

Abstract
Vast amount of research has been recently conducted to discover drugs for efficacious treatment of corona virus disease 2019 (COVID-19). The ambiguity about using Chloroquine/ Hydroxychloroquine to treat this illness was a springboard towards new methods for improving the adequacy of these drugs. The effective treatment of COVID-19 using Zinc complexes as add-on to Chloroquine/ Hydroxychloroquine has received major attention in this context. The current studies have shed a light on molecular docking and molecular dynamics methodologies as powerful techniques in establishing therapeutic strategies to combat COVID-19 pandemic. We are proposing some zinc compounds coordination to Chloroquine/ Hydroxychloroquine in order to enhance their activity. The molecular docking calculations showed that Zn(QC)Cl2(H2O) has the least binding energy -7.70 Kcal /mol then Zn(HQC)Cl2(H2O) -7.54 Kcal /mol. The recorded hydrogen bonds were recognized in the strongest range of H Bond category distances. Identification of binding site interactions revealed that the interaction of Zn(QC)Cl2(H2O)with the protease of COVID-19 results in three hydrogen bonds, while Zn(HQC)Cl2(H2O) exhibited a strong binding to the main protease receptor by forming eight hydrogen bonds. The dynamic behavior of the proposed complexes was revealed by molecular dynamics simulations. The outcomes obtained from Molecular dynamics calculations approved the stability of Mpro-Zn(CQ/HCQ)Cl2H2O systems. These findings recommend Zn (CQ) Cl2H2O and Zn (HCQ) Cl2H2O as potential inhibitors for COVID-19 Mpro.
Keywords: COVID-19, Chloroquine, Hydroxychloroquine, Zinc complexes: molecular docking, Molecular dynamics
Graphical abstract
1. Introduction
The novel coronavirus COVID-19 was informed as pandemic infection in Wuhan city of China at the end of December 2019.The pandemic rapidly spread in the world due to human-human transmission. The reported cases of infected and suspected in different countries exceeded two millions, until April 2020 according to the World Health Organization (WHO) [1]. The crisis has motivated researchers from different fields of sciences towards vaccine finding against this novel disease. For many years, Chloroquine (CQ) was reported as a potential remedy to alleviate exacerbation of pneumonia. Also CQ has a capability of inhibition of autophagy and motivating the apoptosis in malignant cells in broad experimental models [2], [3], [4], [5]. These positive effects nominated CQ to be tested in the treatment of COVID-19 infection [6], [7], [8].
Hydroxychloroquine (HCQ) was first prescribed as anti-malaria drug and it was prove of being useful to treat other diseases such as lupus erythematosus, rheumatoid arthritis. Recently HCQ has been proposed for COVID-19 treatment according to the protocols published during 2020 [9], [10], [11], [12]. The proven acknowledge for the efficiency of CQ/HCQ treatment for COVID- 19 is still questioned and lacking for experimental evidence. Various attempts have been made to improve the therapeutic use for CQ/HCQ including metal complexes addition [13,14]. Zinc element has antiviral effect and could be used to reduce the viral activities of COVID-19. Philip Carlucci et al. suggested using zinc sulfate as add-on therapy to HCQ against COVID-19 [15]. Clinical trials have been made to demonstrate that the improvement of the efficacy of CQ/HCQ against COVID-19 may require Zinc additives [16]. Derwand et al. hypothesized that CQ/HCQ plus zinc supplementation may be more effective in reducing COVID-19 morbidity and mortality than CQ or HCQ in monotherapy [17]. There was a shortage in most of those studies in identifying the synthesis and characterization of CQ/HCQ- zinc complexes. The availability of the chemical and molecular structure of zinc combined to CQ/HCQ may enrich the drug design field in the framework of increasing the potency of CQ/HCQ against COVID-19.
Molecular docking is one of the best techniques in the scientific community for rational design of drugs. Docking addresses the binding between drugs and protein via active sites determination. Molecular dynamics simulations are used in virtual screening the dynamic behavior of protein or protein-molecules complexes. Molecular docking and molecular dynamics aided in understanding the molecular function and biological process of drugs, both have been used extensively in recent time to find an effective vaccine for COVID-19. [18], [19], [20], [21]
In this article, the development of the therapeutic activities of CQ/HCQ against COVID-19 has been based on the incorporation of CQ/HCQ with some zinc structures. The stability of the proposed structures was assessed by molecular dynamic simulations. Molecular docking calculations were carried out to calculate the binding energies and site interactions for inhibition capability evaluation of COVID-19 main protease.
2. Materials and methods
2.1. Ligands and target protein
The optimum structures of all compounds are illustrated in Fig. 1 . CQ and HCQ serve a wide variety of in-vitro activity against viruses and used extensively as antimalarial drugs [22,23]. In 1937, Andersag, Breitner and Jung had synthesized CQ for the first time [24]. HCQ is prepared by substitution the N-diethyl group side chain of CQ by N-hydroxy-ethyl side chain [25,26]. The zinc complexes bind to the similar structure of CQ and HCQ through unsubstituted N atom of pyridine. The complex Zn(CQ/HCQ)Cl could synthesis by adding a solution of ZnCl2 in methanol to CQ/HCQ, the mixture stirred and heated at 60 °C for 1 h then cooled to room temperature [27]. Zn(QC)Cl2(H2O) was synthesized in characterization of new copper/zinc-Chloroquine complexes by Maribel Navarro et al. The compound obtained in high yield from the reaction of ZnCl2 with CQ as an air-stable solid and the work suggested that zinc bound to CQ has a five-coordinated structure [28]
Fig. 1.

The optimum structures of the CQ/HCQ complexes mentioned in the text.
Many studies showed that Mpro (PDB ID 6LU7) the main protease of COVID-19 is the key for its viral replication. This makes it a potent target for potential inhibitor drugs [29,30]. The crystal structure of 6lu7 Mpro was downloaded from Protein Data Bank (PDB: https://www.rcsb.org). The chemical structure of CQ (CID: 2719), HCQ (CID: 3652) were taken from PUBCHEM database.
2.2. Molecular docking
The studied ligands were drawn in the Avogadro molecule editor, and then their stable structure obtained by energy minimization with the MMFF94 Force Field. The 6lu7 protein structure was prepared by removing all water molecules, assignment of Gasteiger partial charges and adding polar hydrogen. Ligand position in Mpro protein was set at grid coordinates x =−11.824, y = 14.735 and z = 74.152. Genetic algorithm (GA) parameters were assigned at 100 GA run and population size of 150. Molecular docking calculations were performed by using Autodock 4.2 with the aid of Auto Dock Tools 1.5.6 [31]. Docked structures and the active sites interaction were visualized by Discovery Studio Visualizer v20.1.0.19295. [32].
2.3. Molecular dynamics simulation
NAnoscale Molecular Dynamics (NAMD) software was used to perform Molecular dynamics simulations [33]. Simulation parameterization specified in topologies and parameter files of ligands and proteins were prepared using the CHARMM-GUI [34]. The CHARMM36 force field was used to parameterize the system which was solvated in TIP3P water solvation box and neutralized by adding sodium chloride ions. Energy-minimization was applied to system and then equilibrated for 200 ps. Subsequently, systems were simulated at constant temperature 310 K with 2 fs time step using NVT ensemble under constant periodic boundary conditions for 100 ns. The simulation analysis and trajectories interpretation were assessed with the aid of Visual Molecular Dynamics program (VMD 1.9.3) [35].
3. Results and discussion
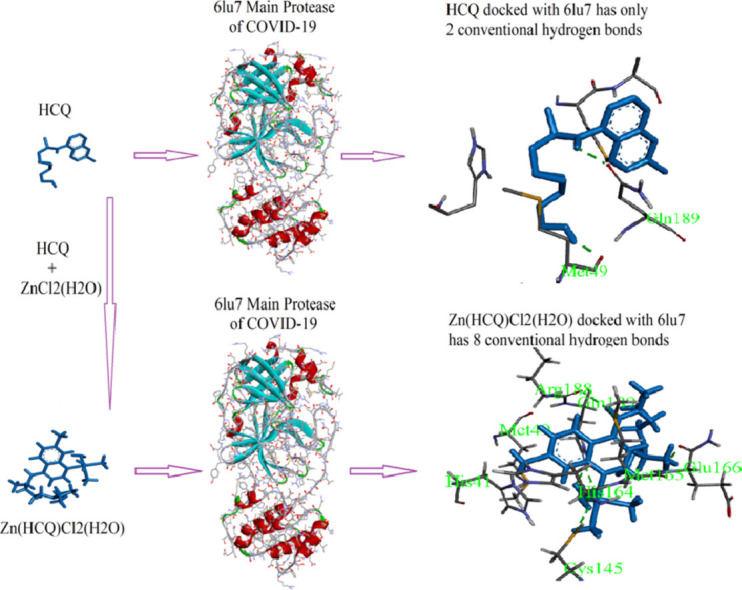
The molecular docking results are recorded in Table 1. Illustrative figures of different orientations for the ligands interaction with protein target are included, the ligand is marked as blue stick model while the protein is displayed as a surface. The more negative binding energy (EB) and smaller value of inhibition constant (Ki) implies best docking score [36]. Hydrogen bonds are a primary contributor factor in supporting the binding affinity of drugs with the receptor. Strong hydrogen bonding interaction represents a high binding capability between ligand and protein [37]. The values of binding energy and inhibition constant for CQ and HCQ are consistent with previous concerned work [38]. There were no commendable findings for Zn(CQ/HCQ)Cl complexes, only insignificant improvement in the binding capabilities than in CQ and HCQ. A strong inhibitory activity achieved by Zn(QC)Cl2(H2O) and Zn(HQC)Cl2(H2O) with binding energies −7.7 and −7.54 Kcal/mol respectively. The considerable decrease in Ki values, classified Zn(QC)Cl2(H2O) and Zn(HQC)Cl2(H2O) as strong potential inhibitors against Mpro activity.
Table 1.
The binding affinity and docked structures of the selected drugs with main protease (6LU7) of COVID-19.
| No | Compound | Docked structure | EB (Kcal/mol) | KI (uM) | Hydrogen bond (Å) |
|---|---|---|---|---|---|
| 1 | CQ |  |
−7.08 | 6.48 | HIS A164 (2.31 Å) |
| 2 | HCQ |  |
−6.87 | 9.15 | GLN A:189 (2.17 Å) MET A:49 (1.99 Å) |
| 3 | Zn(CQ)Cl |  |
−7.09 | 6.38 | HIS A164 (2.14 Å) |
| 4 | Zn(HCQ)Cl |  |
−6.88 | 9.07 | GLU A:166(2.02 Å) HIS A164 (3.06 Å) |
| 5 | Zn(QC)Cl2(H2O) | 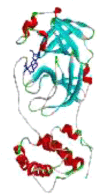 |
−7.7 | 2.27 | GLU A:166(1.76 Å/ 1.95 Å) ARG A:188 (2.06 Å) |
| 6 | Zn(HQC)Cl2(H2O) | 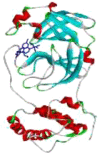 |
−7.54 | 2.99 | GLN A:189 (2.32 Å/ 2.35 Å) HIS A164 (2.19 Å/ 2.27 Å) CYS A:145(2.70 Å) GLU A:166 (1.77 Å) MET A:165 (2.92 Å) ARG A:188 (2.26 Å) |
The 2D binding sites diagram of CQ and HCQ with 6lu7 target is illustrated in Fig. 2 (a and b). CQ demonstrates an average docking by a conventional hydrogen bond with HIS164 residue at 2.31 Å, while HCQ has two hydrogen bonds interactions with MET49 and GLN189 at 1.99 and 2.17 Å respectively. In Fig. 3 (a and b), Zn(CQ)Cl and Zn(HCQ)Cl showed no noteworthy improvement in binding modes than in CQ nor HCQ; Zn(CQ)Cl has one hydrogen bond with HIS 164 amino acid at 2.14 Å while the residues HIS164 and GLU166 are involved in the formation of two hydrogen bonds with Zn(HCQ)Cl at distances 3.06and 2.02 Å. This result is realistic with the very close blind docking results (mentioned in Table 1) between CQ and Zn(CQ)Cl or between HCQ and Zn(HCQ)Cl. Three hydrogen bonds were formed in the interaction between Zn(QC)Cl2(H2O) and Mpro as shown in Fig. 4 a (two hydrogen bonds with GLU166→ 1.76, 1.95 Å and one hydrogen bond with ARG188→2.06 Å). The docking representation of the last complex has shown strong binding activity. As revealed in Fig. 4b, Zn(HQC)Cl2(H2O) was able to form eight hydrogen bonds with GLN189 (2.32, 2.35 Å), CYS145(2.70 Å), HIS164(2.19, 2.27 Å), MET165(2.92 Å), ARG188(2.26 Å) and GLU 166 (1.77 Å) residues. The qualitative aspect of this interaction is that hydrogen bonds are distributed over the sides and center of Zn(HQC)Cl2(H2O), which represents high inhibition efficiency to bind the receptor binding domain. The formed hydrogen bonds were in the categories of strong hydrogen bonds range (1.76–2.35 Å) except two moderate bonds 2.92, 3.06 Å according to hydrogen bonding classification [39].
Fig. 2.

Representation of the interacting sites of SARS-CoV-2 Mpro with CQ (a) and HCQ (b).
Fig. 3.

Representation of the interacting sites of SARS-CoV-2 Mpro with Zn(CQ)Cl (a) and Zn(HCQ)Cl (b).
Fig. 4.

Representation of the interacting sites of SARS-CoV-2 Mpro with Zn(QC)Cl2(H2O) (a) and Zn(HQC)Cl2(H2O) (b).
Fig. 5 showed the RMSD trajectories of Mpro-CQ/HCQ, Mpro-Zn(CQ/HCQ)Cl2 and Mpro-Zn(CQ/HCQ)Cl2(H2O) along with molecular dynamic simulation time. In the monitored RMSD analysis, all trajectories have minor fluctuations after 20 ns and there is no sudden deviation or sliding drift, which suggests no structural changes. Mpro-CQ attained equilibrium after 60 ns with considerable fluctuation compared to the other trajectories, the average RMSD values within of the range of 0.91±0.1 Å. Remarkable change in the stability behavior of Mpro-HCQ docked structure than in Mpro-CQ. The stable trajectory of Mpro-HCQ started at 30 ns with insignificant fluctuations around 0.91± 0.05 Å and last till the end of the simulation time. A similar behavior in RMSD trajectories are observed between Mpro-Zn(CQ)Cl and Mpro-Zn(HCQ)Cl, significant fluctuations around 1.05± 0.07 Å were detected during simulation. The notable deviations in both trajectories have been decayed after 60 ns, however Mpro-Zn(HCQ)Cl showed almost steady RMSD values in the last 10 ns. Mpro-Zn(CQ)Cl2(H2O) made a slight improvement in structural stability by achieving equilibrium after 40 ns with an average RMSD of 1.05 ± 0.04 Å throughout the simulation period. The last complex exhibited the best stabilized mode among other complexes. Mpro-Zn(HCQ)Cl2(H2O) remained converged and stable after only 15 ns and the calculated average RMSD values was found as 1.05 ± 0.03 Å. Smaller deviations reflect that the binding of Zn(HCQ)Cl2(H2O) and Mpro main protease is more stable conformation.
Fig. 5.

The RMSD plots extracted from molecular dynamics simulations for Mpro-complexes.
The effect of zinc ion on CQ and HCQ manifested clearly in the molecular dynamics results of Zn(CQ/HCQ)Cl2(H2O).Stabilized modes were achieved early at 40, 15 ns in Mpro-Zn(CQ/HCQ)Cl2(H2O) than in Mpro-CQ/HCQ at 60, 30 ns. Fluctuations have diminished from ±0.1, ± 0.05 Å in Mpro-CQ/HCQ to ± 0.04, ± 0.03 Å in Mpro-Zn(CQ/HCQ)Cl2(H2O) respectively.
The fluctuations of the Mpro–ligand complexes were explored by calculating the root mean square fluctuation (RMSF). The plot of RMSF for all Mpro-ligand complexes was compiled in Fig. 6 . Large fluctuations can be seen in the structures of Mpro-CQ, Zn (HCQ) Cl and Mpro-Zn (HCQ) Cl during simulation time. The highly fluctuated residue can clearly be noticed for these structures at significantly different positions. The associated average RMSF values were calculated as 0.46, 0.45 and 0.43 Å respectively. There is an overall decline in fluctuations in Mpro-HCQ and Zn(QC/HCQ)Cl2(H2O) complexes in contrast to the previous three structures. The lower RMSF values supplemented by smaller average RMSF values 0.38, 0.42 and 0.37 Å showed that these structures are stable.
Fig. 6.

The RMSF plots extracted from molecular dynamics simulations for Mpro-complexes.
4. Conclusions
On the missing of crucial declaration for an effective drug to cure COVID-19, a lot of endeavors have been made in discovering therapeutic medication to remedy this virus. Among this examined the efficacy of Zinc complexes in combination with CQ/ HCQ against COVID-19. In the present study, some complexes of zinc- CQ/ HCQ were described to target the Mpro main protease of COVID-19. The molecular docking calculations showed that Zn(QC)Cl2(H2O) and Zn(HQC)Cl2(H2O) has the least binding energy. Hydrogen bonds were assigned and recorded in the identification of binding site interactions. The interaction of Zn(QC)Cl2(H2O)with the protease of COVID-19 results in three hydrogen bonds, while Zn(HQC)Cl2(H2O) exhibited a strong binding to the main protease receptor by forming eight hydrogen bonds. In molecular dynamics simulations, the observed analysis of RMSD and RMSF trajectories reported the high binding affinity and structure stability of Mpro-Zn(CQ/HCQ)Cl2H2O complexes. It can conclude that Zn (CQ/HCQ) Cl2H2O indicates a good potential as inhibitors for Mpro of COVID-19. The current work suggests that, more experimental and clinical studies on CQ/HCQ in combination with zinc complexes should be considered in the treatment of COVID-19.
Data availability
The data will be available upon request.
CRediT authorship contribution statement
R.K. Hussein: Conceptualization, Methodology, Software, Investigation, Writing - review & editing. H.M. Elkhair: Writing - review & editing, Data curation, Formal analysis.
Declaration of Competing Interest
I have no conflicts of interest to disclose.
References
- 1.Coronavirus disease . World Health Organization; 2019. (COVID-19) Situation Report–84.https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200413-sitrep-84-covid-19.pdf Available at: cited date April 13, 2020. [Google Scholar]
- 2.Krafts K., Hempelmann E., Skórska-Stania A. From methylene blue to chloroquine: a brief review of the development of an antimalarial therapy. Parasitol. Res. 2012;111(Jul. (1)):1–6. doi: 10.1007/s00436-012-2886-x. [DOI] [PubMed] [Google Scholar]
- 3.Lamoureux F., Zoubeidi A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy. 2013;9(Jul. (7)):1119–1120. doi: 10.4161/auto.24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burikhanov R., et al. Chloroquine-inducible par-4 secretion is essential for tumor cell apoptosis and inhibition of metastasis. Cell Rep. 2017;18(Jan. (2)):508–519. doi: 10.1016/j.celrep.2016.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verschooten L., et al. Autophagy inhibitor chloroquine enhanced the cell death inducing effect of the flavonoid luteolin in metastatic squamous cell carcinoma cells. PLoS ONE. 2012;7(Oct. (10)):e48264. doi: 10.1371/journal.pone.0048264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cortegiani A., Ippolito M., Ingoglia G., Einav S. Chloroquine for COVID-19: rationale, facts, hopes. Crit. Care. 2020;24(Dec. (1)):210. doi: 10.1186/s13054-020-02932-4. s13054-020-02932–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hashem A.M., et al. Therapeutic use of chloroquine and hydroxychloroquine in COVID-19 and other viral infections: a narrative review. Travel Med. Infect. Dis. 2020;35 doi: 10.1016/j.tmaid.2020.101735. May. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kearney J. Chloroquine as a potential treatment and prevention measure for the 2019 novel coronavirus: a review. Med. Pharmacol. 2020 doi: 10.20944/preprints202003.0275.v1. preprintMar. [DOI] [Google Scholar]
- 9.Ali A.S., et al. Optimizing the use of hydroxychloroquine in the management of COVID-19 given its pharmacological profile. J. Pharm. Res. Int. 2020:29–43. doi: 10.9734/jpri/2020/v32i830468. May. [DOI] [Google Scholar]
- 10.Geleris J., et al. Observational study of hydroxychloroquine in hospitalized patients with COVID-19. N. Engl. J. Med. 2020;382(Jun. (25)):2411–2418. doi: 10.1056/NEJMoa2012410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou D., Dai S.-M., Tong Q. COVID-19: a recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J. Antimicrob. Chemother. 2020;75(Jul. (7)):1667–1670. doi: 10.1093/jac/dkaa114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.U. D. Suranagi, H. S. Rehan, and N. Goyal, “Hydroxychloroquine for the management of COVID-19: Hope or Hype? A Systematic review of the current evidence,” p. 31, Apr.2020, doi: 10.1101/2020.04.16.20068205.
- 13.Navarro M., Castro W., Madamet M., Amalvict R., Benoit N., Pradines B. Metal-chloroquine derivatives as possible anti-malarial drugs: evaluation of anti-malarial activity and mode of action. Malar. J. 2014;13(Dec. (1)):471. doi: 10.1186/1475-2875-13-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manning T.J., et al. Structural and some medicinal characteristics of the copper(II)–hydroxychloroquine complex. Bioorg. Med. Chem. Lett. 2013;23(Aug. (15)):4453–4458. doi: 10.1016/j.bmcl.2013.05.041. [DOI] [PubMed] [Google Scholar]
- 15.Carlucci P., Ahuja T., Petrilli C.M., Rajagopalan H., Jones S., Rahimian J. Hydroxychloroquine and azithromycin plus zinc vs hydroxychloroquine and azithromycin alone: outcomes in hospitalized COVID-19 patients. medRxiv. 2020 doi: 10.1101/2020.05.02.20080036. (except HIV/AIDS), preprintMay. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.M. O. Shittu and O. I. Afolami, “Improving the efficacy of chloroquine and hydroxychloroquine against SARS-CoV-2 may require zinc additives - A better synergy for future COVID-19 clinical trials,” Le Infezioni in Medicina. 28 (2020) 192-197 Jun [PubMed]
- 17.Derwand R., Scholz M. Does zinc supplementation enhance the clinical efficacy of chloroquine/hydroxychloroquine to win today's battle against COVID-19? Med. Hypotheses. 2020;142 doi: 10.1016/j.mehy.2020.109815. Sep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Śledź P., Caflisch A. Protein structure-based drug design: from docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018;48:93–102. doi: 10.1016/j.sbi.2017.10.010. Feb. [DOI] [PubMed] [Google Scholar]
- 19.Baildya N., Ghosh N.N., Chattopadhyay A.P. Inhibitory activity of hydroxychloroquine on COVID-19 main protease: an insight from MD-simulation studies. J. Mol. Struct. 2020;1219 doi: 10.1016/j.molstruc.2020.128595. Nov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felsenstein S., Herbert J.A., McNamara P.S., Hedrich C.M. COVID-19: immunology and treatment options. Clin. Immunol. 2020;215 doi: 10.1016/j.clim.2020.108448. Jun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devaux C.A., Rolain J.-M., Raoult D. ACE2 receptor polymorphism: susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J. Microbiol. Immunol. Infect. 2020;53(Jun. (3)):425–435. doi: 10.1016/j.jmii.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rainsford K.D., Parke A.L., Clifford-Rashotte M., Kean W.F. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology. 2015;23(Oct. (5)):231–269. doi: 10.1007/s10787-015-0239-y. [DOI] [PubMed] [Google Scholar]
- 23.Haładyj E., Sikora M., Felis-Giemza A., Olesińska M. Antimalarials–are they effective and safe in rheumatic diseases? Reumatologia/Rheumatology. 2018;56(3):164–173. doi: 10.5114/reum.2018.76904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersag H., Breitner S., Jung H. Process for the preparation of quinoline compounds containing amino groups with basic substituents in the 4-position. German Patent. 1939;683:692. 1939; 683:692. [Google Scholar]
- 25.Evans D., Williamson W.R.N. Chemistry of clinically active anti-inflammatory compounds. In: Williamson WRN (ed) Antiinflammatory compounds. Marcel Dekker; New York: 1987. Chemistry of clinically active anti-inflammatory compounds; pp. 193–302. Williamson WRN (ed) [Google Scholar]
- 26.Somer M, Kallio J, Pesonen U, Pyykkö K, Huupponen R, Scheinin M Influence of hydroxychloroquine on the bioavailability of oral metoprolol. Br. J. Clin. Pharmacol. 2005;49:549–554. doi: 10.1046/j.1365-2125.2000.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obaleye J.A., Caira M.R., Tella A.C. Synthesis, characterization and crystal structure of a polymeric zinc(II) complex containing the antimalarial quinine as ligand. J. Chem. Crystallogr. 2007;37(Sep. (10)):707–712. doi: 10.1007/s10870-007-9236-3. [DOI] [Google Scholar]
- 28.Navarro M., Goitia H., Silva P., Velásquez M., Ojeda L.E., Fraile G. Synthesis and characterization of new copper– and zinc–chloroquine complexes and their activities on respiratory burst of polymorphonuclear leukocytes. J. Inorg. Biochem. 2005;99(Aug. (8)):1630–1636. doi: 10.1016/j.jinorgbio.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Dayer M.R., Taleb-Gassabi S., Dayer M.S. Lopinavir; a potent drug against coronavirus infection: insight from molecular docking study. Arch. Clin. Infect. Dis. 2017;12(Sep. (4)) doi: 10.5812/archcid.13823. [DOI] [Google Scholar]
- 30.Mengist H.M., Fan X., Jin T. Designing of improved drugs for COVID-19: crystal structure of SARS-CoV-2 main protease Mpro. Signal Transduct. Target. Ther. 2020;5(Dec. (1)):67. doi: 10.1038/s41392-020-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris G.M., et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(Dec. (16)):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dassault Systèmes BIOVIA . Dassault Systèmes; San Diego, CA, USA.: 2017. Discovery Studio Modelling Environment, Release 2017. [Google Scholar]
- 33.Phillips J.C., Braun R., Wang W., et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. Dec 2005;26(16):1781. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J., et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016;12(Jan. (1)):405–413. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(Feb. (1)):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 36.Irwin J.J., et al. Automated docking screens: a feasibility study. J. Med. Chem. 2009;52(Sep. (18)):5712–5720. doi: 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pantsar T., Poso A. Binding affinity via docking: fact and fiction. Molecules. 2018;23(Jul. (8)):1899. doi: 10.3390/molecules23081899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheke R.S. The molecular docking study of potential drug candidates showing anti-COVID-19 activity by exploring of therapeutic targets of SARS-CoV-2. EJMO. 2020 doi: 10.14744/ejmo.2020.31503. [DOI] [Google Scholar]
- 39.George A.Jeffrey. Oxford University Press; 1997. An Introduction to Hydrogen Bonding; p. 191. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data will be available upon request.