

Abstract

In recent years, several organocatalytic asymmetric hydroarylations of activated, electron-poor olefins with activated, electron-rich arenes have been described. In contrast, only a few approaches that can handle unactivated, electronically neutral olefins have been reported and invariably require transition metal catalysts. Here we show how an efficient and highly enantioselective catalytic asymmetric intramolecular hydroarylation of aliphatic and aromatic olefins with indoles can be realized using strong and confined IDPi Brønsted acid catalysts. This unprecedented transformation is enabled by tertiary carbocation formation and establishes quaternary stereogenic centers in excellent enantioselectivity and with a broad substrate scope that includes an aliphatic iodide, an azide, and an alkyl boronate, which can be further elaborated into bioactive molecules.
A particularly elegant and atom-economical approach to the catalytic formation of benzylic quaternary stereogenic centers would be via the protonation of a 1,1-disubstituted olefin with a chiral acid, followed by an asymmetric counteranion-directed Friedel–Crafts arylation of the resulting tertiary carbocation (eq 1). However, to the best of our knowledge, such a transformation has not yet been realized.1 This discrepancy is even more remarkable, given that nonasymmetric, catalytic hydroarylations of unactivated olefins with arenes are common carbon–carbon σ bond-forming reactions and are applied on a multimillion-ton scale.2 Previous organocatalytic asymmetric hydroarylations invariably required electronically activated olefins and arenes,3 while transition-metal-catalyzed variants typically relied on covalently bound directing groups.4−8 Notable advances toward less biased substrates in intramolecular asymmetric hydroarylations with indoles using π-Lewis acidic Pt- or Ni-catalysts have been reported by Widenhoefer, Peters, and Cramer.9−11 Additionally, Hartwig and Meek developed enantioselective Ir- and Rh-catalyzed intermolecular hydroarylations using norbornene and 1-arylbutadienes as olefin components, respectively.12,13 However, the catalytic construction of quaternary stereogenic centers via hydroarylation of unactivated olefins has remained elusive to date, using either transition metals or organic catalysts.8b,14 We have recently discovered that strong and confined imidodiphosphorimidate (IDPi) Brønsted acid catalysts can protonate unactivated olefins to engage in highly enantioselective Markovnikov hydroalkoxylations.15,16 Our findings suggested the applicability of this unique activation mode for additional transformations that proceed via carbocation intermediates,17 and an asymmetric hydroarylation of simple olefins became particularly intriguing to us. We now report an intramolecular hydroarylation of unbiased olefins with indoles, furnishing highly enantioenriched tetrahydrocarbazoles possessing a benzylic quaternary stereogenic center (eq 2).
 |
1 |
At the onset of our studies, we subjected 1,1-disubstituted olefin 1a to 2 mol % of various chiral Brønsted acid catalysts at 80 °C for 20 h (Table 1). While weaker acids such as phosphoric acid (CPA) 3 (pKa = 12.7 in MeCN),18 disulfonimide 4 (pKa > 8.4),15b,19,20 imidodiphosphoric acid (IDP) 5 (pKa ca. 11.5),15b,21 iminoimidodiphosphate (iIDP) 6 (pKa ca. 9),15b,22 gave poor or moderate conversions, yields, and enantioselectivities (entries 1–4), IDPi catalyst 7a (pKa = 4.5),20 in contrast, furnished an almost racemic product in 90% yield, suggesting that the key requirement for reactivity is high acidity (entry 5). Installation of biphenyl substituents at the 3,3′-positions of the BINOL backbone, combined with a perfluoronaphthalene-2-sulfonyl “inner core” (IDPi 7c), quickly revealed a significant increase in enantioselectivity (entry 7, 88:12 er). Closer inspection of the crystal structure of catalyst 7c (see Supporting Information) revealed extensive π–π-interactions between the 3,3′-substituents and the aromatic inner core, significantly influencing the shape of the confined, chiral microenvironment. We reasoned that more electron-rich catalyst substituents could potentially stabilize both the π–π-interactions and the critical cationic intermediate. Indeed, with methoxy-substituted catalyst 7d, yields could be increased (entry 8, 93%). Finally, we identified IDPi 7e as the optimal catalyst, which, in addition to the 4′-methoxy group, features two n-hexyl chains in the 3′- and 5′-positions of the biphenyl system, affording the tetrahydrocarbazole 2a in both excellent yield and enantioselectivity (entry 10, 95%, 95:5 er).
Table 1. Reaction Developmenta.

| entry | catalyst | T (°C) | conv. (%) | yield 2a (%) | isom. (%) | er |
|---|---|---|---|---|---|---|
| 1 | 3 | 80 | 41 | 34 | 5 | 54:46 |
| 2 | 4 | 80 | 47 | 30 | trace | 44:56 |
| 3 | 5 | 80 | 13 | <10 | trace | 53:47 |
| 4 | 6 | 80 | 55 | 45 | 9 | 47:53 |
| 5 | 7a | 80 | 95 | 90 | trace | 49:51 |
| 6 | 7b | 80 | full | 93 | — | 56:44 |
| 7 | 7c | 80 | full | 85 | — | 88:12 |
| 8 | 7d | 80 | full | 93 | — | 88:12 |
| 9 | 7e | 80 | full | 93 | — | 90:10 |
| 10b | 7e | 60 | full | 95 | — | 95:5 |
Reactions were performed with substrate 1a (0.02 mmol), catalyst (2 mol %) in methylcyclohexane (CyMe, 0.2 mL); conversions (conv.), yields of 2a, and olefin isomerizations (isom.) were determined by 1H NMR analysis with mesitylene as an internal standard; enantiomeric ratios (er’s) were measured by HPLC. When the reaction was not carried out in the dark, byproducts and lower yields were observed.
48 h.
The outstanding performance of catalyst 7e was then further challenged with several different substrates to explore the scope of the hydroarylation. In general, the reaction tolerates an array of substituents with differing steric and electronic properties, in addition to sensitive functional groups. As displayed in Table 2, irrespective of the alkyl groups attached to the olefins, the corresponding tetrahydrocarbazoles 2a–2g were formed in excellent yields and enantioselectivities. The reaction is not limited to aliphatic olefins, but also the phenyl-substituted olefin 1h gave the corresponding product 2h in 88% yield and 95:5 er. Methyl substituents at any of the benzo-positions (4, 5, 6, and 7) of the indole core did not hinder the recognition with the catalyst, and products 2i–2l formed with equally high enantioselectivities. The annulated substrates, benzo[g]indole and tetrahydrocyclopenta[g]indole, also furnished the desired products 2m and 2n in excellent yields and enantioselectivities. Furthermore, halogen substituents as potential synthetic handles for future modifications, as well as electron-rich alkoxy substituents, were well tolerated (2o–2s). Notably, terminal dioxoborolane-, azido-, and iodo-substitution at the alkyl chain of the olefin fragment gave direct and highly enantioselective access to tetrahydrocarbazoles 2t–2v, illustrating the somewhat unexpected functional group tolerance of our strong acid catalysts. The absolute configuration of boronate product 2t was determined by single-crystal X-ray diffraction, and the configuration of all other products was assigned by analogy.
Table 2. Scope of the Reactiona.
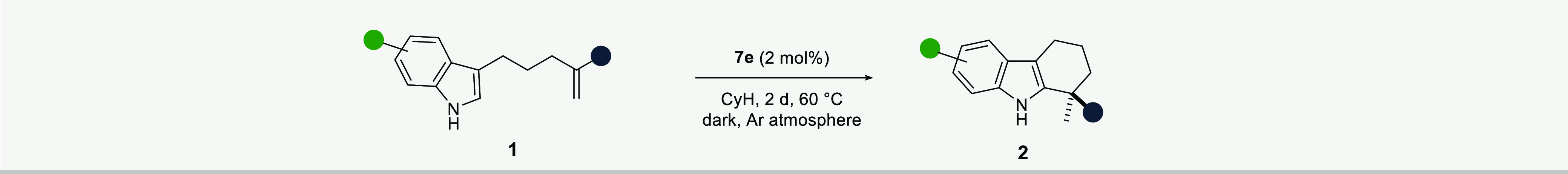

Reactions were carried out with 0.1–0.2 mmol of substrates 1, catalyst 7e (2 mol %) in cyclohexane (0.1 M) at 60 °C unless otherwise noted. Yields are for the isolated compounds. The enantiomeric ratios (er) were determined by HPLC analysis.
Reaction was run at 60 °C for 3 d.
Reaction was run with 3 mol % catalyst at 65 °C for 3 d.
NaBO3·H2O (5 equiv), THF/H2O 1:1, rt, 2 h, 92% yield.
2-Bromonaphthalene (1.05 equiv), Pd(OAc)2 (2.5 mol %), RuPhos (5 mol %), 85 °C, 24 h, 77% yield.
(i) NaH (1.2 equiv), iodoethane (1.2 equiv), 0 °C to rt, DMF, 2 h, 85% yield; (ii) NaBH4 (2 equiv), CoCl2·6H2O (10 mol %), MeOH, 0 °C, 30 min, 84% yield; (iii) NaBH3CN (2 equiv), MeOH, HCHO (37 wt % in H2O), rt, 3 h, 58% yield; (iv) HCl (1 M in Et2O), quant. THF = tetrahydrofuran; DMF = dimethylformamide.
To further demonstrate the synthetic utility of the hydroarylation products, boronate 2t was oxidatively hydrolyzed to the corresponding primary alcohol 2w with complete retention of enantiopurity. Additionally, a C(sp3)–C(sp2) Suzuki–Miyaura cross-coupling with 2-bromonaphthalene provided compound 2x in 77% yield and, once again, without any erosion of enantiopurity. The subjection of azido-functionalized tetrahydrocarbazole 2u to a sequential N-ethylation, azide reduction, and N,N-dimethylation provided the first enantioselective synthesis of ammonium salt 8, in 41% overall yield and with retention of enantiopurity, the racemic mixture of which is being investigated as an antidepressant.23
Keen on understanding the underlying mechanism of our hydroarylation, we performed several control experiments (Scheme 1). Moderately acidic chiral Brønsted acid catalysts such as CPAs have been shown to typically operate via a bifunctional, double-hydrogen bonding mechanism.18b,18c As a consequence, the enantioselectivity and reactivity of CPA-catalyzed reactions involving a nucleophilic indole can massively erode upon N-substitution due to the lack of a recognition and activation element.24 In contrast, we found that N-methylated indole 9 readily converted to the corresponding product under our standard conditions, albeit with a significantly reduced enantioselectivity of 71:29 er (Scheme 1A). This observation is consistent with a bifunctional enantiodiscrimination mechanism involving hydrogen bonding interactions in a putative ion-pairing scenario.
Scheme 1. Mechanistic Study.
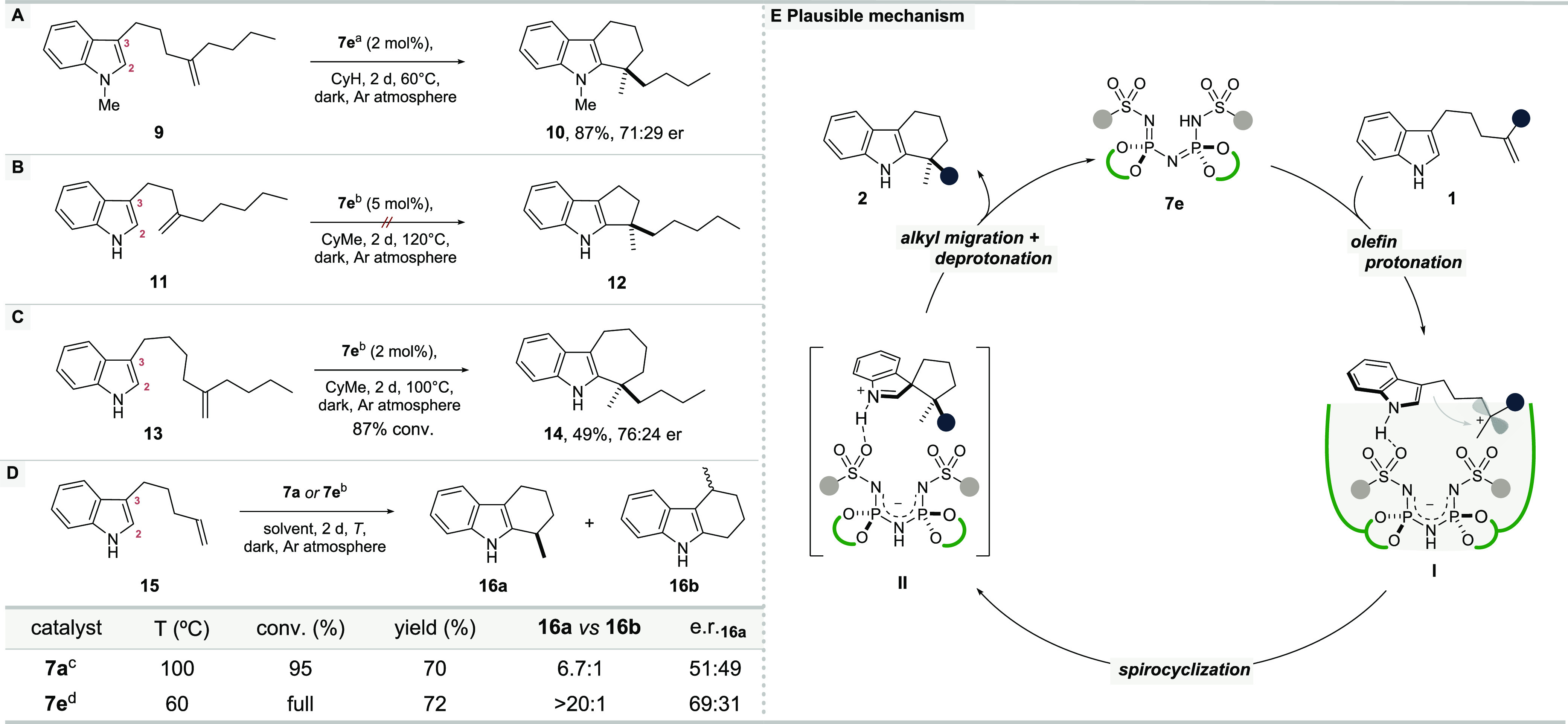
Reaction was carried out with 0.2 mmol of 9, catalyst 7e (2 mol %) in cyclohexane (0.1 M); yield is of for the isolated compound; enantiomeric ratios (er) were measured by HPLC.
Reactions were performed with substrate 11, 13, 15 (0.02 mmol); conversions, yields, and regioisomeric ratios were determined by 1H NMR analysis with mesitylene as an internal standard.
100 °C with 4 mol % 7a in methylcyclohexane; 60 °C with 2 mol % 7a in cyclohexane, trace product.
60 °C with 2 mol % 7e in cyclohexane.
Electrophilic aromatic substitutions of indoles have been shown to operate through a direct reaction at C-2 or the more nucleophilic C-3 position, followed by a migratory opening of a spiroindolenine intermediate.25,26 We approached the important mechanistic question of how the cyclization occurs experimentally in three different ways (Scheme 1B, C, and D). First, removal of one methylene group from the alkyl tether between indole and olefin in substrate 11 would lead to the corresponding tetrahydrocyclopenta[b]indole 12, the formation of which, if nucleophilic attack would proceed directly from C-2, would be expected to be kinetically at least as favorable as the corresponding six-membered ring formation.27 However, even after stirring olefin 11 at 120 °C for 2 d in the presence of 5 mol % of catalyst 7e, product 12 could not be observed, excluding direct reaction at C-2. This result is consistent with the initial C-3 spirocyclization mechanism, which in this specific case is not viable because of the high ring strain of the required four-membered spirocyclic intermediate. Second, addition of a methylene group to the linkage in substrate 13 afforded the corresponding seven-membered hexahydrocyclohepta[b]indole 14 in moderate yield and 76:24 er, consistent with a six-membered spirocyclic intermediate. Third, we subjected terminal olefin 15 to our reaction conditions using IDPi catalysts 7a and 7e, which would undergo cyclization via a secondary carbocation intermediate. Remarkably, both reactions went to full conversion, although catalyst 7a required a slightly elevated temperature to reach completion. Even more interestingly, IDPi 7a provided the tetrahydrocarbazole product as a regioisomeric mixture of 16a/16b = 6.7/1,28 strongly suggesting an initial spirocyclization followed by competitive migration of either methylene or methide fragment. Unfortunately, 1H NMR investigations of the cyclization of substrate 1a under standard conditions (see Supporting Information) did not lead to the detection of a spiroindolenine intermediate, presumably due to a relatively fast migratory process restoring indole aromaticity.
Based on these experiments, we propose a catalytic cycle as depicted in Scheme 1E. Accordingly, the reaction is initiated by a protonation of the olefin to give ion-pair I, followed by a nucleophilic attack of the indole directed by the chiral counteranion, which is coordinating to the substrate via hydrogen bonding to the indole N–H moiety. This spirocyclization gives rise to the spiroindolenine cation in association with the IDPi anion (ion-pair II). Rapid suprafacial and stereoretentive migration of the most electron-rich alkyl fragment then liberates tetrahydrocarbazole 2 and, upon proton transfer, restores catalyst 7e.25b
We speculate that the remarkable substrate scope and functional group tolerance of our system could result from a confinement effect, according to which the catalyst recognizes (or “binds”) the transition state of the cyclization. In contrast, the olefin substituent is mostly positioned outside of the chiral, confined active site, well within the solvent. The transition-state-binding scenario is also typical for even more confined enzymes, with the important difference that our catalyst’s incomplete confinement enables a significant “promiscuity”. Conceptually, these features position our confined acid catalysts in-between small-molecule catalysts on the one end and macromolecular catalysts with a substrate-specific active site on the other end of the spectrum. The peculiar behavior of our confined catalysts has also been displayed in other reactions, most notably in our recent hydroalkoxylation reaction, which has a similar specificity with regard to the reacting subunit, while tolerating a broad range of (functionalized) substituents.16
We have developed a perfectly atom-economical and highly enantioselective catalytic intramolecular hydroarylation of unactivated olefins with indoles. Our reaction is enabled via olefin protonation with highly acidic and confined chiral Brønsted acid catalysts and provides access to valuable enantiopure tetrahydrocarbazoles.29 The previously unattainable activation of 1,1-disubstituted olefin via protonation to the corresponding tertiary carbocation suggests obvious and significant potential for other hydrofunctionalization reactions that provide quaternary stereogenic center-bearing products and related compounds.
Acknowledgments
Generous support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Leibniz Award to B.L. and under Germany’s Excellence Strategy–EXC 2033–390677874–RESOLV, and the European Research Council (ERC, European Union’s Horizon 2020 research and innovation program “C–H Acids for Organic Synthesis, CHAOS” Advanced Grant Agreement No. 694228) and the Alexander von Humboldt Foundation for a fellowship to P.Z. is gratefully acknowledged. We further thank Dr. Markus Leutzsch for NMR studies, Benjamin Mitschke for his help during the preparation of this manuscript, Lukas Münzer for his contributions in initial studies, and several members of the group for crowd reviewing this manuscript. We also thank the technicians of our group and the members of our NMR, MS, X-ray, and chromatography groups for their excellent service.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c12042.
The authors declare no competing financial interest.
Notes
Crystallographic data for compounds 2t (CCDC 2044898) and 7c (CCDC 2044899) can be downloaded free of charge from the Cambridge Crystallographic Data Centre (www.ccdc.cam.ac.uk).
Supplementary Material
References
- Properzi R.; Kaib P. S. J.; Leutzsch M.; Pupo G.; Mitra R.; De C. K.; Song L.; Schreiner P. R.; List B. Catalytic enantiocontrol over a non-classical carbocation. Nat. Chem. 2020, 12, 1174–1179. 10.1038/s41557-020-00558-1. [DOI] [PubMed] [Google Scholar]
- a Olah G. A.; Molnár A.. Hydrocarbon Chemistry; Wiley-Interscience: New York, 2003. [Google Scholar]; b Perego C.; Ingallina P. Combining alkylation and transalkylation for alkylaromatic production. Green Chem. 2004, 6, 274–279. 10.1039/b403277m. [DOI] [Google Scholar]; c Andreatta J. R.; McKeown B. A.; Gunnoe T. B. Transition metal catalyzed hydroarylation of olefins using unactivated substrates: Recent developments and challenges. J. Organomet. Chem. 2011, 696, 305–315. 10.1016/j.jorganchem.2010.09.030. [DOI] [Google Scholar]; d Cai X.; Tohti A.; Ramirez C.; Harb H.; Fettinger J. C.; Hratchian H. P.; Stokes B. J. Dispersion-Controlled Regioselective Acid-Catalyzed Intramolecular Hydroindolation of cis-Methindolylstyrenes To Access Tetrahydrobenzo[cd]indoles. Org. Lett. 2019, 21, 1574–1577. 10.1021/acs.orglett.9b00043. [DOI] [PubMed] [Google Scholar]
- a Paras N. A.; MacMillan D. W. C. New Strategies in Organic Catalysis: The First Enantioselective Organocatalytic Friedel–Crafts Alkylation. J. Am. Chem. Soc. 2001, 123, 4370–4371. 10.1021/ja015717g. [DOI] [PubMed] [Google Scholar]; b You S.-L.; Cai Q.; Zeng M. Chiral Brønsted acid catalyzed Friedel–Crafts alkylation reactions. Chem. Soc. Rev. 2009, 38, 2190–2201. 10.1039/b817310a. [DOI] [PubMed] [Google Scholar]; c Terrasson V.; Marcia de Figueiredo R.; Campagne J. M. Organocatalyzed Asymmetric Friedel–Crafts Reactions. Eur. J. Org. Chem. 2010, 2010, 2635–2655. 10.1002/ejoc.200901492. [DOI] [Google Scholar]; d Correia J. T. M.; List B.; Coelho F. Catalytic Asymmetric Conjugate Addition of Indolizines to α,β-Unsaturated Ketones. Angew. Chem., Int. Ed. 2017, 56, 7967–7970. 10.1002/anie.201700513. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Dong Z.; Ren Z.; Thompson S. J.; Xu Y.; Dong G. Transition-Metal-Catalyzed C–H Alkylation Using Alkenes. Chem. Rev. 2017, 117, 9333–9403. 10.1021/acs.chemrev.6b00574. [DOI] [PubMed] [Google Scholar]; b Coombs J. R.; Morken J. P. Catalytic Enantioselective Functionalization of Unactivated Terminal Alkenes. Angew. Chem., Int. Ed. 2016, 55, 2636–2649. 10.1002/anie.201507151. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Newton C. G.; Wang S.-G.; Oliveira C. C.; Cramer N. Catalytic Enantioselective Transformations Involving C–H Bond Cleavage by Transition-Metal Complexes. Chem. Rev. 2017, 117, 8908–8976. 10.1021/acs.chemrev.6b00692. [DOI] [PubMed] [Google Scholar]
- For selected examples using a directing group in Ir-catalysis, see:; a Aufdenblatten R.; Diezi S.; Togni A. Iridium(I)-Catalyzed Asymmetric Intermolecular Hydroarylation of Norbornene with Benzamide. Monatsh. Chem. 2000, 131, 1345–1350. 10.1007/s007060070014. [DOI] [Google Scholar]; b Shirai T.; Yamamoto Y. Cationic Iridium/S-Me-BIPAM-Catalyzed Direct Asymmetric Intermolecular Hydroarylation of Bicycloalkenes. Angew. Chem., Int. Ed. 2015, 54, 9894–9897. 10.1002/anie.201504563. [DOI] [PubMed] [Google Scholar]; c Grélaud S.; Cooper P.; Feron L. J.; Bower J. F. Branch-Selective and Enantioselective Iridium-Catalyzed Alkene Hydroarylation via Anilide-Directed C–H Oxidative Addition. J. Am. Chem. Soc. 2018, 140, 9351–9356. 10.1021/jacs.8b04627. [DOI] [PubMed] [Google Scholar]; d Shibata T.; Ryu N.; Takano H. Iridium-Catalyzed Intramolecular Enantioselective C-H Alkylation at the C-2 Position of N-Alkenylindoles. Adv. Synth. Catal. 2015, 357, 1131–1135. 10.1002/adsc.201401163. [DOI] [Google Scholar]
- For selected examples using directing group in Rh-catalysis, see:; a Thalji R. K.; Ellman J. A.; Bergman R. G. Highly Efficient and Enantioselective Cyclization of Aromatic Imines via Directed C–H Bond Activation. J. Am. Chem. Soc. 2004, 126, 7192–7193. 10.1021/ja0394986. [DOI] [PubMed] [Google Scholar]; b Harada H.; Thalji R. K.; Bergman R. G.; Ellman J. A. Enantioselective Intramolecular Hydroarylation of Alkenes via Directed C–H Bond Activation. J. Org. Chem. 2008, 73, 6772–6779. 10.1021/jo801098z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ye B.; Cramer N. Chiral Cyclopentadienyl Ligands as Stereocontrolling Element in Asymmetric C–H Functionalization. Science 2012, 338, 504–506. 10.1126/science.1226938. [DOI] [PubMed] [Google Scholar]
- For selected examples using directing group in Co-catalysis, see; a Lee P.-S.; Yoshikai N. Cobalt-Catalyzed Enantioselective Directed C–H Alkylation of Indole with Styrenes. Org. Lett. 2015, 17, 22–25. 10.1021/ol503119z. [DOI] [PubMed] [Google Scholar]; b Pesciaioli F.; Dhawa U.; Oliveira J. C. A.; Yin R.; John M.; Ackermann L. Enantioselective Cobalt(III)-Catalyzed C–H Activation Enabled by Chiral Carboxylic Acid Cooperation. Angew. Chem., Int. Ed. 2018, 57, 15425–15429. 10.1002/anie.201808595. [DOI] [PubMed] [Google Scholar]
- For selected examples Using directing group in other metal catalysis, see:; a Loup J.; Zell D.; Oliveira J. C. A.; Keil H.; Stalke D.; Ackermann L. Asymmetric Iron-Catalyzed C–H Alkylation Enabled by Remote Ligand meta-Substitution. Angew. Chem., Int. Ed. 2017, 56, 14197–14201. 10.1002/anie.201709075. [DOI] [PubMed] [Google Scholar]; b Li G.; Liu Q.; Vasamsetty L.; Guo W.; Wang J. Ruthenium(II)-Catalyzed Asymmetric Inert C–H Bond Activation Assisted by a Chiral Transient Directing Group. Angew. Chem., Int. Ed. 2020, 59, 3475–3479. 10.1002/anie.201913733. [DOI] [PubMed] [Google Scholar]; c Song G.; O W. W. N.; Hou Z. Enantioselective C–H Bond Addition of Pyridines to Alkenes Catalyzed by Chiral Half-Sandwich Rare-Earth Complexes. J. Am. Chem. Soc. 2014, 136, 12209–12212. 10.1021/ja504995f. [DOI] [PubMed] [Google Scholar]
- a Liu C.; Han X.; Wang X.; Widenhoefer R. A. Platinum-Catalyzed Intramolecular Alkylation of Indoles with Unactivated Olefins. J. Am. Chem. Soc. 2004, 126, 3700–3701. 10.1021/ja031814t. [DOI] [PubMed] [Google Scholar]; b Han X.; Widenhoefer R. A. Platinum-Catalyzed Intramolecular Asymmetric Hydroarylation of Unactivated Alkenes with Indoles. Org. Lett. 2006, 8, 3801–3804. 10.1021/ol061441b. [DOI] [PubMed] [Google Scholar]
- Huang H.; Peters R. A Highly Strained Planar-Chiral Platinacycle for Catalytic Activation of Internal Olefins in the Friedel–Crafts Alkylation of Indoles. Angew. Chem., Int. Ed. 2009, 48, 604–606. 10.1002/anie.200804944. [DOI] [PubMed] [Google Scholar]
- Diesel J.; Grosheva D.; Kodama S.; Cramer N. A Bulky Chiral N-Heterocyclic Carbene Nickel Catalyst Enables Enantioselective C–H Functionalizations of Indoles and Pyrroles. Angew. Chem., Int. Ed. 2019, 58, 11044–11048. 10.1002/anie.201904774. [DOI] [PubMed] [Google Scholar]
- Sevov C. S.; Hartwig J. F. Iridium-Catalyzed Intermolecular Asymmetric Hydroheteroarylation of Bicycloalkenes. J. Am. Chem. Soc. 2013, 135, 2116–2119. 10.1021/ja312360c. [DOI] [PubMed] [Google Scholar]
- Marcum J. S.; Roberts C. C.; Manan R. S.; Cervarich T. N.; Meek S. J. Chiral Pincer Carbodicarbene Ligands for Enantioselective Rhodium-Catalyzed Hydroarylation of Terminal and Internal 1,3-Dienes with Indoles. J. Am. Chem. Soc. 2017, 139, 15580–15583. 10.1021/jacs.7b08575. [DOI] [PubMed] [Google Scholar]
- a Lou S.-J.; Mo Z.; Nishiura M.; Hou Z. Construction of All-Carbon Quaternary Stereocenters by Scandium-Catalyzed Intramolecular C–H Alkylation of Imidazoles with 1,1-Disubstituted Alkenes. J. Am. Chem. Soc. 2020, 142, 1200–1205. 10.1021/jacs.9b12503. [DOI] [PubMed] [Google Scholar]; b Shinde V. S.; Mane M. V.; Cavallo L.; Rueping M. Iridium-Catalyzed Enantioselective Hydroarylation of Alkenes through C–H bond Activation: Experiment and Computation. Chem. - Eur. J. 2020, 26, 8308–8313. 10.1002/chem.202001793. [DOI] [PubMed] [Google Scholar]; c Ye B.; Donets P. A.; Cramer N. Chiral Cp-Rhodium(III)-Catalyzed Asymmetric Hydroarylations of 1,1-Disubstituted Alkenes. Angew. Chem., Int. Ed. 2014, 53, 507–511. 10.1002/anie.201309207. [DOI] [PubMed] [Google Scholar]
- a Kaib P. S. J.; Schreyer L.; Lee S.; Properzi R.; List B. Extremely Active Organocatalysts Enable a Highly Enantioselective Addition of Allyltrimethylsilane to Aldehydes. Angew. Chem., Int. Ed. 2016, 55, 13200–13203. 10.1002/anie.201607828. [DOI] [PubMed] [Google Scholar]; b Schreyer L.; Properzi R.; List B. IDPi Catalysis. Angew. Chem., Int. Ed. 2019, 58, 12761–12777. 10.1002/anie.201900932. [DOI] [PubMed] [Google Scholar]
- Tsuji N.; Kennemur J. L.; Buyck T.; Lee S.; Prévost S.; Kaib P. S. J.; Bykov D.; Farès C.; List B. Activation of olefins via asymmetric Brønsted acid catalysis. Science 2018, 359, 1501–1505. 10.1126/science.aaq0445. [DOI] [PubMed] [Google Scholar]
- For examples of substitution reactions proceeding via tertiary carbocations, see:; a Isomura M.; Petrone D. A.; Carreira E. M. Coordination-Induced Stereocontrol over Carbocations: Asymmetric Reductive Deoxygenation of Racemic Tertiary Alcohols. J. Am. Chem. Soc. 2019, 141, 4738–4748. 10.1021/jacs.9b00862. [DOI] [PubMed] [Google Scholar]; b Braun M.; Kotter W. Titanium(IV)-Catalyzed Dynamic Kinetic Asymmetric Transformation of Alcohols, Silyl Ethers, and Acetals under Carbon Allylation. Angew. Chem., Int. Ed. 2004, 43, 514–517. 10.1002/anie.200352128. [DOI] [PubMed] [Google Scholar]; c Zhao W.; Wang Z.; Chu B.; Sun J. Enantioselective Formation of All-Carbon Quaternary Stereocenters from Indoles and Tertiary Alcohols Bearing A Directing Group. Angew. Chem., Int. Ed. 2015, 54, 1910–1913. 10.1002/anie.201405252. [DOI] [PubMed] [Google Scholar]; d Wendlandt A. E.; Vangal P.; Jacobsen E. N. Quaternary Stereocenters via an Enantioconvergent Catalytic SN1 Reaction. Nature 2018, 556, 447–451. 10.1038/s41586-018-0042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; Also see:; e Kikuchi J.; Terada M. Enantioselective Addition Reaction of Azlactones with Styrene Derivatives Catalyzed by Strong Chiral Brønsted Acids. Angew. Chem., Int. Ed. 2019, 58, 8458–8462. 10.1002/anie.201903111. [DOI] [PubMed] [Google Scholar]
- a Kaupmees K.; Tolstoluzhsky N.; Raja S.; Rueping M.; Leito I. On the Acidity and Reactivity of Highly Effective Chiral Brønsted Acid Catalysts: Establishment of an Acidity Scale. Angew. Chem., Int. Ed. 2013, 52, 11569–11572. 10.1002/anie.201303605. [DOI] [PubMed] [Google Scholar]; b Akiyama T. Stronger Brønsted acids. Chem. Rev. 2007, 107, 5744–5758. 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]; c Akiyama T.; Mori K. Stronger Brønsted acids: Recent Progress. Chem. Rev. 2015, 115, 9277–9306. 10.1021/acs.chemrev.5b00041. [DOI] [PubMed] [Google Scholar]
- a García-García P.; Lay F.; García-García P.; Rabalakos C.; List B. A Powerful Chiral Counteranion Motif for Asymmetric Catalysis. Angew. Chem., Int. Ed. 2009, 48, 4363–4366. 10.1002/anie.200901768. [DOI] [PubMed] [Google Scholar]; b James T.; van Gemmeren M.; List B. Development and Applications of Disulfonimides in Enantioselective Organocatalysis. Chem. Rev. 2015, 115, 9388–9409. 10.1021/acs.chemrev.5b00128. [DOI] [PubMed] [Google Scholar]; c Wakchaure V. N.; Obradors C.; List B. Chiral Brønsted Acids Catalyze Asymmetric Additions to Substrates that Are Already Protonated: Highly Enantioselective Disulfonimide-Catalyzed Hantzsch Ester Reductions of NH–Imine Hydrochloride Salts. Synlett 2020, 31, 1707–1712. 10.1055/s-0040-1706413. [DOI] [Google Scholar]; d Zhang Z.; Klussmann M.; List B. Kinetic Study of Disulfonimide-Catalyzed Cyanosilylation of Aldehydes by Using a Method of Progress Rates. Synlett 2020, 31, 1593–1597. 10.1055/s-0040-1707129. [DOI] [Google Scholar]
- Bae H. Y.; Höfler D.; Kaib P. S. J.; Kasaplar P.; De C. K.; Döhring A.; Lee S.; Kaupmees K.; Leito I.; List B. Approaching sub-ppm-level asymmetric organocatalysis of a highly challenging and scalable carbon–carbon bond forming reaction. Nat. Chem. 2018, 10, 888–894. 10.1038/s41557-018-0065-0. [DOI] [PubMed] [Google Scholar]
- Čorić I.; List B. Asymmetric spiroacetalization catalysed by confined Brønsted acids. Nature 2012, 483, 315–319. 10.1038/nature10932. [DOI] [PubMed] [Google Scholar]
- Liu L.; Kaib P. S. J.; Tap A.; List B. A General Catalytic Asymmetric Prins Cyclization. J. Am. Chem. Soc. 2016, 138, 10822–10825. 10.1021/jacs.6b07240. [DOI] [PubMed] [Google Scholar]
- a Pugsley T. A.; Lippmann W. Effects of 1-alkyl-1,2,3,4-tetrahydrocarbazole-1-ethanamines and related compounds, potential antidepressants, on biogenic amine uptake mechanisms. Experientia 1977, 33, 57–59. 10.1007/BF01936753. [DOI] [PubMed] [Google Scholar]; b Asselin A. A.; Humber L. G.; Komlossy J.; Charest M. P. Cycloalkanoindoles. 2. 1-Alkyl-1,2,3,4-tetrahydrocarbazole-1-ethanamines and related compounds. Potential antidepressants. J. Med. Chem. 1976, 19, 792–797. 10.1021/jm00228a011. [DOI] [PubMed] [Google Scholar]
- a Sheng Y.-F.; Gu Q.; Zhang A.-J.; You S.-L. Chiral Brønsted Acid-Catalyzed Asymmetric Friedel–Crafts Alkylation of Pyrroles with Nitroolefins. J. Org. Chem. 2009, 74, 6899–6901. 10.1021/jo9013029. [DOI] [PubMed] [Google Scholar]; b Itoh J.; Fuchibe K.; Akiyama T. Chiral Phosphoric Acid Catalyzed Enantioselective Friedel–Crafts Alkylation of Indoles with Nitroalkenes: Cooperative Effect of 3 Å Molecular Sieves. Angew. Chem., Int. Ed. 2008, 47, 4016–4018. 10.1002/anie.200800770. [DOI] [PubMed] [Google Scholar]; c Mori K.; Wakazawa M.; Akiyama T. Stereoselective construction of all-carbon quaternary center by means of chiral phosphoric acid: highly enantioselective Friedel–Crafts reaction of indoles with β,β-disubstituted nitroalkenes. Chem. Sci. 2014, 5, 1799–1803. 10.1039/C3SC53542H. [DOI] [Google Scholar]
- a Wu Q.-F.; Zheng C.; You S.-L. Enantioselective Synthesis of Spiro Cyclopentane-1,3′-indoles and 2,3,4,9-Tetrahydro-1H-carbazoles by Iridium-Catalyzed Allylic Dearomatization and Stereospecific Migration. Angew. Chem., Int. Ed. 2012, 51, 1680–1683. 10.1002/anie.201107677. [DOI] [PubMed] [Google Scholar]; b Zheng C.; Wu Q.-F.; You S.-L. A Combined Theoretical and Experimental Investigation into the Highly Stereoselective Migration of Spiroindolenines. J. Org. Chem. 2013, 78, 4357–4365. 10.1021/jo400365e. [DOI] [PubMed] [Google Scholar]; c Zheng C.; You S.-L. Exploring the Chemistry of Spiroindolenines by Mechanistically-Driven Reaction Development: Asymmetric Pictet–Spengler-type Reactions and Beyond. Acc. Chem. Res. 2020, 53, 974–987. 10.1021/acs.accounts.0c00074. [DOI] [PubMed] [Google Scholar]
- a Jackson A. H.; Naidoo B.; Smith P. Electrophilic substitution in indoles—IV: The cyclization of indolylbutanol to tetrahydrocarbazole. Tetrahedron 1968, 24, 6119–6129. 10.1016/S0040-4020(01)96344-6. [DOI] [Google Scholar]; b Casnati G.; Dossena A.; Pochini A. Electrophilic substitution in indoles: direct attack at the 2-position of 3-alkylindoles. Tetrahedron Lett. 1972, 13, 5277–5280. 10.1016/S0040-4039(01)85229-1. [DOI] [Google Scholar]
- a Baldwin J. E. Rules for ring closure. J. Chem. Soc., Chem. Commun. 1976, 734–736. 10.1039/c39760000734. [DOI] [Google Scholar]; b Casadei M. A.; Galli C.; Mandolini L. Ring-closure reactions. 22. Kinetics of cyclization of diethyl (ω-Bromoalkyl)malonates in the range of 4- to 21-membered rings. Role of ring strain. J. Am. Chem. Soc. 1984, 106, 1051–1056. 10.1021/ja00316a039. [DOI] [Google Scholar]; c Alabugin I. V.; Gilmore K. Finding the right path: Baldwin “Rules for Ring Closure” and stereoelectronic control of cyclizations. Chem. Commun. 2013, 49, 11246–11250. 10.1039/c3cc43872d. [DOI] [PubMed] [Google Scholar]
- The configuration of 16a was assigned by comparing specific rotation of analogy in ref (25a). 16a 69:31 e.r.; [α]D25 = 8.0 (c 0.1, CHCl3).
- a Kochanowska-Karamyan A. J.; Hamann M. T. Marine Indole Alkaloids: Potential New Drug Leads for the Control of Depression and Anxiety. Chem. Rev. 2010, 110, 4489–4497. 10.1021/cr900211p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tan F.; Cheng H.-G. Catalytic asymmetric synthesis of tetrahydrocarbazoles. Chem. Commun. 2019, 55, 6151–6164. 10.1039/C9CC02486G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.