

Abstract
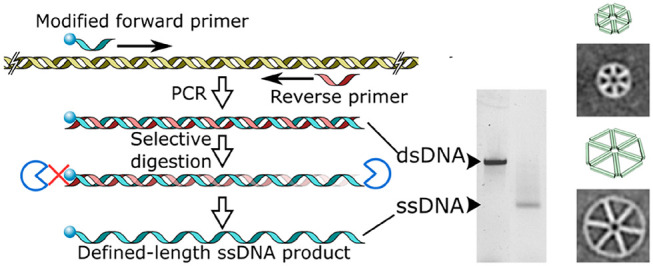
DNA origami nanostructures generally require a single scaffold strand of specific length, combined with many small staple strands. Ideally, the length of the scaffold strand should be dictated by the size of the designed nanostructure. However, synthesizing arbitrary-length single-stranded DNA in sufficient quantities is difficult. Here, we describe a straightforward and accessible method to produce defined-length ssDNA scaffolds using PCR and subsequent selective enzymatic digestion with T7 exonuclease. This approach produced ssDNA with higher yields than other methods and without the need for purification, which significantly decreased the time from PCR to obtaining pure DNA origami. Furthermore, this enabled us to perform true one-pot synthesis of defined-size DNA origami nanostructures. Additionally, we show that multiple smaller ssDNA scaffolds can efficiently substitute longer scaffolds in the formation of DNA origami.
DNA origami nanostructures are produced by thermally annealing a long defined-length single-stranded DNA (ssDNA) scaffold with up to hundreds of complementary small ssDNA staples.1 Whereas the ssDNA staples typically range from 15 to 60 nucleotides (nt) in length, and can therefore be commercially synthesized on a large scale, custom multiple kilobase ssDNA scaffolds are too long for chemical synthesis and are instead made using biological approaches.
The vast majority of DNA origami are constructed using M13mp18 as a folding scaffold, a circular 7249 nt ssDNA viral vector that is produced on a large scale using E. coli infected with the M13 bacteriophage.2 Using this scaffold, however, imposes a maximum size limitation on DNA origami designs, while leaving smaller structures with unpaired scaffold tails. Creating scaffolds with tailor-made lengths opens up the possibility of making DNA origami of any size. To this end, custom-sized ssDNA scaffolds have been made using a wide variety of techniques,3 including the use of phages and helper phages,4 asymmetric polymerase chain reaction (PCR),5 restriction endonucleases (RE) digestion,6 selective digestion of plasmids and PCR products,7,8 rolling circle amplification,9 and even using RNA and dsDNA as a scaffold.10,11 Despite this multitude of available techniques, few have found widespread use, primarily because they often require specialized knowledge and equipment in order to be implemented.
Here, we describe a straightforward method for the production of a wide range of ssDNA scaffold sizes and DNA origami nanostructures using selective enzymatic digestion of dsDNA PCR products by T7 exonuclease. Employing this PCR-based approach, we provide a simple and accessible solution to making defined ssDNA lengths, demonstrate true one-pot synthesis of DNA origami, and also show that multiple smaller ssDNA scaffolds can be combined and easily substitute and function as a traditional longer individual ssDNA scaffold. These advances significantly decrease the time from synthesis to obtaining pure DNA origami, and further open up the potential size range of DNA origami constructs.
Results and Discussion
Production of Defined-Length ssDNA by Selective Digestion of PCR Products
The first of two steps in this one-pot scheme is to make defined-length dsDNA by performing PCR to synthesize a wide range of DNA lengths using conventional PCR enzymes (Figure 1a). No special primer design considerations, which are required for more specialized PCR techniques such as asymmetric PCR (aPCR) and linear-after-the-exponential PCR, have to be taken into account.5,12 There is only a small modification to the forward primer, which contains five sequential phosphorothioate linkages between the first 5′ nucleotides to selectively inhibit exonuclease digestion of the desired scaffold strand later in the production process (Table S1). The PCR template can be derived from both single- and double-stranded DNA sources, such as plasmids, genomic DNA, or custom-designed synthetic gene blocks. This allows the resulting scaffold strand to contain a user-defined nucleotide sequence, free of interfering structural motifs (e.g., strong hairpin or G-rich regions) or potentially undesirable (viral) genetic information.
Figure 1.

Production of ssDNA using T7 exonuclease. (a) Scheme for the production of defined-length ssDNA using T7 exonuclease (T7 exo). (b) Time-resolved digestion kinetics of a dsDNA PCR product using T7 exo. (c) Comparison of different enzyme-based methods for the generation of ssDNA using ApoI RE digestion; aPCR; or PCR followed by selective digestion using either lambda exonuclease (λ exo) or T7 exo.
The second step in the production of tailor-made ssDNA scaffolds is digestion of the unrequired antisense strand, which contains no protective phosphorothioate linkages, using T7 exonuclease. T7 exonuclease reliably catalyzes the 5′ to 3′ removal of nucleotides from dsDNA and acts on both phosphorylated and hydroxyl terminated 5′ ends. The latter is present in the dsDNA products made here with standard synthetic PCR primers. Digestion is simply achieved by adding a small amount of T7 exonuclease and buffer to the completed PCR mixture with subsequent incubation at 25 °C. Time-resolved digestion experiments show that reactions go to completion within 1 h of incubation but can be incubated overnight for convenience with no off-target degradation of sense-DNA (Figure 1b).
Next, we compared our method to other enzyme-based ssDNA production techniques in terms of overall yield and ease of use (Figure 1c, Table S2). For this, we generated scaffolds of around 1250 nucleotides, a size which should be easily synthesized employing any of the techniques discussed here. Using our one-pot method described above, and spin column purification to yield pure product for quantification purposes, we synthesized 8 picomoles (pmol) of a 1229 nt ssDNA scaffold starting from a single standard 50 μL PCR reaction. Making the identical fragment using aPCR (using the same PCR primers, which were designed for aPCR)5 resulted in a final yield of ∼2 pmol. The discrepancy in yields results from the fact that aPCR generates a substantial amount of dsDNA, which necessitates subsequent purification of ssDNA from agarose gels that significantly decreases overall yield.
By digesting single-stranded M13mp18 plasmids using the RE ApoI,6 we generated a 1332 nt ssDNA fragment and a 5917 nt side product, which required purification using gel extraction methods (Figure 1c). Although the yield of this technique is somewhat proportional to the amount of initial M13mp18 ssDNA added, there are limitations in terms of sequence design and scaffold size choices as they rely on existing restriction sites. Furthermore, the resulting two (or more) ssDNA fragments after digestion may be similar in length, prohibiting purification using gel electrophoresis.
Lastly, we compared the synthesis of ssDNA scaffolds made by PCR and subsequent digestion by lambda exonuclease to our approach using T7 exonuclease. These two approaches seem highly similar at first, but result in different purities and yields. In contrast to T7 exonuclease, lambda exonuclease requires the highest purity grade of 5′ phosphorylated antisense primers to work. This phosphorylation requirement has caused the latter enzyme to fall out of favor, as even with HPLC-purified oligonucleotides, this often results in incomplete digestion.13 Furthermore, lambda exonuclease has severely reduced exonuclease activity in standard PCR formulations, even supplemented with its own buffer (Figure 1c). It is therefore necessary to exchange buffers between the individual reactions to ensure a sufficient level of exonuclease activity, and additional agarose gel purification to remove any undigested dsDNA. For the same 1229 nt ssDNA scaffold fragment, this method yielded 0.8 pmol per 50 μL PCR reaction. We can thus conclude that our method using T7 exonuclease-based digestion of dsDNA PCR products generates the highest amounts of ssDNA scaffold and requires minimal experimental design and purification. The ssDNA quantities obtained here can be easily scaled by increasing the number of parallel PCR mixtures run simultaneously.
Folding DNA Origami of Defined Size Using Bespoke ssDNA Scaffolds
To illustrate the wide applicability of the ssDNA production method, multiple DNA origami constructs were designed with different geometries, sizes, and design software. We designed and synthesized an open, sparsely packed double-decker hexagon (DDH) using DAEDALUS (Tables S3–S7),14 and a toroidal shape with a densely packed square lattice structure using caDNAno (Figures S1–S5 and Tables S8–S13).15 These designs span a ssDNA scaffold size range from 1229 to 5904 nt. For the individual DDH DNA origami constructs, agarose gel electrophoresis was used to follow both the ssDNA scaffold fabrication from dsDNA PCR products and subsequent folding of the DNA origami structure (Figure 2a). In all cases, the dsDNA is digested to completion by T7 exonuclease, which causes the appearance of a band with a higher gel mobility corresponding to the ssDNA scaffold. Occasionally, a second, higher, band is observed in this stage as the ssDNA has a propensity to aggregate. However, after addition of the staples to the scaffold and thermal annealing, a band with lower gel mobility appears and all other bands disappear, indicating complete digestion by T7 exonuclease and proper DNA origami folding. TEM imaging of folded DDH and toroidal designs reveals complete folding of all DNA origami constructs with high yield (Figures S6 and S7), and after particle picking, class averages were generated showing excellent resemblance to their computer designed counterparts in terms of size and morphology (Figures 2b and S7).
Figure 2.

Generation of defined-length DNA nanostructures. (a) Gel electrophoresis characterization of the generation of ssDNA scaffold lengths of 1229, 1512, 1872, and 2268 nt from dsDNA using T7 exo, followed by DNA origami folding into DDH designs. (b) Class averages generated after TEM imaging compared to their computer designed counterparts. Green cylinders represent DNA helices. Scale bar is 25 nm for all panels.
From ssDNA to DNA Origami: A True One-Pot Synthesis of DNA Origami
The classical, multistep method to make custom-length DNA origami is by synthesizing ssDNA scaffolds followed by one or more purification steps.3 Next, the DNA origami structure is commonly formed by thermal annealing and subjected to a further round of purification to remove excess staples.16 After PCR and T7 exonuclease digestion to form ssDNA, described above, we reasoned that the subsequent addition of Proteinase K would inactivate T7 exonuclease. This would eliminate the necessity to purify our ssDNA scaffolds before mixing them with staple strands to form DNA origami structures. Thermal annealing of the DNA origami also directly inactivates Proteinase K, preventing any potential downstream interference. After origami folding is complete, excess PCR primers, inactivated enzymes, buffering components, and excess staples can be removed simultaneously in a single purification step using size exclusion spin filters or PEG precipitation.17 Gel electrophoresis and TEM analysis revealed that this workflow yields similar quality DNA origami compared to those made with purified ssDNA scaffolds, and that the inactivation of T7 exonuclease with Proteinase K was crucial to this process as the intact T7 exonuclease degraded the DNA origami (Figure 3). Taken together, this method turns a multistep ssDNA synthesis and DNA origami folding process into a true one-pot synthesis of DNA origami structures, from PCR to product within 1 day.
Figure 3.

True one-pot synthesis of DNA nanostructures using Proteinase K. (a) Gel electrophoresis following origami formation with scaffolds generated using T7 exo, then purified (Purified); folded with active T7 exonuclease (Crude); or after inactivation of T7 exonuclease using Proteinase K (+ ProtK). (b) TEM imaging of origami shows correct folding only after Proteinase K treatment. Scale bars are 50 nm (images) or 20 nm (averages).
Folding Individual DNA Origami Structures from Multiple Scaffold Strands
While determining the maximum length of ssDNA production using T7 exonuclease, we observed that some ssDNA scaffolds degraded within 15 min at 95 °C (Figures 4a and S8), which is a typical incubation temperature used during both PCR and origami thermal annealing.14,15,18 Indeed, this thermal degradation appeared to be proportional to the length of the ssDNA (Figure 4b), which will prohibit synthesis of larger DNA origami structures and may explain previous reports of low yields for large ssDNA scaffolds.19,20 Incubating the same ssDNA scaffolds at either 65 or 40 °C, the temperature range most critical in annealing staples to the scaffold strands,21 resulted in no visible degradation (Figure S8). We therefore avoided high annealing temperatures and initiated folding of all our DNA origamis at 80 °C. Also, although we were able to synthesize ssDNA up to 15,000 nt in length (Table S14), above 5000 nt the yield of ssDNA fell below 1 pmol, which is required for a typical single 50 μL DNA origami folding mixture (Figure 4c).
Figure 4.

Yield of ssDNA is affected by thermal degradation and length. (a) Incubation at 95 °C degrades ssDNA within 15 min. (b) Thermal degradation of ssDNA at 95 °C is proportional to length. (c) Yield of ssDNA decreases for longer scaffolds.
Traditionally, a single ssDNA scaffold is used to fold individual DNA origami structures. We reasoned that we could use multiple short (<4000) ssDNA scaffolds to fold larger individual DNA origami designs, thereby avoiding the thermal degradation of ssDNA and consequently increasing the yield of correctly folded DNA. To test this, we used the 62- or 90-nm-diameter toroidal designs. The 62 nm toroid comprises either a single scaffold of 4096 nt derived from M13mp18 (Tables S8 and S11) or two scaffolds of 2048 and 2068 nt in length derived from the pEGFP-C1 vector and lambda DNA, respectively (Figures 5a,b and S9, Tables S15–S16). The larger 90 nm toroid comprised either one scaffold 5904 nt long, two scaffolds each 2952 nt in length, or three scaffolds of length 1966, 1968, and 1970 nt, all derived from M13mp18 (Figures 5c, S10 and S11; Tables S13, S17, and S18). The individual scaffold strands for the 90 nm toroid scaffolds are found sequentially within the M13mp18 template, and are therefore equivalent to cleaving one long scaffold into 2 or 3 pieces. Consequently, we could use the sample staple strands for both the full-length 90 nm toroid and when split into 2 or 3 scaffold strands (Table S13). This can save substantial amounts of time and money, as staples do not have to be resynthesized upon splitting designs into separate scaffolds.
Figure 5.

Synthesis of multiscaffold DNA nanostructures. (a) Gel electrophoresis of sequential addition of scaffolds to multiscaffold DNA origami. Blue and red dashed boxes are those imaged in panel b. (b) TEM analysis of the 2-scaffold 62-nm-DNA toroid. Scaffold routes within the nanostructure are shown in different colors. (c) TEM analysis of the 3-scaffold 90-nm-DNA toroid. Scale bar represents 20 nm in all panels.
TEM imaging confirmed proper DNA origami structure formation when all scaffold strands were present, while folding mixtures lacking one or more of the required ssDNA scaffold strands resulted in distorted undefined structures (Figures 5b,c and S12). Also, due to shorter fragment generation, ssDNA production significantly increased (0.46 pmol per 5904 nt ssDNA compared to 4 pmol per 1986 nt ssDNA in a single 50 μL PCR reaction), and overall yield of fully folded DNA origami was comparable to their larger single-scaffolded DNA origami counterparts.
Conclusions
Here we have shown a simple and straightforward method for the generation of custom-length DNA origamis using PCR and subsequent enzymatic digestion with T7 exonuclease. Due to the simplicity and ease of scalability of this enzyme-based method, multi-picomolar to low-nanomolar yields can be effectively achieved from combining multiple small reaction volumes. This new method permits the use of any PCR-compatible DNA source as a template and enables true one-pot synthesis of DNA origami, from PCR to product, by inactivation of T7 exonuclease using Proteinase K. Lastly, the novel introduction of using multiple higher-yielding smaller ssDNA scaffolds to fold a single DNA origami leads to similar DNA origami yields compared to single long (>4000 nt) ssDNA scaffolds and makes the field of DNA origami design and synthesis even more versatile.
Acknowledgments
This project was funded by the European Research Council Horizon 2020 program (759517) awarded to THS.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.0c00644.
Experimental procedures, supplementary figures and tables (PDF)
Author Contributions
WEMN, LA and THS designed DNA nanostructures. WEMN performed the experiments. WEMN and THS analyzed the data and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Castro C. E.; Kilchherr F.; Kim D. N.; Shiao E. L.; Wauer T.; Wortmann P.; Bathe M.; Dietz H. (2011) A primer to scaffolded DNA origami. Nat. Methods 8, 221–9. 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- Hong F.; Zhang F.; Liu Y.; Yan H. (2017) DNA Origami: Scaffolds for Creating Higher Order Structures. Chem. Rev. 117, 12584–12640. 10.1021/acs.chemrev.6b00825. [DOI] [PubMed] [Google Scholar]
- Bush J.; Singh S.; Vargas M.; Oktay E.; Hu C. H.; Veneziano R. (2020) Synthesis of DNA Origami Scaffolds: Current and Emerging Strategies. Molecules 25, 3386. 10.3390/molecules25153386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd T. R.; Du R. R.; Huang H.; Wamhoff E. C.; Bathe M. (2019) Bioproduction of pure, kilobase-scale single-stranded DNA. Sci. Rep. 9, 6121. 10.1038/s41598-019-42665-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veneziano R.; Shepherd T. R.; Ratanalert S.; Bellou L.; Tao C.; Bathe M. (2018) In vitro synthesis of gene-length single-stranded DNA. Sci. Rep. 8, 6548. 10.1038/s41598-018-24677-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said H.; Schuller V. J.; Eber F. J.; Wege C.; Liedl T.; Richert C. (2013) M1.3--a small scaffold for DNA origami. Nanoscale 5, 284–90. 10.1039/C2NR32393A. [DOI] [PubMed] [Google Scholar]
- Erkelenz M.; Bauer D. M.; Meyer R.; Gatsogiannis C.; Raunser S.; Sacca B.; Niemeyer C. M. (2014) A facile method for preparation of tailored scaffolds for DNA-origami. Small 10, 73–7. 10.1002/smll.201300701. [DOI] [PubMed] [Google Scholar]
- Han D.; Qi X.; Myhrvold C.; Wang B.; Dai M.; Jiang S.; Bates M.; Liu Y.; An B.; Zhang F.; Yan H.; Yin P. (2017) Single-stranded DNA and RNA origami. Science 358, eaao2648. 10.1126/science.aao2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen F. H.; Goossens E. P.; Tessari M.; Heus H. A. (2015) Enzymatic preparation of multimilligram amounts of pure single-stranded DNA samples for material and analytical sciences. Anal. Biochem. 475, 68–73. 10.1016/j.ab.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Wang P.; Ko S. H.; Tian C.; Hao C.; Mao C. (2013) RNA-DNA hybrid origami: folding of a long RNA single strand into complex nanostructures using short DNA helper strands. Chem. Commun. 49, 5462–4. 10.1039/c3cc41707g. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Han D.; Nangreave J.; Liu Y.; Yan H. (2012) DNA origami with double-stranded DNA as a unified scaffold. ACS Nano 6, 8209–15. 10.1021/nn302896c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez J. A.; Pierce K. E.; Rice J. E.; Wangh L. J. (2004) Linear-after-the-exponential (LATE)-PCR: an advanced method of asymmetric PCR and its uses in quantitative real-time analysis. Proc. Natl. Acad. Sci. U. S. A. 101, 1933–8. 10.1073/pnas.0305476101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marimuthu C.; Tang T. H.; Tominaga J.; Tan S. C.; Gopinath S. C. (2012) Single-stranded DNA (ssDNA) production in DNA aptamer generation. Analyst 137, 1307–15. 10.1039/c2an15905h. [DOI] [PubMed] [Google Scholar]
- Veneziano R.; Ratanalert S.; Zhang K.; Zhang F.; Yan H.; Chiu W.; Bathe M. (2016) Designer nanoscale DNA assemblies programmed from the top down. Science 352, 1534. 10.1126/science.aaf4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas S. M.; Marblestone A. H.; Teerapittayanon S.; Vazquez A.; Church G. M.; Shih W. M. (2009) Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 37, 5001–6. 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenbauer K. F.; Engelhardt F. A. S.; Stahl E.; Hechtl V. K.; Stommer P.; Seebacher F.; Meregalli L.; Ketterer P.; Gerling T.; Dietz H. (2017) How We Make DNA Origami. ChemBioChem 18, 1873–1885. 10.1002/cbic.201700377. [DOI] [PubMed] [Google Scholar]
- Stahl E.; Martin T. G.; Praetorius F.; Dietz H. (2014) Facile and scalable preparation of pure and dense DNA origami solutions. Angew. Chem., Int. Ed. 53, 12735–40. 10.1002/anie.201405991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullis K.; Faloona F.; Scharf S.; Saiki R.; Horn G.; Erlich H. (1986) Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symp. Quant. Biol. 51, 263. 10.1101/SQB.1986.051.01.032. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Chao J.; Pan D.; Liu H.; Huang Q.; Fan C. (2012) Folding super-sized DNA origami with scaffold strands from long-range PCR. Chem. Commun. 48, 6405. 10.1039/c2cc32204h. [DOI] [PubMed] [Google Scholar]
- Marchi A. N.; Saaem I.; Vogen B. N.; Brown S.; LaBean T. H. (2014) Toward larger DNA origami. Nano Lett. 14, 5740–7. 10.1021/nl502626s. [DOI] [PubMed] [Google Scholar]
- Lee Tin Wah J.; David C.; Rudiuk S.; Baigl D.; Estevez-Torres A. (2016) Observing and Controlling the Folding Pathway of DNA Origami at the Nanoscale. ACS Nano 10, 1978–87. 10.1021/acsnano.5b05972. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.