laboratory for their feedback and support.
Funding: This work was supported by the NCI 1R01CA155010 to C.J.W., P01CA206978 to C.J.W., U10CA180861 to C.J.W., Scholar Award from Leukemia and Lymphoma Society to C.J.W., Physician-Scientist Training Award from the Damon Runyon Cancer Research Foundation to P.B., an Amy Strelzer Manasevit Scholar Award from the Be The Match Foundation to P.B., an American Society of Hematology Fellow Scholar Award to P.B., Kay Kendall Leukaemia Fund fellowship to S.H.G., NCI R50CA211482 to S.A.S., the American Cancer Society (PF-17-042-01-LIB) to N.D.M., NCI L30 CA231679-01 to N.D.M, NCI R21 CA216772-01A1 to D.B.K., NCI-SPORE-2P50CA101942-11A1 to D.B.K., NIH 5P30 CA006516 to D.N., NHLBI 1R01HL131768 to P.V.K.
Competing interests: C.J.W. is a co-founder of Neon Therapeutics and member of its scientific advisory board. C.J.W., G.G., D.N. and T.J.K. receive research funding from Pharmacyclics. P.B. reports equity in Agenus, Amgen, Breakbio Corp., Johnson & Johnson, Exelixis, and BioNTech. K.N.N. is currently an employee at Bluebird Bio. T.J.K. has received research funding and/or has served as an advisor to Ascerta/AstraZeneca, Celgene, Genentech/Roche, Gilead, Janssen, Loxo Oncology, Octernal Therapeutics, Pharmacyclics/AbbVie, TG Therapeutics, VelosBio, and Verastem. Cirmtuzumab was developed by T.J.K. and licensed by the University of California to Oncternal Therapeutics, Inc., which has provided stock/options to the university and T.J.K. G.G. receives research funds from IBM. G.G. is an inventor of several bioinformatics-related patents, including patents related to MuTect and ABSOLUTE. S.A.S. previously advised and has received consulting fees from Neon Therapeutics. S.A.S. reported nonfinancial support from Bristol-Myers Squibb, and equity in Agenus Inc., Agios Pharmaceuticals, Breakbio Corp., Bristol-Myers Squibb and NewLink Genetics. N.D.M. serves as a scientific advisor to Immunitas Therapeutics. D.B.K. has previously advised Neon Therapeutics, and has received consulting fees from Neon Therapeutics. D.B.K. owns equity in Aduro Biotech, Agenus Inc., Armata pharmaceuticals, Breakbio Corp., Biomarin Pharmaceutical Inc., Bristol Myers Squibb Com., Celldex Therapeutics Inc., Editas Medicine Inc., Exelixis Inc., Gilead Sciences Inc., IMV Inc., Lexicon Pharmaceuticals Inc., Moderna Inc., Regeneron Pharmaceuticals and Stemline Therapeutics Inc. J.R.B. is a consultant for Abbvie, Acerta, Beigene, Genentech/Roche, Gilead, Juno/Celgene, Kite, Loxo, Novartis, Pfizer, Pharmacyclics, Sunesis, TG Therapeutics and Verastem; received honoraria from Janssen and Teva; received research funding from Gilead, Loxo, Sun and Verastem; and served on data safety monitoring committees for Morphosys and Invectys. D.N. reports stock ownership in Madrigal Pharmaceuticals. J.R. receives research funding from Amgen, Equillium and Kite/Gilead and serves on Data Safety Monitoring Committees for AvroBio and Scientific Advisory Boards for LifeVault Bio, Rheos Medicines, Talaris Therapeutics and TScan Therapeutics. P.V.K. serves on the Scientific Advisory Board to Celsius Therapeutics Inc. The remaining authors declare no competing financial interests.
Footnotes
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. Results of primary analyses are included in Tables S2–S13. Exome, transcriptome, single cell transcriptome, and methylome data are desposited in NCBI’s Database of Genotypes and Phenotype (dbGaP; https://www.ncbi.nlm.nih.gov/gap) under accession number: phs001998.v1.p1.
References and notes
- 1.Jenq RR, van den Brink MRM, Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer, Nat. Rev. Cancer 10, 213–221 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ, Haematological malignancies: at the forefront of immunotherapeutic innovation, Nat. Rev. Cancer 15, 201–215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davids MS, Alyea EP, The evolving role of hematopoietic cell transplantation in chronic lymphocytic leukemia, Curr. Hematol. Malig. Rep 10, 18–27 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Gribben JG, How and when I do allogeneic transplant in CLL, Blood 132, 31–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toffalori C, Zito L, Gambacorta V, Riba M, Oliveira G, Bucci G, Barcella M, Spinelli O, Greco R, Crucitti L, Cieri N, Noviello M, Manfredi F, Montaldo E, Ostuni R, Naldini MM, Gentner B, Waterhouse M, Zeiser R, Finke J, Hanoun M, Beelen DW, Gojo I, Luznik L, Onozawa M, Teshima T, Devillier R, Blaise D, Halkes CJM, Griffioen M, Carrabba MG, Bernardi M, Peccatori J, Barlassina C, Stupka E, Lazarevic D, Tonon G, Rambaldi A, Cittaro D, Bonini C, Fleischhauer K, Ciceri F, Vago L, Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation, Nat. Med 25, 603–611 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, Klco JM, Helton NM, O’Laughlin M, Fronick CC, Fulton RS, Wilson RK, Wartman LD, Welch JS, Heath SE, Baty JD, Payton JE, Graubert TA, Link DC, Walter MJ, Westervelt P, Ley TJ, DiPersio JF, Immune Escape of Relapsed AML Cells after Allogeneic Transplantation, N. Engl. J. Med 379, 2330–2341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, Lukez AP, Avigan D, Chen Y-B, McSweeney P, LeBoeuf NR, Rooney MS, Bowden M, Zhou CW, Granter SR, Hornick JL, Rodig SJ, Hirakawa M, Severgnini M, Hodi FS, Wu CJ, Ho VT, Cutler C, Koreth J, Alyea EP, Antin JH, Armand P, Streicher H, Ball ED, Ritz J, Bashey A, Soiffer RJ, Leukemia and Lymphoma Society Blood Cancer Research Partnership, Ipilimumab for Patients with Relapse after Allogeneic Transplantation, N. Engl. J. Med 375, 143–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prestipino A, Emhardt AJ, Aumann K, O’Sullivan D, Gorantla SP, Duquesne S, Melchinger W, Braun L, Vuckovic S, Boerries M, Busch H, Halbach S, Pennisi S, Poggio T, Apostolova P, Veratti P, Hettich M, Niedermann G, Bartholomä M, Shoumariyeh K, Jutzi JS, Wehrle J, Dierks C, Becker H, Schmitt-Graeff A, Follo M, Pfeifer D, Rohr J, Fuchs S, Ehl S, Hartl FA, Minguet S, Miething C, Heidel FH, Kröger N, Triviai I, Brummer T, Finke J, Illert AL, Ruggiero E, Bonini C, Duyster J, Pahl HL, Lane SW, Hill GR, Blazar BR, von Bubnoff N, Pearce EL, Zeiser R, Oncogenic JAK2 causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms, Sci. Transl. Med 10 (2018), doi: 10.1126/scitranslmed.aam7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathew NR, Baumgartner F, Braun L, O’Sullivan D, Thomas S, Waterhouse M, Müller TA, Hanke K, Taromi S, Apostolova P, Illert AL, Melchinger W, Duquesne S, Schmitt-Graeff A, Osswald L, Yan K-L, Weber A, Tugues S, Spath S, Pfeifer D, Follo M, Claus R, Lübbert M, Rummelt C, Bertz H, Wäsch R, Haag J, Schmidts A, Schultheiss M, Bettinger D, Thimme R, Ullrich E, Tanriver Y, Vuong GL, Arnold R, Hemmati P, Wolf D, Ditschkowski M, Jilg C, Wilhelm K, Leiber C, Gerull S, Halter J, Lengerke C, Pabst T, Schroeder T, Kobbe G, Rösler W, Doostkam S, Meckel S, Stabla K, Metzelder SK, Halbach S, Brummer T, Hu Z, Dengjel J, Hackanson B, Schmid C, Holtick U, Scheid C, Spyridonidis A, Stölzel F, Ordemann R, Müller LP, Sicre-de-Fontbrune F, Ihorst G, Kuball J, Ehlert JE, Feger D, Wagner E-M, Cahn J-Y, Schnell J, Kuchenbauer F, Bunjes D, Chakraverty R, Richardson S, Gill S, Kröger N, Ayuk F, Vago L, Ciceri F, Müller AM, Kondo T, Teshima T, Klaeger S, Kuster B, Kim DDH, Weisdorf D, van der Velden W, Dörfel D, Bethge W, Hilgendorf I, Hochhaus A, Andrieux G, Börries M, Busch H, Magenau J, Reddy P, Labopin M, Antin JH, Henden AS, Hill GR, Kennedy GA, Bar M, Sarma A, McLornan D, Mufti G, Oran B, Rezvani K, Shah O, Negrin RS, Nagler A, Prinz M, Burchert A, Neubauer A, Beelen D, Mackensen A, von Bubnoff N, Herr W, Becher B, Socié G, Caligiuri MA, Ruggiero E, Bonini C, Häcker G, Duyster J, Finke J, Pearce E, Blazar BR, Zeiser R, Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells, Nat. Med 24, 282–291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Böttcher S, Carter SL, Cibulskis K, Mertens D, Sougnez CL, Rosenberg M, Hess JM, Edelmann J, Kless S, Kneba M, Ritgen M, Fink A, Fischer K, Gabriel S, Lander ES, Nowak MA, Döhner H, Hallek M, Neuberg D, Getz G, Stilgenbauer S, Wu CJ, Mutations driving CLL and their evolution in progression and relapse, Nature 526, 525–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leshchiner I, Livitz D, Gainor JF, Rosebrock D, Spiro O, Martinez A, Mroz E, Lin JJ, Stewart C, Kim J, Elagina L, Bozic I, Mino-Kenudson M, Rooney M, Ou S-HI, Wu CJ, Rocco JW, Engelman JA, Shaw AT, Getz G, Comprehensive analysis of tumour initiation, spatial and temporal progression under multiple lines of treatment, Biorxiv doi: 10.1101/508127. [DOI] [Google Scholar]
- 12.Gruber M, Bozic I, Leshchiner I, Livitz D, Stevenson K, Rassenti L, Rosebrock D, Taylor-Weiner A, Olive O, Goyetche R, Fernandes SM, Sun J, Stewart C, Wong A, Cibulskis C, Zhang W, Reiter JG, Gerold JM, Gribben JG, Rai KR, Keating MJ, Brown JR, Neuberg D, Kipps TJ, Nowak MA, Getz G, Wu CJ, Growth dynamics in naturally progressing chronic lymphocytic leukaemia, Nature 570, 474–479 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, Rimm AA, Ringden O, Rozman C, Speck B, Graft-versus-leukemia reactions after bone marrow transplantation, Blood 75, 555–562 (1990). [PubMed] [Google Scholar]
- 14.Ringdén O, Labopin M, Gluckman E, Reiffers J, Vernant JP, Jouet JP, Harrousseau JL, Fiere D, Bacigalupo A, Frassoni F, Gorin NC, Graft-versus-leukemia effect in allogeneic marrow transplant recipients with acute leukemia is maintained using cyclosporin A combined with methotrexate as prophylaxis. Acute Leukemia Working Party of the European Group for Blood and Marrow Transplantation, Bone Marrow Transplant. 18, 921–929 (1996). [PubMed] [Google Scholar]
- 15.Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ, HLA-binding properties of tumor neoepitopes in humans, Cancer Immunol Res 2, 522–529 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, Zhang W, Sougnez C, Cibulskis K, Sidney J, Stevenson K, Ritz J, Neuberg D, Brusic V, Gabriel S, Lander ES, Getz G, Hacohen N, Wu CJ, Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia, Blood 124, 453–462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA, An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors, Nat. Genet 40, 499–507 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zilionis R, Nainys J, Veres A, Savova V, Zemmour D, Klein AM, Mazutis L, Single-cell barcoding and sequencing using droplet microfluidics, Nat. Protoc 12, 44–73 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Barkas N, Petukhov V, Nikolaeva D, Lozinsky Y, Demharter S, Khodosevich K, Kharchenko PV, Joint analysis of heterogeneous single-cell RNA-seq dataset collections, Nat. Methods 16, 695–698 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species, Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan J-B, Zhang K, Chun J, Kharchenko PV, Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis, Nat. Methods 13, 241–244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodzioch M, Lapicka-Bodzioch K, Zapala B, Kamysz W, Kiec-Wilk B, Dembinska-Kiec A, Evidence for potential functionality of nuclearly-encoded humanin isoforms, Genomics 94, 247–256 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC, Humanin peptide suppresses apoptosis by interfering with Bax activation, Nature 423, 456–461 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Swaminathan S, Huang C, Geng H, Chen Z, Harvey R, Kang H, Ng C, Titz B, Hurtz C, Sadiyah MF, Nowak D, Thoennissen GB, Rand V, Graeber TG, Koeffler HP, Carroll WL, Willman CL, Hall AG, Igarashi K, Melnick A, Müschen M, BACH2 mediates negative selection and p53-dependent tumor suppression at the pre-B cell receptor checkpoint, Nat. Med 19, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gómez-Abad C, Pisonero H, Blanco-Aparicio C, Roncador G, González-Menchén A, Martinez-Climent JA, Mata E, Rodríguez ME, Muñoz-González G, Sánchez-Beato M, Leal JF, Bischoff JR, Piris MA, PIM2 inhibition as a rational therapeutic approach in B-cell lymphoma, Blood 118, 5517–5527 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Wang L, Fan J, Francis JM, Georghiou G, Hergert S, Li S, Gambe R, Zhou CW, Yang C, Xiao S, Cin PD, Bowden M, Kotliar D, Shukla SA, Brown JR, Neuberg D, Alessi DR, Zhang C-Z, Kharchenko PV, Livak KJ, Wu CJ, Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemiaGenome, Research 27, 1300–1311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, Stevenson K, Sougnez C, Wang L, Li S, Kotliar D, Zhang W, Ghandi M, Garraway L, Fernandes SM, Livak KJ, Gabriel S, Gnirke A, Lander ES, Brown JR, Neuberg D, Kharchenko PV, Hacohen N, Getz G, Meissner A, Wu CJ, Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia, Cancer Cell 26, 813–825 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Good IJ, Theory of Probability Harold Jeffreys (Third edition, 447 ix pp., Oxford Univ. Press, 84s.) Geophysical Journal International 6, 555–558 (1962). [Google Scholar]
- 29.Hsu J-M, Xia W, Hsu Y-H, Chan L-C, Yu W-H, Cha J-H, Chen C-T, Liao H-W, Kuo C-W, Khoo K-H, Hsu JL, Li C-W, Lim S-O, Chang S-S, Chen Y-C, Ren G-X, Hung M-C, STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion, Nat. Commun 9, 1908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, Tandler C, Mbarga M, Schaefer T, Falcone M, Nievergall E, Dörfel D, Hanns P, Passweg JR, Lutz C, Schwaller J, Zeiser R, Blazar BR, Caligiuri MA, Dirnhofer S, Lundberg P, Kanz L, Quintanilla-Martinez L, Steinle A, Trumpp A, Salih HR, Lengerke C, Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion, Nature 572, 254–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schatton T, Schütte U, Frank NY, Zhan Q, Hoerning A, Robles SC, Zhou J, Hodi FS, Spagnoli GC, Murphy GF, Frank MH, Modulation of T-cell activation by malignant melanoma initiating cells, Cancer Res 70, 697–708 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riches JC, Davies JK, McClanahan F, Fatah R, Iqbal S, Agrawal S, Ramsay AG, Gribben JG, T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production, Blood 121, 1612–1621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herishanu Y, Pérez-Galán P, Liu D, Biancotto A, Pittaluga S, Vire B, Gibellini F, Njuguna N, Lee E, Stennett L, Raghavachari N, Liu P, McCoy JP, Raffeld M, Stetler-Stevenson M, Yuan C, Sherry R, Arthur DC, Maric I, White T, Marti GE, Munson P, Wilson WH, Wiestner A, The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia, Blood 117, 563–574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Dyke DL, Werner L, Rassenti LZ, Neuberg D, Ghia E, Heerema NA, Dal Cin P, Dell Aquila M, Sreekantaiah C, Greaves AW, Kipps TJ, Kay NE, The Dohner fluorescence in situ hybridization prognostic classification of chronic lymphocytic leukaemia (CLL): the CLL Research Consortium experience, Br. J. Haematol 173, 105–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cibulskis K, McKenna A, Fennell T, Banks E, DePristo M, Getz G, ContEst: estimating cross-contamination of human samples in next-generation sequencing data, Bioinformatics 27, 2601–2602 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G, Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples, Nat. Biotechnol 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK, Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs, Bioinformatics 28, 1811–1817 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, Getz G, Oncotator: cancer variant annotation tool, Hum. Mutat 36, E2423–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, Fostel JL, Friedrich DC, Perrin D, Dionne D, Kim S, Gabriel SB, Lander ES, Fisher S, Getz G, Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation, Nucleic Acids Res. 41, e67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G, Discovery and saturation analysis of cancer genes across 21 tumour types, Nature 505, 495–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP, Integrative genomics viewer, Nature Biotechnology 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA, The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data, Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, Lander ES, Meyerson M, Getz G, Absolute quantification of somatic DNA alterations in human cancer, Nat. Biotechnol 30, 413–421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, Sougnez C, Cibulskis K, Kiezun A, Hacohen N, Brusic V, Wu CJ, Getz G, Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes, Nat. Biotechnol 33, 1152–1158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M, NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data, The Journal of Immunology 199, 3360–3368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petukhov V, Guo J, Baryawno N, Severe N, Scadden DT, Samsonova MG, Kharchenko PV, dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments, Genome Biol. 19, 78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL, TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions, Genome Biol. 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Frequencies of mutations, CLL cancer drivers and neoantigens.
Fig. S2. Single cell transcriptome analyses.
Fig. S3. Quantification of single cell gene and HLA protein expression values.
Fig. S4. Comparison of post-HSCT and post-chemotherapy relapses.
Table S1. Clinical characteristics.
Tables S2–S13 are in data file S1:
Table S2. Whole exome sequencing metrics.
Table S3. Somatic single nucleotide variants and insertions/deletions.
Table S4. Somatic copy number alterations (sCNAs) and ABSOLUTE-calculated cancer cell fraction estimates.
Table S5. Clustering- and phylogeny-adjusted subclone-CCFs and driver annotations.
Table S6. Neoantigens predicted per patient.
Table S7. Differentially expressed genes in pre-transplant CLL cells (early versus late).
Table S8. Differentially expressed genes in late relapses (pre- versus post-transplant).
Table S9. Single cell RNA sequencing metrics.
Table S10. Single cell transcriptomes captured per cell subset.
Table S11. Promoter methylation values.
Table S12. Promoter PDR values.
Table S13. Changes in PDR per patient per region.