

Abstract
Lactate dehydrogenase (LDH) catalyzes the conversion of pyruvate to lactate, with concomitant oxidation of reduced nicotinamide adenine dinucleotide as the final step in the glycolytic pathway. Glycolysis plays an important role in the metabolic plasticity of cancer cells and has long been recognized as a potential therapeutic target. Thus, potent, selective inhibitors of LDH represent an attractive therapeutic approach. However, to date, pharmacological agents have failed to achieve significant target engagement in vivo, possibly because the protein is present in cells at very high concentrations. We report herein a lead optimization campaign focused on a pyrazole-based series of compounds, using structure-based design concepts, coupled with optimization of cellular potency, in vitro drug–target residence times, and in vivo PK properties, to identify first-in-class inhibitors that demonstrate LDH inhibition in vivo. The lead compounds, named NCATS-SM1440 (43) and NCATS-SM1441 (52), possess desirable attributes for further studying the effect of in vivo LDH inhibition.
Graphical Abstract
INTRODUCTION
Cancer cells exhibit deregulated metabolic characteristics that are remarkably different from normal cells, demonstrating avidity for glucose uptake and catabolism, ultimately producing lactate and generating adenosine 5′-triphosphate (ATP) through the cytosolic, nonoxidative glycolytic pathway. Glycolysis can be the preferred nicotinamide adenine dinucleotide (NAD+) regeneration and energy production process for cancer cells, even in the presence of oxygen, rather than the more energy-efficient mitochondrial oxidative phosphorylation. This is referred to as aerobic glycolysis, and the preference for aerobic glycolysis is also known as the Warburg effect.1 Although aerobic glycolysis is an inefficient way to generate ATP (compared with oxidative phosphorylation), the rate of ATP generation is rapid. In addition, it is hypothesized that rapidly proliferating cancer cells have adapted this approach to regenerate NAD+ and to support the production of essential cellular building blocks such as amino acids, lipids, and nucleotides needed to support rapid cell growth.2 Indeed, many noncancer cells use a combination of oxidative phosphorylation and glycolysis to achieve the needed metabolic plasticity to serve their biological functions. Although the molecular basis of aberrant cancer metabolism and its role in cancer development and progression have yet to be fully elucidated,3 the Warburg effect and the enzymes in glycolysis have long been recognized as potential targets for the selective killing of cancer cells.
Many glycolytic enzymes are overexpressed in cancer cells, including the lactate dehydrogenase (LDH) enzymes A (LDHA) and B (LDHB).4,5 LDH is a tetrameric protein composed of the products of LDHA (subunit M) and LDHB (subunit H) genes. The tetrameric combination of these gene products generates five LDH isoforms with different combinations of subunits depending on the cell type. The LDH5 (4 M) isoform is the primary form expressed in cancer cells,6 although other isoforms have also been reported. All LDH isoforms catalyze the last step in the glycolytic pathway converting pyruvate to lactate while regenerating NAD+ from reduced nicotinamide adenine dinucleotide (NADH). The lactate produced is then secreted from cells via the monocarboxylate transporter proteins.
Significant evidence exists to support the development of LDH inhibitors as a therapeutic option for cancer treatment. Genetic knockdown of LDHA has been shown to elicit cell death or delayed cell growth in cell lines from colorectal carcinoma (siRNA),7 Burkitt lymphoma (siRNA),8 hepatocellular carcinoma (siRNA),9 pancreatic cancer (shRNA),10 and mouse mammary tumor cells (shRNA).11 For example, mouse mammary tumor cells lacking LDHA implanted as xenografts demonstrated dramatically reduced tumor growth.11 In addition, in a genetically engineered mouse model of non-small-cell lung cancer, induced knockout of mouse LDHA led to the regression of established tumors without serious systemic toxicity.12 In contrast, LDHB knockdown has been reported not to significantly impact tumor cell survival.7
Although the therapeutic potential of LDHA inhibition appears to be substantial, the discovery and development of LDH inhibitors have proven to be challenging. For example, because of the micromolar concentrations of the LDH enzyme in cancer cells, an effective inhibitor will likely need to bind with exceptionally high affinity and also achieve high intracellular concentrations to enable a therapeutic level of target engagement. To date, no inhibitors of LDH which meet these criteria have been reported. The first reported LDH inhibitors came from academic groups (e.g., FX-1113a and NHI-213b) with efforts from biotech14 and pharmaceutical companies15 being disclosed more recently. Molecules from GlaxoSmithKline (GSK)16 and Genentech (GNE-140)17 have demonstrated in vitro inhibition of cellular lactate production and cytotoxicity in cancer cells, but the relatively poor pharmacokinetics of these compounds limited their usefulness for testing of the therapeutic hypothesis in vivo. More recently, LDH inhibitors (e.g., “compound 24c” in Figure 1) that show modest inhibition of MiaPaCa-2 xenograft growth have been described, but target engagement in vivo was not demonstrated.18
Figure 1.

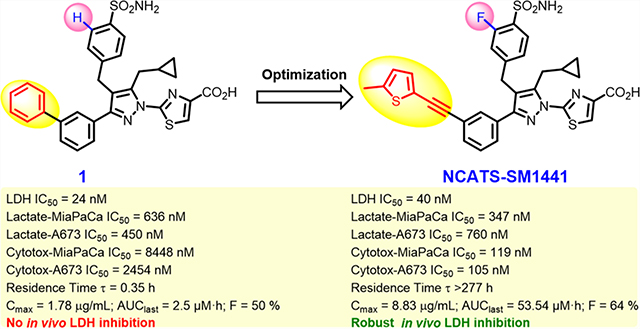
Representative previously described LDH inhibitors and comparison with new leads 43 = NCATS-SM1440 and 52 = NCATS-SM1441. aNamed as NCI-006 in refs 20 and 21. bNamed as NCI-737 in ref 20.
We recently reported the identification of a pyrazole-based hit from quantitative high-throughput screening (qHTS) and used structure-based design to develop nanomolar inhibitors of LDH enzyme activity, exemplified by 1.19 Compound 1 inhibited LDH in highly glycolytic MiaPaCa-2 (human pancreatic cancer) and A673 (human Ewing’s sarcoma) cell lines, demonstrating sub-micromolar suppression of cellular lactate output and inhibition of cell growth. Although 1 showed favorable in vitro ADME properties (e.g., microsomal stability and solubility), its PK profile was not suitable for in vivo use. Herein, we report the optimization of the pyrazole-based chemotype using binding constants (particularly off-rates or drug–target residence time) and cell-based activity to guide improvements in potency while also optimizing the pharmacokinetic properties. These efforts ultimately resulted in lead compounds 43 (NCATS-SM1440) and 52 (NCATS-SM1441) that have enabled studies of the effects of pharmacological LDH inhibition in animal models of Ewing’s sarcoma20 and human pancreatic cancers21 as well as interrogation of a synergistic role in T cell-based immunotherapy.22
CHEMISTRY
A robust linear synthetic strategy was used for the optimization of the cyclopropylmethyl and sulfonamide regions (Schemes 1 and 2). The commercially available carboxylic acids a–l were reacted with 1,2,3-benzotriazole in the presence of thionyl chloride to afford acyl benzotriazole intermediates Ia–l. The reaction of these intermediates (Ia–l) with substituted acetophenones IIa–d, mediated by magnesium bromide ethyl etherate in the presence of Hunig’s base, afforded 1,3-diketone derivatives IIIa–p (Table S3) in good yields. Subsequently, cesium carbonate mediated alkylation of 1,3-diketone intermediates IIIa–p with 4-(bromomethyl)-benzenesulfonamide derivatives IVa–c in dimethyl sulfoxide (DMSO) at room temperature generated advanced intermediates Va–r, as described in our previous paper.19 Initial attempts with tosic acid catalyzed cyclocondensation of 1,3-diketone derivatives IIIa–p with ethyl 2-hydrazinylthiazole-4-carboxylate hydrobromide generated a mixture of both pyrazole regioisomers. We previously reported that the 5-aryl regioisomer is inactive in LDHA assays, so formation of this regioisomer is undesired. The product ratio of the desired isomer was poor (≤10%), particularly when R1 is an electron-donating alkyl group such as cyclopropylmethyl or cyclopropylethyl group which was found to be optimal for activity (e.g., Va–f, Vh–j, and Vn–r). Therefore, it was neither efficient nor practical to use this cyclization protocol to synthesize the quantities of the advanced bromide-containing intermediates VIIa–f, VIIh–j, and VIIn–r needed for late-stage optimization efforts.
Scheme 1.

Syntheses of Intermediates VIa–r and Analogues 1–9a
aReagents and conditions: (a) SOCl2, CH2Cl2, 4 h, and 91–100%; (b) MgBr2·OEt2, iPr2NEt, CH2Cl2, 12 h, and 60–69%; (c) Cs2CO3, DMSO, 1 h, and 55–83%; (d) (i) pyrrolidine (0.5 equiv), TsOH (0.5 equiv), EtOH, reflux, and 1–2 h and (ii) ethyl 2-hydrazinylthiazole-4-carboxylate·HBr and reflux overnight; and (e) LiOH, THF/MeOH/H2O, and 1 h.
Scheme 2.

Syntheses of Analogues 10–32 and 36–89a
aReagents and conditions: (a) [P(t-Bu)3]Pd(crotyl)Cl, DABCO, dioxane, RT, and 1–12 h; (b) [DTBNpP]Pd(crotyl)Cl, DABCO, dioxane, 60 °C, and 12 h; (c) SPhosPd(crotyl)Cl, K3PO4, dioxane/H2O, 100 °C, and 0.5 h; (d) LiOH, THF/MeOH/H2O, and 1 h; and (e) CsF, THF–EtOH, RT, and 2 h.
The ratio of the two regioisomers produced in the reaction depends on the reactivity differences dictated by the electronic environment around the two keto groups. To improve the regioisomeric ratio, we envisioned utilizing these reactivity differences by blocking the undesired reactivity of the 1-keto group via initial formation of an enamine intermediate prior to the addition of the ethyl 2-hydrazinylthiazole-4-carboxylate. Extensive optimization identified improved conditions, in which heating 1,3-diketones Va–r in ethanol in the presence of 0.5 equiv of both pyrrolidine and tosic acid for 1–2 h at reflux, then adding ethyl 2-hydrazinylthiazole-4-carboxylate, and stirring at reflux overnight allowed us to obtain a ~50:50 mixture of the two regioisomers. The desired isomer was isolated as the second peak using reversed-phase (C18) chromatography, as this regioisomer was slightly less polar [as judged by liquid chromatography mass spectrometry (LC–MS) analysis and according to our previous reports.19 Further reaction optimization proved elusive, and the investigation of alternative routes to obtain exclusively the desired isomer were met with limited success. Thus, for all subsequent scale-up and analogue syntheses, we used the above procedure. Compound 1 and analogues 2–9 were obtained by hydrolysis of the intermediates VIIa, VIIg–m, and VIIb–d, respectively, using lithium hydroxide in ethanol and subsequent high-performance liquid chromatography (HPLC) purification.
The synthesis of analogues 11–89 listed in Tables 2–4 was achieved utilizing either Sonogashira or Suzuki coupling of the advanced 3-bromo aryl intermediates VIIe–f, VIIi–j, and VIIn–r, followed by hydrolysis with LiOH (Scheme 2). For the Sonogashira coupling, we developed a robust room-temperature, copper-free protocol catalyzed by commercially available monoligated precatalysts, [P(t-Bu)3] Pd(crotyl)Cl or [DTBNpP] Pd(crotyl)Cl, in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) in dioxane.23 This method allowed us to quickly generate an alkyne library and explore the structure–activity relationship (SAR) around the biphenyl region with access to more complex structures. Moreover, the protocol requires only a minimal work-up that could further facilitate rapid analogue generation during our library synthesis and is amenable to providing large quantities of key intermediates (e.g., VIIIa–d) to support advanced studies on top compounds. Using this method, analogues 14–16, 20–32, 36–43, 48–49, 51–52, 74–75, and 84–89 were synthesized directly from intermediates VIIe–f, VIIi–j, and VIIn–r with the corresponding commercially available terminal alkynes IX and the [P(t-Bu)3] Pd(crotyl)Cl catalyst in dioxane. The syntheses of analogues 33–35 are summarized in Scheme 3. To rapidly explore the SAR with aryl or heteroaryl R substitutions, a slightly modified three-step protocol was used for analogues 44–47, 53–67, and 76–83. The synthesis began with [P(t-Bu)3]Pd(crotyl)Cl-catalyzed Sonogashira coupling of intermediates VIIe–f and VIIi–j with trimethylsilylacetylene, followed by deprotection of the Trimethylsilyl (TMS) group with CsF to afford intermediates VIIIa–d. Further Sonogashira coupling of these alkyne intermediates with the corresponding aryl or heteroaryl bromides X under the same conditions, followed by hydrolysis with LiOH, furnished analogues 44–45, 53–61, 66–67, and 76–79. Analogue 11 (R = H) was obtained upon hydrolysis of intermediate VIIIb using LiOH in a methanol–water mixture. However, the use of the [P(t-Bu)3]Pd(crotyl)Cl catalyst under similar conditions failed to afford the isolable product in the case of more electron-deficient heteroaryl bromides, such as 1,3-thiazolyl or 1,3-oxazolyl bromides. Fortunately, switching to a more active catalyst, [DTBNpP]Pd(crotyl)Cl, with a slightly elevated reaction temperature (60 °C in dioxane) facilitated efficient product formation. Thus, the Sonogashira reaction of intermediates VIIIa–d with the corresponding 1,3-thiazolyl or 1,3-oxazolyl bromides under these conditions in the presence of DABCO, followed by hydrolysis, furnished analogues 46–47, 62–65, and 80–82. Because of the gaseous nature and commercial unavailability of alkyne precursors, we used SPhos precatalyst-catalyzed Suzuki coupling of the corresponding alkynyl trifluoroboronates or pinacol esters, followed by hydrolysis protocol for alkyne analogues 12–13, 17–19, and 50. Finally, Suzuki coupling of the corresponding alkenyl boronates or boronic acids with VIIe–f, using the SPhos Pd(crotyl)Cl precatalyst in the presence of potassium phosphate base at 100 °C and subsequent hydrolysis of the ester group, provided analogues 68–73.
Table 2.
Biochemical and Cellular LDH Inhibition of Analogues 11–49
 | |||||||
|---|---|---|---|---|---|---|---|
| Compd No. |  |
Biochemical LDHA IC50 (nM)a | A673 cells |
MiaPaCa-2 cells |
HEK293 Cells |
||
| lactate inh. IC50 (nM)a | Cytotox. IC50 (nM)a | lactate inh. IC50 (nM)a | Cytotox. IC50 (nM)a | CETSA IC50 (nM)a | |||
| 11 | H | 9 | 902 | 4346 | 660 | 4025 | 253 |
| 12 | CH3 | 11 | 505 | 332 | 450 | 322 | 119 |
| 13 | Et | 14 | 487 | 716 | 470 | 662 | 112 |
| 14 | n-Pr | 24 | 420 | 833 | 361 | 746 | 106 |
| 15 | n-Bu | 50 | 405 | 773 | 298 | 845 | 106 |
| 16 |  |
21 | 284 | 1011 | 487 | 756 | 2053 |
| 17 |  |
16 | 141 | 935 | 137 | 845 | 75 |
| 18 | 3 | 2475 | 1798 | 1659 | 1327 | 577 | |
| 19 | 8 | 769 | 1225 | 525 | 1092 | 119 | |
| 20 | 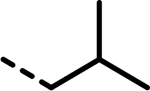 |
54 | 284 | 935 | 487 | 909 | 89 |
| 21 | 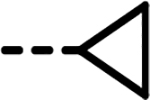 |
9 | 695 | 132 | 554 | 323 | 100 |
| 22 | 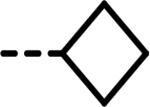 |
23 | 378 | 593 | 428 | 573 | 133 |
| 23 | 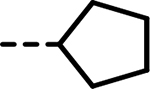 |
22 | 509 | 100 | 333 | 314 | 112 |
| 24 |  |
33 | 450 | 689 | 454 | 593 | 95 |
| 25 |  |
7 | 1931 | 2187 | 1479 | 2178 | 150 |
| 26 |  |
10 | 1052 | 1177 | 898 | 1236 | 84 |
| 27 |  |
7 | 1324 | 2647 | 1047 | 2263 | 126 |
| 28 | 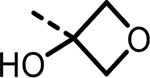 |
6 | 12241 | 3587 | 4873 | 2994 | 1499 |
| 29 | 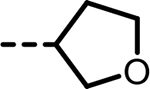 |
7 | 1268 | 973 | 831 | 1224 | 89 |
| 30 | 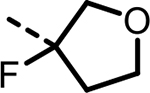 |
1 | 1056 | 1054 | 867 | 882 | 201 |
| 31 | 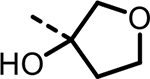 |
5 | 5937 | 2635 | 3871 | 1738 | 815 |
| 32 |  |
15 | 12241 | 9052 | 8910 | 17302 | 2579 |
| 33 | CN | 15 | 4010 | >30000 | 4401 | >30000 | 474 |
| 34 | CF3 | 450 | >30000 | >30000 | >30000 | >30000 | 14005 |
| 35 | CF2H | 15 | 937 | 1482 | 741 | 1327 | 163 |
| 36 | 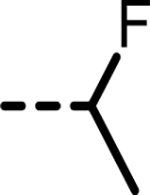 |
14 | 800 | 1134 | 822 | 1020 | 127 |
| 37 | 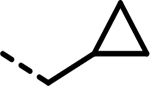 |
31 | 234 | 1016 | 259 | 1030 | 89 |
| 38 | 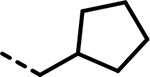 |
202 | 487 | 939 | 424 | 848 | 178 |
| 39 | 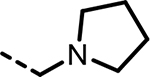 |
8 | 1086 | 973 | 1007 | 1102 | 267 |
| 40 | 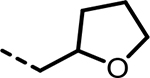 |
9 | 933 | 1380 | 937 | 1225 | 178 |
| 41 | 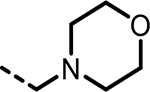 |
107 | 15349 | 5834 | 18852 | 18061 | 5045 |
| 42 | 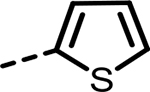 |
32 | 438 | 265 | 307 | 268 | 89 |
| 43 (NCATS-SM1440) | 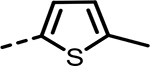 |
57 | 873 | 119 | 403 | 257 | 106 |
| 44 | 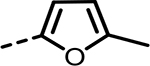 |
47 | 544 | 528 | 547 | 552 | 79 |
| 45 | 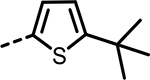 |
713 | 529 | 743 | 507 | 665 | 10602 |
| 46 | 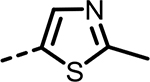 |
33 | 937 | 569 | 867 | 488 | - |
| 47 | 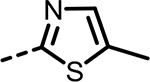 |
52 | 1324 | 803 | 1603 | 909 | 170 |
| 48 | 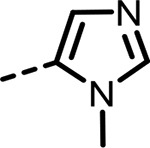 |
8 | 1597 | 1542 | 1066 | 1387 | 299 |
| 49 | 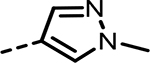 |
9 | 1603 | 593 | 1141 | 619 | 189 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay using a dose response in the 1536-well format. (n = 2 for lactate and n = 3 for cytotoxicity, CETSA, and biochemical LDHA.)
Table 4.
Biochemical and Cellular LDH Inhibition of Compounds 68–73
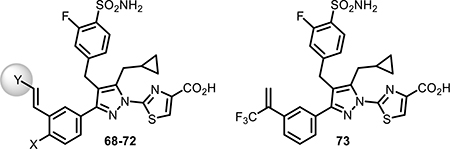 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd No. |  |
X | Biochemical LDHA IC50 (nM)a | A673 cells |
MiaPaCa-2 cells |
HEK293 Cells |
||
| lactate inh IC50 (nM)a | Cytotox IC50 (nM)a | lactate inh IC50 (nM)a | Cytotox IC50 (nM)a | CETSA IC50 (nM)a | ||||
| 68 | 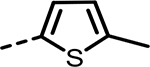 |
H | 52 | 357 | 452 | 331 | 471 | 158 |
| 69 | 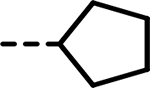 |
F | 74 | 371 | 488 | 149 | 454 | 108 |
| 70 | 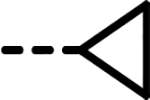 |
F | 15 | 320 | 833 | 231 | 837 | 134 |
| 71 | CH3 | F | 11 | 405 | 614 | 321 | 528 | 126 |
| 72 | 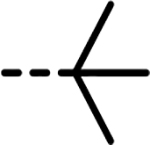 |
F | 59 | 361 | 2263 | 255 | 1899 | 112 |
| 73 | - | - | 39 | 713 | 16023 | 972 | 17376 | 398 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay using a dose response in the 1536-well format. (n = 2 for lactate and n = 3 for cytotoxicity, CETSA, and biochemical LDHA.)
Scheme 3.

Syntheses of Analogues 33–35a
aReagents and conditions: (a) Dess–Martin periodinane, DCM, RT, and 2 h; (b) NaN3, CF3SO3H, ACN, RT, and 12 h; (c) (CH3)3Sn–OH, dichloroethane, MW, 110 °C, and 1 h; (d) Deoxo-Fluor, DCM, RT, and 12 h; (e) P(t-Bu)3.HBF4, ([PdCl(allyl)]2, dioxane, 80 °C, and 4 h; and (f) LiOH, THF/MeOH/H2O, and 1 h.
RESULTS AND DISCUSSION
In our previous work, we described structure-guided optimization of a weakly active qHTS hit into 1, a potent, selective LDH inhibitor suitable for probing LDH function in cells.19 Although 1 showed nanomolar inhibition of LDHA in the biochemical activity assay and was able to achieve reasonable cellular potency, additional optimization was required to identify highly potent molecules with PK properties suitable for in vivo studies. Here, we report a focused lead optimization effort that has identified compounds with good activity in highly glycolytic MiaPaCa-2 and A673 cell lines and with improved pharmacokinetic properties.
We previously showed that the thiazole carboxylic acid moiety forms a critical interaction with the active site R168 and could not be replaced by other groups. The 4-benzyl sulfonamide group likewise forms critical hydrogen bond interactions with Asp140, Glu191, and Ile141 that contribute significantly to the potency. Further, the cyclopropylmethyl group forms an important π stacking interaction with Tyr238 and was found to be a key substituent for conferring cellular potency. However, previous modifications of the benzyl sulfonamide or replacements for the cyclopropylmethyl group had been limited, and thus, the potential to improve potency and residence time had not been fully explored. Additionally, in our previous cocrystal structure,19 the binding pose shows that the biphenyl does not fully occupy the hydrophobic pocket in this region, suggesting an opportunity to introduce other lipophilic groups in a region that appeared to be the most accommodating for further SAR optimization. Therefore, the lead optimization efforts described here have focused on these three regions of the compounds in this series.
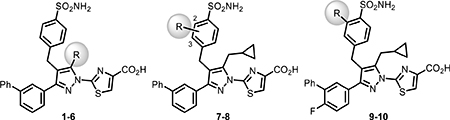
Based on our published crystal structures of compounds in this series bound to LDHA, the SAR campaign began with more extensive investigations of the cyclopropylmethyl group. Initially, a difluoro group was introduced into the cyclopropyl ring (2), anticipating an additional hydrogen bonding interaction with the phenol of residue Tyr238 (see Figure 2). Although analogue 2 maintained the biochemical potency (24 vs 25 nM), it showed reduced cellular potency and cytotoxicity compared to 1 (A673 lactate = 1423 vs 450 nM, A673 cytotox = 4642 vs 2450 nM; MiaPaCa-2 lactate = 1721 vs 636 nM, cytotox = 10,660 vs 8448 nM). To extend deeper into this binding site, the cyclopropylethyl (3) analogue was made. This group resulted in reduced biochemical LDHA activity and offered only marginal improvement in cellular potency. This trend appeared to be general, as it replicated across numerous matched pairs of analogues (see 43 vs 74, 52 vs 75, 54 vs 77, 56 vs 78, 57 vs 79, 63 vs 80, 64 vs 81, 65 vs 82, and 67 vs 83 in Tables 2–4). Replacing the cyclopropylmethyl group with other fluorinated alkyl groups (4–6) diminished potency in the biochemical assay and drastically reduced the cellular activity compared to 1. Adding a branched substitution to the methylene group (84, 85, and 89) or adding substitution on the cyclopropyl ring (86–88) further reduced both biochemical and cellular potencies, relative to 75. The primary focus was to gain cellular potency, yet moieties other than cyclopropylmethyl or cyclopropylethyl failed to improve the activity in both lactate and cytotoxicity cellular assays. Therefore, the preferred cyclopropylmethyl group was utilized in subsequent analogues. We next explored fluorine substitution at the 2- (7) or 3-positions (8) of the benzenesulfonamide ring. Analogue 7, with a 2-fluorobenzene sulfonamide substitution, showed a similar biochemical potency to 1 and a slightly decreased activity in the lactate assay but exhibited an improvement in cytotoxicity. The 3-fluoro analogue, 8, exhibited a significant decrease in activity across all assays. Initially, we were unable to rationalize these observations, as neither fluorine group appeared positioned to contribute an additional specific interaction with LDH, based on the previous crystal structure. However, crystal structures with 2-fluoro-containing analogues (23, 52 Figure 2) clearly show a preferred orientation of the ring, with fluorine occupying a position pointed into the protein and away from the space occupied by the biphenyl or alkyne substitutions. Based on the previously reported correlation between the binding kinetics and the cellular potency, we subsequently found that the 2-fluoro substitution improves the off-rate of the molecule (Table 6), which might explain the enhanced cytotoxic effects in MiaPaCa-2 and A673 cellular assays. Therefore, the 2-fluoro group was retained as an optimal substitution to evaluate further modifications of the lead molecule.
Figure 2.

Crystal structure of LDHA bound to inhibitor 23 (A, pdb code 6Q0D) and 52 (B, pdb code 6Q13). LDHA is shown in ribbon (blue) and key residues in the active site are shown in green. Small-molecule inhibitors are shown in sticks with salmon- and magenta-colored carbons.
Table 6.
koff and Residence Time (τ) Data Correlation to Cell Activity for Selected Analogues
| A673 cells |
MiaPaCa-2 cells |
||||||
|---|---|---|---|---|---|---|---|
| cpd. | KD (nM)a | residence time τ (h) | off ratea koff (×104) | lactate inh IC50 (nM)a | cytotox IC50 (nM) | lactate inh IC50 (nM)a | cytotox IC50 (nM)a |
| 1 | 0.11 | 0.35 | 8 | 450 | 2454 | 636 | 8448 |
| 23 | 0.0193 | >277 | 0.01 | 509 | 100 | 333 | 314 |
| 38 | 0.1235 | >277 | 0.01 | 487 | 939 | 424 | 848 |
| 43 | 0.0667 | >277 | 0.01 | 873 | 119 | 403 | 257 |
| 52 | 0.0634 | >277 | 0.01 | 760 | 105 | 367 | 347 |
KD and koff were determined via SPR on a Mass-1 instrument from Sierra Sensors and residence time τ was calculated as 1/koff (s−1).
Previous SAR19 suggested that the 3-biphenyl moiety provided significant enzyme and cellular potency. However, this functional group failed to provide the pharmacokinetic profile needed for evaluation in in vivo studies. An initial assessment showed no clear SAR trend regarding the presence of a 4-fluoro group (7 and 10, Table 1). Consequently, we employed both 4-fluoro and 4-H for further modifications of the 3-phenyl group in this region. In addition to extensive SAR in the biphenyl series, to be published in due course, we explored alkynes and alkenes as phenyl isosteres. We rationalized that alkynes, with a linear conformation and minimal steric bulk, could present the terminal groups to a hydrophobic region in the enzyme and potentially provide tighter binding. In an initial SAR exploration, a simple terminal alkyne 11 (Table 2) retained biochemical activity relative to 10 (Table 1), with only modest loss in potency toward lactate output (<2 fold) and cytotoxicity (<3 fold) in both cell lines. We thus explored a variety of substitutions on the alkyne to better understand the SAR in this hydrophobic space in LDH (Tables 2 and 3). Initially, we explored the terminal alkyne region with numerous alkyl and cycloalkyl groups, which could occupy the hydrophobic pocket. Most changes were tolerated, with steadily improved cell-based activities noted as the size of the alkyl group increases (e.g., 16–17 and 20). Introducing electron-withdrawing polar groups, particularly terminal –CN (33) and –CF3 groups (34) which are strong hydrogen bond acceptors, markedly reduced cellular activity. A similar trend of lower cell potency was observed for analogues 18–19 and 35–36, which also possess electron-withdrawing groups capable of hydrogen bond interactions. Introducing cycloalkyl groups onto the terminal alkyne region resulted in very potent compounds in the biochemical assay, but the activity in cellular assays was more variable. Groups such as cyclopropyl (21), cyclobutyl (22), and cyclopentyl (23 and 51) showed promising potency increases in the cellular assays. When a methylene linker was introduced (37 vs 21 and 38 vs 23), the potency in the lactate inhibition assay increased or remained similar, while less potency was observed in the cytotoxicity assays. Interestingly, when the cycloalkyl groups contained hydrogen bond acceptor or donor atoms, biochemical potency was maintained, but cellular activities significantly dropped (24–32 and 39–41 in Table 2), consistent with our previous observations.
Table 1.
Biochemical and Cellular LDH Inhibition of 2–10 with Comparator 1
 | |||||||
|---|---|---|---|---|---|---|---|
| Compd No. |  |
Biochemical LDHA IC50 (nM)a | A673 cells |
MiaPaCa-2 cells |
HEK293 Cells |
||
| lactate inh IC50 (nM)a | Cytotox. IC50 (nM)a | lactate inh. IC50 (nM)a | Cytotox. IC50 (nM)a | CETSA IC50(nM)a | |||
| 1 | 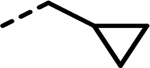 |
24 | 450 | 2454 | 636 | 8448 | - |
| 2 | 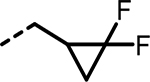 |
25 | 1423 | 4642 | 1721 | 1066 | 501 |
| 3 | 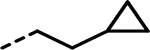 |
36 | 509 | 1959 | 804 | 6195 | 727 |
| 4 | 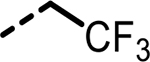 |
274 | 9719 | 148b | 19400 | 3168b | 15849 |
| 5 | 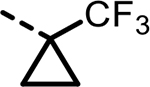 |
4104 | 20186 | >30000 | 12791 | >30000 | 303 |
| 6 | 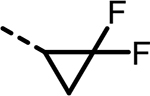 |
36 | 1861 | 24440 | 4760 | 17706 | 2529 |
| 7 | 2-F | 25 | 613 | 1144 | 867 | 3376 | 150 |
| 8 | 3-F | 229 | 4465 | 11471 | 6465 | 5747 | - |
| 9 | H | 23 | 835 | 2647 | 1224 | 8448 | 401 |
| 10 | F | 23 | 465 | 1344 | 585 | 3416 | 141 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay using a dose response in the 1536-well format. (n = 2 for lactate and n = 3 for cytotoxicity, CETSA, and biochemical LDHA.)
Efficacy < 20% with curve class 4; therefore, it should be considered as inactive.
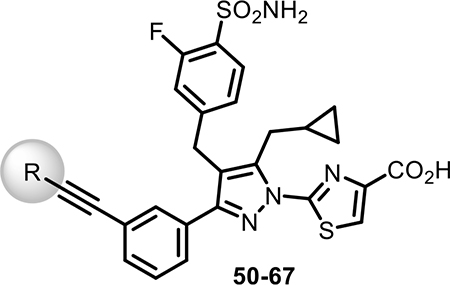
Table 3.
Biochemical and Cellular LDH Inhibition of Analogues 50–67
 | |||||||
|---|---|---|---|---|---|---|---|
| Compd No. |  |
Biochemical LDHA IC50 (nM)a | A673 cells |
MiaPaCa-2 cells |
HEK293 Cells |
||
| lactate inh. IC50 (nM)a | Cytotox IC50 (nM)a | lactate inh. IC50 (nM)a | Cytotox IC50 (nM)a | CETSA IC50 (nM)a | |||
| 50 | CH3 | 9 | 713 | 695 | 525 | 619 | 150 |
| 51 | 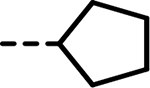 |
47 | 298 | 419 | 281 | 377 | 89 |
| 52 (NCATS-SM1441) | 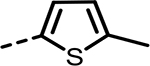 |
40 | 760 | 105 | 367 | 347 | 119 |
| 53 | 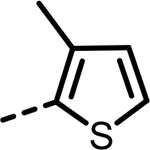 |
101 | 390 | 419 | 417 | 452 | 119 |
| 54 | 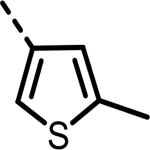 |
87 | 494 | 359 | 657 | 332 | 95 |
| 55 | 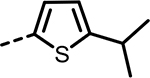 |
385 | 296 | 528 | 203 | 590 | 179 |
| 56 | 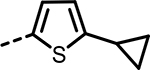 |
201 | 403 | 332 | 434 | 342 | 183 |
| 57 | 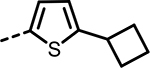 |
169 | 333 | 347 | 491 | 905 | 270 |
| 58 | 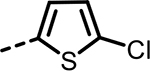 |
167 | 218 | 452 | 408 | 556 | 113 |
| 59 | 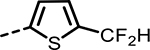 |
154 | 345 | 528 | 286 | 528 | 134 |
| 60 | 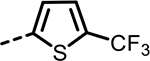 |
434 | 485 | 833 | 337 | 803 | 113 |
| 61 | 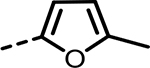 |
36 | 450 | 285 | 432 | 296 | 168 |
| 62 | 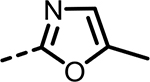 |
12 | 525 | 552 | 450 | 671 | 119 |
| 63 | 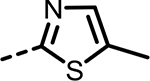 |
17 | 527 | 309 | 487 | 360 | 90 |
| 64 | 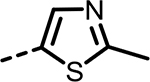 |
43 | 713 | 532 | 720 | 641 | 38 |
| 65 | 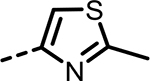 |
13 | 1174 | 347 | 1180 | 471 | 101 |
| 66 | 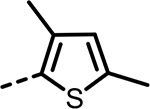 |
137 | 345 | 528 | 218 | 533 | 201 |
| 67 |  |
235 | 161 | 716 | 218 | 810 | 299 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay using a dose response in the 1536-well format. (n = 2 for lactate and n = 3 for cytotoxicity, CETSA, and biochemical LDHA.)
As the SAR indicated a preference for lipophilic groups at the terminal position of the alkyne, we synthesized analogue 42, which replaced the terminal alkyl group with a 2-thienyl group. Though analogue 42 was less potent in the biochemical assay (34 nM), it showed improved cellular potency (Table 2) in lactate (438 nM in A673 and 307 nM in MiaPaca-2) and cytotoxicity assays (265 nM in A673 and 268 nM in MiaPaca-2) compared to 10. As an unsubstituted thienyl moiety could be metabolically labile,24 we incorporated a 5-methyl thienyl group, which retained a similar potency in the cellular assays (43 and 52, which are also named as NCATS-SM1440 and NCATS-SM1441, respectively). Replacing the methyl group of thiophene with other alkyl groups (45 and 55–57) maintained the potency; however, bulkier substituents such as a t-butyl group decreased the potency (45). Moving the 5-methyl group to the 3-position of the thiophene resulted in a similar cellular potency (53), whereas switching to the 5-methylthien-3-yl substitution pattern (54 and 77) slightly decreased the activity. A 3,5- (66) or 2,5-dimethyl substitution pattern (67 and 83) on thiophene further improved the cellular potency. Interestingly, introduction of halogens onto 5-alkylthiophene significantly reduced the cellular potency (58–60). Replacing thiophene with similarly sized heterocycles was well tolerated. For instance, 5-methylfuran (44 and 61) showed similar inhibition of lactate production and cytotoxicity in both cell types compared to the thiophene analogues (43 and 52). Introducing methylthiazole (46–47, 63–65, and 80–82) or methyloxazole (62) also retained the biochemical and cellular potencies. The more hydrophilic methyl imidazole (48) or methylpyrazole (49) analogues showed good biochemical potency but less favorable cellular effects in lactate and cytotoxicity assays.
We also examined the replacement of the alkyne linker (68–73, Table 4) with an alkene motif. This slightly diminished the cellular potency despite low nanomolar biochemical activity. The alkene analogues 68 and 69 showed marginally inferior cellular potency compared to the corresponding alkyne analogues 52 and 23. A similar trend was also observed for other matched pairs, including for alkene analogue 70 versus alkyne 21, 71 versus 12, and 72 versus 17.
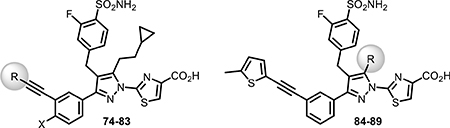
Recalling the improved cellular potency of earlier analogues with the ethylcyclopropane pyrazole substitution, we synthesized analogues 74–83 (Table 5) combining this substitution with optimal groups from the terminal alkyne region. As summarized in Table 5, analogues 74–83 showed a noteworthy improvement in cellular potency when compared to their corresponding cyclopropylmethyl analogues mentioned above.
Table 5.
Biochemical and Cellular LDH Inhibition of Analogues 74–89
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd No. |  |
X | Biochem LDHA IC50 (nM)a | A673 cells |
MiaPaCa-2 cells |
HEK293 Cells |
||
| lactate inh IC50 (nM)a | Cytotox IC50 (nM)a | lactate inh IC50 (nM)a | Cytotox IC50 (nM)a | CETSA IC50 (nM)a | ||||
| 74 |  |
F | 202 | 390 | 218 | 197 | 226 | 200 |
| 75 |  |
H | 117 | 174 | 187 | 172 | 171 | 152 |
| 76 | 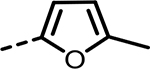 |
H | 52 | 547 | 137 | 417 | 137 | 95 |
| 77 | 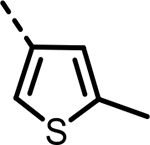 |
H | 160 | 434 | 197 | 254 | 223 | 142 |
| 78 | 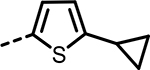 |
H | 306 | 434 | 320 | 387 | 265 | 189 |
| 79 | 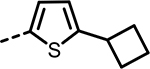 |
H | 636 | 572 | 571 | 379 | 496 | 299 |
| 80 | 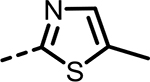 |
H | 33 | 364 | 180 | 417 | 173 | 84 |
| 81 | 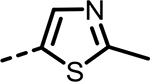 |
H | 24 | 434 | 218 | 334 | 218 | 17756 |
| 82 | 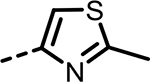 |
H | 24 | 300 | 219 | 253 | 219 | 90 |
| 83 | 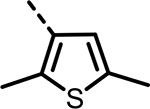 |
H | 487 | 162 | 402 | 188 | 402 | 299 |
| 84 | 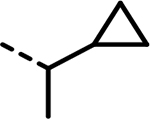 |
- | 874 | 685 | 968 | 1027 | 9350 | 708 |
| 85 | 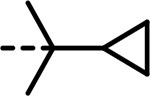 |
- | 4540 | 18264 | 5899 | 5144 | 7427 | 10751 |
| 86 | 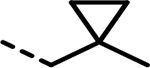 |
- | 307 | 346 | 590 | 409 | 1049 | 251 |
| 87 | 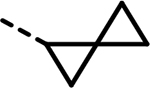 |
- | 122 | 485 | 526 | 459 | 469 | 175 |
| 88 | 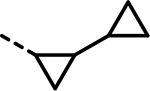 |
- | 1174 | 364 | 4686 | 515 | 5258 | 1027 |
| 89 | 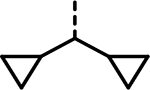 |
- | 5270 | 1864 | 26351 | 1219 | 20931 | 3183 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay using a dose response in the 1536-well format. (n = 2 for lactate and n = 3 for cytotoxicity, CETSA, and biochemical LDHA.)
In our previous report,19 we described several crystal structures with similar analogues bound to LDHA, establishing the binding mode of this series of compounds within the LDHA catalytic site and helping to guide our lead optimization campaign toward the identification of compounds with improved potency. As part of the optimization campaign, we obtained cocrystal structures of analogues 23 and 52 bound to LDHA in the presence of cofactor NADH (Figure 2). Analogues 23 and 52 present similar binding poses as the previously reported molecules, with the carboxylate making a clearly defined salt bridge interaction with Arg168 and a hydrogen bond with Thr247. Additionally, the cyclopropylmethyl group and the cyclopropylethyl group both maintain a potential pseudo π–π interaction between the cyclopropyl group and active-site tyrosine Tyr238; this substitution was found to be vital for potent cellular activity. As demonstrated previously, the 4-benzyl sulfonamide motif makes well-defined H-bonding interactions with the side chains of Asp140 and Glu191 as well as with the main chain nitrogens of Asp140, Ile141, and Glu191 to confer potent biochemical and cellular activities. Further, the cyclopentylalkyne in analogue 23 and 5-methyl thienyl alkyne in analogue 52 presented similar binding poses compared to the biphenyl ortholog. The linear rigid confirmation of the alkynes enables the presentation of moieties that offer increased hydrophobic interactions with LDHA while offering lower lipophilicity compared with a phenyl ring. It is unclear from the crystal structure exactly how some of these variations improve cellular potency. For example, the 2-fluoro moiety on the benzenesulfonamide ring is oriented in the same way in both structures. Although this group does occupy a small subpocket in the enzyme, no specific interactions with the protein are evident. The changed binding kinetics that this substitution produces (vide infra) may instead result from subtle electronic modulation of the hydrogen bonding interactions, which ultimately provided better cellular potency. These efforts demonstrate that a multiparameter optimization strategy that utilizes a structure-based design combined with targeted improvement of residence time can elicit significant improvements in cellular potency, at least in the context of LDHA inhibitors.
All of the synthesized analogues were profiled for their biochemical activity against both LDH isozymes, LDHA and LDHB, and several compounds were tested against a related “off-target” dehydrogenase, malate dehydrogenase (MDH). All analogues possess nearly identical potency against LDHB compared to LDHA (Table S1) and thus display a highly significant correlation coefficient (Figure S1). Recent reports allude to the need for a pan LDH inhibitor in order to elicit a cellular phenotype,25 highlighting a possible advantage of this profile. All the analogues showed little or no biochemical activity against MDH, with over 2500-fold selectivity for LDHA/LDHB (Table S1).
While compounds inactive in the biochemical assay elicit no cellular activity, indicating the on-target nature of the observed effects, we noted only a marginal correlation between the biochemical LDHA inhibition and inhibition of cellular lactate production throughout this optimization campaign. This poor correlation may have hampered prior efforts to identify cell-active LDHA inhibitors. The unusual trend and poor correlation between biochemical and cellular potency may be due to the disparity in the concentration of LDHA in the biochemical assay (2 nM) versus the extremely high concentration of intracellular LDH (estimated to be in the range of 2–17 μM) in glycolytic tumor cells. Furthermore, the nature of the active site preferentially selects for highly lipophilic compounds with an acidic moiety such as carboxylic acid, which can introduce additional permeability and protein binding challenges that might reduce cellular efficacy and thus influence the correlation. Though structure-guided optimization was crucial in driving the cellular potency, it was also difficult to explain the drastically improved cytotoxic effects for certain analogues as a result of subtle structural changes during our optimization.
To address such challenges, we reasoned that a highly potent compound with a long drug–target residence time might more effectively inhibit the cellular LDH activity. To better understand the reason for improved cellular activity emerging from relatively small structural changes, we complemented the structure-guided SAR with data from a surface plasmon resonance (SPR) binding assay to analyze the top cell-active compounds, in search of compounds with longer off-rates. A summary of SPR data and the corresponding cell data is outlined in Table 6 for compounds 23, 38, 43, and 52. All analogues showed more potent binding affinities and longer off-rates (koff) compared to 1. Analysis of a larger data set across the series (including unpublished data) suggests that the compounds with long off-rates and residence times (τ) [calculated as 1/koff (s–1)] offer high cellular potency in both the lactate and cytotoxicity assays. The most potent compounds exhibited off-rates that exceeded the detection limit of the assay, 10–6 s–1. Comparing the data presented here with our previously published results suggests that the 2-fluoro group on the sulfonamide phenyl ring and the alkyne substitution both positively contribute to longer residence times, leading to improved binding affinity and more potent cellular activity.
A good correlation was observed for the IC50 values for our compounds in MiaPaCa-2 and A673 cell lines in both lactate secretion and cytotoxicity assays (Figure S2); however, the correlation between inhibition of lactate secretion and cytotoxic effect within the same cell line was poor (Figure S3). As such, a superior lactate inhibition profile did not always translate into improved cytotoxic effects for some compounds. The reason for such discrepancy is unclear but may reflect the differences in the two assay conditions. For example, in the lactate secretion assay, lactate output was measured after 30 min exposure to compound, while cytotoxicity was assessed after 48 h of incubation with compound. It is possible that the ability of compounds to display higher cytotoxic effects depends on a longer residence time along with higher lactate inhibition.
Given the potential for the inhibitors to alter lactate production or viability due to unidentified off-target activity, we evaluated our LDHA inhibitors in a recently developed split Nano Luciferase (NLuc) high-throughput cellular thermal shift assay (SplitLuc-CETSA) to obtain concentration-dependent measurements of intracellular target engagement. We have previously demonstrated the applicability and performance of our SplitLuc CETSA assay to detect LDHA target engagement, in both 384- and 1536-well plate formats, for a set of well-validated inhibitors.26 Accordingly, we evaluated the majority of analogues utilizing the SplitLuc-CETSA technique in HEK293T cells (Tables 1–5), where LDHA was appended with a C-terminal 15-amino acid HiBiT fusion tag. A majority of the active compounds in the lactate assay showed cellular binding and thermal stabilization of LDHA in the SplitLuc assay. In general, we observed a good correlation between CETSA IC50 and lactate IC50 (Figure S4), though we used different cell lines in the two assays. Our lead compounds 42 and 53, along with many other compounds in the lactate assay, demonstrated stabilization of LDHA within cells. Though biochemically active compounds tended to show good CETSA activity, no clear correlation emerged between the cellular stabilization of LDHA via CETSA and the biochemical IC50. As we stated before, a similar disconnect was observed between biochemical and lactate IC50. As such biochemical LDHA screening appears to be a poor predictor of cellular effects in this series, the SplitLuc-CETSA cell-based screening platform provides a new avenue for HTS screening in a cellular context, particularly for LDHA.
To assess the impact of LDH inhibitors on the glycolytic pathway, the glycolysis stress test (GST) was performed in A673 cells (Figure 3). Glycolytic flux is assessed by measuring the extracellular acidification rate (ECAR) of the media that results from glycolysis-dependent proton production by the cells. As shown in Figure 3, after a stable baseline is recorded, the cells were treated with compounds (43 and 52-Figure 3A,B) at increasing concentrations and the ECAR is measured for approximately 50 min before injection of glucose, which results in a substantial stimulation of glycolytic flux and proton production. After three further measurements, oligomycin is injected to inhibit mitochondrial oxidative phosphorylation, which results in a compensatory increase in ECAR that is then suppressed to basal levels by the hexokinase inhibitor 2-deoxyglucose. From this experiment, two glycolysis-dependent parameters can be obtained; the basal glucose-dependent ECAR (glycolysis Figure 3C) and the maximal glycolytic capacity (shown in Figure 3B). As anticipated, as LDH inhibition increases with the concentration of compounds 43 or 52, the reserve biochemical capacity of LDH within the glycolytic pathway is exceeded, resulting in a depletion of NAD+ and, ultimately, inhibition of the entire pathway, as evidenced by decreased ECAR.
Figure 3.

LDH inhibitor-dependent suppression of glycolytic flux in A673 cells. The GST was performed in A673 cells to measure the ECAR over time; cellular basal ECAR was measured and then compound 43 (panel A) or 52 (panel B) was injected at increasing concentrations. After 48 min, subsequent injections of glucose (10 mM; glycolysis), oligomycin (1 μg/mL) (O; reaching maximal glycolytic capacity), and 2-deoxyglucose (50 mM) (2-DG; inhibition of glycolysis) were made. (C) Quantification of the glycolysis (ECAR after glucose injection minus basal ECAR) and (D) quantification of the glycolytic capacity of the LDHA inhibitors, 43 and 52, in a dose–response manner in comparison with compounds 1 and 23. Data represent the mean ± standard error of the mean, n = 4–6 per group. All LDH inhibitors completely suppressed both basal and maximal glycolysis between 1 and 3 μM, with 43 and 52 being the most potent and 23 the least.
Compounds in the series were assessed for their tier 1 ADME profile using stability in rat liver microsomes (RLM), PAMPA permeability, and aqueous solubility at pH 7.4 (data shown in Table S2). Most compounds showed good aqueous kinetic solubility, presumably enabled by the free carboxylic acid on the thiazole and the benzyl sulfonamide on the 4-position of the pyrazole. However, when additional hydrophilic groups are introduced in the alkyne region (e.g., 26, 30, 39, 47–48, 65, and 80 in Table S2), the solubility significantly diminished. The RLM stability varied depending primarily on the structure of the terminal alkyne substitutions. Analogues with n-alkyl groups longer than 2-carbon linkers in the terminal alkyne group (e.g., 14 and 15; Table S2) were metabolized quickly, likely because of CYP-mediated oxidation. This metabolism was blocked with t-butyl (17), i-Pr (16), hydroxy (18), or cyclobutyl (22) replacements (Table S2). As anticipated, a similar trend was observed for analogues 20 and 21, with iso-butyl and cyclopropyl groups, respectively. Importantly, all the analogues with cycloalkyl or heterocycles on the terminal alkyne exhibited high microsomal stabilities of >30 min, the highest estimable T1/2 from a single-point (15 min) measurement in our assay. The PAMPA permeability was low [(1 – 10) × 10–6 cm/s]27 in most cases, presumably because of the presence of a carboxylic acid group. Our representative compounds 43 and 52 showed low permeability in the PAMPA-blood–brain barrier (BBB) assay (Table 7). Notably, a combination of cyclopropylethyl group on the 5-position of pyrazole and methyl thiazole in the terminal alkyne (80–82; Table S2) slightly improves the permeability. A broader assessment of the in vitro ADME profile for representative compounds 43 and 52 is summarized in Table 7. In time course studies, both representative compounds showed excellent multispecies metabolic profiles in hepatocytes (T1/2 > 150 min), cytosol (T1/2 > 120 min), plasma (T1/2 > 240 min), and liver microsomes (T1/2 > 120 min). The analogues showed very high plasma protein binding in mouse and human plasma, likely because of the presence of both carboxylic acid and sulfonamide moieties. Both compounds only showed CYP inhibition and induction at higher concentrations. Further, the compounds were not active in hERG (patch-clamp assay) and Ames profiling assays. No significant phase I and II metabolites were detected in our in vitro metabolic study even in the presence of GSH, which is consistent with the prolonged half-life in mouse hepatocytes. Further, both compounds showed less than 2-fold PXR and AhR activation at a 10 μM.
Table 7.
Summary of in Vitro ADME Profiles for 43 and 52
| profiling assays | 43 (NCATS-SM1440) | 52 (NCATS-SM1440) |
|---|---|---|
| liver microsomal T1/2 (human, mouse, and rat) | >120 min | >120 min |
| liver cytosolic T1/2 (human, mouse, and rat) | >120 min | >120 min |
| metabolic stability (hepatocytes-rat, dog, and mouse) | >150 min | >150 min |
| plasma stability T1/2 | >240 min | >240 min |
| PAMPA permeability | 10.4 × 10−6 (cm/s) | 2.2 × 10−6 (cm/s) |
| PAMPA-BBB permeability | 16 × 10−6 (cm/s) | 9.6 × 10−6 (cm/s) |
| aq solubility at pH 7.4 | >70 μwg/mL | >70 μg/mL |
| plasma protein binding (Fu = % fraction unbound) | 0 (human), 0.7 (mouse) | 0 (human), 4.4 (mouse) |
| CYP450 inhibition (isozyme) | 2C8: 80% @10 μM, 2B6: 46% @10 μM, 2C9: 45% @10 μM | 2C8: 7.5 μM, 2B6: 14 μM |
| metabolite ID (in vitro) | no significant phase I or II metabolites | no significant phase I or II metabolites |
| CYP induction | no induction at 10 μM | no induction at 10 μM |
| PXR and AhR Activation | <2-fold activation up to 10 μM | <2-fold activation up to 10 μM |
| reactive metabolite formation | no GSH adducts formed | no GSH adducts formed |
| hERG (patch clamp) | >10 μM | >10 μM |
| Ames | negative | negative |
Aqueous kinetic solubility (PBS buffer) and PAMPA permeability, liver microsomal & cytosolic stability studies, were conducted at NCATS. Mouse plasma stability studies were conducted at Pharmaron Inc. and involved five-time points. The microsomal stability data [mouse liver microsomes (MLM), human liver microsomes (HLM), and mouse hepatocytes] were conducted at QuintaraBio and represent the stability in the presence of NADPH and UDPGA. The parent compound was monitored at five-time points.
Encouraged by the improved cellular potency and in vitro ADME profiles, we evaluated top compounds for mouse PK properties following IV and PO administration (summarized in Table 8). Representative analogues (43–44, 46–47, 52, 61, 69, and 74–75) showed improved mouse PK properties compared to 1. IV administration at 10 mg/kg for selected alkyne analogues showed a terminal half-life of ~5 h or more, far superior to 0.85 h for 1. Thus, both the use of the substituted alkyne as a phenyl isostere and adding 2-fluoro in the sulfonamide region seem to help improve the in vivo half-life. Moreover, most of these analogues, except for 46, also showed reduced clearance. The clearance increased steadily as the steady-state volume of distribution (Vss) increased, as noted for several matched pairs in the series of analogues 44, 46–47, 61, and 69. Despite the presence of the carboxylic acid functional group, favorable systemic exposure was achieved for several of these compounds, with plasma concentrations in the range of cellular IC50 values following PO dosing at 40 or 50 mg/kg. Introducing more lipophilic groups, such as 5-methyl thienyl (43 and 52), 5-methyl furyl (44 and 61), or cyclopentyl (69) in the alkyne region, significantly improved the bioavailability (74% for 43; 106% for 44; 64% for 52, and 83% for 69) presumably because of higher lipophilicity leading to better permeability and absorption. Analogues 43 (AUClast = 31.39 μg/mL h and Cmax of 5.26 μg/mL at 40 mg/kg) and 52 (AUClast = 53.54 μg/mL h and Cmax of 8.83 μg/mL) possessed the best systemic exposure in this series. Relative to these molecules, replacing the cyclopropylmethyl group with cyclopropylethyl in the 5-position of the pyrazole and keeping 5-methyl thienyl in the alkyne region (analogues 76 and 77 with 37 and 42%, respectively) significantly reduced the bioavailability and systemic exposure. A similar trend was manifested for analogues 44 and 46 that contain methyl thiazole groups at the terminal alkyne. Overall, our SAR study shows that the 5-methylthienyl group in the alkyne region is key for improved pharmacokinetic properties. Thus, analogues 43 and 52 possess suitable PK profiles for evaluation in efficacy models.
Table 8.
Pharmacokinetic Profiles of Top Analogues in CD1 Micea
| compd | route | dose | Cl (mL/min/kg) | T1/2 (h) | bCmax (μg/mL) | AUClast (μg/mL·h) | Vss (L/kg) | F (%) |
|---|---|---|---|---|---|---|---|---|
| 43 | IV | 10 | 16.1 | 5.2 | 41.79 | 10.57 | 1.79 | |
| PO | 40 | 2.78 | 5.26 | 31.39 | 73.8 | |||
| 44 | IV | 10 | 75 | 5.1 | 4.77 | 2.32 | 10.3 | |
| PO | 50 | 10.7 | 1.26 | 13.52 | 106 | |||
| 46 | IV | 10 | 97 | 5.75 | 5.5 | 1.82 | 13.9 | |
| PO | 50 | 3.2 | 1.82 | 3.44 | 38 | |||
| 47 | IV | 10 | 128 | 4.9 | 3.32 | 1.36 | 17.9 | |
| PO | 50 | 3.54 | 2.03 | 2.65 | 39 | |||
| 52 | IV | 10 | 10.4 | 5.3 | 33.82 | 16.53 | 1.7 | |
| PO | 50 | 2.6 | 8.83 | 53.54 | 64 | |||
| 61 | IV | 10 | 72.3 | 5 | 4.57 | 2.28 | 12.5 | |
| PO | 50 | 3.7 | 2.28 | 10.17 | 89 | |||
| 69 | IV | 10 | 74.7 | 5.5 | 6.44 | 2.81 | 13.3 | |
| PO | 50 | 3.1 | 1.2 | 9.22 | 83 | |||
| 74 | IV | 10 | 74.7 | 6.5 | 35.33 | 6.65 | 2.5 | |
| PO | 50 | 3.2 | 4.73 | 12.24 | 37 | |||
| 75 | IV | 10 | 18.6 | 5.8 | 58.23 | 9.6 | 1.47 | |
| PO | 50 | 3.7 | 6.85 | 20.03 | 42 |
Values calculated from drug concentration in plasma following IV or PO dosing. n = 3, 8 time points taken over 24 h. Compounds were formulated as a solution in PBS with 1.1 equiv of NaOH (final pH 7–8).
Cmax = C0 (t = 0) for IV administration. All pharmacokinetic studies were conducted at Pharmaron, Inc.
Despite the long-standing interest in drugging LDH, efforts have thus far failed to achieve sub-micromolar cellular potency while also achieving optimal PK properties. Having identified the best-in-class lead compounds in terms of cellular potency and PK profile, we evaluated the top compounds in a mouse tumor model using A673 cells to demonstrate LDH inhibition in vivo (Figure 4). An IV administration at 50 mg/kg of 43 and 52 significantly reduced the LDH activity in tumors analyzed at 1 and 6 h postdose. These two analogues have high exposure and low clearance rates compared to analogues 47, 69, and 74. In mice dosed with 52, 80% inhibition of tumor LDH activity was observed at 6 h postdose, compared to approximately 60% inhibition with 43. Although both 43 and 52 possess good pharmacokinetic properties, 52 has a slightly higher Cmax and lower clearance, which correlates with its superior in vivo LDH inhibition profile. 56 and 74 showed moderate inhibition of LDH activity in tumors, and 14, 47, 69, and 71 have less effects. 56 suppressed tumor LDH activity below 40% of control at 1 h postdose but failed to maintain significant inhibition at 6 h possibly because of a higher clearance. The diminished activity of analogues 47, 69, and 74 can be attributed to a less optimal PK profile having reduced exposure and higher clearance compared to 43 and 52 (Table 8). These data suggest an excellent correlation of in vivo LDH inhibition with the pharmacokinetic profile, which is vital to demonstrate sustained in vivo LDH inhibition. It is important to note that analogues 42 and 53 are the first LDH inhibitors to demonstrate sustained target engagement in vivo. Of note, a clear correlation is evident between the cellular concentrations necessary to affect glycolytic flux and the concentrations achieved in vivo at doses that demonstrate target engagement. Consistent with these data, both compounds showed efficacy in sarcoma20 and pancreatic21 xenograft models at similar doses.
Figure 4.

In vivo target engagement and tumor LDH activity upon administration of compounds in mice bearing A673 flank xenograft tumors. Mice received a single IV injection of LDHA inhibitors 14, 43, 47, 52, 56, 69, and 71 at 50 mg/kg. At the time of sacrifice, the samples of tumor and plasma were collected and flash-frozen in liquid nitrogen. Compound levels in plasma and tumor were determined by LC–MS/MS, and LDHA activity was measured in tumor lysates.
In order to develop an LDH inhibitor suitable for in vivo experiments, a diverse set of parameters were used to inform design decisions, with a focus on improving cellular potency, establishing a long residence time in the binding pocket, and identifying optimal PK properties. In our case, structure-guided optimization coupled with long residence time helped to identify lead compounds with nanomolar cellular potency. Ultimately, improvement in PK attributes allowed us to demonstrate the first notable inhibition of LDH in tumors following dosing of mice harboring A673 xenografts. We identified critical structural features in each region of the molecule that contributed to improvements in the properties summarized in Figure 5. In short, the carboxylic acid on the thiazole ring and the 4-benzyl sulfonamide are keystone interactions critical for biochemical and cellular potency. Meanwhile, cyclopropylmethyl or cyclopropylethyl substitution on the pyrazole was crucial for cellular potency. The alkyne linker in the biphenyl region and the 2-fluoro group on benzenesulfonamide provide a longer off-rate that further improves the cellular activity. Heterocycles, such as 5-methylthienyl, 5-methylfuryl, or thiazolyl groups distal to the alkyne, contribute to improved cellular potency and, most importantly, improved in vitro ADME and in vivo PK properties.
Figure 5.

SAR summary and essential structural moieties contributing to the cellular potency, binding affinity, and PK properties of analogue 52 (NCATS-SM1441) as an example.
CONCLUSIONS
Pharmaceutical companies and researchers in academia have invested significant efforts to identify small-molecule inhibitors of LDH. Such attempts appear to have shown limited success in discovering potent and drug-like inhibitors that can be utilized to validate LDH as a drug target in vivo, a necessary step toward advancing such a molecule toward the clinic. We describe a lead optimization campaign, aided by crystallography as well as the assessment of residence time, that has resulted in LDH inhibitors with improved cellular potency, in vitro ADME, and in vivo PK properties. We report the first LDH inhibitors showing direct target engagement in human tumor xenografts. Through a systematic SAR campaign, we identified optimal structural features in each region of the molecule that contributed to slow off-rates and improved PK properties, parameters which we found to be significant determinants of improved cellular potency and in vivo target engagement. We report compounds with enhanced cellular potency and properties that enable modulation of LDH activity in vivo, thus opening the door for studies to evaluate LDH as a pharmaceutical target in cancer models such as Ewing’s sarcoma20 or pancreatic cancers.21
EXPERIMENTAL SECTION
Chemistry.
General Methods.
All air- or moisture-sensitive reactions were performed under a positive pressure of nitrogen or argon with oven-dried glassware. Anhydrous solvents and bases such as dichloromethane (DCM), N,N-dimethylformamide, acetonitrile, ethanol, DMSO, dioxan, and DABCO were purchased from Sigma-Aldrich. Palladium catalysts were purchased from Johnson Matthey and used as such. Preparative purification was performed on a Waters semipreparative HPLC system using a Phenomenex Luna C18 column (5 μm, 30 × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10–50% acetonitrile over 8 min was used during the purification. Fraction collection was triggered by UV detection (220 nm). The analytical analysis was performed on an Agilent LC–MS (Agilent Technologies, Santa Clara, CA). Method 1: A 7 min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 min run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 μm, 3 × 75 mm) was used at a temperature of 50 °C. Method 2: A 3 min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 min run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 μm, 3 × 100 mm) was used at a temperature of 50 °C. Purity determination was performed using an Agilent Diode Array Detector for both method 1 and method 2. The mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. Chemical shifts are reported in parts per million with an undeuterated solvent (DMSO-d6 at 2.49 ppm) as an internal standard for DMSO-d6 solutions. All of the analogues tested in the biological assays have purity greater than 95% based on both analytical methods. High-resolution mass spectrometry was performed on an Agilent 6210 time-of-flight LC–MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
General Procedure for the Synthesis of Acylbenzotriazole Derivatives Ia–I (Procedure A).
These compounds were prepared as described previously.19 To the solution of 1H-benzo[d][1,2,3]-triazole (476 g, 3995 mmol, 4 equiv) in DCM (600 mL) was added thionyl chloride (72.9 mL, 999 mmol, 1 equiv), and the reaction mixture was stirred at room temperature for 0.5 h. After cooling in an ice–water bath (for larger scale, the cooling was necessary because of the exothermic reaction. If the reaction mixture formed a thick precipitate that was difficult to stir, then more DCM was added), carboxylic acid (999 mmol, 1 equiv) was carefully added and the mixture was stirred for 6 h. The reaction was filtered, and the filter cake was washed with DCM. To the filtrate was added bicarbonate solution slowly, and the resulting mixture was stirred for 30 min and then transferred to a separatory funnel. The organic layer was subsequently washed with bicarbonate solution and brine solution. The organic layer was dried under sodium sulfate and concentrated to provide a thick oil. The crude product was purified on a CombiFlash system using a 340 g silica column eluting with 0–20% ethyl acetate in hexanes over 10 column volumes. The first peak was collected, concentrated, and dried to get pure products Ia–l as oil, which eventually solidifies into white solid in 91–100% yield.
General Procedure for the Synthesis of 1-Aryl-3-substituted Propane-1,3-diones III (Procedure B).
To a mixture of appropriate acetophenone IIa–d (51.0 mmol, 1 equiv), magnesium bromide diethyl etherate (32.9 g, 127 mmol, 2.5 equiv), and the corresponding acylbenzotriazole derivative Ia–l (76 mmol, 1.5 equiv in CH2Cl2) (100 mL) was added Hunig’s base (26.7 mL, 153 mmol, 3 equiv) slowly (cooling is necessary for large scale), and the mixture was stirred at rt for 12 h. The reaction mixture was cooled in an ice bath and quenched with 1 M HCl. The product was extracted with CH2Cl2, and the organic layer was subsequently washed with brine. After drying the organic layer with MgSO4, the crude product was purified on an Isco flash system using a 220 g gold column eluting with 0–30% ethyl acetate over 20 column volumes in hexanes to afford a yellow oil after removing the solvent in 60–69% yield required intermediates III.
General Procedure for the Alkylation of 1-Aryl-3-substituted Propane-1,3-diones Va–r (Procedure C).
1-Aryl-3-substituted propane-1,3-dione (35.9 mmol, 1 equiv) and cesium carbonate (14.05 g, 43.1 mmol, 1.2 equiv) in DMSO (25 mL) were stirred at room temperature for 10 min. The appropriate 4-(bromomethyl)-benzenesulfonamide IVa–c (43.1 mmol, 1.2 equiv) was added in one portion, and the reaction mixture was further stirred at room temperature for another 1–2 h. The resulting mixture was diluted with large excess of ethyl acetate and filtered through Celite to remove any solid impurities. The filtrate was washed with saturated ammonium chloride (3×) and then with brine. The organic layer was dried with Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified directly on a flash system using a 220 g silica column eluting with 20–60% ethyl acetate in hexanes over 16 column volumes to afford pure products Va–r in 55–83% yield.
General Procedure for the Cyclization of 1-Aryl-2,3-disubstituted Propane-1,3-diones To Obtain VIa–r and VIIa–r (Procedure D).
To a mixture of the appropriate 1-aryl-2, 3-disubstituted propane-1,3-dione (2.24 mmol, 1 equiv) in ethanol were added pyrrolidine (0.5 equiv) and tosic acid (0.5 equiv), and the mixture was heated at reflux for 1–2 h. The reaction mixture was removed from the heating block, and then ethyl 2-hydrazinylthiazole-4-carboxylate hydrogen bromide (0.600 g, 2.24 mmol, 1 equiv) was added in one portion. The reaction was again heated at reflux for 12 h. The reaction mixture was concentrated, and the residue was taken up in DCM and loaded to a prepacked silica loading cartridge. The product was purified on a flash system using a 100 g silica column eluting with 20–40% ethyl acetate in hexanes to get an inseparable 1:1 mixture of regioisomers in a combined yield of 77–83%. The product mixture was then taken up in DMSO and injected onto a C18 gold column and eluted with 60–100% ACN–water (contains 0.1% TFA). The second peak (the desired isomers VIa–r) was pooled, and most of the ACN was removed under reduced pressure. The solid formed was collected by filtration, washed with water, and air-dried to get pure compounds VIa–r in 21–36% yield.
General Procedure for Sonogashira Coupling Using the P(t-Bu)3]Pd(crotyl)Cl Catalyst (Procedure E).
A mixture of ethyl 2-(3-(3-bromo-4H/fluoro phenyl)-5-(substituted)-4-(3/4-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate (1 mmol), P(t-Bu)3]Pd(crotyl)Cl precatalyst Pd-162 (5 mol %), and DABCO (2 mmol, 2 equiv) in dioxan (0.5 M concentration) was bubbled with argon for 5 min, then alkyne (1.5 mmol, 1.5 equiv) was added, and the mixture was stirred at room temperature overnight. After completion of the reaction, a silica-bound palladium scavenger was added and the mixture was stirred at room temperature for 1 h, then diluted with ethyl acetate, and filtered through a pad of Celite. The filtrate was concentrated, and the residue was purified on an Isco flash system eluting with 20–40% ethyl acetate in hexanes over 20 column volumes to obtain pure products (NOTE: the above reaction also works with 10 mol % tri-tert-butylphosphonium tetrafluoroborate and 5 mol % allylpalladium chloride dimer under the same conditions).
General Procedure for Sonogashira Coupling Using the [DTBNpP]Pd(crotyl)Cl Catalyst (Procedure F).
A mixture of ethyl 2-(3-(3-bromo-4H/fluoro phenyl)-5-(substituted)-4-(3/4-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate (1 mmol), DTBNpP]Pd(crotyl)Cl precatalyst Pd-163, and DABCO (2 mmol, 2 equiv) in dioxane (0.5 M concentration) was bubbled with argon for 5 min, alkyne (1.5 mmol, 1.5 equiv) was added, and the reaction mixture was stirred at 60 °C overnight. After completion of the reaction, a silica-bound palladium scavenger was added and the mixture was stirred at room temperature for 1 h, then diluted with ethyl acetate, and filtered through a pad of Celite. The filtrate was concentrated, and the residue was purified on an Isco flash system eluting with 20–100% ethyl acetate in hexanes over 25 column volumes to obtain pure products.
General Procedure for Suzuki Coupling (Procedure G).
To a mixture of ethyl 2-(3-(3-bromo-4-substitutedphenyl)-5-(cyclopropylmethyl)-4-(3-substituted-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carbox-ylate (0.2 mmol, 1 equiv), potassium phosphate (0.4 mmol, 2 equiv), SPhos Pd(crotyl)Cl precatalyst Pd-172 (2.5 mol %), and the appropriate boronic acid/pinacol ester/trifluoroboronate in a Biotage microwave vial were added dioxane (2 mL) and water (0.5 mL). The reaction mixture was bubbled with argon for few minutes, then capped, and stirred at 100 °C in a preheated heating block for 0.5 h. Upon completion of the reaction, the reaction mixture was cooled and stirred with a metal scavenger for 1 h. The reaction mixture was then diluted with ethyl acetate and filtered through a pad of Celite. The filtrate was concentrated and purified on an Isco flash system using a silica column eluting with 20–40% ethyl acetate in hexanes to get pure products.
General Procedure for the Hydrolysis of Esters To Synthesize Analogues 1–89 (Procedure H).
The corresponding ethyl ester intermediates were suspended in tetrahydrofuran (THF)–MeOH (3/2; 5 mL) and treated with 1.5 M solution (5 equiv) of aqueous lithium hydroxide. The reaction mixture was stirred for 1 h, and the solvent was removed by forced air and acidified with 1 M hydrochloric acid. The crude material was taken up in DMSO and purified on a preparative HPLC.
2-(3-([1,1′-Biphenyl]-3-yl)-5-((2,2-difluorocyclopropyl)methyl)-4-(4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (2).
This compound was synthesized using general procedures A, B, C, D, and H via intermediates Ib through VIIg. LC–MS retention time: (method 1) = 6.071 min and (method 2) = 3.483 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.30 (s, 1H), 7.77–7.67 (m, 4H), 7.63–7.50 (m, 2H), 7.49–7.41 (m, 4H), 7.39–7.28 (m, 5H), 4.19 (s, 2H), 3.45–3.35 (m, 2H), 2.27–2.11 (m, 1H), 1.56–1.30 (m, 2H); HRMS (ESI) m/z: (M + Na)+ calcd for C30H24F2N4NaO4S2, 629.1099; found, 629.1122.
2-(3-([1,1′-Biphenyl]-3-yl)-5-(2-cyclopropylethyl)-4-(4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (3).
This compound was synthesized using general procedures A, B, C, D, and H via intermediates Ic through VIIh. LC–MS retention time: (method 1) = 6.355 min and (method 2) = 3.598 min; 1H NMR (400 MHz, DMSO-d6): δ 13.12 (s, 1H), 8.28 (s, 1H), 7.79–7.65 (m, 4H), 7.61 (dt, J = 7.7, 1.4 Hz, 1H), 7.57–7.48 (m, 1H), 7.52–7.40 (m, 4H), 7.44–7.33 (m, 2H), 7.38–7.29 (m, 4H), 4.18 (s, 2H), 3.30–3.22 (m, 2H), 1.45 (q, J = 7.3 Hz, 2H), 0.87–0.67 (m, 1H), 0.36–0.25 (m, 2H), 0.17–0.09 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.79, 152.52, 144.58, 144.09, 142.23, 140.38, 139.40, 132.38, 129.51, 129.01, 128.26, 127.73, 126.95, 126.57, 126.53, 126.00, 125.87, 125.68, 117.02, 33.32, 28.37, 24.81, 15.23, 15.20, 10.62, 4.19; HRMS (ESI) m/z: (M + H)+ calcd for C31H29N4O4S2, 585.1625; found, 585.1604.
2-(3-([1,1′-Biphenyl]-3-yl)-4-(4-sulfamoylbenzyl)-5-(2,2,2-trifluoroethyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (4).
This compound was synthesized using general procedures A, B, C, D, and H via intermediates Id through VIIk. LC–MS retention time: (method 1) = 5.991 min and (method 2) = 3.588 min; 1H NMR (400 MHz, DMSO-d6): δ 13.24 (s, 1H), 8.33 (s, 1H), 7.74–7.62 (m, 5H), 7.50 (td, J = 7.6, 0.7 Hz, 1H), 7.43 (d, J = 4.3 Hz, 4H), 7.39–7.28 (m, 5H), 4.69 (q, J = 10.5 Hz, 2H), 4.30 (s, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C28H22F3N4O4S2, 599.1029; found, 599.1043.
2-(3-([1,1′-Biphenyl]-3-yl)-4-(4-sulfamoylbenzyl)-5-(1-(trifluoromethyl)cyclopropyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (5).
This compound was synthesized using general procedures A, B, C, D, and H via intermediates Ie through VIIl. LC–MS retention time: (method 1) = 6.031 min and (method 2) = 3.61 min; 1H NMR (400 MHz, DMSO-d6): δ 13.11 (s, 1H), 8.34 (s, 1H), 7.76–7.70 (m, 2H), 7.69–7.62 (m, 3H), 7.52–7.45 (m, 1H), 7.45–7.27 (m, 9H), 4.37 (s, 2H), 1.79–1.09 (m, 5H); HRMS (ESI) m/z: (M + H)+ calcd for C30H24F3N4O4S2, 625.1186; found, 625.1215.
2-(3-([1,1′-Biphenyl]-3-yl)-5-(2,2-difluorocyclopropyl)-4-(4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (6).
This compound was synthesized using general procedures A, B, C, D, and H via intermediates If through VIIm. LC–MS retention time: (method 1) = 5.858 min and (method 2) = 3.553 min; 1H NMR (400 MHz, DMSO-d6): δ 13.20–12.91 (m, 1H), 8.34 (s, 1H), 7.79–7.66 (m, 4H), 7.62–7.50 (m, 2H), 7.49–7.40 (m, 4H), 7.39–7.27 (m, 5H), 4.22 (s, 2H), 3.29–3.23 (m, 1H), 2.25–2.04 (m, 1H), 1.85–1.63 (m, 1H); HRMS (ESI) m/z: (M + H)+ calcd for C29H23F2N4O4S2, 593.1123; found, 593.114.
2-(3-([1,1′-Biphenyl]-3-yl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (7).
This compound was synthesized using general procedures A, B, C, D, and H via intermediates Ia through IVb and VIIb. LC–MS retention time: (method 1) = 6.133 min and (method 2) = 3.641 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.30 (s, 1H), 7.73–7.65 (m, 3H), 7.64–7.58 (m, 3H), 7.52 (t, J = 7.7 Hz, 1H), 7.49–7.41 (m, 4H), 7.40–7.33 (m, 1H), 7.26–7.07 (m, 2H), 4.20 (s, 2H), 3.19 (d, J = 6.9 Hz, 2H), 1.34–1.04 (m, 1H), 0.44–0.29 (m, 2H), 0.29–0.17 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H26FN4O4S2, 589.1374; found, 589.1363.
2-(3-([1,1′-Biphenyl]-3-yl)-5-(cyclopropylmethyl)-4-(2-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (8).
This compound was synthesized using general procedures A, B, C, D, and H via intermediates Ia through IVc and VIIc. LC–MS retention time: (method 1) = 6.234 min and (method 2) = 3.576 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.31 (s, 1H), 7.73–7.64 (m, 2H), 7.62–7.56 (m, 2H), 7.53 (ddd, J = 7.6, 4.2, 1.2 Hz, 2H), 7.51–7.40 (m, 6H), 7.40–7.33 (m, 1H), 7.21 (t, J = 7.8 Hz, 1H), 4.14 (s, 2H), 3.20 (d, J = 6.9 Hz, 2H), 1.27–1.06 (m, 1H), 0.40–0.31 (m, 2H), 0.27–0.20 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H26FN4O4S2, 589.1374; found, 589.1394.
2-(5-(Cyclopropylmethyl)-3-(6-fluoro-[1,1′-biphenyl]-3-yl)-4-(4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (9).
This compound was synthesized using general procedures A, B, C, D, and H via intermediates Ia through IVa and VIId. LC–MS retention time: (method 1) = 6.297 min and (method 2) = 3.534 min; 1H NMR (400 MHz, DMSO-d6): δ 11.56 (s, 1H), 6.68 (s, 1H), 6.11 (d, J = 8.1 Hz, 2H), 6.03 (ddd, J = 8.7, 4.7, 2.3 Hz, 1H), 5.96 (dd, J = 7.6, 2.3 Hz, 1H), 5.88–5.66 (m, 10H), 2.56 (s, 2H), 1.56 (d, J = 6.9 Hz, 2H), –0.37 to –0.53 (m, 1H), –1.28 (dt, J = 8.5, 2.8 Hz, 2H), –1.35 to –1.42 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H26FN4O4S2, 589.1374; found, 589.1364.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(6-fluoro-[1,1′-biphenyl]-3-yl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (10).
This compound was synthesized from coupling the advanced intermediate VIIf (0.78 mmol) with phenylboronic acid utilizing the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.073 min and (method 2) = 3.54 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.30 (s, 1H), 7.72–7.62 (m, 2H), 7.60 (s, 2H), 7.55 (dd, J = 7.6, 2.3 Hz, 1H), 7.50–7.35 (m, 6H), 7.19 (dd, J = 11.3, 1.6 Hz, 1H), 7.08 (dd, J = 8.1, 1.6 Hz, 1H), 4.18 (s, 2H), 3.24–3.11 (m, 2H), 1.24–1.10 (m, 1H), 0.45–0.31 (m, 2H), 0.29–0.17 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.77, 161.19, 151.50, 144.67, 144.47, 134.41, 129.72, 128.70, 128.67, 128.64, 128.57, 128.53, 128.29, 128.14, 126.13, 123.64, 116.95, 116.72, 116.55, 116.45, 116.23, 28.23, 28.14, 15.18, 10.32, 4.49; HRMS (ESI) m/z: (M + H)+ calcd for C30H25F2N4O4S2, 607.128; found, 607.129.
2-(5-(Cyclopropylmethyl)-3-(3-ethynyl-4-fluorophenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (11).
This compound was synthesized from coupling the advanced intermediate VIIf (3.14 mmol, 2 g) with trimethylsilylacetylene utilizing the general Sonogashira coupling procedure E. The product (3.05 mmol, 2 g) obtained was deprotected while stirring with CsF (0.51 g, 3.36 mmol, 1.1 equiv) in THF/ethanol (11 mL/4 mL) at room temperature for 2 h. The reaction mixture was diluted with ethyl acetate and filtered through a Celite pad. The crude product after removing the solvent was purified in an Isco flash system using a silica column eluting with 20–40% ethyl acetate in hexanes to get pure TMS deprotected product VIIIb in 90% yield over two steps. LC–MS retention time: (method 2) = 3.72 (M + H)+ = 583. 1H NMR (400 MHz, DMSO-d6): δ 8.38 (d, J = 1.2 Hz, 1H), 7.75–7.60 (m, 3H), 7.57 (s, 2H), 7.39–7.31 (m, 1H), 7.14 (dd, J = 11.3, 1.7 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 4.55 (s, 1H), 4.32 (qd, J = 7.1, 1.1 Hz, 2H), 4.16 (s, 2H), 3.16 (d, J = 6.9 Hz, 2H), 1.32 (td, J = 7.1, 1.1 Hz, 2H), 1.23–1.08 (m, 1H), 0.39–0.31 (m, 2H), 0.25 (dt, J = 5.1, 1.4 Hz, 2H). A portion of VIIIb was hydrolyzed using general procedure H to obtain analogue 11. LC–MS retention time: (method 1) = 5.653 min and (method 2) = 3.66 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.74–7.59 (m, 3H), 7.57 (s, 2H), 7.35 (t, J = 9.1 Hz, 1H), 7.14 (dd, J = 11.4, 1.6 Hz, 1H), 7.04 (dd, J = 8.1, 1.6 Hz, 1H), 4.55 (s, 1H), 4.16 (s, 2H), 3.16 (d, J = 6.9 Hz, 2H), 1.22–1.04 (m, 1H), 0.43–0.26 (m, 2H), 0.26–0.11 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C26H21F2N4O4S2, 555.0967; found, 555.0966.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(prop-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (12).
This compound was synthesized from coupling the advanced intermediate VIIf (0.314 mmol, 0.2 g) with potassium trifluoro(prop-1-yn-1-yl)borate (0.069 g, 0.471 mmol) using the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.928 min and (method 2) = 3.518 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.30 (s, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.59 (d, J = 6.6 Hz, 3H), 7.53 (ddd, J = 8.6, 5.0, 2.3 Hz, 1H), 7.30 (dd, J = 9.5, 8.6 Hz, 1H), 7.14 (dd, J = 11.4, 1.6 Hz, 1H), 7.04 (dd, J = 8.2, 1.6 Hz, 1H), 4.14 (s, 2H), 3.16 (s, 2H), 2.09 (s, 3H), 1.20–1.06 (m, 1H), 0.39–0.29 (m, 2H), 0.25–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C27H23F2N4O4S2, 569.1123; found, 569.1116.
2-(3-(3-(But-1-yn-1-yl)-4-fluorophenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (13).
This compound was synthesized from coupling the advanced intermediate VIIf (0.196 mmol, 0.125 g) with 2-(1-butyn-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (0.294 mmol, 0.053 g) using the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.118 min and (method 2) = 3.605 min; 1H NMR (400 MHz, DMSO-d6): δ 8.00 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.60–7.47 (m, 4H), 7.34–7.23 (m, 1H), 7.15 (dd, J = 11.3, 1.6 Hz, 1H), 7.04 (dd, J = 8.1, 1.6 Hz, 1H), 4.14 (s, 2H), 3.17 (d, J = 6.9 Hz, 2H), 2.45 (t, J = 7.5 Hz, 2H), 1.15 (s, 1H), 1.14–1.07 (m, 1H), 0.38–0.29 (m, 2H), 0.24–0.17 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C28H25F2N4O4S2, 583.128; found, 583.1267.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(pent-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (14).
This compound was synthesized from coupling the advanced intermediate VIIf (0.196 mmol, 0.125 g) with 1-pentyne (0.294 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.246 min and (method 2) = 3.718 min; 1H NMR (400 MHz, DMSO-d6): δ 13.18 (s, 1H), 8.30 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.62–7.51 (m, 4H), 7.35–7.25 (m, 1H), 7.15 (dd, J = 11.3, 1.6 Hz, 1H), 7.04 (dd, J = 8.1, 1.6 Hz, 1H), 4.14 (s, 2H), 3.24–3.07 (m, 4H), 2.44 (t, J = 6.9 Hz, 2H), 1.56 (h, J = 7.2 Hz, 2H), 1.26–1.06 (m, 1H), 0.99 (t, J = 7.4 Hz, 3H), 0.39–0.29 (m, 2H), 0.27–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C29H27F2N4O4S2, 597.1436; found, 597.144.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(hex-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (15).
This compound was synthesized from coupling the advanced intermediate VIIf (0.196 mmol, 0.125 g) with 1-hexyne (0.294 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.511 min and (method 2) = 3.644 min; 1H NMR (400 MHz, DMSO-d6): δ 13.18 (s, 1H), 8.30 (s, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.62–7.50 (m, 4H), 7.30 (dd, J = 9.4, 8.6 Hz, 1H), 7.15 (dd, J = 11.3, 1.6 Hz, 1H), 7.04 (dd, J = 8.1, 1.6 Hz, 1H), 4.14 (s, 2H), 3.23–3.10 (m, 3H), 2.46 (t, J = 6.9 Hz, 2H), 1.59–1.37 (m, 4H), 1.20–1.06 (m, 1H), 0.91 (t, J = 7.3 Hz, 3H), 0.38–0.29 (m, 2H), 0.25–0.16 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H29F2N4O4S2, 611.1593; found, 611.1572.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(3-methylbut-1-yn-1-yl)-phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (16).
This compound was synthesized from coupling the advanced intermediate VIIf (0.235 mmol, 0.15 g) with 3-methyl-1-butyne (0.353 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.274 min and (method 2) = 3.563 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.30 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.60–7.50 (m, 4H), 7.30 (t, J = 9.0 Hz, 1H), 7.16 (dd, J = 11.4, 1.6 Hz, 1H), 7.05 (dd, J = 8.2, 1.6 Hz, 1H), 4.14 (s, 2H), 3.17 (d, J = 6.9 Hz, 2H), 2.84 (hept, J = 6.8 Hz, 1H), 1.22 (d, J = 6.9 Hz, 6H), 1.14 (ddt, J = 9.9, 7.5, 4.8 Hz, 1H), 0.39–0.28 (m, 2H), 0.27–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C29H27F2N4O4S2, 597.1436; found, 597.1424.
2-(5-(Cyclopropylmethyl)-3-(3-(3,3-dimethylbut-1-yn-1-yl)-4-fluorophenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (17).
This compound was synthesized from coupling the advanced intermediate VIIf (0.314 mmol, 0.2 g) with 2-(3,3-dimethylbut-1-yn-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (0.471 mmol) utilizing the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.679 min and (method 2) = 3.754 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.30 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.58 (s, 2H), 7.62–7.53 (m, 1H), 7.50 (dd, J = 6.9, 2.3 Hz, 1H), 7.29 (dd, J = 9.4, 8.7 Hz, 1H), 7.17 (dd, J = 11.4, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 4.15 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 1.29 (s, 9H), 1.20–1.07 (m, 1H), 0.39–0.29 (m, 2H), 0.27–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H29F2N4O4S2, 611.1593; found, 611.1598.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(4-hydroxybut-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (18).
This compound was synthesized from coupling the advanced intermediate VIIf (0.235 mmol, 0.15 g) with potassium (4-((tert-butyldimethylsilyl)oxy)but-1-yn-1-yl)-trifluoroborate (0.353 mmol) utilizing the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.211 min and (method 2) = 3.266 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.30 (s, 1H), 7.69–7.60 (m, 2H), 7.58 (s, 2H), 7.54 (ddd, J = 8.7, 5.0, 2.3 Hz, 1H), 7.30 (dd, J = 9.4, 8.7 Hz, 1H), 7.14 (dd, J = 11.4, 1.6 Hz, 1H), 7.04 (dd, J = 8.2, 1.6 Hz, 1H), 4.91 (d, J = 6.0 Hz, 1H), 4.15 (s, 2H), 3.59 (q, J = 6.5 Hz, 2H), 3.17 (d, J = 6.8 Hz, 2H), 2.60 (t, J = 6.8 Hz, 2H), 1.19–1.06 (m, 1H), 0.39–0.28 (m, 2H), 0.26–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C28H25F2N4O4S2, 599.1229; found, 599.123.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(3-methoxyprop-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (19).
This compound was synthesized from coupling the advanced intermediate VIIf (0.235 mmol, 0.15 g) with 2-(3-methoxyprop-1-yn-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (0.353 mmol) utilizing the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.782 min and (method 2) = 3.465 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.29 (s, 1H), 7.70–7.56 (m, 5H), 7.40–7.31 (m, 1H), 7.15 (dd, J = 11.4, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 4.36 (s, 2H), 4.16 (s, 2H), 3.33 (s, 3H), 3.17 (d, J = 6.4 Hz, 2H), 1.32–1.01 (m, 1H), 0.39–0.28 (m, 2H), 0.26–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C28H25F2N4O4S2 599.1229; found, 599.1232.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(4-methylpent-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (20).
This compound was synthesized from coupling the advanced intermediate VIIf (0.235 mmol, 0.15 g) with isobutylacetylene (0.353 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.536 min and (method 2) = 3.787 min; 1H NMR (400 MHz, DMSO-d6): δ 13.18 (s, 1H), 8.30 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.61–7.52 (m, 4H), 7.31 (ddd, J = 9.3, 8.0, 1.2 Hz, 1H), 7.15 (dd, J = 11.4, 1.6 Hz, 1H), 7.04 (dd, J = 8.1, 1.6 Hz, 1H), 4.14 (s, 2H), 3.16 (d, J = 6.8 Hz, 2H), 2.36 (d, J = 6.4 Hz, 2H), 1.85 (dp, J = 13.1, 6.6 Hz, 1H), 1.19–1.08 (m, 1H), 0.99 (d, J = 6.7 Hz, 6H), 0.38–0.28 (m, 2H), 0.26–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H29F2N4O4S2, 611.1593; found, 611.1608.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(4-methylpent-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (21).
This compound was synthesized from coupling the advanced intermediate VIIf (0.235 mmol, 0.15 g) with ethynylcyclopropane (0.353 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.026 min and (method 2) = 3.491 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.60–7.49 (m, 4H), 7.28 (t, J = 9.0 Hz, 1H), 7.15 (dd, J = 11.3, 1.6 Hz, 1H), 7.04 (dd, J = 8.2, 1.6 Hz, 1H), 4.14 (s, 2H), 3.16 (d, J = 6.6 Hz, 2H), 1.59 (tt, J = 8.2, 5.0 Hz, 1H), 1.14 (ddt, J = 14.8, 7.7, 3.7 Hz, 1H), 0.98–0.86 (m, 2H), 0.81–0.72 (m, 2H), 0.39–0.28 (m, 2H), 0.31–0.18 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.75, 161.12, 159.28, 156.76, 150.92, 147.40, 147.33, 144.58, 144.45, 132.35, 129.40, 128.89, 128.80, 128.49, 126.17, 123.66, 116.76, 116.40, 116.18, 115.96, 111.92, 111.76, 100.05, 68.25, 28.10, 15.24, 15.08, 10.30, 8.65, 4.47; HRMS (ESI) m/z: (M + Na)+ calcd for C29H24F2N4NaO4S2, 617.1099; found, 617.1126.
2-(3-(3-(Cyclobutylethynyl)-4-fluorophenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (22).
This compound was synthesized from coupling the advanced intermediate VIIf (0.275 mmol, 0.175 g) with ethynylcyclobutane (0.412 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.392 min and (method 2) = 3.698 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.60–7.51 (m, 4H), 7.30 (t, J = 8.9 Hz, 1H), 7.16 (dd, J = 11.4, 1.6 Hz, 1H), 7.05 (dd, J = 8.2, 1.6 Hz, 1H), 4.15 (s, 2H), 3.38–3.25 (m, 1H), 3.17 (d, J = 6.7 Hz, 2H), 2.38–2.25 (m, 2H), 2.21–2.06 (m, 2H), 2.05–1.81 (m, 2H), 1.14 (ddd, J = 12.7, 7.5, 5.0 Hz, 1H), 0.39–0.28 (m, 2H), 0.31–0.18 (m, 2H); HRMS (ESI) m/z: (M + Na)+ calcd for C30H26F2N4NaO4S2, 631.1256; found, 631.1285.
2-(3-(3-(Cyclopentylethynyl)-4-fluorophenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (23).
This compound was synthesized from coupling the advanced intermediate VIIf (3.14 mmol, 2 g) with cyclopentyl acetylene (4.39 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.319 min and (method 2) = 3.772 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.60–7.51 (m, 4H), 7.34–7.24 (m, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.05 (dd, J = 8.2, 1.6 Hz, 1H), 4.14 (s, 2H), 3.17 (d, J = 6.9 Hz, 2H), 2.89 (p, J = 7.2 Hz, 1H), 2.04–1.92 (m, 1H), 1.76–1.51 (m, 4H), 1.19–1.08 (m, 1H), 0.39–0.26 (m, 2H), 0.29–0.18 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 167.15, 161.75, 161.12, 159.31, 150.90, 147.33, 144.61, 144.46, 132.27, 129.56, 129.41, 128.94, 128.85, 128.52, 128.26, 126.17, 123.64, 116.70, 116.39, 116.22, 116.17, 116.00, 112.00, 72.79, 33.33, 30.08, 28.10, 24.64, 15.30, 15.23, 15.09, 10.30, 4.48; HRMS (ESI) m/z: (M + H)+ calcd for C31H29F2N4O4S2, 623.1593; found, 623.1584.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((1-fluorocyclopentyl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (24).
To a mixture of triethylamine trihydrofluoride (0.049 mL, 0.300 mmol) and tetraethylammonium (TEA) (0.021 mL, 0.150 mmol) in DCM (1 mL) at –78 °C was added XtalFluor-M28 (0.055 g, 0.225 mmol), followed by ethyl 2-(5-(cyclopropylmethyl)-3-(4-fluoro-3-((1-hydroxycyclopentyl)ethynyl)-phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate (prepared from the intermediate VIIf and 1-ethynyl-1-cyclopentanol using the Sonogashira coupling procedure E) (0.1 g, 0.150 mmol). The reaction mixture was allowed to attain room temperature and stirred overnight. The reaction was quenched with aqueous sodium bicarbonate solution and extracted twice using ethyl acetate. The organic layer was concentrated, and the crude product was purified in an Isco flash system eluting with 10–50% ethyl acetate in hexanes over 20 column volumes. The intermediate obtained was subsequently hydrolyzed using general procedure H. LC–MS retention time: (method 1) = 6.706 min and (method 2) = 3.739 min; (sufficient amount of material was not available to run NMR); LCMS m/z: (M + H)+ calcd for C31H28F3N4O4S, 641.15; found, 641.2.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((1-hydroxycyclopentyl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (25).
This compound was synthesized from coupling the advanced intermediate VIIf (0.345 mmol, 0.22 g) with 1-ethynyl-1-cyclopentanol (0.449 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.576 min and (method 2) = 2.651 min; 1H NMR (400 MHz, DMSO-d6): δ 8.11 (s, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.62–7.53 (m, 2H), 7.43 (s, 2H), 7.36–7.27 (m, 1H), 7.15 (dd, J = 11.4, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 5.41 (s, 1H), 4.15 (s, 2H), 3.20–3.13 (m, 2H), 1.97–1.62 (m, 8H), 1.12 (tt, J = 11.6, 4.6 Hz, 1H), 0.38–0.27 (m, 2H), 0.29–0.17 (m, 2H); HRMS (ESI) m/z: (M + Na)+ calcd for C31H28F2N4NaO5S2 661.1361; found, 661.1376.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((3-methyloxetan-3-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (26).
This compound was synthesized from coupling the advanced intermediate VIIf (0.196 mmol, 0.125 g) with 3-ethynyl-3-methyloxetane (0.235 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.598 min and (method 2) = 3.496 min; 1H NMR (400 MHz, DMSO-d6): δ 13.19 (s, 1H), 8.31 (s, 1H), 7.70–7.61 (m, 1H), 7.65–7.56 (m, 4H), 7.38–7.29 (m, 1H), 7.17 (dd, J = 11.3, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 4.74 (d, J = 5.5 Hz, 2H), 4.45 (d, J = 5.6 Hz, 2H), 4.16 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 1.64 (s, 3H), 1.22–1.09 (m, 1H), 0.40–0.29 (m, 2H), 0.27–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H27F2N4O5S2, 625.1385; found, 625.1388.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((3-fluorooxetan-3-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (27).
To a mixture of triethylamine trihydrofluoride (0.458 mmol, 0.075 mL) and TEA (0.229 mmol, 0.032 mL) in DCM (1 mL) at –78 °C was added XtalFluor-M (0.344 mmol, 0.084 g), followed by ethyl 2-(5-(cyclopropylmethyl)-3-(4-fluoro-3-((3-hydroxyoxetan-3-yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate (prepared from the intermediate VIIf and 3-ethynyloxetan-3-ol using the Sonogashira coupling procedure E) (0.125 g, 0.187 mmol). The reaction mixture was allowed to attain room temperature and stirred overnight. The reaction was quenched with aqueous sodium bicarbonate solution and extracted twice using ethyl acetate. The organic layer was concentrated, and the crude product was purified in an Isco flash system using a 12 g silica column eluting with 20–40% ethyl acetate in hexanes over 20 column volumes. The intermediate obtained was subsequently hydrolyzed using general procedure H. LC–MS retention time: (method 1) = 5.767 min and (method 2) = 3.413 min; 1H NMR (400 MHz, DMSO-d6): δ 13.19 (s, 1H), 8.32 (s, 1H), 7.78 (dd, J = 6.7, 2.3 Hz, 1H), 7.70 (ddd, J = 8.7, 5.1, 2.3 Hz, 1H), 7.64 (t, J = 7.9 Hz, 1H), 7.59 (s, 2H), 7.41 (t, J = 9.0 Hz, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.04 (dd, J = 8.1, 1.6 Hz, 1H), 5.01–4.94 (m, 1H), 4.94–4.88 (m, 2H), 4.87 (dd, J = 7.9, 1.1 Hz, 1H), 4.17 (s, 2H), 3.18 (t, J = 6.1 Hz, 2H), 1.22–1.07 (m, 1H), 0.40–0.29 (m, 2H), 0.31–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C29H24F3N4O5S2, 629.1135; found, 629.1147.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((3-hydroxyoxetan-3-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (28).
This compound was synthesized from coupling the advanced intermediate VIIf (0.314 mmol, 0.2 g) with 1–3-ethynyloxetan-3-ol (0.392 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 4.877 min and (method 2) = 3.231 min; 1H NMR (400 MHz, DMSO-d6): δ 13.14 (br, 1H), 8.20 (s, 1H), 7.72 (dd, J = 6.9, 2.3 Hz, 1H), 7.69–7.50 (m, 4H), 7.41–7.32 (m, 1H), 7.15 (dd, J = 11.4, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 6.71 (s, 1H), 4.84–4.72 (m, 2H), 4.65–4.57 (m, 2H), 4.17 (s, 2H), 3.17 (d, J = 6.5 Hz, 2H), 1.20–1.06 (m, 1H), 0.40–0.29 (m, 2H), 0.26–0.17 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C29H25F2N4O6S2, 627.1178; found, 627.1181.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((tetrahydrofuran-3-yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (29).
This compound was synthesized from coupling the advanced intermediate VIIf (0.196 mmol, 0.125 g) with 3–3-ethynyltetrahydrofuran (0.235 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.816 min and (method 2) = 3.478 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.61–7.55 (m, 4H), 7.35–7.28 (m, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 4.15 (s, 2H), 3.97 (dd, J = 8.1, 7.3 Hz, 1H), 3.88–3.71 (m, 2H), 3.60 (dd, J = 8.1, 6.5 Hz, 1H), 3.17 (d, J = 6.9 Hz, 2H), 2.34–2.21 (m, 1H), 1.94 (ddt, J = 12.1, 7.8, 6.5 Hz, 1H), 1.14 (dddd, J = 13.1, 11.9, 5.1, 3.5 Hz, 1H), 0.40–0.31 (m, 2H), 0.28–0.16 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H27F2N4O5S2, 625.1385; found, 625.1377.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((3-fluorotetrahydrofuran-3-yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (30).
To a mixture of triethylamine trihydrofluoride (0.374 mmol, 0.061 mL) and TEA (0.187 mmol, 0.026 mL) in DCM (1 mL) at –78 °C was added XtalFluor-M (0.280 mmol, 0.068 g), followed by ethyl 2-(5-(cyclopropylmethyl)-3-(4-fluoro-3-((3-hydroxytetrahydrofuran-3-yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate (prepared from the intermediate VIIf and 3-ethynyloxetan-3-ol using the Sonogashira coupling procedure E) (0.125 g, 0.187 mmol). The reaction mixture was allowed to attain room temperature and stirred overnight. The reaction was quenched with aqueous sodium bicarbonate solution and extracted twice using ethyl acetate. The organic layer was concentrated, and the crude product was purified in an Isco flash system using a 12 g silica column eluting with 20–40% ethyl acetate in hexanes over 30 column volumes. The intermediate obtained was subsequently hydrolyzed using general procedure H. LC–MS retention time: (method 1) = 5.587 min and (method 2) = 3.489 min; 1H NMR (400 MHz, DMSO-d6): δ 13.19 (s, 1H), 8.31 (s, 1H), 7.72–7.62 (m, 3H), 7.59 (s, 2H), 7.39 (t, J = 9.3 Hz, 1H), 7.17 (dd, J = 11.4, 1.6 Hz, 1H), 7.04 (dd, J = 8.1, 1.6 Hz, 1H), 4.24–4.13 (m, 3H), 3.97 (dd, J = 8.5, 5.8 Hz, 2H), 3.87 (ddd, J = 32.4, 10.7, 0.5 Hz, 1H), 3.18 (d, J = 7.0 Hz, 2H), 2.68–2.52 (m, 1H), 2.49–2.30 (m, 1H), 1.14 (dddd, J = 13.4, 8.1, 4.9, 1.9 Hz, 1H), 0.41–0.31 (m, 2H), 0.27–0.17 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H26F3N4O5S2, 643.1291; found, 643.1288.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((3-hydroxytetrahydrofuran-3-yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (31).
This compound was synthesized from coupling the advanced intermediate VIIf (0.392 mmol, 0.25 g) with 3-ethynyltetrahydrofuran-3-ol (0.471 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 4.929 min and (method 2) = 3.262 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.29 (s, 1H), 7.78–7.52 (m, 5H), 7.34 (t, J = 9.0 Hz, 1H), 7.15 (dd, J = 11.5, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 5.94 (s, 1H), 4.16 (s, 2H), 4.00–3.74 (m, 4H), 3.17 (dd, J = 5.9, 1.8 Hz, 2H), 2.28–2.15 (m, 2H), 1.19–1.06 (m, 1H), 0.37–0.30 (m, 2H), 0.30–0.14 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H27F2N4O6S2, 641.1332; found, 641.1335.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(piperidin-4-ylethynyl)-phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (32).
This compound was synthesized from coupling the advanced intermediate VIIf (0.627 mmol, 0.4 g) with tert-butyl 4-ethynylpiperidine-1-carboxylate (0.94 mmol) using the general Sonogashira coupling procedure E and subsequent Boc deprotection with TFA, followed by hydrolysis using general procedure H. LC–MS retention time: (method 1) = 4.466 min and (method 2) = 3.009 min; 1H NMR (400 MHz, DMSO-d6): δ 13.1 (br, 1H), 8.28 (s, 1H), 7.73–6.96 (m, 8H), 5.73 (s, 1H), 4.12 (s, 2H), 3.79 (s, 2H), 3.18 (dq, J = 16.7, 6.8, 5.5 Hz, 4H), 3.00 (qd, J = 8.8, 3.4 Hz, 4H), 2.40 (d, J = 6.8 Hz, 1H), 1.99 (ddq, J = 11.1, 6.9, 4.4, 3.8 Hz, 2H), 1.74 (ddq, J = 13.5, 9.1, 4.3 Hz, 2H), 1.09 (t, J = 6.7 Hz, 1H), 1.01–0.86 (m, 1H), 0.35–0.13 (m, 4H); HRMS (ESI) m/z: (M + H)+ calcd for C31H30F2N5O4S2, 638.1702; found, 638.1703.
2-(3-(3-(Cyanoethynyl)-4-fluorophenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (33).
To a solution of ethyl 2-(5-(cyclopropylmethyl)-3-(4-fluoro-3-(3-hydroxyprop-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate (0.5 g, 0.816 mmol, 1 equiv) in DCM (5 mL) was added Dess-Martin periodinane (0.519 g, 1.224 mmol, 1.5 equiv), and the reaction was stirred at room temperature for 2 h. The reaction was extracted with ethyl acetate, and the organic layer was washed with 1 M HCl and brine. The organic layer was concentrated, and the crude product was purified in an Isco flash system using a 12 g silica column eluting with 20–100% ethyl acetate in hexanes over 20 column volumes to obtain 260 mg of the intermediate ethyl 2-(5-(cyclopropylmethyl)-3-(4-fluoro-3-(3-oxoprop-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate. LC–MS retention time: (method 1) = 5.721 min; m/z: (M + H)+ for C29H25F2N4O5S2, 611.2.
To the above intermediate (0.1 g, 0.164 mmol, 1 equiv) and sodium azide (0.016 g, 0.246 mmol, 1.5 equiv) in ACN (1 mL) was added triflic acid (0.044 mL, 0.491 mmol, 3 equiv) at rt, and the mixture was then stirred overnight. The reaction was extracted with ethyl acetate, and the organic layer was washed with water, bicarbonate solution, and brine. The organic layer was concentrated, and the crude product was purified in an Isco flash system using a 4 g silica column eluting with 20–80% ethyl acetate in hexanes over 20 column volumes to obtain 32 mg of the intermediate ethyl 2-(3-(3-(cyanoethynyl)-4-fluorophenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate. LC–MS retention time: (method 1) = 6.965 min; m/z: (M + H)+ for C29H24F2N5O4S2, 608.
The above intermediate ethyl 2-(3-(3-(cyanoethynyl)-4-fluorophenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate (0.015 g, 0.025 mmol, 1 equiv) and hydroxytrimethylstannane (8.93 mg, 0.049 mmol, 2 equiv) in dichloroethane (1 mL) were heated in a microwave at 110 °C for 1 h. The solvent was removed by forced air, and the residue was taken up in 1 mL of DMSO. The crude product was purified in prep HPLC. LC–MS retention time: (method 1) = 5.721 min and (method 2) = 3.41 min; (sufficient amount of material was not available to run NMR); HRMS (ESI) m/z: (M + H)+ calcd for C27H20F2N5O4S2, 580.0919; found, 580.0935.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(3,3,3-trifluoroprop-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (34).
A mixture of VIIf (0.157 mmol, 0.1 g), tri(tert-butylphosphonium)tetrafluoroborate (0.016 mmol, 4.55 mg), and allylpalladium chloride dimer (7.84 μmol, 2.84 mg) in dioxane was bubbled with argon, and then tributyl(3,3,3-trifluoroprop-1-yn-1-yl)stannane (0.196 mmol, 0.083 g) was added. The vial was capped and stirred at 80 °C for 4 h. LC–MS showed only 15% conversion. [NOTE: a further attempt to modify the conditions did not improve the yield and the reaction did not reproduce well under the same conditions]. The reaction was diluted with DCM and stirred sequentially with KF and palladium scavenger. The product obtained after filtration was directly hydrolyzed using general procedure H to obtain a minute amount of the pure product. LC–MS retention time: (method 1) = 5.543 min and (method 2) = 3.484 min; (sufficient amount of material was not available to run NMR); LCMS m/z: (M + H)+ calcd for C27H20F5N4O4S2, 623.08; found: 623.1.
2-(5-(Cyclopropylmethyl)-3-(3-(3,3-difluoroprop-1-yn-1-yl)-4-fluorophenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (35).
To a solution of ethyl 2-(5-(cyclopropylmethyl)-3-(4-fluoro-3-(3-oxoprop-1-yn-1-yl)phenyl)-4(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate (0.115 mmol, 0.07 g) in DCM (3 mL) was added deoxofluor (0.229 mmol, l0.042 mL), and the mixture was stirred overnight at room temperature. The reaction was quenched with aqueous sodium bicarbonate solution and extracted twice using ethyl acetate. The organic layer was concentrated, and the crude product was purified in an Isco flash system using a 6 g silica column eluting with 20–40% ethyl acetate in hexanes over 20 column volumes. The intermediate obtained was subsequently hydrolyzed using general procedure H. LC–MS retention time: (method 1) = 5.896 min and (method 2) = 3.434 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.30 (s, 1H), 7.85 (dd, J = 6.7, 2.3 Hz, 1H), 7.74 (ddd, J = 8.7, 5.1, 2.3 Hz, 1H), 7.64 (t, J = 7.9 Hz, 1H), 7.57 (s, 2H), 7.44 (t, J = 9.1 Hz, 1H), 7.18–7.10 (m, 1H), 7.07–7.00 (m, 1H), 4.18 (s, 2H), 3.17 (d, J = 7.0 Hz, 2H), 1.21–1.06 (m, 1H), 0.39–0.28 (m, 2H), 0.27–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C27H21F4N4O4S2 605.0935; found, 605.0949.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(3-fluorobut-1-yn-1-yl)-phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (36).
Following the general procedure E, the reaction of VIIf (0.275 g, 0.431 mmol) with 2-trimethylsilyloxy-3butyne (0.604 mmol, 0.105 mL) and subsequent deprotection of the TMS group with 2 equiv of K2CO3 in methanol provided the intermediate ethyl 2-(5-(cyclopropylmethyl)-3-(4-fluoro-3-(3-hydroxybut-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylate. To a mixture of the above intermediate (0.207 mmol, 0.13 g), triethylamine trihydrofluoride (0.415 mmol, 0.068 mL), and TEA (0.207 mmol, 0.029 mL) in DCM (1 mL) was added XtalFluor-M (0.311 mmol, 0.076 g) at –78 °C, and the reaction mixture was stirred overnight at room temperature. The reaction was quenched with aqueous sodium bicarbonate solution and extracted twice using ethyl acetate. The organic layer was concentrated, and the crude product was purified in an Isco flash system using a 6 g silica column eluting with 20–40% ethyl acetate in hexanes over 20 column volumes. The intermediate obtained was subsequently hydrolyzed using general procedure H to afford a white solid. LC–MS retention time: (method 1) = 5.811 min and (method 2) = 3.519 min; 1H NMR (400 MHz, DMSO-d6): δ 13.30–13.07 (m, 1H), 8.31 (s, 1H), 7.71–7.62 (m, 3H), 7.58 (s, 2H), 7.38 (t, J = 8.9 Hz, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.04 (dd, J = 8.2, 1.6 Hz, 1H), 5.83–5.53 (m, 1H), 4.16 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 1.62 (dd, J = 23.0, 6.6 Hz, 3H), 1.26–1.04 (m, 1H), 0.41–0.31 (m, 2H), 0.27–0.18 (m, 2H).; HRMS (ESI) m/z: (M + H)+ calcd for C28H24F3N4O4S2, 601.1186; found, 601.1207.
2-(5-(Cyclopropylmethyl)-3-(3-(3-cyclopropylprop-1-yn-1-yl)-4-fluorophenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (37).
This compound was synthesized from coupling the advanced intermediate VIIf (0.196 mmol, 0.125 g) with prop-2-yn-1-ylcyclopropane (0.294 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.013 min and (method 2) = 3.671 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.61–7.51 (m, 4H), 7.31 (dd, J = 9.4, 8.5 Hz, 1H), 7.15 (dd, J = 11.3, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 4.15 (s, 2H), 3.16 (d, J = 6.9 Hz, 2H), 2.53 (d, J = 5.9 Hz, 2H), 1.13 (ddt, J = 9.7, 7.8, 2.9 Hz, 1H), 1.07–0.94 (m, 1H), 0.54–0.42 (m, 2H), 0.39–0.27 (m, 2H), 0.30–0.18 (m, 4H); HRMS (ESI) m/z: (M + H)+ calcd for C30H27F2N4O4S2, 609.1436; found, 609.1449.
2-(3-(3-(3-Cyclopentylprop-1-yn-1-yl)-4-fluorophenyl)-5-(cyclo-propylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (38).
This compound was synthesized from coupling the advanced intermediate VIIf (0.196 mmol, 0.125 g) with 1–3-cyclopentyl-1-propyne (0.294 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 8.045 min and (method 2) = 3.87 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.30 (s, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.58 (s, 2H), 7.61–7.51 (m, 2H), 7.30 (dd, J = 9.4, 8.6 Hz, 1H), 7.14 (dd, J = 11.3, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 5.75 (s, 2H), 4.15 (s, 2H), 3.16 (d, J = 6.8 Hz, 2H), 2.47 (d, J = 6.7 Hz, 2H), 2.15–2.03 (m, 1H), 1.84–1.71 (m, 2H), 1.69–1.46 (m, 3H), 1.39–1.23 (m, 2H), 1.20–1.06 (m, 1H), 0.38–0.28 (m, 2H), 0.30–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C32H31F2N4O4S2, 637.1749; found, 637.1761.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(3-(pyrrolidin-1-yl)prop-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (39).
This compound was synthesized from coupling the advanced intermediate VIIf (0.196 mmol, 0.125 g) with 1–1-(prop-2-yn-1-yl)pyrrolidine HCl (0.235 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 4.49 min and (method 2) = 2.999 min; 1H NMR (400 MHz, DMSO-d6): δ 13.20 (s, 1H), 8.32 (s, 1H), 7.76–7.58 (m, 5H), 7.40 (t, J = 9.0 Hz, 1H), 7.14 (dd, J = 11.3, 1.6 Hz, 1H), 7.04 (dd, J = 8.2, 1.6 Hz, 1H), 4.40 (s, 2H), 4.16 (s, 2H), 3.28 (s, 6H), 3.17 (d, J = 6.9 Hz, 2H), 1.96 (s, 4H), 1.27–1.04 (m, 1H), 0.39–0.28 (m, 2H), 0.27–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C31H30F2N5O4S2, 638.1702; found, 638.1692.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(3-(tetrahydrofuran-2-yl)prop-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (40).
This compound was synthesized from coupling the advanced intermediate VIIf (0.196 mmol, 0.125 g) with 2-(prop-2-yn-1-yl)tetrahydrofuran (0.235 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.98 min and (method 2) = 3.412 min; (sufficient amount of material was not available to run NMR); HRMS (ESI) m/z: (M + Na)+ calcd for C31H28F2N4NaO5S2, 661.1361; found, 661.137.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(3-morpholinoprop-1-yn-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (41).
This compound was synthesized from coupling the advanced intermediate VIIf (0.627 mmol, 0.4 g) with 4-(prop-2-yn-1-yl)morpholine (1.882 mmol) using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 4.391 min and (method 2) = 3.008 min; 1H NMR (400 MHz, DMSO-d6): δ 12.84 (s, 1H), 8.18 (s, 1H), 7.65 (t, J = 7.9 Hz, 2H), 7.60 (s, 2H), 7.57–7.47 (m, 1H), 7.37 (t, J = 9.0 Hz, 1H), 7.13–7.01 (m, 2H), 3.83 (s, 2H), 3.68–3.38 (m, 4H), 2.68–2.32 (m, 6H), 0.98–0.91 (m, 1H), 0.43–0.40 (m, 2H), 0.15–0.11 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C31H30F2N5O5S2 654.1651; found, 654.1672.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(thiophen-2-ylethynyl)-phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (42).
This compound was synthesized from coupling the advanced intermediate VIIf with either 4,4,5,5-tetramethyl-2-(thiophen-2-ylethynyl)-1,3,2-dioxaborolane following general Suzuki procedure G or 2-ethynylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.472 min and (method 2) = 3.687 min; 1H NMR (400 MHz, DMSO-d6): δ 13.18 (s, 1H), 8.31 (s, 1H), 7.80–7.71 (m, 2H), 7.70–7.60 (m, 2H), 7.58 (s, 2H), 7.49 (dd, J = 3.7, 1.2 Hz, 1H), 7.39 (dd, J = 9.4, 8.7 Hz, 1H), 7.21–7.13 (m, 2H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 4.18 (s, 2H), 3.18 (d, J = 7.0 Hz, 2H), 1.20–1.09 (m, 1H), 0.40–0.29 (m, 2H), 0.27–0.19 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 166.99, 161.75, 161.10, 160.26, 150.78, 144.62, 144.48, 133.54, 131.98, 130.07, 129.92, 129.55, 129.41, 128.63, 128.50, 127.97, 126.20, 123.71, 120.96, 116.89, 116.44, 116.24, 110.74, 85.57, 28.11, 15.16, 10.31, 4.47; HRMS (ESI) m/z: (M + H)+ calcd for C30H23F2N4O4S3, 637.0844; found, 637.0856.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(thiophen-2-ylethynyl)-phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (43).
This compound was synthesized from coupling the advanced intermediate VIIf with 2-ethynyl-5-methylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H (a detailed scale-up procedure is provided in the Supporting Information). LC–MS retention time: (method 1) = 6.133 min and (method 2) = 3.694 min; 1H NMR (400 MHz, DMSO-d6): δ 13.14 (s, 1H), 8.29 (s, 1H), 7.71 (dd, J = 6.9, 2.3 Hz, 1H), 7.66–7.54 (m, 4H), 7.35 (dd, J = 9.4, 8.7 Hz, 1H), 7.26 (dd, J = 3.6, 0.5 Hz, 1H), 7.14 (dd, J = 11.3, 1.6 Hz, 1H), 7.03 (dd, J = 8.1, 1.6 Hz, 1H), 6.83 (dq, J = 3.6, 1.0 Hz, 1H), 4.15 (s, 2H), 3.15 (d, J = 6.9 Hz, 2H), 2.47–2.45 (m, 3H), 1.20–1.06 (m, 1H), 0.38–0.29 (m, 2H), 0.24–0.15 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.58, 160.94, 159.18, 150.73, 147.20, 147.12, 144.47, 144.44, 143.30, 133.59, 131.80, 129.81, 129.73, 129.50, 129.35, 128.51, 128.48, 128.39, 126.25, 125.90, 123.59, 123.56, 118.47, 116.75, 116.28, 116.23, 116.07, 116.02, 110.99, 84.86, 27.99, 14.90, 10.15, 4.34; HRMS (ESI) m/z: (M + H)+ calcd for C31H25F2N4O4S3, 651.1001; found, 651.1027.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((5-methylfuran-2-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (44).
This compound was synthesized from coupling the advanced intermediate VIIIb with 2-bromo-5-methylfuran using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.202 min and (method 2) = 3.593 min; 1H NMR (400 MHz, DMSO-d6): δ 13.23 (s, 1H), 8.30 (d, J = 3.2 Hz, 1H), 7.75 (dd, J = 6.8, 2.3 Hz, 1H), 7.70–7.58 (m, 4H), 7.39 (t, J = 9.0 Hz, 1H), 7.16 (d, J = 11.3 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 6.88 (d, J = 3.3 Hz, 1H), 6.29–6.23 (m, 1H), 4.17 (s, 2H), 3.17 (d, J = 6.9 Hz, 2H), 2.33 (s, 3H), 1.14 (t, J = 7.6 Hz, 1H), 0.34 (dd, J = 8.0, 1.8 Hz, 2H), 0.23 (t, J = 4.9 Hz, 2H); 13C NMR (101 MHz, DMSO-d6): δ 167.00, 161.74, 161.10, 159.28, 156.76, 154.84, 150.77, 147.29, 144.61, 144.47, 133.57, 131.70, 130.13, 128.67, 128.49, 126.22, 123.73, 118.45, 116.89, 116.48, 116.43, 116.28, 116.22, 110.55, 110.40, 107.99, 85.97, 28.11, 28.02, 15.20, 15.19, 13.51, 10.30, 4.47; HRMS (ESI) m/z: (M + H)+ calcd for C31H25F2N4O5S2, 635.1229; found, 635.124.
2-(3-(3-((5-(tert-Butyl)thiophen-2-yl)ethynyl)-4-fluorophenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (45).
This compound was synthesized from coupling the advanced intermediate VIIIb with 2-bromo-5-(tert-butyl)thiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 7.143 min and (method 2) = 3.959 min; 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.32 (s, 1H), 7.73 (dd, J = 6.9, 2.3 Hz, 1H), 7.70–7.58 (m, 2H), 7.57 (s, 2H), 7.42–7.33 (m, 1H), 7.29 (d, J = 3.8 Hz, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 6.92 (d, J = 3.7 Hz, 1H), 4.17 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 1.36 (s, 9H), 1.25–1.07 (m, 1H), 0.40–0.29 (m, 2H), 0.27–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C34H31F2N4O5S3, 693.147; found, 693.1484.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((2-methylthiazol-5-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (46).
This compound was synthesized from coupling the advanced intermediate VIIIb with 5-bromo-2methylthiazole using the general Sonogashira coupling procedure F and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.093 min and (method 2) = 3.556 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.32 (s, 1H), 8.01 (s, 1H), 7.77 (dd, J = 6.8, 2.3 Hz, 1H), 7.70–7.61 (m, 2H), 7.58 (s, 2H), 7.40 (t, J = 9.0 Hz, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 4.18 (s, 2H), 3.18 (d, J = 6.8 Hz, 2H), 2.70 (s, 3H), 1.14 (s, 1H), 0.40–0.29 (m, 2H), 0.31–0.19 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 168.13, 161.75, 161.09, 160.28, 159.28, 156.76, 150.71, 147.44, 147.30, 144.63, 144.48, 132.09, 129.55, 129.40, 128.67, 128.50, 126.23, 123.74, 116.91, 116.52, 116.38, 116.31, 116.23, 110.35, 87.96, 84.71, 28.10, 19.00, 15.20, 10.31, 4.48; HRMS (ESI) m/z: (M + H)+ calcd for C30H24F2N5O4S3, 652.0953; found, 652.097.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((5-methylthiazol-2-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (47).
This compound was synthesized from coupling the advanced intermediate VIIIb with 2-bromo-5methylthiazole using the general Sonogashira coupling procedure F and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.063 min and (method 2) = 3.595 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.32 (s, 1H), 7.87 (dd, J = 6.8, 2.3 Hz, 1H), 7.73–7.62 (m, 3H), 7.58 (s, 2H), 7.43 (t, J = 9.0 Hz, 1H), 7.16 (d, J = 11.2 Hz, 1H), 7.06 (dd, J = 8.2, 1.6 Hz, 1H), 4.19 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 2.52 (s, 3H), 1.14 (dh, J = 13.0, 6.6 Hz, 1H), 0.34 (dt, J = 8.3, 2.9 Hz, 2H), 0.23 (t, J = 4.9 Hz, 2H); 13C NMR (101 MHz, DMSO-d6): δ 163.16, 161.75, 161.09, 160.63, 156.75, 150.65, 147.35, 144.62, 144.57, 144.47, 142.38, 137.77, 132.38, 131.25, 131.16, 129.54, 128.81, 128.78, 128.49, 126.25, 123.75, 116.97, 116.63, 116.44, 116.23, 109.67, 109.51, 87.66, 85.43, 28.11, 27.99, 15.31, 15.28, 15.24, 15.21, 11.66, 10.31, 4.47; HRMS (ESI) m/z: (M + H)+ calcd for C30H24F2N5O4S3, 652.0953; found, 652.097.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((1-methyl-1H-imidazole-5-yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (48).
This compound was synthesized from coupling the advanced intermediate VIIf with 5-ethynyl-1-methyl-1H-imidazole using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 4.583 min and (method 2) = 2.868 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.32 (s, 1H), 7.88 (s, 1H), 7.72 (dd, J = 6.8, 2.3 Hz, 1H), 7.70–7.62 (m, 2H), 7.59 (s, 2H), 7.45–7.36 (m, 2H), 7.18 (dd, J = 11.4, 1.6 Hz, 1H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 4.18 (s, 2H), 3.69 (d, J = 0.5 Hz, 3H), 3.18 (d, J = 6.9 Hz, 2H), 1.22–1.07 (m, 1H), 0.40–0.29 (m, 2H), 0.27–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H25F2N6O4S2, 635.1344; found, 635.1345.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-((1-methyl-1H-pyrazol-4yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1yl)thiazole-4-carboxylic Acid (49).
This compound was synthesized from coupling the advanced intermediate VIIf with 4-ethynyl-1methyl-1H-pyrazole using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC– MS retention time: (method 1) = 5.607 min and (method 2) = 2.612 min; 1H NMR (400 MHz, DMSO-d6): δ 13.20 (s, 1H), 8.26 (s, 1H), 8.11 (d, J = 0.6 Hz, 1H), 7.72 (d, J = 0.7 Hz, 1H), 7.70–7.62 (m, 2H), 7.62–7.54 (m, 3H), 7.35 (dd, J = 9.4, 8.6 Hz, 1H), 7.17 (dd, J = 11.4, 1.6 Hz, 1H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 4.16 (s, 2H), 3.86 (s, 3H), 3.17 (d, J = 6.9 Hz, 2H), 1.30–1.06 (m, 1H), 0.39–0.30 (m, 2H), 0.33–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H25F2N6O4S2, 635.1344; found, 635.1345.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3(prop-1-yn-1-yl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (50).
This compound was synthesized from coupling the advanced intermediate VIIe (0.202 mmol, 0.125 g) with potassium trifluoro(prop-1-yn-1-yl)borate (0.303 mmol) using the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.653 min and (method 2) = 3.535 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.27 (s, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.59–7.52 (m, 3H), 7.52–7.45 (m, 1H), 7.42–7.32 (m, 2H), 7.14 (dd, J = 11.5, 1.6 Hz, 1H), 7.05 (dd, J = 8.2, 1.6 Hz, 1H), 4.15 (s, 2H), 3.20–3.13 (m, 2H), 2.04 (s, 3H), 1.12 (dd, J = 12.2, 7.1 Hz, 1H), 0.38–0.28 (m, 2H), 0.30–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C27H24FN4O4S2, 551.1218; found, 551.1208.
2-(3-(3-(Cyclopentylethynyl)phenyl)-5-(cyclopropylmethyl)-4-(3fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (51).
This compound was synthesized from coupling the advanced intermediate VIIe with cyclopentyl acetylene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.386 min and (method 2) = 3.68 min; 1H NMR (400 MHz, DMSO-d6): δ 13.18 (s, 1H), 8.31 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.58 (s, 2H), 7.55–7.47 (m, 2H), 7.41–7.32 (m, 2H), 7.16 (dd, J = 11.4, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 4.15 (s, 2H), 3.17 (d, J = 6.8 Hz, 2H), 2.90–2.78 (m, 1H), 2.08–1.87 (m, 2H), 1.78– 1.65 (m, 2H), 1.69–1.50 (m, 4H), 1.19–1.05 (m, 1H), 0.39–0.28 (m, 2H), 0.31–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C31H30FN4O4S2, 605.1687; found, 605.1707.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3((5-methylthiophen-2-yl)-ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole4-carboxylic Acid (52).
This compound was synthesized from coupling the advanced intermediate VIIe with 2-ethynyl-5-methylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time = 6.402 min (M + H)+ = 632. 1H NMR (400 MHz, DMSOd6): δ 13.16 (s, 1H), 8.32 (d, J = 0.9 Hz, 1H), 7.71–7.62 (m, 2H), 7.58 (d, J = 6.6 Hz, 3H), 7.52 (dt, J = 7.7, 1.4 Hz, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.24 (d, J = 3.6 Hz, 1H), 7.16 (d, J = 11.3 Hz, 1H), 7.06 (dd, J = 8.1, 1.5 Hz, 1H), 6.83 (dt, J = 3.5, 1.2 Hz, 1H), 4.18 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 2.47 (d, J = 1.1 Hz, 3H), 1.21–1.09 (m, 1H), 0.35 (dt, J = 8.1, 2.8 Hz, 2H), 0.31–0.19 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.70, 161.11, 159.24, 156.73, 151.55, 147.42, 147.34, 144.53, 144.45, 142.72, 133.19, 132.20, 131.08, 129.80, 129.50, 129.36, 129.26, 128.46, 127.59, 126.24, 126.11, 123.67, 123.64, 122.51, 119.11, 116.86, 116.36, 116.14, 91.67, 83.60, 28.10, 15.01, 10.27, 4.45. HRMS (ESI) m/z: (M + H)+ calcd for C31H26FN4O4S3, 633.1095; found, 633.1086.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3((3-methylthiophen-2-yl)-ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole4-carboxylic Acid (53).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-3-methylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.488 min and (method 2) = 3.679 min; 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.31 (s, 1H), 7.71–7.62 (m, 2H), 7.65–7.58 (m, 1H), 7.58–7.51 (m, 4H), 7.46 (td, J = 7.7, 0.6 Hz, 1H), 7.17 (dd, J = 11.3, 1.6 Hz, 1H), 7.08 (dd, J = 8.1, 1.6 Hz, 1H), 7.02 (dd, J = 5.1, 0.5 Hz, 1H), 4.19 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 2.31 (s, 3H), 1.20–1.07 (m, 1H), 0.39–0.29 (m, 2H), 0.31–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C31H26FN4O4S3, 633.1095; found, 633.1099.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3((5-methylthiophen-3-yl)-ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole4-carboxylic Acid (54).
This compound was synthesized from coupling the advanced intermediate VIIIa with 3-bromo-2-methylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.516 min and (method 2) = 3.691 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.71– 7.61 (m, 3H), 7.58 (s, 2H), 7.58 (dt, J = 7.6, 1.5 Hz, 1H), 7.50 (dt, J = 7.7, 1.4 Hz, 1H), 7.44 (td, J = 7.6, 0.6 Hz, 1H), 7.18 (dd, J = 11.3, 1.6 Hz, 1H), 7.07 (dd, J = 8.2, 1.6 Hz, 1H), 6.95 (p, J = 1.1 Hz, 1H), 4.18 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 2.46 (d, J = 1.1 Hz, 3H), 1.22–1.08 (m, 1H), 0.40–0.29 (m, 2H), 0.27–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C31H26FN4O4S3, 633.1095; found, 633.1099.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-isopropylthiophen-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (55).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-5-isopropylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 7.071 min and (method 2) = 3.835 min; 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.31 (s, 1H), 7.71–7.62 (m, 2H), 7.62–7.55 (m, 1H), 7.56 (s, 2H), 7.51 (dt, J = 7.7, 1.4 Hz, 1H), 7.45 (td, J = 7.7, 0.6 Hz, 1H), 7.25 (d, J = 3.6 Hz, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.07 (dd, J = 8.2, 1.6 Hz, 1H), 6.88 (dd, J = 3.7, 1.0 Hz, 1H), 4.18 (s, 2H), 3.25–3.10 (m, 3H), 1.29 (d, J = 6.8 Hz, 6H), 1.21–1.09 (m, 1H), 0.40–0.29 (m, 2H), 0.31–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C33H30FN4O4S3, 661.1408; found, 661.1427.
2-(5-(Cyclopropylmethyl)-3-(3-((5-cyclopropylthiophen-2-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (56).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-5-cyclopropylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.769 min and (method 2) = 3.771 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17–13.12 (m, 1H), 8.31 (s, 1H), 7.70–7.62 (m, 2H), 7.61–7.53 (m, 1H), 7.56 (s, 2H), 7.51 (dt, J = 7.7, 1.4 Hz, 1H), 7.44 (td, J = 7.7, 0.6 Hz, 1H), 7.22 (d, J = 3.7 Hz, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 6.82 (dd, J = 3.7, 0.7 Hz, 1H), 4.18 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 2.17 (ttd, J = 8.3, 4.9, 0.7 Hz, 1H), 1.20–1.08 (m, 1H), 1.11–0.98 (m, 2H), 0.79–0.69 (m, 2H), 0.39–0.29 (m, 2H), 0.31–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C33H28FN4O4S3, 659.1251; found, 659.127.
2-(3-(3-((5-Cyclobutylthiophen-2-yl)ethynyl)phenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (57).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-5-cyclobutylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 7.119 min and (method 2) = 3.893 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.70–7.61 (m, 2H), 7.61–7.55 (m, 3H), 7.52 (dt, J = 7.7, 1.4 Hz, 1H), 7.48–7.40 (m, 1H), 7.26 (d, J = 3.6 Hz, 1H), 7.16 (dd, J = 11.4, 1.6 Hz, 1H), 7.07 (dd, J = 8.1, 1.6 Hz, 1H), 6.88 (dd, J = 3.7, 0.9 Hz, 1H), 4.18 (s, 2H), 3.79–3.65 (m, 1H), 3.18 (d, J = 6.9 Hz, 2H), 2.45–2.32 (m, 2H), 2.19–2.04 (m, 2H), 2.08–1.88 (m, 1H), 1.91–1.78 (m, 1H), 1.21–1.09 (m, 1H), 0.41–0.29 (m, 2H), 0.31–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C34H30FN4O4S3, 673.1408; found, 673.1391.
2-(3-(3-((5-Chlorothiophen-2-yl)ethynyl)phenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (58).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-5-chlorothiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.776 min and (method 2) = 3.844 min; 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.31 (s, 1H), 7.74–7.52 (m, 6H), 7.46 (td, J = 7.8, 0.6 Hz, 1H), 7.34 (d, J = 3.9 Hz, 1H), 7.21–7.12 (m, 2H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 4.18 (s, 2H), 3.22–3.14 (m, 2H), 1.29–0.98 (m, 1H), 0.40–0.27 (m, 2H), 0.30–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H23ClFN4O4S3, 653.0548; found, 653.0566.
2-(5-(Cyclopropylmethyl)-3-(3-((5-(difluoromethyl)thiophen-2-yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (59).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-5-(difluoromethyl)thiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.315 min and (method 2) = 3.682 min; 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.31 (s, 1H), 7.75 (td, J = 1.7, 0.6 Hz, 1H), 7.70–7.58 (m, 2H), 7.58 (dt, J = 7.7, 1.4 Hz, 1H), 7.56 (s, 2H), 7.52–7.42 (m, 3H), 7.22–7.13 (m, 1H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 4.19 (s, 2H), 3.19 (d, J = 6.9 Hz, 2H), 1.22–1.08 (m, 1H), 0.40–0.27 (m, 2H), 0.30–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C31H24F3N4O4S3, 669.0906; found, 669.0909.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-(trifluoromethyl)thiophen-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (60).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-5-(trifluoromethyl)thiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.897 min and (method 2) = 3.837 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.76 (dp, J = 3.4, 1.2 Hz, 2H), 7.70–7.61 (m, 2H), 7.64–7.57 (m, 1H), 7.56 (s, 2H), 7.54 (dq, J = 3.9, 1.3 Hz, 1H), 7.49 (td, J = 7.8, 0.6 Hz, 1H), 7.17 (dd, J = 11.4, 1.6 Hz, 1H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 4.19 (s, 2H), 3.19 (d, J = 6.9 Hz, 2H), 1.21–1.09 (m, 1H), 0.40–0.29 (m, 2H), 0.32–0.20 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C31H23F4N4O4S3, 687.0812; found, 687.0807.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylfuran-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (61).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-5-methylfuran using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.261 min and (method 2) = 3.591 min; 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.32 (s, 1H), 7.70 (td, J = 1.7, 0.6 Hz, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.62–7.56 (m, 1H), 7.57 (s, 2H), 7.53 (dt, J = 7.7, 1.4 Hz, 1H), 7.46 (td, J = 7.7, 0.6 Hz, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 6.81 (dd, J = 3.3, 0.6 Hz, 1H), 6.23 (dq, J = 3.1, 1.0 Hz, 1H), 4.18 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 2.32 (dd, J = 1.0, 0.5 Hz, 3H), 1.22–1.07 (m, 1H), 0.39–0.29 (m, 2H), 0.31–0.19 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.76, 161.16, 154.33, 151.53, 147.46, 144.59, 144.47, 134.00, 132.31, 131.05, 129.65, 129.39, 128.49, 127.88, 126.20, 123.73, 121.98, 117.70, 116.92, 116.41, 116.20, 107.85, 92.48, 80.50, 28.12, 15.34, 15.28, 15.19, 13.50, 10.30, 4.48; HRMS (ESI) m/z: (M + H)+ calcd for C31H26FN4O5S2, 617.1323; found, 617.1341.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methyloxazol-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (62).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-5-methyloxazole using the general Sonogashira coupling procedure F and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.764 min and (method 2) = 3.499 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.32 (s, 1H), 7.85 (td, J = 1.7, 0.6 Hz, 1H), 7.72–7.61 (m, 3H), 7.56 (s, 2H), 7.51 (td, J = 7.8, 0.6 Hz, 1H), 7.15 (dd, J = 11.3, 1.6 Hz, 1H), 7.10–7.03 (m, 2H), 4.20 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 2.37 (d, J = 1.2 Hz, 3H), 1.20–1.07 (m, 1H), 0.39–0.29 (m, 2H), 0.27–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H25FN5O5S2, 618.1276; found, 618.1261.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylthiazol-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (63).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-5-methylthiazole using the general Sonogashira coupling procedure F and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.875 min and (method 2) = 3.491 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.29 (s, 1H), 7.81 (td, J = 1.7, 0.6 Hz, 1H), 7.70–7.60 (m, 4H), 7.56 (s, 2H), 7.50 (td, J = 7.8, 0.6 Hz, 1H), 7.19–7.12 (m, 1H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 4.20 (s, 2H), 3.28 (d, J = 10.6 Hz, 2H), 3.21–3.14 (m, 2H), 1.17–1.09 (m, 1H), 0.39–0.30 (m, 2H), 0.27–0.19 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.75, 161.14, 151.39, 147.36, 145.12, 144.61, 144.47, 142.14, 137.19, 132.41, 131.77, 130.37, 129.50, 128.78, 128.49, 126.22, 123.73, 121.04, 116.97, 116.41, 92.09, 83.00, 28.10, 15.19, 11.64, 10.31, 4.48; HRMS (ESI) m/z: (M + H)+ calcd for C30H25FN5O4S3, 634.1047; found, 634.1056.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((2-methylthiazol-5-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (64).
This compound was synthesized from coupling the advanced intermediate VIIIa with 5-bromo-2-methylthiazole using the general Sonogashira coupling procedure F and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.804 min and (method 2) = 3.468 min; 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.31 (s, 1H), 7.96 (s, 1H), 7.72 (d, J = 1.7 Hz, 1H), 7.73–7.59 (m, 2H), 7.59–7.52 (m, 3H), 7.47 (t, J = 7.8 Hz, 1H), 7.16 (dd, J = 11.3, 1.6 Hz, 1H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 4.18 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 2.69 (s, 3H), 2.07 (s, 1H), 1.20–1.07 (m, 1H), 0.40–0.27 (m, 2H), 0.30–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H25FN5O4S3, 634.1047; found, 634.1046.
2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((2-methylthiazol-4-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (65).
This compound was synthesized from coupling the advanced intermediate VIIIa with 4-bromo-2-methylthiazole using the general Sonogashira coupling procedure F and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.741 min and (method 2) = 3.437 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.32 (s, 1H), 7.91 (s, 1H), 7.73 (t, J = 1.7 Hz, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.62–7.53 (m, 4H), 7.47 (t, J = 7.7 Hz, 1H), 7.16 (dd, J = 11.4, 1.6 Hz, 1H), 7.07 (dd, J = 8.2, 1.6 Hz, 1H), 4.19 (s, 2H), 3.18 (d, J = 6.9 Hz, 2H), 2.68 (s, 3H), 1.23–1.03 (m, 1H), 0.43–0.30 (m, 2H), 0.30–0.16 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H25FN5O4S3, 634.1047; found, 634.1041.
2-(5-(Cyclopropylmethyl)-3-(3-((3,5-dimethylthiophen-2-yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (66).
This compound was synthesized from coupling the advanced intermediate VIIIa with 2-bromo-3,5-dimethylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.749 min and (method 2) = 3.834 min; 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.31 (s, 1H), 7.70–7.63 (m, 2H), 7.60 (dt, J = 7.6, 1.5 Hz, 1H), 7.56 (s, 2H), 7.51 (dt, J = 7.7, 1.4 Hz, 1H), 7.45 (td, J = 7.7, 0.6 Hz, 1H), 7.17 (dd, J = 11.4, 1.6 Hz, 1H), 7.07 (dd, J = 8.1, 1.6 Hz, 1H), 6.72 (dt, J = 1.4, 0.7 Hz, 1H), 4.18 (s, 2H), 3.17 (d, J = 6.9 Hz, 2H), 2.42 (d, J = 1.1 Hz, 3H), 2.23 (s, 3H), 1.19–1.07 (m, 1H), 0.39–0.31 (m, 2H), 0.26–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C32H28FN4O4S3, 647.1251; found, 647.1229.
2-(5-(Cyclopropylmethyl)-3-(3-((2,5-dimethylthiophen-3-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (67).
This compound was synthesized from coupling the advanced intermediate VIIIa with 3-bromo-2,5-dimethylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.787 min and (method 2) = 3.777 min; 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.31 (s, 1H), 7.67 (t, J = 8.0 Hz, 1H), 7.64–7.57 (m, 2H), 7.58 (s, 2H), 7.51 (dt, J = 7.7, 1.5 Hz, 1H), 7.45 (td, J = 7.6, 0.7 Hz, 1H), 7.18 (dd, J = 11.3, 1.6 Hz, 1H), 7.08 (dd, J = 8.1, 1.6 Hz, 1H), 6.77 (q, J = 1.2 Hz, 1H), 4.18 (s, 2H), 3.17 (d, J = 6.9 Hz, 2H), 2.44 (d, J = 0.7 Hz, 3H), 2.38 (t, J = 0.9 Hz, 3H), 1.20–1.09 (m, 1H), 0.39–0.28 (m, 2H), 0.27–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C32H28FN4O4S3, 647.1251; found, 647.124.
(E)-2-(5-(Cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-(2-(5-methylthiophen-2-yl)vinyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (68).
This compound was synthesized from coupling the advanced intermediate VIIe with (E)-4,4,5,5-tetramethyl-2-(2-(5-methylthiophen-2-yl)vinyl)-1,3,2-dioxaborolane using the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.619 min and (method 2) = 3.753 min; 1H NMR (400 MHz, DMSO-d6): δ 13.14 (s, 1H), 8.30 (s, 1H), 7.70–7.62 (m, 2H), 7.58–7.51 (m, 3H), 7.43 (dt, J = 7.7, 1.5 Hz, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.23 (dd, J = 16.2, 0.7 Hz, 1H), 7.17 (dd, J = 11.4, 1.6 Hz, 1H), 7.08 (dd, J = 8.1, 1.6 Hz, 1H), 6.99 (d, J = 3.5 Hz, 1H), 6.83–6.72 (m, 2H), 4.19 (s, 2H), 3.19 (d, J = 6.9 Hz, 2H), 2.45 (d, J = 1.1 Hz, 3H), 1.21–1.08 (m, 1H), 0.40–0.29 (m, 2H), 0.32–0.19 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.79, 161.24, 152.52, 144.46, 144.42, 140.00, 139.28, 137.16, 132.14, 129.15, 128.49, 127.52, 126.57, 126.40, 126.34, 126.11, 125.90, 124.93, 123.68, 122.90, 116.83, 116.37, 116.16, 28.13, 15.31, 10.35, 4.49; HRMS (ESI) m/z: (M + H)+ calcd for C31H28FN4O4S3, 635.1251; found, 635.1258.
(E)-2-(3-(3-(2-Cyclopentylvinyl)-4-fluorophenyl)-5-(cyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (69).
This compound was synthesized from coupling the advanced intermediate VIIf with (E)-(2-cyclopentylvinyl)boronic acid using the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.444 min and (method 2) = 3.792 min; 1H NMR (400 MHz, DMSO-d6): δ 13.18 (s, 1H), 8.30 (s, 1H), 7.67 (t, J = 7.9 Hz, 1H), 7.58 (s, 2H), 7.60–7.54 (m, 1H), 7.49 (ddd, J = 8.6, 5.0, 2.3 Hz, 1H), 7.27–7.15 (m, 2H), 7.07 (dd, J = 8.1, 1.6 Hz, 1H), 6.43 (dd, J = 16.0, 1.1 Hz, 1H), 6.10 (dd, J = 16.0, 8.0 Hz, 1H), 4.16 (s, 2H), 3.17 (d, J = 6.9 Hz, 2H), 2.63–2.52 (m, 1H), 1.85–1.49 (m, 4H), 1.33 (ddt, J = 14.1, 11.9, 5.0 Hz, 2H), 1.22–1.07 (m, 1H), 0.39–0.28 (m, 2H), 0.31–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C31H31F2N4O4S2, 625.1749; found, 625.1744.
(E)-2-(5-(Cyclopropylmethyl)-3-(3-(2-cyclopropylvinyl)-4-fluorophenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (70).
This compound was synthesized from coupling the advanced intermediate VIIf with (E)-2-(2-cyclopropylvinyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane using the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.229 min and (method 2) = 3.694 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.30 (s, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.59 (s, 2H), 7.53 (dd, J = 7.4, 2.3 Hz, 1H), 7.44 (ddd, J = 8.6, 5.0, 2.2 Hz, 1H), 7.25–7.12 (m, 2H), 7.05 (dd, J = 8.2, 1.6 Hz, 1H), 6.53 (d, J = 15.9 Hz, 1H), 5.66 (dd, J = 15.9, 9.4 Hz, 1H), 4.15 (s, 2H), 3.16 (d, J = 6.7 Hz, 2H), 1.61 (dddd, J = 12.8, 9.4, 8.0, 4.7 Hz, 1H), 1.20–1.08 (m, 1H), 0.87–0.75 (m, 2H), 0.56–0.48 (m, 2H), 0.38–0.28 (m, 2H), 0.30–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C29H27F2N4O4S2, 597.1436; found, 597.1425.
(E)-2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(prop-1-en-1-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4carboxylic Acid (71).
This compound was synthesized from coupling the advanced intermediate VIIf with (E)-prop-1-en-1-ylboronic acid using the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.188 min and (method 2) = 3.502 min; 1H NMR (400 MHz, DMSO-d6): δ 13.17 (s, 1H), 8.30 (s, 1H), 7.67 (t, J = 7.9 Hz, 1H), 7.60 (s, 2H), 7.54 (dd, J = 7.4, 2.3 Hz, 1H), 7.45 (ddd, J = 8.6, 5.0, 2.3 Hz, 1H), 7.26–7.14 (m, 2H), 7.06 (dd, J = 8.1, 1.6 Hz, 1H), 6.47 (dq, J = 15.9, 1.7 Hz, 1H), 6.13 (dq, J = 16.0, 6.6 Hz, 1H), 4.14 (s, 2H), 3.18 (d, J = 7.0 Hz, 2H), 1.85 (dd, J = 6.6, 1.7 Hz, 3H), 1.22–1.08 (m, 1H), 0.40–0.29 (m, 2H), 0.31–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C27H25F2N4O4S2, 571.128; found, 571.1279.
(E)-2-(5-(Cyclopropylmethyl)-3-(3-(3,3-dimethylbut-1-en-1-yl)-4-fluorophenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (72).
This compound was synthesized from coupling the advanced intermediate VIIf with (E)-(3,3-dimethylbut-1-en-1-yl)boronic acid using the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.048 min and (method 2) = 3.696 min; 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.30 (d, J = 0.8 Hz, 1H), 7.67 (t, J = 7.9 Hz, 1H), 7.58 (dd, J = 7.3, 2.3 Hz, 1H), 7.56 (s, 2H), 7.51 (ddd, J = 8.5, 5.0, 2.3 Hz, 1H), 7.28–7.15 (m, 2H), 7.07 (dd, J = 8.1, 1.6 Hz, 1H), 6.36 (d, J = 16.4 Hz, 1H), 6.18 (d, J = 16.3 Hz, 1H), 4.17 (s, 2H), 3.17 (d, J = 6.9 Hz, 2H), 1.24–1.10 (m, 1H), 1.07 (d, J = 1.0 Hz, 9H), 0.39–0.28 (m, 2H), 0.31–0.19 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C30H31F2N4O4S2, 613.1749; found, 613.1728.
2-(5-(Cyclopropylmethyl)-3-(4-fluoro-3-(3,3,3-trifluoroprop-1-en-2-yl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (73).
This compound was synthesized from coupling the advanced intermediate VIIf with 4,4,6,6-tetramethyl-2-(3,3,3-trifluoroprop-1-en-2-yl)-1,3,2-dioxaborinane using the general Suzuki coupling procedure G and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 5.911 min and (method 2) = 3.538 min; 1H NMR (400 MHz, DMSO-d6): δ 13.20 (s, 1H), 8.26 (s, 1H), 7.71–7.60 (m, 2H), 7.59 (s, 2H), 7.51 (dd, J = 7.2, 2.3 Hz, 1H), 7.39 (dd, J = 10.0, 8.6 Hz, 1H), 7.12 (dd, J = 11.3, 1.6 Hz, 1H), 7.03 (dd, J = 8.1, 1.6 Hz, 1H), 6.32 (dt, J = 2.1, 1.0 Hz, 1H), 5.97 (s, 1H), 4.14 (s, 2H), 3.17 (d, J = 6.9 Hz, 2H), 1.24–1.07 (m, 1H), 0.39–0.31 (m, 2H), 0.29–0.18 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C27H22F5N4O4S2, 625.0997; found, 625.099.
2-(5-(2-Cyclopropylethyl)-3-(4-fluoro-3-((5-methylthiophen-2yl)ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (74).
This compound was synthesized from coupling the advanced intermediate VIIj with 2-ethynyl-5-methylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.859 min and (method 2) = 3.779 min. 1H NMR (400 MHz, DMSO-d6): δ 13.12 (s, 1H), 8.29 (s, 1H), 7.73 (dd, J = 6.9, 2.3 Hz, 1H), 7.67 (t, J = 7.9 Hz, 1H), 7.61 (ddd, J = 8.7, 5.0, 2.3 Hz, 1H), 7.58 (s, 2H), 7.39 (dd, J = 9.4, 8.7 Hz, 1H), 7.29 (dd, J = 3.6, 0.5 Hz, 1H), 7.19 (dd, J = 11.3, 1.6 Hz, 1H), 7.07 (dd, J = 8.1, 1.6 Hz, 1H), 6.85 (dt, J = 3.4, 1.0 Hz, 1H), 4.17 (s, 2H), 3.29–3.20 (m, 2H), 2.48 (d, J = 1.1 Hz, 3H), 1.43 (q, J = 7.3 Hz, 2H), 0.73 (ddt, J = 10.2, 7.5, 3.7 Hz, 1H), 0.36–0.26 (m, 2H), 0.16–0.08 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.75, 160.93, 160.21, 159.32, 156.81, 150.73, 147.44, 147.36, 144.69, 144.57, 143.50, 133.80, 131.92, 130.01, 129.93, 129.60, 129.46, 128.59, 126.45, 125.98, 123.71, 118.51, 116.83, 116.46, 116.24, 110.94, 88.55, 84.99, 33.27, 27.98, 24.73, 15.07, 10.55, 4.18; HRMS (ESI) m/z: (M + H)+ calcd for C32H27F2N4O4S3, 665.1157; found, 665.1174.
2-(5-(2-Cyclopropylethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylthiophen-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (75).
This compound was synthesized from coupling the advanced intermediate VIIi with 2-ethynyl-5-methylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.713 min and (method 2) = 3.838 min. 1H NMR (400 MHz, DMSO-d6): δ 13.12 (s, 1H), 8.29 (s, 1H), 7.72–7.63 (m, 2H), 7.57 (s, 2H), 7.56 (ddt, J = 17.6, 7.7, 1.4 Hz, 2H), 7.46 (td, J = 7.7, 0.6 Hz, 1H), 7.24 (dd, J = 3.6, 0.5 Hz, 1H), 7.19 (dd, J = 11.3, 1.6 Hz, 1H), 7.07 (dd, J = 8.1, 1.6 Hz, 1H), 6.83 (dt, J = 3.4, 1.1 Hz, 1H), 4.17 (s, 2H), 3.29–3.21 (m, 2H), 2.47 (d, J = 1.1 Hz, 3H), 1.43 (q, J = 7.3 Hz, 2H), 0.81–0.67 (m, 1H), 0.36–0.26 (m, 2H), 0.16–0.06 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 161.79, 160.96, 159.32, 156.80, 151.49, 144.72, 144.66, 142.80, 133.26, 132.28, 131.14, 129.88, 129.57, 129.43, 129.37, 128.60, 127.71, 126.31, 125.86, 123.69, 122.57, 119.12, 116.85, 116.42, 116.21, 91.71, 83.65, 33.27, 28.06, 24.74, 15.06, 10.56, 4.18; HRMS (ESI) m/z: (M + H)+ calcd for C32H28FN4O4S3 647.1251; found, 647.1272.
2-(5-(2-Cyclopropylethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylfuran-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (76).
This compound was synthesized from coupling the advanced intermediate VIIIc with 2-bromo-5-methylfuran using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.449 min and (method 2) = 3.533 min. 1H NMR (400 MHz, DMSO-d6): δ 13.32 (s, 1H), 8.47 (s, 1H), 7.87 (d, J = 7.5 Hz, 2H), 7.77 (s, 3H), 7.75–7.62 (m, 2H), 7.37 (d, J = 11.2 Hz, 1H), 7.26 (d, J = 8.1 Hz, 1H), 6.99 (s, 1H), 6.41 (s, 1H), 4.36 (s, 2H), 2.69 (s, 2H), 1.67–1.56 (m, 2H), 0.93 (s, 1H), 0.50–0.31 (m, 4H); 13C NMR (101 MHz, DMSO-d6): δ 161.76, 160.99, 154.34, 151.45, 147.44, 144.67, 144.58, 132.34, 131.06, 129.70, 129.44, 128.59, 127.95, 125.98, 123.73, 122.01, 117.70, 116.88, 116.43, 107.85, 92.48, 80.52, 33.26, 28.04, 24.75, 13.50, 10.56, 4.18; HRMS (ESI) m/z: (M + H)+ calcd for C32H28FN4O5S2, 631.148; found, 631.1486.
2-(5-(2-Cyclopropylethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylthiophen-3-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (77).
This compound was synthesized from coupling the advanced intermediate VIIIc with 3-bromo-2-methylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.722 min and (method 2) = 3.751 min. 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H), 8.29 (s, 1H), 7.72–7.61 (m, 3H), 7.59 (s, 2H), 7.57 (dt, J = 7.6, 1.5 Hz, 1H), 7.55–7.49 (m, 1H), 7.53–7.42 (m, 1H), 7.20 (dd, J = 11.3, 1.6 Hz, 1H), 7.08 (dd, J = 8.2, 1.6 Hz, 1H), 6.95 (p, J = 1.1 Hz, 1H), 4.17 (s, 2H), 3.29–3.21 (m, 2H), 2.46 (d, J = 1.1 Hz, 3H), 1.44 (dt, J = 10.3, 7.1 Hz, 2H), 0.82–0.67 (m, 1H), 0.36–0.26 (m, 2H), 0.16–0.08 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C32H28FN4O4S3, 647.1251; found, 647.1237.
2-(5-(2-Cyclopropylethyl)-3-(3-((5-cyclopropylthiophen-2-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (78).
This compound was synthesized from coupling the advanced intermediate VIIIc with 2-bromo-5-cyclopropylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 8.339 min and (method 2) = 3.842 min. 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H), 8.28 (s, 1H), 7.72–7.60 (m, 2H), 7.58 (s, 2H), 7.60–7.49 (m, 2H), 7.46 (t, J = 7.7 Hz, 1H), 7.25–7.15 (m, 2H), 7.07 (dd, J = 8.2, 1.6 Hz, 1H), 6.82 (dd, J = 3.7, 0.7 Hz, 1H), 4.17 (s, 2H), 3.29–3.21 (m, 2H), 2.17 (tt, J = 8.3, 5.0 Hz, 1H), 1.43 (q, J = 7.3 Hz, 2H), 1.11–0.98 (m, 2H), 0.81–0.65 (m, 3H), 0.36–0.25 (m, 2H), 0.16–0.08 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C34H30FN4O4S3, 673.1408; found, 673.1392.
2-(3-(3-((5-Cyclobutylthiophen-2-yl)ethynyl)phenyl)-5-(2-cyclopropylethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (79).
This compound was synthesized from coupling the advanced intermediate VIIIc with 2-bromo-5-cyclobutylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 7.29 min and (method 2) = 4.251 min. 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H), 8.28 (s, 1H), 7.72–7.64 (m, 2H), 7.62–7.54 (m, 3H), 7.53 (dt, J = 7.7, 1.4 Hz, 1H), 7.46 (td, J = 7.7, 0.6 Hz, 1H), 7.26 (d, J = 3.6 Hz, 1H), 7.19 (dd, J = 11.4, 1.6 Hz, 1H), 7.08 (dd, J = 8.1, 1.6 Hz, 1H), 6.88 (dd, J = 3.7, 0.9 Hz, 1H), 4.17 (s, 2H), 3.72 (dqt, J = 9.0, 8.0, 1.0 Hz, 1H), 3.29–3.21 (m, 2H), 2.47–2.32 (m, 2H), 2.19–2.04 (m, 2H), 2.07–1.88 (m, 1H), 1.91–1.78 (m, 1H), 1.43 (q, J = 7.4 Hz, 2H), 0.74 (ddd, J = 12.6, 8.5, 5.0 Hz, 1H), 0.36–0.24 (m, 2H), 0.16–0.08 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C35H32FN4O4S3, 687.1564; found, 687.1558.
2-(5-(2-Cyclopropylethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3((5-methylthiazol-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (80).
This compound was synthesized from coupling the advanced intermediate VIIIc with 2-bromo-5-methylthiazole using the general Sonogashira coupling procedure F and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.096 min and (method 2) = 3.581 min. 1H NMR (400 MHz, DMSO-d6): δ 13.14 (s, 1H), 8.28 (s, 1H), 7.80 (td, J = 1.7, 0.6 Hz, 1H), 7.71–7.62 (m, 4H), 7.58 (s, 2H), 7.56–7.47 (m, 1H), 7.18 (dd, J = 11.4, 1.5 Hz, 1H), 7.08 (dd, J = 8.2, 1.6 Hz, 1H), 4.19 (s, 2H), 3.25 (s, 2H), 2.51 (s, 3H), 1.43 (q, J = 7.5 Hz, 2H), 0.78–0.69 (m, 1H), 0.36–0.27 (m, 2H), 0.16–0.08 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C31H27FN5O4S3, 648.1204; found, 648.1202.
2-(5-(2-Cyclopropylethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((2-methylthiazol-5-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (81).
This compound was synthesized from coupling the advanced intermediate VIIIc with 5-bromo-2-methylthiazole using the general Sonogashira coupling procedure F and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.039 min and (method 2) = 3.566 min. 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H), 8.28 (s, 1H), 7.96 (s, 1H), 7.74–7.54 (m, 7H), 7.48 (td, J = 7.7, 0.6 Hz, 1H), 7.19 (dd, J = 11.3, 1.6 Hz, 1H), 7.07 (dd, J = 8.1, 1.6 Hz, 1H), 4.18 (s, 2H), 3.24 (d, J = 8.2 Hz, 2H), 2.69 (s, 3H), 1.43 (q, J = 7.4 Hz, 2H), 0.74 (td, J = 7.5, 3.8 Hz, 1H), 0.36–0.27 (m, 2H), 0.16–0.08 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 167.55, 161.76, 160.97, 156.79, 151.40, 146.95, 144.69, 144.57, 132.33, 131.34, 130.07, 129.43, 128.58, 128.19, 125.98, 123.71, 122.00, 116.93, 116.88, 116.22, 94.67, 79.87, 33.27, 28.04, 24.74, 18.97, 10.56, 4.18; HRMS (ESI) m/z: (M + H)+ calcd for C31H27FN5O4S3, 648.1204; found, 648.1191.
2-(5-(2-Cyclopropylethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((2-methylthiazol-4-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (82).
This compound was synthesized from coupling the advanced intermediate VIIIc with 4-bromo-2-methylthiazole using the general Sonogashira coupling procedure F and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.017 min and (method 2) = 3.538 min. 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H), 8.30 (s, 1H), 7.92 (s, 1H), 7.73 (dt, J = 1.8, 1.0 Hz, 1H), 7.68 (t, J = 7.9 Hz, 1H), 7.59 (s, 2H), 7.64–7.54 (m, 2H), 7.53–7.44 (m, 1H), 7.19 (dd, J = 11.4, 1.6 Hz, 1H), 7.08 (dd, J = 8.1, 1.6 Hz, 1H), 4.18 (s, 2H), 3.29–3.21 (m, 2H), 2.68 (s, 3H), 1.43 (q, J = 7.3 Hz, 2H), 0.81–0.67 (m, 1H), 0.38–0.26 (m, 2H), 0.16–0.08 (m, 2H); 13C NMR (101 MHz, DMSO-d6): δ 165.85, 161.76, 160.99, 151.47, 147.46, 144.68, 144.58, 132.34, 131.51, 130.19, 129.44, 129.43, 128.60, 128.05, 125.97, 124.77, 123.75, 122.17, 116.88, 116.44, 116.23, 87.46, 84.70, 33.26, 28.06, 24.76, 18.74, 10.57, 4.18; HRMS (ESI) m/z: (M + H)+ calcd for C31H27FN5O4S3, 648.1204; found, 648.1229.
2-(5-(2-Cyclopropylethyl)-3-(3-((2,5-dimethylthiophen-3-yl)-ethynyl)phenyl)-4-(3-fluoro-4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (83).
This compound was synthesized from coupling the advanced intermediate VIIIc with 3-bromo-2,5-dimethylthiophene using the general Sonogashira coupling procedure E and subsequent hydrolysis using general procedure H. LC–MS retention time: (method 1) = 6.982 min and (method 2) = 3.833 min. 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H), 8.28 (s, 1H), 7.68 (t, J = 7.9 Hz, 1H), 7.64–7.56 (m, 2H), 7.59 (s, 2H), 7.52 (dt, J = 7.7, 1.5 Hz, 1H), 7.46 (td, J = 7.6, 0.7 Hz, 1H), 7.21 (dd, J = 11.3, 1.6 Hz, 1H), 7.09 (dd, J = 8.1, 1.6 Hz, 1H), 6.77 (q, J = 1.1 Hz, 1H), 4.17 (s, 2H), 3.29–3.20 (m, 2H), 2.44 (d, J = 0.7 Hz, 3H), 2.38 (t, J = 0.9 Hz, 3H), 1.44 (q, J = 7.3 Hz, 2H), 0.81–0.67 (m, 1H), 0.36–0.25 (m, 2H), 0.16–0.08 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C33H30FN4O4S3, 661.1408; found, 661.1414.
2-(5-(1-Cyclopropylethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylthiophen-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (84).
This compound was synthesized using general procedures A, B, C, D, E, and H via intermediates If through VIIm. LC–MS retention time: (method 1) = 6.581 min and (method 2) = 3.785 min. 1H NMR (400 MHz, DMSO-d6): δ 13.14 (s, 1H), 8.32 (s, 1H), 7.73–7.63 (m, 2H), 7.59–7.49 (m, 4H), 7.44 (td, J = 7.7, 0.6 Hz, 1H), 7.23 (dd, J = 3.6, 0.5 Hz, 1H), 7.17 (dd, J = 11.4, 1.6 Hz, 1H), 7.07 (dd, J = 8.1, 1.6 Hz, 1H), 6.83 (dt, J = 3.4, 1.0 Hz, 1H), 4.25 (d, J = 3.2 Hz, 2H), 2.47 (d, J = 1.1 Hz, 3H), 1.37 (d, J = 7.2 Hz, 3H), 0.54–0.46 (m, 2H), 0.29–0.08 (m, 4H); HRMS (ESI) m/z: (M + H)+ calcd for C32H28FN4O4S3, 647.1251; found, 647.1268.
2-(5-(2-Cyclopropylpropan-2-yl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylthiophen-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (85).
This compound was synthesized using general procedures A, B, C, D, E, and H via intermediates Ig through VIIo. LC–MS retention time: (method 1) = 6.425 min and (method 2) = 3.741 min. 1H NMR (400 MHz, DMSO-d6): δ 13.27 (s, 1H), 8.58 (s, 1H), 7.74 (t, J = 8.0 Hz, 1H), 7.59 (s, 2H), 7.57–7.47 (m, 2H), 7.47–7.36 (m, 2H), 7.27–7.15 (m, 2H), 7.09 (dd, J = 8.1, 1.6 Hz, 1H), 6.81 (dt, J = 3.4, 1.1 Hz, 1H), 4.30 (s, 2H), 2.46 (dd, J = 1.1, 0.4 Hz, 3H), 1.28 (tt, J = 8.3, 5.8 Hz, 1H), 1.19 (s, 6H), 0.29–0.15 (m, 4H); HRMS (ESI) m/z: (M + H)+ calcd for C33H30FN4O4S3, 661.1408; found, 661.14.
2-(4-(3-Fluoro-4-sulfamoylbenzyl)-5-((1-methylcyclopropyl)-methyl)-3-(3-((5-methylthiophen-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (86).
This compound was synthesized using general procedures A, B, C, D, E, and H via intermediates Ih through VIIn. LC–MS retention time: (method 1) = 6.694 min and (method 2) = 3.751 min. 1H NMR (400 MHz, DMSO-d6): δ 13.16 (s, 1H), 8.32 (s, 1H), 7.70 (td, J = 1.7, 0.6 Hz, 1H), 7.64 (t, J = 7.9 Hz, 1H), 7.63–7.56 (m, 1H), 7.55 (s, 2H), 7.50 (dt, J = 7.8, 1.4 Hz, 1H), 7.43 (td, J = 7.7, 0.6 Hz, 1H), 7.24 (dd, J = 3.6, 0.5 Hz, 1H), 7.17–7.00 (m, 2H), 6.83 (dt, J = 3.4, 1.1 Hz, 1H), 4.21 (s, 2H), 3.45 (s, 2H), 2.48 (d, J = 1.1 Hz, 3H), 1.00 (s, 3H), 0.37–0.26 (m, 2H), 0.20–0.08 (m, 2H); HRMS (ESI) m/z: (M + H)+ calcd for C32H28FN4O4S3, 647.1251; found, 647.127.
2-(4-(3-Fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylthiophen-2-yl)ethynyl)phenyl)-5-(spiro[2.2]pentan-1-yl)-1H-pyrazol-1-yl)-thiazole-4-carboxylic Acid (87).
This compound was synthesized using general procedures A, B, C, D, E, and H via intermediates Ii through VIIp. LC–MS retention time: (method 1) = 6.604 min and (method 2) = 3.769 min. 1H NMR (400 MHz, DMSO-d6): δ 13.19 (s, 1H), 8.33 (d, J = 4.1 Hz, 1H), 7.74–7.38 (m, 7H), 7.24 (ddd, J = 3.5, 2.7, 0.5 Hz, 1H), 7.20–7.01 (m, 2H), 6.83 (dq, J = 3.4, 1.1 Hz, 1H), 4.34–4.12 (m, 2H), 2.77–2.58 (m, 1H), 2.47 (s, 3H), 1.65–1.08 (m, 2H), 0.77–0.09 (m, 3H); HRMS (ESI) m/z: (M + H)+ calcd for C32H26FN4O4S3, 645.1095; found, 645.1123.
2-(5-([1,1′-Bi(cyclopropan)]-2-yl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylthiophen-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (88).
This compound was synthesized using general procedures A, B, C, D, E, and H via intermediates Iq through VIIk. LC–MS retention time: (method 1) = 7.018 min and (method 2) = 3.871 min. 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H), 8.30 (s, 1H), 7.74–7.65 (m, 2H), 7.62–7.52 (m, 4H), 7.47 (td, J = 7.7, 0.6 Hz, 1H), 7.24 (dd, J = 3.6, 0.5 Hz, 1H), 7.20 (dd, J = 11.4, 1.6 Hz, 1H), 7.08 (dd, J = 8.2, 1.6 Hz, 1H), 6.83 (dt, J = 3.4, 1.0 Hz, 1H), 4.14 (s, 2H), 2.78–2.63 (m, 1H), 2.47 (d, J = 1.1 Hz, 3H), 1.80 (ddq, J = 75.6, 14.6, 7.6 Hz, 2H), 0.78 (ddt, J = 13.1, 8.2, 4.2 Hz, 1H), 0.55 to –0.05 (m, 4H); HRMS (ESI) m/z: (M + H)+ calcd for C33H28FN4O4S3, 659.1251; found, 659.1282.
2-(5-(Dicyclopropylmethyl)-4-(3-fluoro-4-sulfamoylbenzyl)-3-(3-((5-methylthiophen-2-yl)ethynyl)phenyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic Acid (89).
This compound was synthesized using general procedures A, B, C, D, E, and H via intermediates Il through VIIr. LC–MS retention time: (method 1) = 6.68 min and (method 2) = 3.741 min. 1H NMR (400 MHz, DMSO-d6): δ 13.11 (s, 1H), 8.32 (s, 1H), 7.72–7.63 (m, 2H), 7.63–7.54 (m, 3H), 7.51 (dt, J = 7.7, 1.4 Hz, 1H), 7.43 (td, J = 7.7, 0.6 Hz, 1H), 7.23 (dd, J = 3.6, 0.5 Hz, 1H), 7.18–7.11 (m, 1H), 7.05 (d, J = 7.8 Hz, 1H), 6.83 (dt, J = 3.4, 1.1 Hz, 1H), 4.26 (d, J = 11.1 Hz, 2H), 2.47 (d, J = 1.1 Hz, 3H), 1.96–1.14 (m, 3H), 0.73 to –0.19 (m, 8H); HRMS (ESI) m/z: (M + H)+ calcd for C34H30FN4O4S3, 673.1408; found, 673.1422.
Biological Assays.
LDH and MDH biochemical assays, cellular lactate production assay, and cytotoxicity assays were performed as previously reported19 and also provided in the Supporting Information.
Split Nano Luciferase Cellular Thermal Shift Assay.
The SplitLuc CETSA was performed in 1536-well plates as previously described.23 Briefly, HEK293T cells were transiently transfected with plasmid encoding a CMV-driven LDHA open-reading frame with a carboxy-terminal 86b fusion tag (GS[HiBiT]GS). Cells were transfected in a T175 flask for 24 h using 2.3 × 107 cells, 52.5 μg of plasmid DNA, and 105 μL of Lipofectamine 2000. Cells were lifted, resuspended at 5 × 105 cells/mL (DPBS with CaCl2 and MgCl2 plus 1 g/L glucose), and reseeded into 1536-well cyclic olefin white plates (Aurora, cyclic olefin polymer, cat# EWB041000A) using a Multidrop Combi at 2500 cells per well (5 μL volume). Compounds (23 nL) were added to cells using a pin tool (Wako Automation) and incubated for 1 h at 37 °C. Plates were heated to 63.5 °C for 7.5 min using a custom-machined copper heat block fitted with an internal type-T thermocouple and controlled by a Watlow temperature controller. The plates were removed from the heat block and cooled to room temperature. One microliter of 6% NP40 was added per well and plates were incubated for 30 min to allow cell lysis, followed by addition of 3 μL of substrate containing 11S (final concentration 100 nM) and furimazine (final concentration 0.5×; from Promega 50× stock). The plates were centrifuged and analyzed for luminescence intensity using a ViewLux reader equipped with clear filters. Luminescence values were normalized to an unheated control sample.
Glycolytic Stress Test Assay.
A673 cells were cultured in Dulbecco’s modified Eagle’s medium (ATCC Catalog no. 302002) supplemented with fetal bovine serum (10%). The cells were plated into a XF96 cell culture microplate in the above-mentioned medium and maintained in a 5% CO2 incubator at 37 °C for 24 h prior to the experiments. On the day of the assay, the compounds were diluted to the appropriate concentration in freshly prepared assay media (Seahorse basic DMEM with 2 mM glutamine, pH 7.4 at 37 °C). The media in the plate with cells were then changed to assay media and maintained in a non-CO2 incubator at 37 °C for 1 h prior to the assay. The Seahorse XF GST was conducted by injecting the LDH inhibitors and then, at 48 min, subsequent injections of glucose (10 mM final concentration), oligomycin (1 μg/mL final concentration), and 2-deoxyglucose (2-DG; 50 mM final concentration) as described previously.29
PAMPA Permeability Assay.
The stirring double-sink PAMPA method patented by pION Inc. (Billerica, MA) was employed to determine the permeability of compounds via PAMPA passive diffusion. The PAMPA lipid membrane consisted of an artificial membrane of a proprietary lipid mixture and dodecane (Pion Inc.), optimized to predict gastrointestinal tract passive diffusion permeability, and immobilized on a plastic matrix of a 96-well “donor” filter plate placed above a 96-well “acceptor” plate. A pH 7.4 solution was used in both donor and acceptor wells. The test articles, stocked in 10 mM DMSO solutions, were diluted to 0.05 mM in aqueous buffer (pH 7.4), and the concentration of DMSO was 0.5% in the final solution. During the 30 min permeation period at room temperature, the test samples in the donor compartment were stirred using the Gutbox technology (Pion Inc.) to reduce the unstirred water layer. The test article concentrations in the donor and acceptor compartments were measured using a UV plate reader (Nano Quant, Infinite 200 PRO, Tecan Inc., Männedorf, Switzerland). Permeabilitÿ calculations were performed using Pion Inc. software and were expressed in units of 10–6 cm/s.
BBB-PAMPA Permeability Assay.
The double-sink PAMPA method was utilized for the BBB permeability assay as described previously.30 The PAMPA-BBB-1 lipid (PN 110672, Pion) membrane consisted of 10% (w/v) porcine lipid brain extract dissolved in a proprietary phospholipid mixture and alkane. Like the PAMPA assay, test articles initially stocked at 10 mM DMSO solutions were diluted to 0.05 mM in an aqueous buffer (pH 7.4). The test samples were stirred using the Gutbox technology at a 60 μM aqueous boundary layer setting for 1 h at room temperature. The concentrations of compounds in the donor and acceptor compartments were measured against a reference and blank samples. The membrane permeability calculations were performed using PAMPA Evolution Software (version 3.8) from Pion Inc. and were expressed in units of 10–6 cm/s.
Kinetic Solubility Test Assay.
Pion’s patented μSOL assay for kinetic solubility determination was used. In this assay, the classical saturation shake flask solubility method was adapted to a 96-well microtiter plate format, and a cosolvent method with n-propanol as the reference compound was utilized. Test compounds were prepared in 10 mM DMSO solutions (45 μL) and diluted with the cosolvent to a final drug concentration of 150 μM in the aqueous solution (pH 7.4). The samples were incubated at room temperature for 6 h to achieve equilibrium. The samples were then filtered to remove any precipitate formed. The concentration of the compound in the filtrate was measured by UV absorbance. A reference drug concentration of 17 μM was used for the quantitation of unknown drug concentration in the filtrate. Spectroscopically pure 1-propanol was used as a cosolvent to suppress precipitation in the reference solutions. The kinetic solubility (μg/mL) was calculated using the μSOL Evolution software.
RLM Stability Assay.
Single time point microsomal stability was determined in a 96-well HTS format. Sample preparation was automated using a Tecan EVO 200 robot. A high-resolution LC–MS (Thermo Q Exactive) instrument was used to measure the percentage of the compound remaining after incubation using a previously described method.31 Six standard controls were tested in each run: buspirone and propranolol (for short half-life), loperamide and diclofenac (for short to medium half-life), and carbamazepine and antipyrine (for long half-life). Briefly, the incubation consisted of 0.5 mg/mL microsomal protein, 1.0 μM drug concentration, and an NADPH regeneration system (containing 0.650 mM NADP+, 1.65 mM glucose 6-phosphate, 1.65 mM MgCl2, and 0.2 unit/mL G6PDH) in 100 mM phosphate buffer at pH 7.4. The incubation was carried out at 37 °C for 15 min. The reaction was quenched by adding 555 μL of acetonitrile (~1:2 ratio) containing 0.28 μM albendazole (internal standard). Sample acquisition and data analysis were done using a previously described method.29
Mouse Pharmacokinetic Studies.
All PK studies were conducted by Pharmaron. Male CD1 mice (sourced from Si Bei Fu Laboratory Animal Technology Co. Ltd), approximately 6–8 weeks of age and a weight of approximately 20–30 g, were dosed with compounds at 10 mg/kg (IV) or 40 or 50 mg/kg (PO). The formulation (0.1 M NaOH in PBS, adjusted with 1 N HCl to pH 7–8.5) was prepared on the day of dosing. Each dosing cohort had three mice and plasma was collected at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, and 24 h postdose for IV administration and 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, and 24 h for PO administration. Approximately 0.025 mL of blood was collected via the dorsal metatarsal vein at each time point. Blood samples were then transferred into plastic microcentrifuge tubes containing heparin-Na as an anticoagulant. The samples were then centrifuged at 4000g for 5 min at 4 °C to obtain plasma. Plasma samples were then stored in polypropylene tubes, quickly frozen, and maintained at –75 °C until analyzed by LC–MS/MS. The following pharmacokinetic parameters were measured: T1/2, C0, Cmax, Tmax, CL, Vd, AUClast, and F. Animals were also monitored during the in-life phase by once daily cage side observations, any adverse clinical signs are noted as part of the PK report.
In Vivo LDHA Activity in Tumor Mouse Model.
Two million A673 cells were injected SQ into each female athymic nude mouse from Taconic (CrTac:NCr-Foxn1nu). The mice were divided into four treatment groups of 16 mice each. Each group received a single IV injection of 50 mg/kg of an LDHA inhibitor. Four mice from each group were sacrificed at 1, 3, 6, and 24 h after dosing. The inhibitors were dissolved in a standard PBS-based vehicle. At the time of sacrifice, frozen samples of tumor and plasma were collected and compound levels were determined through LC–MS/MS measurement against standard curves of each compound. Frozen tumor samples were also collected for LDHA activity measurements as follows. Briefly, 10–50 mg of frozen tumors was pulverized in liquid nitrogen, followed by the addition of 10 volumes of PBS at pH 7.4 containing 0.1% Triton ×−100 and incubated on ice for 1 h. The samples were clarified by centrifugation and cleared tumor lysates (10–20 μg) were measured by UV–vis spectrometry for LDH activity in LDH assay buffer containing 10 mM sodium pyruvate (Sigma-Aldrich, St. Louis, MO). The reactions were initiated by the addition of 15 mM NADH (final concentration 0.3 mM) and monitored for the oxidation of NADH at 340 nM at 37 °C.
Supplementary Material
ACKNOWLEDGMENTS
This project has been funded in whole or in part with Federal Funds from the National Cancer Institute, National Institutes of Health, under the contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. G.R., D.J.U., B.T.M., X.H., K.R.B., S.-M.Y., T.D.L., D.M.C., M.J.H., D.T., Y.F., A.S., A.J., and D.J.M. also gratefully acknowledge the funding from the Intramural Research Program, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH). The authors wish to thank Sam Michael and Richard Jones for automation support; Paul Shinn, Misha Itkin, Zina Itkin, and Danielle van Leer for the assistance with compound management; Christopher LeClair Heather Baker and Elizabeth Fernandez for analytical chemistry and purification support; and Xin Xu for in vitro ADME data. They would also like to thank Genentech for the generous donation of GNE140 for our studies, David Myszka at Biosensor Tools for conducting the SPR experiments, and Pharmaron Inc. for conducting the pharmacokinetic studies.
ABBREVIATIONS
- ACN
acetonitrile
- CETSA
cellular thermal shift assay
- LDH
lactate dehydrogenase
- MeOH
methanol
- MW
microwave
- NCGC
NCATS Chemical Genomics Center
- PAMPA
parallel artificial membrane permeability assay
- PK
pharmacokinetics
- qHTS
quantitative high-throughput screening
- SARs
structure–activity relationships
- SPR
surface plasmon resonance
- TBAF
tetra-n-butylammonium fluoride
- TFAF
trifluoroacetic acid
- THF
tetrahydrofuran
Footnotes
The authors declare no competing financial interest.
All animal studies included as part of this manuscript were performed in accordance with institutional guidelines as defined by the Institutional Animal Care and Use Committee (IACUC).
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00916.
Correlation between LDHA and LDHB; correlation of A673 lactate versus MiaPaCa-2 lactate and A673 cytotoxicity versus MiaPaCa-2; correlation of A673 lactate versus A673 and MiaPaCa-2 lactate versus MiaPaCa-2; MiaPaCa-2 72h viability versus LDHA CETSA binding activity; original LCMS, proton, fluorine, carbon NMR for 43, 52, and key compounds; experimental procedures for representative scale up of lead compounds and key intermediates; and spectroscopic data (PDF)
Molecular formula strings and PDB ID codes of 6Q0D (23) and 6Q13 (52). Authors will release the atomic coordinates and experimental data upon article publication (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00916
Contributor Information
Ganesha Rai, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Daniel J. Urban, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Bryan T. Mott, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States
Xin Hu, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Shyh-Ming Yang, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Gloria A. Benavides, Mitochondrial Medicine Laboratory, Department of Pathology, University of Alabama at Birmingham, Birmingham, Alabama 35294, United States
Michelle S. Johnson, Mitochondrial Medicine Laboratory, Department of Pathology, University of Alabama at Birmingham, Birmingham, Alabama 35294, United States
Giuseppe L. Squadrito, Mitochondrial Medicine Laboratory, Department of Pathology, University of Alabama at Birmingham, Birmingham, Alabama 35294, United States
Kyle R. Brimacombe, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States
Tobie D. Lee, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States
Dorian M. Cheff, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States
Hu Zhu, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Mark J. Henderson, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Katherine Pohida, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Gary A. Sulikowski, Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37232, United States.
David M. Dranow, Beryllium Discovery Corp., Bainbridge Island, Washington 98110, United States
Md Kabir, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Pranav Shah, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Elias Padilha, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Dingyin Tao, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland, 20850, United States.
Yuhong Fang, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Plamen P. Christov, Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37232, United States
Kwangho Kim, Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37232, United States.
Somnath Jana, Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37232, United States.
Pavan Muttil, College of Pharmacy, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131, United States.
Tamara Anderson, College of Pharmacy, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131, United States.
Nitesh K. Kunda, College of Pharmacy, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131, United States.
Helen J. Hathaway, College of Pharmacy, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131, United States
Donna F. Kusewitt, Dept of Pathology, University of New Mexico Cancer Center, Albuquerque, New Mexico 87131, United States
Nobu Oshima, Urologic Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland 20892, United States.
Murali Cherukuri, Urologic Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland 20892, United States.
Douglas R. Davies, Beryllium Discovery Corp., Bainbridge Island, Washington 98110, United States
Jeffrey P. Norenberg, College of Pharmacy, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131, United States
Larry A. Sklar, Dept of Pathology, University of New Mexico Cancer Center, Albuquerque, New Mexico 87131, United States
William J. Moore, NExT Program Support, Applied/Developmental Research Directorate, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland 21702, United States
Chi V. Dang, Abramson Cancer Center, Abramson Family Cancer Research Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States; Ludwig Institute for Cancer Research, New York, New York 10017, United States
Gordon M. Stott, NExT Program Support, Applied/Developmental Research Directorate, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland 21702, United States
Leonard Neckers, Urologic Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland 20892, United States.
Andrew J. Flint, NExT Program Support, Applied/Developmental Research Directorate, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland 21702, United States
Victor M. Darley-Usmar, Mitochondrial Medicine Laboratory, Department of Pathology, University of Alabama at Birmingham, Birmingham, Alabama 35294, United States
Anton Simeonov, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Alex G. Waterson, Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37232, United States
Ajit Jadhav, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Matthew D. Hall, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United State.
David J. Maloney, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
REFERENCES
- (1).Warburg O; Posener K; Negelein E Ueber den stoffwechsel der tumoren. Biochem. Z 1924, 152, 319–344. [Google Scholar]
- (2).Vander Heiden MG; Cantley LC; Thompson CB Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vander Heiden MG Exploiting tumor metabolism: challenges for clinical translation. J. Clin. Invest. 2013, 123, 3648–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yu Y; Liao M; Liu R; Chen J; Feng H; Fu Z Overexpression of lactate dehydrogenase-A in human intrahepatic cholangiocarcinoma: its implication for treatment. World J. Surg. Oncol. 2014, 12, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Rong Y; Wu W; Ni X; Kuang T; Jin D; Wang D; Lou W Lactate dehydrogenase A is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumor Biol. 2013, 34, 1523–1530. [DOI] [PubMed] [Google Scholar]
- (6).Rani R; Kumar V Recent update on human lactate dehydrogenase enzyme 5 (hLDH5) inhibitors: a promising approach for cancer chemotherapy. J. Med. Chem. 2016, 59, 487–496. [DOI] [PubMed] [Google Scholar]
- (7).Allison SJ; Knight JRP; Granchi C; Rani R; Minutolo F; Milner J; Phillips RM Identification of LDH-A as a therapeutic target for cancer cell killing via (i) p53/NAD(H)-dependent and (ii) p53-independent pathways. Oncogenesis 2014, 3, No. e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Le A; Cooper CR; Gouw AM; Dinavahi R; Maitra A; Deck LM; Royer RE; Vander Jagt DL; Semenza GL; Dang CV Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sheng SL; Liu JJ; Dai YH; Sun XG; Xiong XP; Huang G Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012, 279, 3898–3910. [DOI] [PubMed] [Google Scholar]
- (10).Boudreau A; Purkey HE; Hitz A; Robarge K; Peterson D; Labadie S; Kwong M; Hong R; Gao M; Del Nagro C; Pusapati R; Ma S; Salphati L; Pang J; Zhou A; Lai T; Li Y; Chen Z; Wei B; Yen I; Sideris S; McCleland M; Firestein R; Corson L; Vanderbilt A; Williams S; Daemen A; Belvin M; Eigenbrot C; Jackson PK; Malek S; Hatzivassiliou G; Sampath D; Evangelista M; O’Brien T Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [DOI] [PubMed] [Google Scholar]
- (11).Fantin VR; St-Pierre J; Leder P Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [DOI] [PubMed] [Google Scholar]
- (12).Xie H; Hanai J.-i.; Ren J-G; Kats L; Burgess K; Bhargava P; Signoretti S; Billiard J; Duffy KJ; Grant A; Wang X; Lorkiewicz PK; Schatzman S; Bousamra M 2nd; Lane AN; Higashi RM; Fan TWM; Pandolfi PP; Sukhatme VP; Seth P. Targeting lactate dehydrogenase–a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metabol. 2014, 19, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Le A; Cooper CR; Gouw AM; Dinavahi R; Maitra A; Deck LM; Royer RE; Vander Jagt DL; Semenza GL; Dang CV Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Granchi C; Roy S; Giacomelli C; Macchia M; Tuccinardi T; Martinelli A; Lanza M; Betti L; Giannaccini G; Lucacchini A; Funel N; León LG; Giovannetti E; Peters GJ; Palchaudhuri R; Calvaresi EC; Hergenrother PJ; Minutolo F Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [DOI] [PubMed] [Google Scholar]
- (14).Kohlmann A; Zech SG; Li F; Zhou T; Squillace RM; Commodore L; Greenfield MT; Lu X; Miller DP; Huang W-S; Qi J; Thomas RM; Wang Y; Zhang S; Dodd R; Liu S; Xu R; Xu Y; Miret JJ; Rivera V; Clackson T; Shakespeare WC; Zhu X; Dalgarno DC Fragment growing and linking lead to novel nanomolar lactate dehydrogenase inhibitors. J. Med. Chem. 2013, 56, 1023–1040. [DOI] [PubMed] [Google Scholar]
- (15).Ward RA; Brassington C; Breeze AL; Caputo A; Critchlow S; Davies G; Goodwin L; Hassall G; Greenwood R; Holdgate GA; Mrosek M; Norman RA; Pearson S; Tart J; Tucker JA; Vogtherr M; Whittaker D; Wingfield J; Winter J; Hudson K Design and synthesis of novel lactate dehydrogenase A inhibitors by fragment-based lead generation. J. Med. Chem. 2012, 55, 3285–3306. [DOI] [PubMed] [Google Scholar]
- (16).Billiard J; Dennison JB; Briand J; Annan RS; Chai D; Colón M; Dodson CS; Gilbert SA; Greshock J; Jing J; Lu H; McSurdy-Freed JE; Orband-Miller LA; Mills GB; Quinn CJ; Schneck JL; Scott GF; Shaw AN; Waitt GM; Wooster RF; Duffy KJ Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Canc. Metabol. 2013, 1, 19–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Fauber BP; Dragovich PS; Chen J; Corson LB; Ding CZ; Eigenbrot C; Giannetti AM; Hunsaker T; Labadie S; Liu Y; Liu Y; Malek S; Peterson D; Pitts K; Sideris S; Ultsch M; VanderPorten E; Wang J; Wei B; Yen I; Yue Q Identification of 2-amino-5-aryl-pyrazines as inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2013, 23, 5533–5539. [DOI] [PubMed] [Google Scholar]; (b) Dragovich PS; Fauber BP; Corson LB; Ding CZ; Eigenbrot C; Ge H; Giannetti AM; Hunsaker T; Labadie S; Liu Y; Malek S; Pan B; Peterson D; Pitts K; Purkey HE; Sideris S; Ultsch M; VanderPorten E; Wei B; Xu Q; Yen I; Yue Q; Zhang H; Zhang X Identification of substituted 2-thio-6-oxo-1,6-duhydropyrimidines as inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2013, 23, 3186–3194. [DOI] [PubMed] [Google Scholar]
- (18).Zhou Y; Tao P; Wang M; Xu P; Lu W; Lei P; You Q Development of novel human lactate dehydrogenase A inhibitors: high-throughput screening, synthesis, and biological evaluations. Eur. J. Med. Chem. 2019, 177, 105–115. [DOI] [PubMed] [Google Scholar]
- (19).Rai G; Brimacombe KR; Mott BT; Urban DJ; Hu X; Yang S-M; Lee TD; Cheff DM; Kouznetsova J; Benavides GA; Pohida K; Kuenstner EJ; Luci DK; Lukacs CM; Davies DR; Dranow DM; Zhu H; Sulikowski G; Moore WJ; Stott GM; Flint AJ; Hall MD; Darley-Usmar VM; Neckers LM; Dang CV; Waterson AG; Simeonov A; Jadhav A; Maloney DJ Discovery and optimization of potent, cell-active pyrazole-based inhibitors of lactate dehydrogenase (LDH). J. Med. Chem. 2017, 60, 9184–9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yeung C; Gibson AE; Issaq SH; Oshima N; Baumgart JT; Edessa LD; Rai G; Urban DJ; Johnson MS; Benavides GA; Squadrito GL; Yohe ME; Lei H; Eldridge S; Hamre J; Dowdy T; Ruiz-Rodado V; Lita A; Mendoza A; Shern JF; Larion M; Helman LJ; Stott GM; Krishna MC; Hall MD; Darley-Usmar V; Neckers LM; Heske CM Targeting glycolysis through inhibition of lactate dehydrogenase impairs tumor growth in preclinical models of Ewing sarcoma. Cancer Res. 2019, 79, 5060–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Oshima N; Ishida R; Kishimoto S; Beebe K; Brender JR; Yamamoto K; Urban D; Rai G; Johnson MS; Benavides G; Squadrito GL; Crooks D; Jackson J; Joshi A; Mott BT; Shrimp JH; Moses MA; Lee M-J; Yuno A; Lee TD; Hu X; Anderson T; Kusewitt D; Hathaway HH; Jadhav A; Picard D; Trepel JB; Mitchell JB; Stott GM; Moore W; Simeonov A; Sklar LA; Norenberg JP; Linehan WM; Maloney DJ; Dang CV; Waterson AG; Hall M; Darley-Usmar VM; Krishna MC; Neckers LM Dynamic imaging of LDH inhibition in tumors reveals rapid in vivo metabolic rewiring and vulnerability to combination therapy. Cell Rep. 2020, 30, 1798–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Quon E; Hart ML; Sullivan LB Redox debt leads to metabolic bankruptcy in tumors. Trends Cancer 2020, 6, 359–361. [DOI] [PubMed] [Google Scholar]
- (22).Hermans D; Gautam S; García-Cañaveras JC; Gromer D; Mitra S; Spolski R; Li P; Christensen S; Nguyen R; Lin J-X; Oh J; Du N; Veenbergen S; Fioravanti J; Ebina-Shibuya R; Bleck C; Neckers LM; Rabinowitz JD; Gattinoni L; Leonard WJ Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8+ T cell stemness and antitumor immunity. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pohida K; Maloney DJ; Mott BT; Rai G Room-temperature, copper-free Sonogashira reactions facilitated by air-stable, monoligated precatalyst [DTBNpP] Pd(crotyl)Cl. ACS Omega 2018, 3, 12985–12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dalvie DK; Kalgutkar AS; Khojasteh-Bakht SC; Obach RS; O’Donnell JP Biotransformation reactions of five-membered aromatic heterocyclic rings. Chem. Res. Toxicol. 2002, 15, 269–299. [DOI] [PubMed] [Google Scholar]
- (25).Purkey HE; Robarge K; Chen J; Chen Z; Corson LB; Ding CZ; DiPasquale AG; Dragovich PS; Eigenbrot C; Evangelista M; Fauber BP; Gao Z; Ge H; Hitz A; Ho Q; Labadie SS; Lai KW; Liu W; Liu Y; Li C; Ma S; Malek S; O’Brien T; Pang J; Peterson D; Salphati L; Sideris S; Ultsch M; Wei B; Yen I; Yue Q; Zhang H; Zhou A Cell active hydroxylactam inhibitors of human lactate dehydrogenase with oral bioavailability in mice. ACS Med. Chem. Lett. 2016, 7, 896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Martinez NJ; Asawa RR; Cyr MG; Zakharov A; Urban DJ; Roth JS; Wallgren E; Klumpp-Thomas C; Coussens NP; Rai G; Yang S-M; Hall MD; Marugan JJ; Simeonov A; Henderson MJ A widely-applicable high-throughput cellular thermal shift assay (CETSA) using split Nano Luciferase. Sci. Rep. 2018, 8, 9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sun H; Nguyen K; Kerns E; Yan Z; Yu KR; Shah P; Jadhav A; Xu X Highly predictive and interpretable models for PAMPA permeability. Bioorg. Med. Chem. 2017, 25, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).L’Heureux A; Beaulieu F; Bennett C; Bill DR; Clayton S; LaFlamme F; Mirmehrabi M; Tadayon S; Tovell D; Couturier M Aminodifluorosulfinium salts: selective fluorination reagents with enhanced thermal stability and ease of handling. J. Org. Chem. 2010, 75, 3401–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hill BG; Benavides GA; Lancaster JR; Ballinger S; Dell’Italia L; Zhang J; Darley-Usmar VM Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tsinman O; Tsinman K; Sun N; Avdeef A Physicochemical selectivity of the BBB microenvironment governing passive diffusion matching with a porcine brain lipid extract artificial membrane permeability model. Pharm. Res. 2011, 28, 337–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Di L; Kerns EH; Gao N; Li SQ; Huang Y; Bourassa JL; Huryn DM Experimental design on single-time-point high-throughput microsomal stability assay. J. Pharm. Sci. 2004, 93, 1537–1544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.