

Abstract
Non-model microorganisms often possess complex phenotypes that could be important for the future of biofuel and chemical production. They have received significant interest the last several years, but advancement is still slow due to the lack of a robust genetic toolbox in most organisms. Typically, “domestication” of a new non-model microorganism has been done on an ad hoc basis, and historically, it can take years to develop transformation and basic genetic tools. Here, we review the barriers and solutions to rapid development of genetic transformation tools in new hosts, with a major focus on Restriction-Modification systems, which are a well-known and significant barrier to efficient transformation. We further explore the tools and approaches used for efficient gene deletion, DNA insertion, and heterologous gene expression. Finally, more advanced and high-throughput tools are now being developed in diverse non-model microbes, paving the way for rapid and multiplexed genome engineering for biotechnology.
Keywords: Non-model microbes, Genetics, Genetic tools, Transformation, Metabolic engineering, Synthetic biology
Introduction
The world’s energy and chemical demand is ever-increasing, and currently, the demand for fuels and chemicals is primarily met with fossil fuels. Fossil fuels like petroleum not only provide the raw materials to make liquid fuel for the transportation sector, they also provide the building blocks needed for a wide variety of heavily utilized chemicals and materials, including plastics, perfumes, paints, fertilizer, and detergents [1]. However, fossil-derived carbon is inherently unsustainable, and a promising alternative is microbial conversion of renewable feedstocks. Microorganisms often have the ability to make drop-in or functional replacements for the fuels and chemical building blocks used today, but metabolic engineering is typically required to reach the titers, rates, and yields needed for a commercial process as well as to diversify the chemicals that can be produced.
Currently, model organisms like Saccharomyces cerevisiae and E. coli are most commonly used for metabolic engineering research and are often used for industrial biochemical production. Model organisms are attractive because they are industrially robust, decades of research have enabled deep understanding of the organisms, and they have a large genetic toolbox to enable rapid and simple modifications. However, engineering of synthetic pathways into these organisms is often most successful with short heterologous pathways, while long synthetic and heterologous pathways are generally not as robust and can cause a large metabolic burden [2]. To overcome the challenges with current industrial biochemical production and to make next-generation fuels and chemicals profitable, recent research has focused on non-model organisms that natively have phenotypes of interest for each unique feedstock and processing method [3].
Industrial bioproduction of fuels and chemicals initially focused on first generation feedstocks such as starch and sucrose. These feedstocks are easy to breakdown and convert, but potential competition with food crops, limited greenhouse gas emission benefits, and environmental sustainability concerns makes these feedstocks problematic [4]. Alternate feedstocks such as the sugars and aromatics in lignocellulosic biomass [5], sunlight or renewable electricity coupled with CO2 fixation [6, 7], syngas [8], and waste plastic [9] have the potential to be cheaper. These feedstocks are also more environmentally friendly sources of carbon for fuel and chemical production, but bioconversion of these substrates is much more challenging. However, many non-model microbes often have evolved to utilize these feedstocks efficiently, making them potentially attractive platforms for bioconversion. Unfortunately, many of the organisms with these native capabilities have poorly characterized metabolisms and are challenging to modify genetically, hindering metabolic engineering efforts.
Other traits potentially beneficial to industrial processing methods include tolerance to low pH, tolerance to high salt, or the ability to grow at high temperatures. These traits could streamline chemical production by reducing contamination, reducing the need for pH control and buffering, and increasing enzymatic reaction speed. These abilities are multigene, complex phenotypes that are difficult or impossible to engineer into model organisms. The more widespread use of non-model organisms could leverage these phenotypes to usher in a new era of biotechnology for biofuel and chemical production.
Two major barriers to the use of non-model organisms for metabolic engineering include a lack of genetic tools and limited knowledge of the organism’s physiology. Traditionally, physiological characterization of a new organism was time-consuming and labor-intensive, with much of the new knowledge generated through biochemistry and characterization of substrate utilization and product formation. It was difficult to gain additional in-depth physiological information to uncover cell metabolism and regulation [2, 10]. However, genomic knowledge has become easier to acquire due to the rise of next-generation sequencing techniques and the dramatic drop in the cost of genome sequencing [11, 12]. Using basic genomic knowledge, the groundwork can quickly be laid for systems-level biological analyses, or “omics” in non-model microbes. Enabling tools like transcriptomics and proteomics are straightforward to apply to new microorganisms, and they assist in addressing fundamental questions and provide measurements for all cellular components [10]. The cost and time associated with these tools are also dropping, and the major challenge is now turning these large datasets into usable knowledge. Integration of multi-omics data to models helps give a more comprehensive dataset and helps to link the genotypes associated with the complex phenotypes of interest [13, 14]. Having comprehensive knowledge of a microorganisms’ metabolic pathways and flux is important to successful metabolic engineering [15]. With the increased availability of affordable -omics analyses and DNA synthesis, the major remaining barrier to widespread adoption and “domestication” of non-model microbes is the lack of robust genetic tools.
Barriers to genetic modification and enabling transformation
In order to develop non-model organisms for bioprocessing, genetic modification is typically needed. The development of genetic tools requires the ability to efficiently transform DNA into a target organism. There are four primary barriers to overcome for successful transformation of bacteria: cellular uptake of foreign DNA, evasion of native immune systems that degrade foreign DNA, selection for transformants, and stable maintenance of foreign DNA by the microbial host. Each poses a challenge, and the largest barrier is often the evasion of host defense systems.
DNA entry into the cell
The first challenge, introducing DNA into cells, requires DNA to get past one or two membranes, as well as other physical barriers, such as peptidoglycan, to reach the cytoplasm. Depending on the organism, a variety of techniques can be utilized, including commonly used methods such as electroporation, conjugation, protoplast transformation, and natural competence. For electroporation, an electrical field is applied to a mixture of DNA and washed cells, which opens holes in the membrane, and enables the uptake of DNA [16, 17]. Electroporation works broadly across phylogenetic groups [18–23], including in diverse bacteria, eukaryotes, and archaea, and therefore is often the first method attempted by researchers trying to transform a new organism. Conjugation enables DNA transfer through direct cell to cell contact from a donor cell to the recipient. This technique has been used in diverse organisms using a donor E. coli strain carrying broad host range conjugation machinery [24–26]. DNA is transported into the cell as single stranded DNA (ssDNA), which is usually highly recombinogenic, making it an especially good method of transformation when homologous recombination is desired [27–29]. Conjugation can be more challenging into microorganisms that have a vastly different optimal growth conditions than E. coli, such as halophiles or thermophilic anaerobes. In protoplast transformation, the bacterial cell wall is degraded through the introduction of lysozyme. Removal of the cell wall allows for cells to uptake DNA without a barrier. This is a technique that has seen wide use the actinomycetes [30–33]. Natural competence, on the other hand, is a mechanism driven by suite of genes that allows some bacteria to uptake DNA from their environment [34], and this imported DNA is also single stranded. Transformation using this technique is often relatively simple in strains containing the natural competence genes, where the strain of interest is incubated with DNA under conditions where the competence genes are expressed. Prominent examples of hosts that use natural competence for transformation include the Firmicute Bacillus subtilis and the Gammaproteobacterium Acinetobacter baylyi [35, 36]. In strains that are not naturally competent, conjugation and electroporation are the most common methods of transformation, with electroporation often enabling the highest transformation efficiencies [37]. Though these three techniques are the most commonly used, there are many less commonly used techniques reported in the literature, including phage-enabled transfer [38], chemically induced competence [39], sonoporation [40], biolistic bombardment [41], liposome-mediated fusion [42], and nanofiber piercing [43]. Because these methods are not used as widely, their application to new organisms is relatively underexplored.
Host defense systems
Prokaryotic organisms have evolved multiple defenses against foreign DNA, which can present a major barrier to DNA transformation. Restriction-Modification (RM) systems act as innate immune systems and are one of the primary systems used by prokaryotes to protect themselves against foreign DNA. Cells recognize and degrade DNA that is methylated differentially from that of its own DNA [44]. Almost 90% of prokaryotes encode RM systems, with most encoding two or more [45]. Microbes often also encode adaptive immune systems, including clustered regularly interspaced short palindromic repeats (CRISPR). CRISPR systems natively protect prokaryotes against foreign DNA, for instance from bacteriophage. Viral derived DNA sequences are acquired during an infection and inserted into the chromosome where they may be transcribed to prevent against repetitive infection [46]. Native CRISPR systems are unlikely to pose a major barrier to DNA transformation in most cases because the specific sequences acquired by the host CRISPR system are unlikely to be present in the DNA that is being transformed. Therefore, CRISPR will only be further discussed in this review in relation to their use as genetic tools. Several other defense systems have been discovered, but they are rare and/or poorly characterized. These include abortive infection (Abi), BREX, Dnd, Dpd, DISARM, pAgos, and others [47, 48]. The impact of these systems on genetic transformation is currently unknown, but not yet observed to present major barriers.
To enable genetic modification, transformed DNA must evade the native immune systems of the host. RM systems are the most important immune system that DNA needs to evade, and many studies have shown the importance of this for successful transformation (e.g., 49–58). Of the four classes of RM systems, Types I, II, and III typically comprised two primary components, a restriction enzyme and a methyltransferase. The methyltransferase modifies a specific base within a specific motif throughout the host chromosome so that the genome is protected from restriction enzyme cleavage, with modifications including 6-methyladenine (m6A), 4-methylcytosine (m4C), or 5-methylcytosine (m5C). The restriction enzyme cleaves the same motif in unmethylated DNA that enters the cell. Type IV systems, on the other hand, only consist of a nuclease that cleaves methylated DNA, with the recognized motifs different than those targeted by native methyltransferases from Types I, II, and III. The sequence specificity of Type IV systems is typically poorly characterized and is a ripe area for future research.
To evade RM systems, the first step is to identify the methylated motifs within a cell. Microbial methylation sites are most commonly determined through genome sequencing with single molecule real-time (SMRT) sequencing on the PacBio platform [59, 60]. SMRT sequencing routinely identifies m6A and m4C motifs and, with a significantly lower efficiency, m5C motifs through kinetic delays in nucleotide incorporation at modified bases during sequencing. While eukaryotic methylome analysis often employs whole genome bisulfite sequencing (WGBS) to detect m5C motifs, it has rarely been used in bacteria. However, robust detection of m5C is critical for complete methylome analysis [50]. Other emerging techniques, such as nanopore sequencing, may also be used for methylome analysis [61, 62]. New England Biolabs (NEB) has created a database, REBASE, of all known RM systems (both experimentally determined and computationally predicted) and publicly available microbial methylomes to help in the identification of RM systems [63]. Not all methyltransferases are associated with restriction enzymes; they can also play a regulatory role in DNA replication, gene expression, and DNA mismatch repair [64]. However, the methylated motifs reveal the maximum number of sites that could be subject to restriction in a given host.
Once the methylated motifs are identified, several methods have been used to evade the corresponding RM systems. One approach is transforming DNA that lacks the identified motifs, or mutating DNA so that it no longer contains the motif [50, 65, 66]. Recently, software has been designed to aid in the process of eliminating restriction sites [67]. This can often be done for uncommon motifs, but it cannot always be used for shorter, more common motifs, like four base pair recognition sequences that may exist in plasmid origins of replication or other DNAs that require a specific sequence, such as those needed for homologous recombination.
Another way to overcome RM systems is by methylating DNA of interest in the same way as the target host prior to transformation, such that the organism does not recognize it as foreign. The most common approach to achieving methylation is to express the target organism’s restriction-associated methyltransferases in E. coli, and then isolate the DNA of interest from this E. coli strain to properly methylate it prior to transformation. Early pioneering work demonstrated transformation of Clostridium acetobutylicum by first methylating plasmid DNA in E. coli with the methyltransferase from B. subtilis phage phi3T [49]. This approach was expanded with a method called plasmid artificial modification (PAM), which introduced all the methyltransferases from a given strain into E. coli using plasmid-based expression, followed by isolation of the DNA of interest out of the PAM host to properly methylate it prior to transformation of the target organism [68]. More recently, informed by methylome analysis, only those methyltransferases identified as important were introduced to the E. coli chromosome to more stably express the enzymes and to only mimic the methylome data.
DNA can also be properly methylated through in vitro methylation [69]. Some enzymes are commercially available, which can be especially useful for 4-base motifs that are targeted by commercially available methyltransferases. For those motifs that are not methylated by commercially available enzymes, cell-free extracts can be used, as demonstrated in Helicobacter pylori [70] and Saccharopolyspora spinosa [71], to increase transformation efficiency.
RM systems can also be partially evaded by the transformation mechanism chosen. Often, electroporation is unsuccessful when using improperly methylated DNA and, in these cases, using conjugation can yield successful transformation. The conjugation machinery transfers single stranded DNA (ssDNA) that can avoid restriction enzyme cleavage until it has time to become properly methylated [72]. Similarly, natural competence also imports ssDNA and can reduce the impact of restriction systems [73].
Another important consideration when transforming a new bacterial strain is the presence of Type IV restriction systems, which degrade methylated DNA motifs. Researchers typically isolate plasmid DNA from E. coli prior to transformation into their desired host, and commonly used laboratory E. coli strains encode two major DNA methylases, dam and dcm, targeting G(m6A)TC and C(m5C)WGG, respectively. Many potential target strains encode dam, either as part of an RM system or as a housekeeping methyltransferase, and a few encode dcm, but many do not. Therefore, it is important to isolate plasmid DNA from an appropriate E. coli genetic background, using strains deleted for dam and/or dcm when the target organism does not methylate the same site. This prevents unnecessary DNA methylation and subsequent degradation by Type IV systems, which has been demonstrated in many strains [74–76]. It is also important to utilize an E. coli strain that lacks the native Type IV systems (mcrA, mcrBC, and mrr) when expressing methyltransferases. Methylation of motifs recognized by the native type IV systems would cause E. coli to restrict its own chromosome, which would kill the E. coli strain.
RM systems are frequently acquired via horizontal gene transfer [48], and therefore methods to improve transformation in one strain typically do not directly translate to closely related strains. These systems tend to be hyper-variable within an environmental community of closely related strains, such that if a phage happens to avoid one cell’s restriction systems and becomes methylated like that host, it does not eliminate the entire population [45]. Therefore, methylome analysis and RM system evasion must be developed for each desired host, even if they are strains of the same species.
Maintaining and selecting for DNA
Once DNA has entered the cell and evaded degradation, it needs to be maintained during cell division, and transformed cells must be selected from the untransformed cells that constitute the majority of the population. DNA can be maintained either through autonomous replication or through chromosomal integration. Plasmid-based autonomous replication is typically the best way to achieve the highest transformation efficiency for a new microbe and is the most commonly used method to demonstrate initial transformation. For metabolic engineering, however, plasmid-based gene expression has drawbacks, such as high copy number artifacts, variable plasmid copy number, the need for continuous antibiotic selection, and a high metabolic burden. Therefore, chromosomal integration is also an important tool, and different approaches to developing DNA integration genetic tools will be discussed in more detail below.
Development of a plasmid-based DNA maintenance system relies on identification of an origin of replication that functions in the target organism. The most common vectors used for cloning and replication in E. coli use origins such as pUC, ColE1, and p15a, each with varying copy numbers [77], but often these origins do not replicate in non-model bacteria. Therefore, origins that function in diverse bacteria are needed. Broad host range plasmids are capable of transfer and maintenance in bacteria from different phylogenetic subgroups, examples include pBBR1, pRK2, pBC1, and many others [78]. Plasmid libraries have been formed around the broad host-range origins of replication and used in a variety of organisms [79]. Alternatively, origins can come from native plasmids of the strain or close relatives [80, 81]. If none are present, replicating plasmids may be constructed by cloning the chromosomal origin of replication to create a mini-chromosome [82, 83].
Next, one must select for cells that were transformed and eliminate cells that were not. The most common positive selectable markers are antibiotic resistance genes (Table 1). To determine the marker(s) most likely to work, the minimum inhibitory concentration (MIC) is determined by exposing the host bacteria to increasing levels of a panel of antibiotics. This is most easily done in liquid growth medium, but higher concentrations of antibiotic may be needed for selection on agar plates. Another consideration when choosing antibiotic resistance markers is compatibility with an organism’s growth conditions. For example, when working with thermophiles, one should focus on antibiotics that are more stable at higher temperatures like thiamphenicol and kanamycin [84] and thermotolerant selectable markers [85]. A less commonly used alternative to antibiotics is nutritional selection. In this case, an auxotrophic strain is generated by deletion of an essential nutrient gene and transformed with a plasmid encoding the missing biosynthesis gene(s). The resulting transformants are selected on media lacking the target nutrient [86, 87]. Libraries of modular plasmids combining antibiotic resistance markers, origins of replication, multiple cloning sites (MCS), and other genetic parts have been developed for different classes of organisms, and they are very useful for the rapid testing of genetic parts [88–90].
Table 1.
Commonly used antibiotics and corresponding selectable markers
| Antibiotic | Marker | Class |
|---|---|---|
| Kanamycin/neomycin | neo | Aminoglycoside |
| Chloramphenicol/thiamphenicol | cat | Chloramphenicol |
| Erythromycin | ermB, ermF | Macrolide |
| Ampicillin/carbenicillin | bla | Beta-lactam |
| Tetracycline | tetA | Tetracycline |
| Bleomycin | ble | Glycopeptide |
| Gentamicin | aacC1 | Aminoglycoside |
| Streptomycin and spectinomycin | aadA | Aminoglycoside |
| Zeocin | zeo | Glycopeptide |
| Apramycin | apr | Aminoglycoside |
| Thiostrepton | tsr | Cyclic oligopeptide |
| Puromycin | pac | Aminonucleoside |
| Hygromycin | hph | Aminoglycoside |
| Nourseothricin | nat | Aminoglycoside |
Native antibiotic resistance levels can vary widely between strains. Therefore, an MIC experiment should be performed with each target organism to determine the level of antibiotic needed for selection.
Chromosome modification tools in non-model microorganisms
After demonstration of initial transformation, more advanced tools can be developed to enable efficient genome editing and ultimately high-throughput strain engineering. Genome editing can allow for gene deletions, insertion of heterologous pathways, point mutations, and altered gene regulation. Enabling genome integration and deletion tools facilitates rational metabolic engineering, where competing production pathways can be eliminated, and new pathways can be stably introduced. Development of more sophisticated tools is critical to the engineering of non-model organisms for high rates and titers of desired products.
Homologous recombination
Homologous recombination is a naturally occurring mechanism essential for DNA repair in bacteria, and it can be leveraged to rationally introduce modifications to an organism’s chromosome. The most commonly deployed method uses a non-replicating plasmid-based technique to create a scar-less mutation [91]. Typically, this is mediated by nuclease–helicase complex, RecBCD, and the single stranded DNA-binding DNA repair protein, RecA [92]. To use homologous recombination for gene deletion, a plasmid is required that contains a selectable maker, a counter-selectable marker, and DNA that is homologous to the upstream and downstream regions of the gene targeted for deletion (Fig. 1a). The lengths of homologous DNA regions are often around 500–1000 bp each. After transformation of the plasmid into the microbe, the plasmid recombines into the chromosome at one region of homology to integrate the entire plasmid, which is selected via the positive selectable marker contained on the plasmid. A second recombination event will resolve the merodiploid into either the parent chromosome or a gene deletion. If the first recombination event is repeated, the strain reverts to wild type, but if the recombination event occurs in the second region of homology, the targeted gene is deleted. When there is no fitness defect associated with the deletion, the frequency of deletion should be approximately 50%, which can be easily screened via PCR.
Fig. 1.
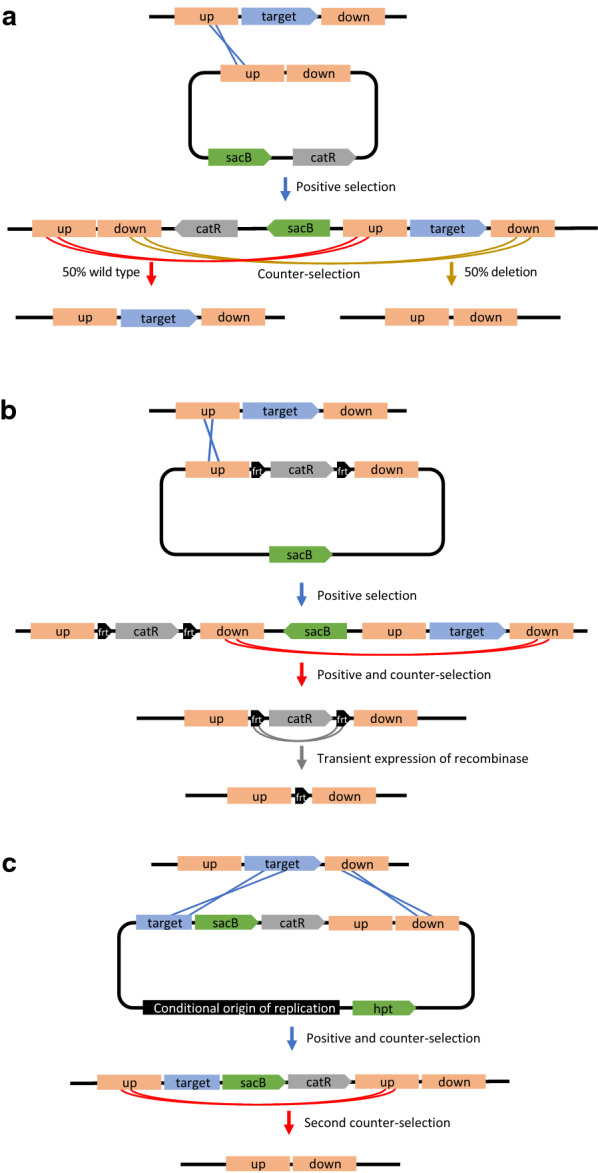
Methods using homologous recombination for gene deletions, with details for each method described in the text. a Basic homologous recombination using a non-replicating plasmid where 50% of colonies will be the desired deletion. b Homologous recombination using the positive selectable marker between the homology arms, flanked with recombination sites, and c homologous recombination using a replicating vector and three homology regions. Orange, homology regions, with “up” representing the DNA region upstream of the target gene, and “down” representing the DNA region downstream of the target gene; blue, target gene for deletion; green, counter-selectable markers; gray, positive selectable marker; crossed lines, sites of recombination
Selection for resolution of the merodiploid requires a counter-selectable marker, which allows selection for loss of DNA. Numerous counter-selections have been demonstrated (Table 2), including sacB in Gram-negative bacteria and pyrF and hpt in Gram-positive bacteria [91, 93–96]. Some of the markers are less desirable than others, though. Markers such as sacB often do not function well in Gram-positive bacteria [93]. There are also markers such as pyrF that can act as both a selectable and counter-selectable marker making them more flexible [97–99], but in many cases, they also result in auxotrophy, requiring growth on minimal medium for prototrophic selection of transformants. Many genes used as counter-selectable markers are natively encoded in the host genome. In these cases, the native copy must first be deleted before it can be used as a counter-selectable marker. However, the presence of a native counter-selectable marker also presents an opportunity for metabolic engineering, as it can serve as a simple site for insertion of heterologous DNA with a direct selection for gene replacement. Importantly, inactivating mutations (e.g., frameshifts, active site mutations, etc.) in counter-selectable markers have the same phenotype as recombinants and arise spontaneously, resulting in a background of cells that do not contain the targeted deletion. Therefore, counter-selections often require more screening to find correct clones relative to positive selection. In the absence of reliable counter-selectable markers, screens can be used, but this is much less efficient and should be avoided if possible [100].
Table 2.
Commonly used counter-selectable markers and associated selections for homologous recombination in bacteria
| Marker | Counterselection | Reference |
|---|---|---|
| sacB | Sucrose | [101] |
| upp | 5-Fluorouracil | [102] |
| hpt | 8-Azahypoxanthine, others | [103] |
| tdk | 5-Fluorodeoxyuridine | [104] |
| pyrF/ura3a | 5-Fluoroorotic acid | [105] |
| pheS*(A294G) | p-Chloro-phenylalanine | [106] |
| codA | 5-Fluorocytosine | [107] |
| Inducible mazF | N/A; expression is toxic | [95] |
| galK | 2-Deoxygalactose | [108] |
| apt | 2-Fluoroadenine | [109] |
| rpsL(strA) | Streptomycin | [110] |
| tetARa | Fusaric acid | [111] |
| thyA | Trimethoprim | [112] |
| Inducible ccdB | Expression is toxic | [113] |
| oroP | 5-Fluoroorotate | [114] |
| pta-ack | Chloroacetate | [115] |
aCan be used as a positive selectable marker with uracil prototrophy and tetracycline resistance, respectively
Another approach is to place the antibiotic resistance marker between the homology arms, allowing for direct selection of the gene deletion (Fig. 1b) [116]. This approach is especially useful when the mutant phenotype is deleterious, which would make the approach in Fig. 1a challenging. This approach can also work in the absence of a counter-selectable marker when using conjugation or natural competence and selecting directly for the double-recombination event that replaces the gene target with the antibiotic resistance gene. Often the antibiotic resistance marker is flanked by recombination sites, like frt or lox, so that the marker can be removed after integration into the chromosome by introduction of the corresponding recombinase, like Flp or Cre [116–118].
The frequency of homologous recombination varies widely in bacteria and in some cases is a rare event, making the transformation efficiency of integrating plasmids multiple orders of magnitude lower than for replicating plasmids. Therefore, replicating plasmid-based homologous recombination methods can be an attractive approach when the combination of transformation efficiency and homologous recombination in the target organism is low. However, because the plasmid replicates autonomously, it can be challenging to select chromosomal modifications; therefore, alternate approaches are needed.
One option is to use a conditional origin of replication, such as a temperature-sensitive plasmid. This allows the transformation event and the homologous recombination event to be uncoupled, where the plasmid can be transformed at a permissive temperature, generating a whole population of cells that contain the plasmid. Upon moving the culture to the non-permissive temperature, one can select for the cells that have performed recombination. Selection then can proceed as in Fig. 1a.
Alternatively, introduction of a third region of homology and a second counter-selectable marker can allow for selection of each recombination event and plasmid loss (Fig. 1c) [20]. In this approach, the replicating plasmid is first transformed into the strain. Then, while maintaining selection for the antibiotic resistance gene, selection is performed against the counter-selectable marker that is on the plasmid backbone. This selects for two recombination events to insert the antibiotic resistance onto the chromosome and simultaneous plasmid backbone loss. The resulting merodiploid can then be resolved by selecting against the second counter-selectable marker, resulting in the desired deletion.
Homologous recombination based genetic tools have been used in a variety of non-model organisms to increase biochemical production and enable non-native carbon catabolism though the deletion of competing pathways and insertion of heterologous genes [119–122].
Recombineering
Methods to increase the efficiency of homologous recombination have been developed, often called recombination-mediated genetic engineering, or recombineering [123, 124]. By increasing the recombination efficiency, construction of gene deletions, insertions, and point mutations is more efficient. Recombineering uses proteins like those derived from the lambda bacteriophage Red complex, Beta, Exo, and Gam, to integrate single stranded and linear DNA into the genome, often using short homology arms to guide recombination. Recombineering is widely used in E. coli, and the genes associated with lambda Red have also been demonstrated to function in other proteobacteria, like Pseudomonas putida [125]. Recombineering can be difficult to develop in non-model organisms because lambda Red genes are often not functional in those hosts. Considerable effort has gone towards identifying genes analogous to lambda Red and the recombination protein RecT in phylogenetically diverse organisms [126, 127]. Using this approach, recombineering has been demonstrated in a variety of other strains, including Clostridium acetobutylicum [128], Lactococcus lactis [129], Clostridium thermocellum [130], and others [126, 131, 132]. The recombinases can then be used to enable ssDNA recombination for a recombination techniques often requires 300–1000 bp of homology for efficient recombination, while recombineering techniques can shorten this to as few as 20 bp.
Random DNA insertion
Non-homology-based approaches are also useful when engineering non-model microbes. One such approach is transposition. Transposons allow random integration of DNA segments throughout the genome with no required homology. Therefore, transposition is traditionally used to create a library of insertional disruptions that can be screened for particular phenotypes. These phenotypes can then be correlated with genotypes by identifying the site of insertion [133]. Commonly used transposons include Tn5, which inserts mostly randomly but is known to favor “hotspots”, and Himar1, which can insert randomly between any TA dinucleotide [134, 135]. Recently, random transposon mutagenesis has been combined with high-throughput sequencing for genome-scale analyses, called TnSeq [136]. Further combination with randomly barcoded transposons simplifies reverse genetics, where a pooled library of barcoded insertional mutants can be created, and the bar codes can be mapped to the genome to identify disrupted genes [137]. A barcoded library can be an incredibly powerful tool for genome-scale analyses. A list of putatively essential genes can be compiled by identifying the genes that were not disrupted in the library. Furthermore, genes related to phenotypes such as solvent and organic acid resistance, product formation, and carbon assimilation can be identified by finding barcodes that increase or decrease in abundance under selective conditions [136, 138, 139].
Transposons can also be used to insert heterologous pathways into bacteria [140], which have been applied to non-model microbes such as C. ljungdahlii for acetone production [141] and Acidothiobacillus ferroxidans for isobutyric acid biosynthesis [142]. Transposons have also been combined with the Cre-lox site-specific recombination system to insert a landing pad, followed by site-specific insertion of heterologous pathways [143]. Transposons are especially useful for application in organisms that lack other genetic tools such as homologous recombination for integrating DNA into the chromosome. Integrating DNA randomly has its drawbacks, however. The transposon often integrates into coding regions of the genome, which can interfere with the fitness of the strain, and identifying the location of transposition requires additional effort. Random integration also does not enable comparison of a construct across genetic backgrounds because the transposition event occurs in different loci, which can result in differing levels of gene expression for the construct [144].
Site-specific DNA integration
Site-specific recombinases have been a valuable tool for DNA insertions and excisions where the enzyme catalyzes a recombination event between two specific DNA sequences, sometimes called attachment (att) sites. The most commonly used site-specific recombinases are the Cre and Flp that enable a reversible recombination (integration or excision) reaction at identical sites, loxP and frt, respectively [145]. Both systems have been used to remove antibiotic resistance markers by integrating a cassette into the chromosome with loxP or frt flanking the marker followed by transient expression of the corresponding recombinase [123]. For example, Flp/frt is widely used in the E. coli lambda Red system to remove the antibiotic resistance marker, and it has also been used in a variety of non-model organisms, including thermophiles, methanotrophs, and biofuel producing Clostridium species [94, 116, 122, 146]. One drawback to using Cre and Flp, however, is that the loxP or frt scar left behind can cause genetic instability when multiple modifications are stacked into a single strain, leaving behind multiple, identical copies of the scar that can all serve as future substrates for the recombinase. Using mutated sites can help overcome the genome instability [147, 148].
A second group enables a site-specific recombination event at non-identical attachment sites, typically called attB (bacterial) and attP (phage), reflective of the fact that these recombinases often derive from bacteriophage that lysogenize their hosts. Recombination creates two new sites, attL (left) and attR (right). The integration event is unidirectional because the attL and attR are not substrates for recombination, so the recombinase cannot excise the construct from the genome [149]. Some unidirectional recombinases, including the lambda integrase, require host machinery to function, which limits use outside of their native host or very close relatives. Lambda phage also has relatively large attachment sites of 350–450 bp each. Alternatively, the large serine recombinases, as exemplified by the archetypical ΦC31 integrase, can function in a broad-host range because they do not require host machinery. This has led to their development as genetic tools in a wide variety of microbial hosts, especially in the actinomyces [150–153]. Due to their utility for rapid and high efficiency integration of DNA, they have been used to test genetic libraries at single copy on the chromosome, such as promoter libraries [150], and to integrate heterologous DNA [154–156]. Genetic tools developed with serine integrases can rely on native or pseudo-att sites that are present in the genome [157] or non-native att sites that have been previously integrated into the genome [154, 158, 159]. One of the most commonly used integrases is ΦC31, which was identified from Streptomyces and is commonly used in a DNA integration system in this genus [28, 153]. Multiple orthogonal recombinases have been identified and characterized [149, 153], setting the stage for them to be used much more broadly. Another type of site-specific recombinase includes transposases that target highly conserved sequences like Tn7 and Tn1545 [144]. Tn7 has been demonstrated to function in more than 20 bacterial species. It enables integration into the highly conserved attTn7 site downstream of the glmS gene. This makes the use of site-specific transposons more desirable for some applications than the random transposons for integration of heterologous constructs [144].
CRISPR/Cas
A recent and potentially universal tool for engineering non-model microbes uses clustered regularly interspaced palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins. Native CRISPR–Cas systems comprise a CRISPR array containing spacers separated by short repeats. Spacers become incorporated into the array when the microorganism encounters invading DNA to enable the microorganism to recognize and attack that same sequence when they encounter it again [160]. Many of the systems cleave DNA at this sequence using a Cas nuclease that is directed by a guide RNA (gRNA). Cas9 was first identified in Streptococcus pyogenes (spCas9), and it introduces a double-stranded break (DSB) at a specific sequence guided by a sgRNA adjacent to a protospacer adjacent motif (PAM) [161, 162]. Natively, the gRNA is derived from the CRISPR array, and in the case of the most utilized type II Cas nuclease, Cas9, the array is transcribed and processed (crRNA) with trans-acting RNA (tracrRNA) into a single guide RNA (sgRNA)[163]. In heterologous systems, the sgRNA can be transcribed as a single RNA, simplifying the number of genetic parts needed for engineering. If a template containing the desired modification is also included, the DSB can be repaired, resulting in an edited genome.
CRISPR–Cas systems have been leveraged in a wide variety of organisms to introduce scarless point mutations, insertions, and deletions in the genome. The most widely used CRISPR nuclease is spCas9 because, unlike many of the other Cas proteins, it is a single enzyme that only requires the introduction of a synthetic gRNA and the Cas9 nuclease to enable genome editing. CRISPR-spCas9 was first introduced as a genome engineering tool for bacteria in E. coli [162, 164] and has now been implemented in a wide variety of species using a plasmid for the repair template and an organism’s native homologous recombination machinery, as recently reviewed in [162]. Cas9-based editing has been further developed by combination with recombineering machinery to enhance the efficiency of DSB repair, and allowing use of a linear recombineering template with homology to the edited region [164, 165]. The combination of CRISPR and recombineering can increase the rate of mutation close to 100% due to the lethality of the DSB. Functionally, this makes CRISPR–Cas a potent and targetable counter-selectable marker [166]. The combination of recombineering and CRISPR has been used in many proteobacteria, but it can take substantial effort to transfer to organisms where transformation efficiency is low and recombineering tools do not yet exist.
The nuclease spCas9 is most commonly used for genome editing across many types of bacteria. However, Cas9 expression can be toxic in a variety of hosts [159, 167]. Toxicity can be overcome by placing the gene under an inducible promoter so that it is only expressed when needed [168], but this is only possible in organisms where inducible promoters have been developed. Toxicity can also be overcome in some organisms by mutating one of the active sites, resulting in Cas9n, so that it can only nick one strand of DNA rather than creating a DSB [169, 170]. Another issue with the commonly used Cas9 enzymes is that they are unable to function in thermophiles; therefore, thermophilic Cas9 enzymes have been identified and implemented along with thermophilic recombineering machinery for engineering thermophiles [130]. Although spCas9 is the most widely used nuclease, other CRISPR–Cas systems have been identified to overcome problems with spCas9. These nucleases also have different cleavage patterns and target different PAM sequences. Cpf1, a member of the Cas12a group, has been used in several organisms for genome editing, including those where spCas9 did not function [159, 171, 172]. Native CRISPR systems have also been leveraged for genome editing [173, 174]. There is significant interest in CRISPR based tools for non-model organisms, and it has been used wildly and in more depth elsewhere [161, 162, 175, 176].
CRISPR/Cas for gene regulation
While CRISPR–Cas9 has been widely demonstrated for DNA insertions and deletions, it can also be used for gene regulation by silencing or activating transcription using a catalytically inactive Cas9 (dCas9). In this case, the dCas9 is unable to act as a nuclease but is still able to bind DNA at a targeted sequence. In CRISPR interference (CRISPRi), a gRNA-targeted dCas9 binds to either the promoter of the desired gene or the open reading frame (ORF), therefore blocking either transcription initiation or elongation and inhibiting gene expression (177). This enables gene knockdowns in a regulated and reversible way, and expression of multiple gRNAs can allow multiplexing of gene knockdowns [178]. Researchers have developed CRISPRi tools in a wide variety of non-model microorganisms to both study the activity of essential genes and to knockdown expression of competing pathways to increase production of fuels and chemicals [179–182].
While CRISPR can be a powerful genome engineering and gene regulation tool, it is often difficult to optimize in non-model bacteria. Therefore, research has gone into developing a modular CRISPRi system that can be deployed across phylogenetically diverse bacteria. Mobile-CRISPRi has demonstrated the gene knockdown efficacy in a diverse set of pathogenic microbes, though this system has yet to be used in organisms of interest for industrial use [183]. While CRISPRi represses transcription, dCas9 can also be used to activate transcription via CRISPR activation (CRISPRa). In CRISPRa the dCas9 is fused to a transcriptional activator so that when dCas9 binds upstream of a gene it recruits RNA polymerase leading to increased transcription. While CRISPRa has been used in E. coli using both PolII [184] and SoxS [185], this tool is still in the early stages of being applied to other prokaryotes [186].
Gene expression tools in non-model microorganisms
The ability to reliably fine-tune gene expression is an important synthetic biology tool and can play an important role in increasing biofuel and chemical yields from engineered pathways. Transcription and translation levels can be controlled by several different components including promoters, riboswitches, ribosome binding sites (RBS), and terminators. Genes can be constitutively expressed, where protein production levels are largely determined by the strengths of the promoter and RBS, or genes can be regulated where expression can be turned “on” or “off” by inducible promoters and riboswitches. Reporter genes can provide an easily assayed output to help determine the impact of different genetic parts on expression levels, and they also having applications in the development of biosensors for the detection and control metabolite levels during bioconversion. Developing these gene expression tools for non-model microbes is critical to enabling rational metabolic pathway optimization for improved product formation.
Constitutive gene expression
The simplest way to modulate gene transcription is through the evaluation of promoters with varying activity levels. Often, strong characterized promoters from model organisms, like Ptac from E. coli, are used but they do not always confer robust gene expression in non-native hosts. Identifying and implementing native promoters can overcome this issue. Strong promoters often drive expression of genes in central metabolism and identifying those genes in a target organism may help in identifying useful promoters. RNAseq can be employed on cells grown under desired culture conditions [187–189], where genes with high transcript levels may be indicative of a strong promoter. Further characterization of the identified promoters through a time course study under various growth conditions may identify those that are constitutive [189].
Overexpression with strong promoters is not always desirable, especially if an intermediate in the pathway is toxic or if a protein is membrane-bound. Furthermore, when many genes need to be expressed, the metabolic burden of protein production can become substantial [190]. Therefore, the development of a promoter library with a range of promoter strengths is also useful. Typically, the closer the − 35 and − 10 sequences are to consensus, the stronger the promoter is. Mutating each of the bases in the sequence and varying the space between them has been shown to vary promoter strength [191, 192]. Mutated promoters can also be obtained through error-prone PCR or through site-directed mutagenesis [188]. Promoter libraries have been characterized in a wide variety of non-model organisms [193–196]. Other regions that impact gene expression and can be varied include the UP element and the ribosome binding site (RBS). UP elements are sequences upstream of the − 35 box in the promoter that bind to the alpha subunit of the RNA polymerase, greatly increasing expression levels [197]. While underexplored for many non-model microbes, UP elements can bring an additional boost to expression when very high transcription levels are desired [198–200]. The RBS recruits the ribosome and it is an important control point for translation initiation. The RBS is commonly used to tune gene expression through alterations of the sequence and the space between the sequence and the start codon. Libraries of RBSs have been created in combination with promoters to fine-tune gene expression for fuel and chemical production [195, 201, 202]. An RBS calculator has been created to aid in the creation of RBS libraries and to predict translation initiation rates [203].
Regulated gene expression
The ability to regulate gene expression is an important tool for both strain engineering and bioproduction. Applications include expression of a toxic gene (e.g., Cas9) for a short period [168, 204], balancing of metabolic flux with a biosensor [205], or turning on a production pathway once cell growth has reached stationary phase to reduce the metabolic burden [206–208]. They are also important to determine whether a gene is essential under the conditions being tested. One mechanism of creating inducible promoters involves the use of transcription factors. Well-characterized promoters from E. coli such as lactose- and arabinose-inducible promoters have been transferred, sometimes with mutations to enable functionality, to a wide variety of other organisms, including species of Clostridium, Pseudomonas, Bacillus, and Ralstonia [79, 209]. Other inducers have also been commonly used such as tetracycline [210], xylose [211], and nisin [94]. Newer promoters such as the Jungle Express expression system for proteobacteria [212], and less commonly used ones such as laminaribiose [213] also have the potential to be useful for engineering various non-model organisms. However, transferring well characterized promoters into non-model bacteria can still be challenging. Many result in “leaky” expression or lack regulatory proteins, such as repressors or activators, not present in heterologous hosts. In addition, even if regulatory proteins are transferred with the promoter, transport of the inducer molecule may be an issue.
Gene expression can also be regulated post-transcriptionally. Riboswitches are regulatory mRNA elements in the 5′ untranslated region that are capable of regulating gene expression through small molecule-induced structural switching [214]. They add a layer of regulation, but are less explored for the engineering of non-model microbes relative to model organisms like E. coli. In this mechanism, mRNA forms two distinct shapes depending on the presence of a specific small molecule, which either blocks or allows translation. Examples of the use of riboswitches for engineering of non-model microbes include theophylline for inducible production of biofuels and other compounds [215], lysine to balance flux for competing pathways [214, 216], and flavonoids such as naringenin for increased production of flavonoids [217]. Recently, thermophilic riboswitches have also been identified and used to regulate biofuel production [218]. Riboswitches are particularly beneficial for regulating gene expression in non-model microbes because they function independently of host-associated proteins and without the need for organism-specific promoters. For example, a set of theophylline riboswitches have been demonstrated to function in a many species including multiple cyanobacteria, Mycobacterium, and Streptomyces. These switches have also proven to be superior for regulation of gene expression when compared to traditional IPTG-inducible promoters [215].
Terminators
Transcriptional terminators in E. coli have been well characterized and are an important tool for gene expression. Terminators can impact mRNA stability and expression levels of adjacent genes [219] Simple terminators that do not require additional termination factors have been used widely in diverse genera. Some terminators, like those from bacteriophage T7, can be inefficient at termination, causing read through on multigene constructs. However, several commonly used terminators contain repeats that increase the stability of transcriptional pausing and increase reliability, though this can cause issues when using the same terminator repeatedly use within a single construct [173]. Efficient and reliable control of multigene constructs for metabolic engineering of bacteria therefore requires several well characterized terminators. Libraries [174] and terminator prediction programs [175] have been developed, but terminator research in non-model bacteria lags behind other parts for gene expression like promoters.
Reporter genes
Reporter genes encode proteins that can be tracked or quantified, often visually or spectrophotometrically. They have many applications in flow cytometry, microscopy, protein localization, and microbial co-cultures, and they are particularly useful when screening promoter and gene expression libraries. Enzymes can act as reporter genes, including common ones such as lacZ (β-galactosidase) and uidA/gusA (β-glucuronidase), where a substrate (e.g., X-gal) is cleaved to generate a colored compound, and enzyme activity is proportional to enzyme abundance. Other enzymes include chloramphenicol acetyltransferase (catP) [220] and alcohol dehydrogenase (adhE) [221, 222]. Determining enzymatic activity can be laborious, which makes these enzymatic reporter genes less useful, and they are typically used only as end-point assays [223].
Genes encoding fluorescent proteins are the most commonly used type of reporter gene because they can provide real-time measurements of gene expression. In aerobic microbes, several different genes encoding fluorescent protein have been engineered, including the green fluorescent protein, gfp, and the red fluorescent protein mCherry. Each of the fluorescent proteins has different characteristics from color to brightness to stability [224]. These genes have been used in a wide variety of applications. The fluorescent genes are used in transcriptional fusions to indicate the strength of promoters and expression vectors during bacterial cell growth [191, 225]. The simplicity and non-invasive nature of fluorescent proteins allow easier tracking of the dynamics of gene expression and allow much higher throughput. This higher throughput enables the use of these genes as biosensors by fusing the fluorescent reporter to regulated promoters that respond to the presence of metabolites, such as the target product molecule, metabolic intermediate, or toxic compound [226–228]. One caveat is that reporters are only a proxy for gene expression, and the expression level of heterologous genes can depend on the exact genetic context, especially if mRNA secondary structure or stability changes. These fluorescent reporter genes are incredibly useful in aerobic bacteria because they require O2 to form the fluorescent chromophore.
Many biotechnology-relevant organisms are strict anaerobes, where O2-dependent reporters can be more challenging to use. The use of fluorescent proteins requires that anaerobic cultures be brought into an aerobic environment, which can be lethal, and the cells often need to be washed in a time-consuming process. Therefore, they cannot be used in real-time growth experiments anaerobically. Flavin-binding fluorescent proteins (FbFPs) do not require O2 for fluorescence, making them potentially useful tools for anaerobes. Examples of FbFPs include iLOV, BsFbFP, PpFbFP, and EcFbFP which have been demonstrated in a variety of organisms including many species of clostridia [229]. However, FbFPs are not as bright as aerobic fluorescent genes, which make them more difficult to quantify. To overcome the limitation of FbFPs, a fluorescence-activating and absorption-shifting tag (FAST) protein has been developed. FAST relies on the presence of an exogenously added ligand that is only fluorescent when bound to FAST. Two color signals have been developed, a green yellow (YFAST) signal where 4-hydroxy-3-methylbenzylidine-rhodanine (HMBR) binds FAST and a red signal (rFAST) where 4-hydroxy-3,5-dimethoxybenzylidene-rhodanine (HBR3,5DOM) binds. FAST produces a fluorescence signal similar to that of GFP and has recently been used in a few organisms [230, 231]. This tool has the potential to revolutionize the use of reporter genes in anaerobic microbes. This includes the ability to monitor real-time gene expression, screen promoter libraries, study protein localization, and develop other high-throughput tools such as biosensors.
Conclusion
We are entering an era of both rational and systematic design of genetic tools for the metabolic engineering of diverse non-model bacteria. Transformation methods that overcome native RM systems via targeted DNA methylation and the utilization of libraries of genetic parts is enabling the manipulation of numerous new hosts to harness their native complex phenotypes. Here we outline an approach to “domesticate” non-model organisms by rapidly developing a genetic toolbox to enable both rational and untargeted metabolic engineering. As more bacteria become genetically tractable and more tools are established in these hosts, advanced genome engineering tools like CRISPR–Cas, biosensors, and phage recombinases will further accelerate metabolic engineering efforts. The ability to fine-tune metabolic pathways in organisms that have beneficial, complex phenotypes will enable engineering for increased titer, rate, and yield for a range of important products to build a biobased, sustainable future.
Acknowledgements
Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the U.S. DOE under contract DE-AC05-00OR22725.
This manuscript has been authored by UT-Battelle, LLC, under Contract No. DE-AC0500OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for the United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Authors’ contributions
LAR and AMG wrote the manuscript. Both authors read and approved the final manuscript.
Funding
This work was in part supported by the Center for Bioenergy Innovation, U.S. DOE Bioenergy Research Center, supported by the Office of Biological and Environmental Research in the DOE Office of Science. This work was in part supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, Genomic Science Program under Award Number DE-SC0019401. Funding was provided in part by the U.S. Department of Energy Office of Energy Efficiency and Renewable Energy the Bioenergy Technologies Office via the Agile BioFoundry project. The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Naik SN, Goud VV, Rout PK, Dalai AK. Production of first and second generation biofuels: a comprehensive review. Renew Sust Energ Rev. 2010;14(2):578–597. doi: 10.1016/j.rser.2009.10.003. [DOI] [Google Scholar]
- 2.Czajka J, Wang Q, Wang Y, Tang YJ. Synthetic biology for manufacturing chemicals: constraints drive the use of non-conventional microbial platforms. Appl Microbiol Biotechnol. 2017;101(20):7427–7434. doi: 10.1007/s00253-017-8489-9. [DOI] [PubMed] [Google Scholar]
- 3.Gowen CM, Fong SS. Applications of systems biology towards microbial fuel production. Trends Microbiol. 2011;19(10):516–524. doi: 10.1016/j.tim.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Sims R, Taylor M. From 1st-to 2nd-generation biofuel technologies: an overview of current industry and RD&D activities. IEA Bioenergy. 2008.
- 5.Olson DG, McBride JE, Shaw AJ, Lynd LR. Recent progress in consolidated bioprocessing. Curr Opin Biotechnol. 2012;23(3):396–405. doi: 10.1016/j.copbio.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 6.Knoot CJ, Ungerer J, Wangikar PP, Pakrasi HB. Cyanobacteria: Promising biocatalysts for sustainable chemical production. J Biol Chem. 2018;293(14):5044–5052. doi: 10.1074/jbc.R117.815886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karthikeyan R, Singh R, Bose A. Microbial electron uptake in microbial electrosynthesis: a mini-review. J Ind Microbiol Biotechnol. 2019;46(9–10):1419–1426. doi: 10.1007/s10295-019-02166-6. [DOI] [PubMed] [Google Scholar]
- 8.Devarapalli M, Atiyeh HK. A review of conversion processes for bioethanol production with a focus on syngas fermentation. Biofuel Res J. 2015;2(3):268–280. doi: 10.18331/BRJ2015.2.3.5. [DOI] [Google Scholar]
- 9.Pathak VM, Navneet. Review on the current status of polymer degradation: a microbial approach. Bioresour Bioprocess. 2017;4(1):15.
- 10.Armengaud J, Trapp J, Pible O, Geffard O, Chaumot A, Hartmann EM. Non-model organisms, a species endangered by proteogenomics. J Proteomics. 2014;105:5–18. doi: 10.1016/j.jprot.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Eldem V, Zararsiz G, Taşçi T, Duru YBIP, Bakir Y, Erkan M. Transcriptome analysis for non-model organism: current status and best practices. Applications of RNA-Seq and omics strategies—from microorganisms to human health. 2017.
- 12.Ellegren H. Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol. 2014;29(1):51–63. doi: 10.1016/j.tree.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Joyce AR, Palsson BO. The model organism as a system: Integrating ‘omics’ data sets. Nat Rev Mol Cell Biol. 2006;7(3):198–210. doi: 10.1038/nrm1857. [DOI] [PubMed] [Google Scholar]
- 14.Fondi M, Liò P. Multi -omics and metabolic modelling pipelines: Challenges and tools for systems microbiology. Microbiol Res. 2015;171:52–64. doi: 10.1016/j.micres.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Yan Q, Fong SS. Challenges and advances for genetic engineering of non-model bacteria and uses in consolidated bioprocessing. Front Microbiol. 2017 doi: 10.3389/fmicb.2017.02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calvin N, Hanawalt P. High-efficiency transformation of bacterial cells by electroporation. J Bacteriol. 1988;170(6):2796–2801. doi: 10.1128/JB.170.6.2796-2801.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dower WJ, Miller JF, Ragsdale CW. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988;16(13):6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liebl W, Bayerl A, Schein B, Stillner U, Schleifer KH. High efficiency electroporation of intact Corynebacterium glutamicum cells. FEMS Microbiol Lett. 1989;65(3):299–303. doi: 10.1111/j.1574-6968.1989.tb03677.x. [DOI] [PubMed] [Google Scholar]
- 19.Brigidi P, De Rossi E, Bertarini ML, Riccardi G, Matteuzzi D. Genetic transformation of intact cells of Bacillus subtilis by electroporation. FEMS Microbiol Lett. 1990;67(1–2):135–138. doi: 10.1111/j.1574-6968.1990.tb13850.x. [DOI] [PubMed] [Google Scholar]
- 20.Olson DG, Lynd LR. Transformation of Clostridium thermocellum by electroporation. Methods Enzymol. 2012;510:317–330. doi: 10.1016/B978-0-12-415931-0.00017-3. [DOI] [PubMed] [Google Scholar]
- 21.Iwasaki K, Uchiyama H, Yagi O, Kurabayashi T, Ishizuka K, Takamura Y. Transformation of Pseudomonas putida by electroporation. Biosci Biotechnol Biochem. 1994;58(5):851–854. doi: 10.1271/bbb.58.851. [DOI] [PubMed] [Google Scholar]
- 22.Benatuil L, Perez JM, Belk J, Hsieh C-M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23(4):155–159. doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- 23.Hileman T, Santangelo T. Genetics techniques for Thermococcus kodakarensis. Front Microbiol. 2012;3(195):195. doi: 10.3389/fmicb.2012.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trieu-Cuot P, Carlier C, Martin P, Courvalin P. Plasmid transfer by conjugation from Escherichia coli to Gram-positive bacteria. FEMS Microbiol Lett. 1987;48(1–2):289–294. doi: 10.1111/j.1574-6968.1987.tb02558.x. [DOI] [Google Scholar]
- 25.Mazodier P, Petter R, Thompson C. Intergeneric conjugation between Escherichia coli and Streptomyces species. J Bacteriol. 1989;171(6):3583–3585. doi: 10.1128/JB.171.6.3583-3585.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tominaga Y, Ohshiro T, Suzuki H. Conjugative plasmid transfer from Escherichia coli is a versatile approach for genetic transformation of thermophilic Bacillus and Geobacillus species. Extremophiles. 2016;20(3):375–381. doi: 10.1007/s00792-016-0819-9. [DOI] [PubMed] [Google Scholar]
- 27.Matsushima P, Broughton MC, Turner JR, Baltz RH. Conjugal transfer of cosmid DNA from Escherichia coli to Saccharopolyspora spinosa: Effects of chromosomal insertions on macrolide A83543 production. Gene. 1994;146(1):39–45. doi: 10.1016/0378-1119(94)90831-1. [DOI] [PubMed] [Google Scholar]
- 28.Bierman M, Logan R, O'Brien K, Seno ET, Nagaraja Rao R, Schoner BE. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene. 1992;116(1):43–49. doi: 10.1016/0378-1119(92)90627-2. [DOI] [PubMed] [Google Scholar]
- 29.Baltz RH, Hosted TJ. Molecular genetic methods for improving secondary-metabolite production in actinomycetes. Trends Biotechnol. 1996;14(7):245–250. doi: 10.1016/0167-7799(96)10034-2. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto H, Maurer KH, Hutchinson CR. Transformation of Streptomyces erythraeus. J Antiobiot. 1986;39(9):1304–1313. doi: 10.7164/antibiotics.39.1304. [DOI] [PubMed] [Google Scholar]
- 31.Hopwood DA. Genetic manipulation of Streptomyces. A laboratory manual. Endeavour. 1985 doi: 10.1016/0160-9327(87)90187-6. [DOI] [Google Scholar]
- 32.Katsumata R, Ozaki A, Oka T, Furuya A. Protoplast transformation of glutamate-producing bacteria with plasmid DNA. J Bacteriol. 1984;159(1):306–311. doi: 10.1128/JB.159.1.306-311.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bibb MJ, Ward JM, Hopwood DA. Transformation of plasmid DNA into Streptomyces at high frequency. Nature. 1978;274(5669):398–400. doi: 10.1038/274398a0. [DOI] [PubMed] [Google Scholar]
- 34.Blokesch M. Natural competence for transformation. Curr Biol. 2016;26(21):R1126–R1130. doi: 10.1016/j.cub.2016.08.058. [DOI] [PubMed] [Google Scholar]
- 35.Dubnau D. Genetic competence in Bacillus subtilis. Microbiol Rev. 1991;55(3):395–424. doi: 10.1128/MR.55.3.395-424.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vaneechoutte M, Young DM, Ornston LN, De Baere T, Nemec A, Van Der Reijden T, et al. Naturally transformable Acinetobacter sp. strain ADP1 belongs to the newly described species Acinetobacterbaylyi. Appl Environ Microbiol. 2006;72(1):932–936. doi: 10.1128/AEM.72.1.932-936.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pyne ME, Bruder M, Moo-Young M, Chung DA, Chou CP. Technical guide for genetic advancement of underdeveloped and intractable Clostridium. Biotechnol Adv. 2014;32(3):623–641. doi: 10.1016/j.biotechadv.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 38.Thomason LC, Costantino N, Court DL. E. coli genome manipulation by P1 transduction. Curr Protocols Mol Biol. 2007 doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- 39.Mandel M, Higa A. Calcium-dependent bacteriophage DNA infection. J Mol Biol. 1970;53(1):159–162. doi: 10.1016/0022-2836(70)90051-3. [DOI] [PubMed] [Google Scholar]
- 40.Song Y, Hahn T, Thompson IP, Mason TJ, Preston GM, Li G, et al. Ultrasound-mediated DNA transfer for bacteria. Nucleic Acids Res. 2007;35(19):129. doi: 10.1093/nar/gkm710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shark KB, Smith FD, Harpending PR, Rasmussen JL, Sanford JC. Biolistic transformation of a prokaryote, Bacillus megaterium. Appl Environ Microbiol. 1991;57(2):480–485. doi: 10.1128/AEM.57.2.480-485.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawata Y, Yano S, Kojima H. Escherichia coli can be transformed by a liposome-mediated lipofection method. Biosci Biotechnol Biochem. 2003;67(5):1179–1181. doi: 10.1271/bbb.67.1179. [DOI] [PubMed] [Google Scholar]
- 43.Wilharm G, Lepka D, Faber F, Hofmann J, Kerrinnes T, Skiebe E. A simple and rapid method of bacterial transformation. J Microbiol Methods. 2010;80(2):215–216. doi: 10.1016/j.mimet.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 44.Arber W, Linn S. DNA modification and restriction. Annu Rev Biochem. 1969;38:467–500. doi: 10.1146/annurev.bi.38.070169.002343. [DOI] [PubMed] [Google Scholar]
- 45.Vasu K, Nagaraja V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol Mol Biol Rev. 2013;77(1):53. doi: 10.1128/MMBR.00044-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 47.Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018;359(6379):4120. doi: 10.1126/science.aar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernheim A, Sorek R. The pan-immune system of bacteria: antiviral defence as a community resource. Nat Rev Microbiol. 2020;18(2):113–119. doi: 10.1038/s41579-019-0278-2. [DOI] [PubMed] [Google Scholar]
- 49.Mermelstein LD, Papoutsakis ET. In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Applied Env Microbiol. 1993;59(4):1077–1081. doi: 10.1128/AEM.59.4.1077-1081.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riley LA, Ji L, Schmitz RJ, Westpheling J, Guss AM. Rational development of transformation in Clostridium thermocellum ATCC 27405 via complete methylome analysis and evasion of native restriction-modification systems. J Ind Microbiol Biotechnol. 2019 doi: 10.1007/s10295-019-02218-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chung D, Farkas J, Westpheling J. Overcoming restriction as a barrier to DNA transformation in Caldicellulosiruptor species results in efficient marker replacement. Biotechnol Biofuels. 2013;6(1):82. doi: 10.1186/1754-6834-6-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mermelstein LD, Welker NE, Bennett GN, Papoutsakis ET. Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824. Bio/Technology. 1992;10(2):190–195. doi: 10.1038/nbt0292-190. [DOI] [PubMed] [Google Scholar]
- 53.Chung D, Farkas J, Huddleston JR, Olivar E, Westpheling J. Methylation by a unique α-class N4-cytosine methyltransferase is required for DNA transformation of Caldicellulosiruptor bescii DSM6725. PLoS ONE. 2012;7(8):e43844. doi: 10.1371/journal.pone.0043844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang G, Wang W, Deng A, Sun Z, Zhang Y, Liang Y, et al. A mimicking-of-DNA-methylation-patterns pipeline for overcoming the restriction barrier of bacteria. PLoS Genet. 2012;8(9):e1002987. doi: 10.1371/journal.pgen.1002987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Monk IR, Shah IM, Xu M, Tan M-W, Foster TJ. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio. 2012;3(2):e00277-11. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ando T, Xu Q, Torres M, Kusugami K, Israel DA, Blaser MJ. Restriction-modification system differences in Helicobacter pylori are a barrier to interstrain plasmid transfer. Mol Microbiol. 2000;37(5):1052–1065. doi: 10.1046/j.1365-2958.2000.02049.x. [DOI] [PubMed] [Google Scholar]
- 57.Chen Q, Fischer JR, Benoit VM, Dufour NP, Youderian P, Leong JM. In vitro CpG methylation increases the transformation efficiency of Borrelia burgdorferi strains harboring the endogenous linear plasmid lp56. J Bacteriol. 2008;190(24):7885–7891. doi: 10.1128/JB.00324-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cox KL, Baltz RH. Restriction of bacteriophage plaque formation in Streptomyces spp. J Bacteriol. 1984;159(2):499–504. doi: 10.1128/JB.159.2.499-504.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rhoads A, Au KF. PacBio sequencing and its applications. Genom Proteom Bioinform. 2015;13(5):278–289. doi: 10.1016/j.gpb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7(6):461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Urich MA, Nery JR, Lister R, Schmitz RJ, Ecker JR. MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat Protoc. 2015;10(3):475–483. doi: 10.1038/nprot.2014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, et al. Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods. 2017;14(4):411–413. doi: 10.1038/nmeth.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE--a database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015;43(Database issue):D298–9. [DOI] [PMC free article] [PubMed]
- 64.Mohapatra SS, Biondi EG. DNA methylation in prokaryotes: regulation and function. In: Krell T, editor. Cellular ecophysiology of microbe. Cham: Springer International Publishing; 2017. pp. 1–21. [Google Scholar]
- 65.Rachkevych N, Sybirna K, Boyko S, Boretsky Y, Sibirny A. Improving the efficiency of plasmid transformation in Shewanella oneidensis MR-1 by removing ClaI restriction site. J Microbiol Methods. 2014;99:35–37. doi: 10.1016/j.mimet.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 66.Kim JY, Wang Y, Park MS, Ji GE. Improvement of transformation efficiency through in vitro methylation and SacII site mutation of plasmid vector in Bifidobacterium longum MG1. J Microbiol Biotechnol. 2010;20(6):1022–1026. doi: 10.4014/jmb.1003.03014. [DOI] [PubMed] [Google Scholar]
- 67.Johnston CD, Cotton SL, Rittling SR, Starr JR, Borisy GG, Dewhirst FE, et al. Systematic evasion of the restriction-modification barrier in bacteria. Proc Natl Acad Sci. 2019;116(23):11454–11459. doi: 10.1073/pnas.1820256116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yasui K, Kano Y, Tanaka K, Watanabe K, Shimizu-Kadota M, Yoshikawa H, et al. Improvement of bacterial transformation efficiency using plasmid artificial modification. Nucleic Acids Res. 2009;37(1):e3. doi: 10.1093/nar/gkn884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jennert KCB, Tardif C, Young DI, Young M. Gene transfer to Clostridium cellulolyticum ATCC 35319. Microbiology. 2000;146(12):3071–3080. doi: 10.1099/00221287-146-12-3071. [DOI] [PubMed] [Google Scholar]
- 70.Donahue JP, Israel DA, Peek RM, Jr, Blaser MJ, Miller GG. Overcoming the restriction barrier to plasmid transformation of Helicobacter pylori. Mol Microbiol. 2000;37(5):1066–1074. doi: 10.1046/j.1365-2958.2000.02036.x. [DOI] [PubMed] [Google Scholar]
- 71.Matsushima P, Baltz RH. Transformation of Saccharopolyspora spinosa protoplasts with plasmid DNA modified in vitro to avoid host restriction. Microbiology. 1994;140(1):139–143. doi: 10.1099/13500872-140-1-139. [DOI] [Google Scholar]
- 72.Purdy D, O'Keeffe TA, Elmore M, Herbert M, McLeod A, Bokori-Brown M, et al. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol Microbiol. 2002;46(2):439–452. doi: 10.1046/j.1365-2958.2002.03134.x. [DOI] [PubMed] [Google Scholar]
- 73.Mell JC, Redfield RJ. Natural competence and the evolution of DNA uptake specificity. J Bacteriol. 2014;196(8):1471. doi: 10.1128/JB.01293-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guss AM, Olson DG, Caiazza NC, Lynd LR. Dcm methylation is detrimental to plasmid transformation in Clostridium thermocellum. Biotechnol Biofuels. 2012;5(1):30. doi: 10.1186/1754-6834-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kolek J, Sedlar K, Provaznik I, Patakova P. Dam and Dcm methylations prevent gene transfer into Clostridium pasteurianum NRRL B-598: Development of methods for electrotransformation, conjugation, and sonoporation. Biotechnol Biofuels. 2016;9(1):14. doi: 10.1186/s13068-016-0436-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.González-Cerón G, Miranda-Olivares OJ, Servín-González L. Characterization of the methyl-specific restriction system of Streptomyces coelicolor A3(2) and of the role played by laterally acquired nucleases. FEMS Microbiol Lett. 2009;301(1):35–43. doi: 10.1111/j.1574-6968.2009.01790.x. [DOI] [PubMed] [Google Scholar]
- 77.Baker TA, Wickner SH. Genetics and enzymology of DNA replication in Escherichia coli. Ann Rev Genet. 1992;26:447–477. doi: 10.1146/annurev.ge.26.120192.002311. [DOI] [PubMed] [Google Scholar]
- 78.Jain A, Srivastava P. Broad host range plasmids. FEMS Microbiol Lett. 2013;348(2):87–96. doi: 10.1111/1574-6968.12241. [DOI] [PubMed] [Google Scholar]
- 79.Bi C, Su P, Müller J, Yeh Y-C, Chhabra SR, Beller HR, et al. Development of a broad-host synthetic biology toolbox for Ralstonia eutropha and its application to engineering hydrocarbon biofuel production. Microb Cell Fact. 2013;12(1):107. doi: 10.1186/1475-2859-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chung D, Cha M, Farkas J, Westpheling J. Construction of a stable replicating shuttle vector for Caldicellulosiruptor species: Use for extending genetic methodologies to other members of this genus. PLoS ONE. 2013;8(5):e62881-e. doi: 10.1371/journal.pone.0062881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu D, Pakrasi HB. Exploring native genetic elements as plug-in tools for synthetic biology in the cyanobacterium Synechocystis sp. PCC 6803. Microb Cell Fact. 2018;17(1):48. doi: 10.1186/s12934-018-0897-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee S-W, Browning GF, Markham PF. Development of a replicable oriC plasmid for Mycoplasma gallisepticum and Mycoplasma imitans, and gene disruption through homologous recombination in M. gallisepticum. Microbiology. 2008;154(9):2571–2580. doi: 10.1099/mic.0.2008/019208-0. [DOI] [PubMed] [Google Scholar]
- 83.Zakrzewska-Czerwińska J, Majka J, Schrempf H. Minimal requirements of the Streptomyces lividans 66 oriC region and its transcriptional and translational activities. J Bacteriol. 1995;177(16):4765. doi: 10.1128/JB.177.16.4765-4771.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peteranderl R, Shotts EB, Jr, Wiegel J. Stability of antibiotics under growth conditions for thermophilic anaerobes. Appl Environ Microbiol. 1990;56(6):1981–1983. doi: 10.1128/AEM.56.6.1981-1983.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoseki J, Yano T, Koyama Y, Kuramitsu S, Kagamiyama H. Directed evolution of thermostable kanamycin-resistance gene: a convenient selection marker for Thermus thermophilus. J Biochem. 1999;126(5):951–956. doi: 10.1093/oxfordjournals.jbchem.a022539. [DOI] [PubMed] [Google Scholar]
- 86.Vidal L, Pinsach J, Striedner G, Caminal G, Ferrer P. Development of an antibiotic-free plasmid selection system based on glycine auxotrophy for recombinant protein overproduction in Escherichia coli. J Biotechnol. 2008;134(1–2):127–136. doi: 10.1016/j.jbiotec.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 87.Schneider JC, Jenings AF, Mun DM, McGovern PM, Chew LC. Auxotrophic markers pyrF and proC can replace antibiotic markers on protein production plasmids in high-cell-density Pseudomonas fluorescens fermentation. Biotechnol Progress. 2005;21(2):343–348. doi: 10.1021/bp049696g. [DOI] [PubMed] [Google Scholar]
- 88.Heap JT, Pennington OJ, Cartman ST, Minton NP. A modular system for Clostridium shuttle plasmids. J Microbiol Methods. 2009;78(1):79–85. doi: 10.1016/j.mimet.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 89.Wicke N, Radford D, Verrone V, Wipat A, French CE. BacilloFlex: a modular DNA assembly toolkit for Bacillus subtilis synthetic biology. bioRxiv. 2017 doi: 10.1101/185108. [DOI] [Google Scholar]
- 90.Anthony WE, Carr RR, DeLorenzo DM, Campbell TP, Shang Z, Foston M, et al. Development of Rhodococcus opacus as a chassis for lignin valorization and bioproduction of high-value compounds. Biotechnol Biofuels. 2019;12(1):192. doi: 10.1186/s13068-019-1535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Song CW, Lee J, Lee SY. Genome engineering and gene expression control for bacterial strain development. Biotechnol J. 2015;10(1):56–68. doi: 10.1002/biot.201400057. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, Buchholz F, Muyrers JPP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet. 1998;20(2):123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- 93.Reyrat JM, Pelicic V, Gicquel B, Rappuoli R. Counterselectable markers: Untapped tools for bacterial genetics and pathogenesis. Infect Immun. 1998;66(9):4011–4017. doi: 10.1128/IAI.66.9.4011-4017.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Van Zyl WF, Dicks LMT, Deane SM. Development of a novel selection/counter-selection system for chromosomal gene integrations and deletions in lactic acid bacteria. BMC Mol Bio. 2019;20(1):10. doi: 10.1186/s12867-019-0127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang X-Z, Yan X, Cui Z-L, Hong Q, Li S-P. mazF, a novel counter-selectable marker for unmarked chromosomal manipulation in Bacillus subtilis. Nucleic Acids Res. 2006;34(9):e71-e. doi: 10.1093/nar/gkl358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dubeau M-P, Ghinet MG, Jacques P-E, Clermont N, Beaulieu C, Brzezinski R. Cytosine deaminase as a negative selection marker for gene disruption and replacement in the genus Streptomyces and other actinobacteria. Appl Environ Microbiol. 2009;75(4):1211–1214. doi: 10.1128/AEM.02139-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boeke JD, La Croute F, Fink GR. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet. 1984;197(2):345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- 98.Tripathi SA, Olson DG, Argyros DA, Miller BB, Barrett TF, Murphy DM, et al. Development of pyrF-based genetic system for targeted gene deletion in Clostridium thermocellum and creation of a pta mutant. Appl Environ Microbiol. 2010;76(19):6591–6599. doi: 10.1128/AEM.01484-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Galvao TC, de Lorenzo V. Adaptation of the yeast URA3 selection system to gram-negative bacteria and generation of a ∆betCDE Pseudomonas putida strain. Appl Environ Microbiol. 2005;71(2):883–892. doi: 10.1128/AEM.71.2.883-892.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Khasanov FK, Zvingila DJ, Zainullin AA, Prozorov AA, Bashkirov VI. Homologous recombination between plasmid and chromosomal DNA in Bacillus subtilis requires approximately 70 bp of homology. Mol Gen Genet. 1992;234(3):494–497. doi: 10.1007/BF00538711. [DOI] [PubMed] [Google Scholar]
- 101.Gay P, Le Coq D, Steinmetz M, Berkelman T, Kado CI. Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J Bacteriol. 1985;164(2):918–921. doi: 10.1128/JB.164.2.918-921.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fabret C, Ehrlich D, Noirot P. A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol Microbiol. 2002;46(1):25–36. doi: 10.1046/j.1365-2958.2002.03140.x. [DOI] [PubMed] [Google Scholar]
- 103.Pritchett MA, Zhang JK, Metcalf WW. Development of a markerless genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic archaea. Appl Environ Microbiol. 2004;70(3):1425. doi: 10.1128/AEM.70.3.1425-1433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Argyros DA, Tripathi SA, Barrett TF, Rogers SR, Feinberg LF, Olson DG, et al. High ethanol titers from cellulose by using metabolically engineered thermophilic, anaerobic microbes. Appl Environ Microbiol. 2011;77(23):8288–8294. doi: 10.1128/AEM.00646-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Husson RN, James BE, Young RA. Gene replacement and expression of foreign DNA in mycobacteria. J Bacteriol. 1990;172(2):519–524. doi: 10.1128/JB.172.2.519-524.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kast P. pKSS–a second-generation general purpose cloning vector for efficient positive selection of recombinant clones. Gene. 1994;138(1–2):109–114. doi: 10.1016/0378-1119(94)90790-0. [DOI] [PubMed] [Google Scholar]
- 107.van der Geize R, de Jong W, Hessels GI, Grommen AWF, Jacobs AAC, Dijkhuizen L. A novel method to generate unmarked gene deletions in the intracellular pathogen Rhodococcus equi using 5-fluorocytosine conditional lethality. Nucleic Acids Res. 2008;36(22):e151-e. doi: 10.1093/nar/gkn811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ueki T, Inouye S, Inouye M. Positive-negative KG cassettes for construction of multi-gene deletions using a single drug marker. Gene. 1996;183(1–2):153–157. doi: 10.1016/S0378-1119(96)00546-X. [DOI] [PubMed] [Google Scholar]
- 109.Zhang C, She Q, Bi H, Whitaker RJ. The apt/6-methylpurine counterselection system and its applications in genetic studies of the hyperthermophilic archaeon Sulfolobus islandicus. Appl Environ Microbiol. 2016;82(10):3070–3081. doi: 10.1128/AEM.00455-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dean D. A plasmid cloning vector for the direct selection of strains carrying recombinant plasmids. Gene. 1981;15(1):99–102. doi: 10.1016/0378-1119(81)90108-6. [DOI] [PubMed] [Google Scholar]
- 111.Maloy SR, Nunn WD. Selection for loss of tetracycline resistance by Escherichia coli. J Bacteriol. 1981;145(2):1110–1111. doi: 10.1128/JB.145.2.1110-1111.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stacey KA, Simson E. Improved method for the isolation of the thymine-requiring mutants of Escherichia coli. J Bacteriol. 1965;90(2):554–555. doi: 10.1128/JB.90.2.554-555.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bernard P, Gabant P, Bahassi EM, Couturier M. Positive-selection vectors using the F plasmid ccdB killer gene. Gene. 1994;148(1):71–74. doi: 10.1016/0378-1119(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 114.Solem C, Defoor E, Jensen PR, Martinussen J. Plasmid pCS1966, a new selection/counterselection tool for lactic acid bacterium strain construction based on the oroP gene, encoding an orotate transporter from Lactococcus lactis. Appl Environ Microbiol. 2008;74(15):4772. doi: 10.1128/AEM.00134-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shaw AJ, Covalla SF, Hogsett DA, Herring CD. Marker removal system for Thermoanaerobacterium saccharolyticum and development of a markerless ethanologen. Appl Environ Microbiol. 2011;77(7):2534–2536. doi: 10.1128/AEM.01731-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marx CJ, Lidstrom ME. Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques. 2002;33(5):1062–1067. doi: 10.2144/02335rr01. [DOI] [PubMed] [Google Scholar]
- 117.Lee S-H, Kim HJ, Shin Y-A, Kim KH, Lee SJ. Single crossover-mediated markerless genome engineering in Clostridium acetobutylicum. J Microbiol Biotechnol. 2016;26(4):725–729. doi: 10.4014/jmb.1512.12012. [DOI] [PubMed] [Google Scholar]
- 118.Tan X, Liang F, Cai K, Lu X. Application of the FLP/FRT recombination system in cyanobacteria for construction of markerless mutants. Appl Microbiol Biotechnol. 2013;97(14):6373–6382. doi: 10.1007/s00253-013-4837-6. [DOI] [PubMed] [Google Scholar]
- 119.Tian L, Papanek B, Olson DG, Rydzak T, Holwerda EK, Zheng T, et al. Simultaneous achievement of high ethanol yield and titer in Clostridium thermocellum. Biotechnol Biofuels. 2016;9(1):116. doi: 10.1186/s13068-016-0528-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tian L, Conway PM, Cervenka ND, Cui J, Maloney M, Olson DG, et al. Metabolic engineering of Clostridium thermocellum for n-butanol production from cellulose. Biotechnol Biofuels. 2019;12(1):186. doi: 10.1186/s13068-019-1524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Basen M, Geiger I, Henke L, Muller V. A genetic system for the thermophilic acetogenic bacterium Thermoanaerobacter kivui. Appl Enviorn Microbiol. 2018 doi: 10.1128/AEM.02210-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li Z, Xiong B, Liu L, Li S, Xin X, Li Z, et al. Development of an autotrophic fermentation technique for the production of fatty acids using an engineered Ralstonia eutropha cell factory. J Ind Microbiol Biotechnol. 2019;46(6):783–790. doi: 10.1007/s10295-019-02156-8. [DOI] [PubMed] [Google Scholar]
- 123.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97(12):6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci. 2001;98(12):6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Luo X, Yang Y, Ling W, Zhuang H, Li Q, Shang G. Pseudomonas putida KT2440 markerless gene deletion using a combination of λ Red recombineering and Cre/loxP site-specific recombination. FEMS Microbiol Lett. 2016;363(4):fnw014. doi: 10.1093/femsle/fnw014. [DOI] [PubMed] [Google Scholar]
- 126.Sun Z, Deng A, Hu T, Wu J, Sun Q, Bai H, et al. A high efficiency recombineering system with PCR-based ssDNA in Bacillus subtilis mediated by the native phage recombinase GP35. Appl Microbiol Biotechnol. 2015;99:5151–5162. doi: 10.1007/s00253-015-6485-5. [DOI] [PubMed] [Google Scholar]
- 127.van Kessel JC, Hatfull GF. Recombineering in Mycobacterium tuberculosis. Nat Methods. 2007;4(2):147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- 128.Dong H, Tao W, Gong F, Li Y, Zhang Y. A functional recT gene for recombineering of Clostridium. J Bacteriol. 2014;173:65–67. doi: 10.1016/j.jbiotec.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 129.van Pijkeren JP, Britton RA. High efficiency recombineering in lactic acid bacteria. Nucleic Acids Res. 2012;40(10):e76. doi: 10.1093/nar/gks147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Walker JE, Lanahan AA, Zheng T, Toruno C, Lynd LR, Cameron JC, et al. Development of both type I-B and type II CRISPR/Cas genome editing systems in the cellulolytic bacterium Clostridium thermocellum. Metab Eng Commun. 2020;10:e00116. doi: 10.1016/j.mec.2019.e00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Corts AD, Thomason LC, Gill RT, Gralnick JA. A new recombineering system for precise genome-editing in Shewanella oneidensis strain MR-1 using single-stranded oligonucleotides. Sci Rep. 2019;9(1):39. doi: 10.1038/s41598-018-37025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.van Kessel JC, Marinelli LJ, Hatfull GF. Recombineering mycobacteria and their phages. Nat Rev Microbiol. 2008;6(11):851–857. doi: 10.1038/nrmicro2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269(5222):400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- 134.Lampe DJ, Churchill ME, Robertson HM. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 1996;15(19):5470–5479. doi: 10.1002/j.1460-2075.1996.tb00930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Simon R, Priefer U, Pühler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology. 1983;1(9):784. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 136.van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods. 2009;6(10):767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wetmore KM, Price MN, Waters RJ, Lamson JS, He J, Hoover CA, et al. Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. mBio. 2015;6(3):e00306-15. doi: 10.1128/mBio.00306-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Calero P, Jensen SI, Bojanovič K, Lennen RM, Koza A, Nielsen AT. Genome-wide identification of tolerance mechanisms toward p-coumaric acid in Pseudomonas putida. Biotechnol Bioeng. 2018;115(3):762–774. doi: 10.1002/bit.26495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Curtis PD. Essential genes predicted in the genome of Rubrivivax gelatinosus. J Bacteriol. 2016;198(16):2244–2250. doi: 10.1128/JB.00344-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yomano LP, York SW, Zhou S, Shanmugam KT, Ingram LO. Re-engineering Escherichia coli for ethanol production. Biotechnol Lett. 2008;30(12):2097–2103. doi: 10.1007/s10529-008-9821-3. [DOI] [PubMed] [Google Scholar]
- 141.Philipps G, de Vries S, Jennewein S. Development of a metabolic pathway transfer and genomic integration system for the syngas-fermenting bacterium Clostridium ljungdahlii. Biotechnol Biofuels. 2019;12(1):112. doi: 10.1186/s13068-019-1448-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Inaba Y, Banerjee I, Kernan T, Banta S. Transposase-mediated chromosomal integration of exogenous genes in Acidithiobacillus ferrooxidans. Appl Environ Microbiol. 2018;84(21):e01381–e1418. doi: 10.1128/AEM.01381-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang G, Zhao Z, Ke J, Engel Y, Shi Y-M, Robinson D, et al. CRAGE enables rapid activation of biosynthetic gene clusters in undomesticated bacteria. Nat Microbiol. 2019;4(12):2498–2510. doi: 10.1038/s41564-019-0573-8. [DOI] [PubMed] [Google Scholar]
- 144.Choi K-H, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, et al. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2005;2(6):443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 145.Groth AC, Calos MP. Phage integrases: biology and applications. J Mol Biol. 2004;335(3):667–678. doi: 10.1016/j.jmb.2003.09.082. [DOI] [PubMed] [Google Scholar]
- 146.Chou Y-C, Linger J, Yang S, Zhang M. Genetic engineering and improvement of a Zymomonas mobilis for arabinose utilization and its performance on pretreated corn stover hydrolyzate. Journal Biotechnol Biomater. 2015;5(NREL/JA-5100–64639).
- 147.Lambert JM, Bongers RS, Kleerebezem M. Cre-lox-based system for multiple gene deletions and selectable-marker removal in Lactobacillus plantarum. Appl Environ Microbiol. 2007;73(4):1126–1135. doi: 10.1128/AEM.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Langer SJ, Ghafoori AP, Byrd M, Leinwand L. A genetic screen identifies novel non-compatible loxP sites. Nucleic Acids Res. 2002;30(14):3067–3077. doi: 10.1093/nar/gkf421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Brown WRA, Lee NCO, Xu Z, Smith MCM. Serine recombinases as tools for genome engineering. Methods. 2011;53(4):372–379. doi: 10.1016/j.ymeth.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 150.Elmore JR, Furches A, Wolff GN, Gorday K, Guss AM. Development of a high efficiency integration system and promoter library for rapid modification of Pseudomonas putida KT2440. Metab Eng Comm. 2017;5:1–8. doi: 10.1016/j.meteno.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Combes P, Till R, Bee S, Smith MC. The Streptomyces genome contains multiple pseudo-attB sites for the (phi)C31-encoded site-specific recombination system. J Bacteriol. 2002;184(20):5746–5752. doi: 10.1128/JB.184.20.5746-5752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Guss AM, Rother M, Zhang JK, Kulkarni G, Metcalf WW. New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea. 2008;2(3):193–203. doi: 10.1155/2008/534081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Baltz RH. Streptomyces temperate bacteriophage integration systems for stable genetic engineering of actinomycetes (and other organisms) J Ind Microbiol Biotechnol. 2012;39(5):661–672. doi: 10.1007/s10295-011-1069-6. [DOI] [PubMed] [Google Scholar]
- 154.Huang H, Chai CS, Yang S, Jiang WH, Gu Y. Phage serine integrase-mediated genome engineering for efficient expression of chemical biosynthetic pathway in gas-fermenting Clostridium ljungdahlii. Metab Eng. 2019;52:293–302. doi: 10.1016/j.ymben.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 155.Ko B, D’Alessandro J, Douangkeomany L, Stumpf S, deButts A, Blodgett J. Construction of a new integrating vector from actinophage ϕOZJ and its use in multiplex Streptomyces transformation. J Ind Microbiol Biotechnol. 2019 doi: 10.1007/s10295-019-02246-7. [DOI] [PubMed] [Google Scholar]
- 156.Baltz RH. Combinatorial biosynthesis of cyclic lipopeptide antibiotics: a model for synthetic biology to accelerate the evolution of secondary metabolite biosynthetic pathways. ACS Synth Biol. 2014;3(10):748–758. doi: 10.1021/sb3000673. [DOI] [PubMed] [Google Scholar]
- 157.Bilyk B, Luzhetskyy A. Unusual site-specific DNA integration into the highly active pseudo-attB of the Streptomyces albus J1074 genome. Appl Microbiol Biotechnol. 2014;98(11):5095–5104. doi: 10.1007/s00253-014-5605-y. [DOI] [PubMed] [Google Scholar]
- 158.Zhang JJ, Moore BS, Tang X. Engineering Salinispora tropica for heterologous expression of natural product biosynthetic gene clusters. Appl Microbiol Biotechnol. 2018;102(19):8437–8446. doi: 10.1007/s00253-018-9283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jiang Y, Qian F, Yang J, Liu Y, Dong F, Xu C, et al. CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun. 2017;8:15179. doi: 10.1038/ncomms15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 161.Javed MR, Noman M, Shahid M, Ahmed T, Khurshid M, Rashid MH, et al. Current situation of biofuel production and its enhancement by CRISPR/Cas9-mediated genome engineering of microbial cells. Microbiol Res. 2019;219:1–11. doi: 10.1016/j.micres.2018.10.010. [DOI] [PubMed] [Google Scholar]
- 162.Vento JM, Crook N, Beisel CL. Barriers to genome editing with CRISPR in bacteria. J Ind Microbiol Biotechnol. 2019;46(9–10):1327–1341. doi: 10.1007/s10295-019-02195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR–Cas systems. Nat Biotechnol. 2013;31(3):233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pyne ME, Moo-Young M, Chung DA, Chou CP. Coupling the CRISPR/Cas9 system with lambda Red recombineering enables simplified chromosomal gene replacement in Escherichia coli. Appl Environ Microbiol. 2015;81(15):5103–5114. doi: 10.1128/AEM.01248-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Aparicio T, de Lorenzo V, Martínez-García E. CRISPR/Cas9-based counterselection boosts recombineering efficiency in Pseudomonas putida. Biotechnol J. 2018;13(5):e1700161. doi: 10.1002/biot.201700161. [DOI] [PubMed] [Google Scholar]
- 167.Cho S, Choe D, Lee E, Kim SC, Palsson B, Cho BK. High-level dCas9 expression induces abnormal cell morphology in Escherichia coli. ACS Synth Biol. 2018;7(4):1085–1094. doi: 10.1021/acssynbio.7b00462. [DOI] [PubMed] [Google Scholar]
- 168.Wasels F, Jean-Marie J, Collas F, Lopez-Contreras AM, Lopes FN. A two-plasmid inducible CRISPR/Cas9 genome editing tool for Clostridium acetobutylicum. J Microbiol Methods. 2017;140:5–11. doi: 10.1016/j.mimet.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 169.Li K, Cai D, Wang Z, He Z, Chen S. Development of an efficient genome editing tool in Bacillus licheniformis using CRISPR–Cas9 nickase. Appl Environ Microbiol. 2018;84(6):e02608–e2617. doi: 10.1128/AEM.02608-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ran FA, Hsu Patrick D, Lin C-Y, Gootenberg Jonathan S, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zetsche B, Gootenberg Jonathan S, Abudayyeh Omar O, Slaymaker Ian M, Makarova Kira S, Essletzbichler P, et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR–Cas System. Cell. 2015;163(3):759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ungerer J, Pakrasi HB. Cpf1 is a versatile tool for CRISPR genome editing across diverse species of cyanobacteria. Sci Rep. 2016;6(1):1–9. doi: 10.1038/srep39681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pyne ME, Bruder MR, Moo-Young M, Chung DA, Chou CP. Harnessing heterologous and endogenous CRISPR–Cas machineries for efficient markerless genome editing in Clostridium. Sci Rep. 2016;6(1):1–15. doi: 10.1038/s41598-016-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hidalgo-Cantabrana C, Goh YJ, Pan M, Sanozky-Dawes R, Barrangou R. Genome editing using the endogenous type I CRISPR–Cas system in Lactobacillus crispatus. Proc Natl Acad Sci. 2019;116(32):15774–15783. doi: 10.1073/pnas.1905421116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mougiakos I, Bosma EF, de Vos WM, van Kranenburg R, van der Oost J. Next generation prokaryotic engineering: The CRISPR–Cas toolkit. Trends Biotechnol. 2016;34(7):575–587. doi: 10.1016/j.tibtech.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 176.Mougiakos I, Bosma EF, Ganguly J, van der Oost J, van Kranenburg R. Hijacking CRISPR–Cas for high-throughput bacterial metabolic engineering: advances and prospects. Curr Opin Biotechnol. 2018;50:146–157. doi: 10.1016/j.copbio.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 177.Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc. 2013;8(11):2180–2196. doi: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yao L, Cengic I, Anfelt J, Hudson EP. Multiple gene repression in cyanobacteria using CRISPRi. ACS Synth Biol. 2016;5(3):207–212. doi: 10.1021/acssynbio.5b00264. [DOI] [PubMed] [Google Scholar]
- 179.Cleto S, Jensen JVK, Wendisch VF, Lu TK. Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi) ACS Synth Biol. 2016;5(5):375–385. doi: 10.1021/acssynbio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Huang C-H, Shen CR, Li H, Sung L-Y, Wu M-Y, Hu Y-C. CRISPR interference (CRISPRi) for gene regulation and succinate production in cyanobacterium S. elongatus PCC 7942. Microb Cell Fact. 2016;15(1):196. doi: 10.1186/s12934-016-0595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Liu X, Gallay C, Kjos M, Domenech A, Slager J, van Kessel SP, et al. High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol Syst Biol. 2017;13(5):931. doi: 10.15252/msb.20167449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tao W, Lv L, Chen GQ. Engineering Halomonas species TD01 for enhanced polyhydroxyalkanoates synthesis via CRISPRi. Microb Cell Fact. 2017;16(1):48. doi: 10.1186/s12934-017-0655-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Peters JM, Koo B-M, Patino R, Heussler GE, Hearne CC, Qu J, et al. Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nat Microbiol. 2019;4(2):244–250. doi: 10.1038/s41564-018-0327-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR–Cas system. Nucleic Acids Res. 2013;41(15):7429–7437. doi: 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Dong C, Fontana J, Patel A, Carothers JM, Zalatan JG. Synthetic CRISPR–Cas gene activators for transcriptional reprogramming in bacteria. Nat Commun. 2018;9(1):2489. doi: 10.1038/s41467-018-04901-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liu Y, Wan X, Wang B. Engineered CRISPRa enables programmable eukaryote-like gene activation in bacteria. Nat Commun. 2019;10(1):3693. doi: 10.1038/s41467-019-11479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Yang Y, Shen W, Huang J, Li R, Xiao Y, Wei H, et al. Prediction and characterization of promoters and ribosomal binding sites of Zymomonas mobilis in system biology era. Biotechnol Biofuels. 2019;12(1):52. doi: 10.1186/s13068-019-1399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jin L-Q, Jin W-R, Ma Z-C, Shen Q, Cai X, Liu Z-Q, et al. Promoter engineering strategies for the overproduction of valuable metabolites in microbes. Appl Microbiol Biotechnol. 2019;103(21):8725–8736. doi: 10.1007/s00253-019-10172-y. [DOI] [PubMed] [Google Scholar]
- 189.Luo Y, Zhang L, Barton KW, Zhao H. Systematic identification of a panel of strong constitutive promoters from Streptomyces albus. ACS Synth Biol. 2015;4(9):1001–1010. doi: 10.1021/acssynbio.5b00016. [DOI] [PubMed] [Google Scholar]
- 190.Glick BR. Metabolic load and heterologous gene expression. Biotechnol Adv. 1995;13(2):247–261. doi: 10.1016/0734-9750(95)00004-A. [DOI] [PubMed] [Google Scholar]
- 191.Siegl T, Tokovenko B, Myronovskyi M, Luzhetskyy A. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab Eng. 2013;19:98–106. doi: 10.1016/j.ymben.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 192.Rytter JV, Helmark S, Chen J, Lezyk MJ, Solem C, Jensen PR. Synthetic promoter libraries for Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2014;98(6):2617–2623. doi: 10.1007/s00253-013-5481-x. [DOI] [PubMed] [Google Scholar]
- 193.Mordaka PM, Heap JT. Stringency of synthetic promoter sequences in Clostridium revealed and circumvented by tuning promoter library mutation rates. ACS Synth Biol. 2018;7(2):672–681. doi: 10.1021/acssynbio.7b00398. [DOI] [PubMed] [Google Scholar]
- 194.Yang G, Jia D, Jin L, Jiang Y, Wang Y, Jiang W, et al. Rapid generation of universal synthetic promoters for controlled gene expression in both gas-fermenting and saccharolytic Closdridim species. ACS Synth Biol. 2017;6(9):1672–1678. doi: 10.1021/acssynbio.7b00155. [DOI] [PubMed] [Google Scholar]
- 195.Englund E, Liang F, Lindberg P. Evaluation of promoters and ribosome binding sites for biotechnological applications in the unicellular cyanobacterium Synechocystis sp. PCC 6803. Sci Rep. 2016;6(1):36640. doi: 10.1038/srep36640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zobel S, Benedetti I, Eisenbach L, de Lorenzo V, Wierckx N, Blank LM. Tn7-based device for calibrated heterologous gene expression in Pseudomonas putida. ACS Synth Biol. 2015;4(12):1341–1351. doi: 10.1021/acssynbio.5b00058. [DOI] [PubMed] [Google Scholar]
- 197.Ross W, Gosink KK, Salomon J, Igarashi K, Zou C, Ishihama A, et al. A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science. 1993;262(5138):1407. doi: 10.1126/science.8248780. [DOI] [PubMed] [Google Scholar]
- 198.Presnell KV, Flexer-Harrison M, Alper HS. Design and synthesis of synthetic UP elements for modulation of gene expression in Escherichia coli. Synth Syst Biotechnol. 2019;4(2):99–106. doi: 10.1016/j.synbio.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ross W, Aiyar SE, Salomon J, Gourse RL. Escherichia coli promoters with UP elements of different strengths: modular structure of bacterial promoters. J Bacteriol. 1998;180(20):5375–5383. doi: 10.1128/JB.180.20.5375-5383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Phan TTP, Nguyen HD, Schumann W. Development of a strong intracellular expression system for Bacillus subtilis by optimizing promoter elements. J Biotechnol. 2012;157(1):167–172. doi: 10.1016/j.jbiotec.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 201.Zhang B, Zhou N, Liu Y-M, Liu C, Lou C-B, Jiang C-Y, et al. Ribosome binding site libraries and pathway modules for shikimic acid synthesis with Corynebacterium glutamicum. Microb Cell Factor. 2015;14:71. doi: 10.1186/s12934-015-0254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sun T, Miao L, Li Q, Dai G, Lu F, Liu T, et al. Production of lycopene by metabolically-engineered Escherichia coli. Biotechnol Lett. 2014;36(7):1515–1522. doi: 10.1007/s10529-014-1543-0. [DOI] [PubMed] [Google Scholar]
- 203.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27(10):946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Altenbuchner J. Editing of the Bacillus subtilis genome by the CRISPR–Cas9 system. Appl Environ Microbiol. 2016;82(17):5421–5427. doi: 10.1128/AEM.01453-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Morgan S-A, Nadler DC, Yokoo R, Savage DF. Biofuel metabolic engineering with biosensors. Curr Opin Chem Biol. 2016;35:150–158. doi: 10.1016/j.cbpa.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lo T-M, Chng SH, Teo WS, Cho H-S, Chang MW. A two-layer gene circuit for decoupling cell growth from metabolite production. Cell Syst. 2016;3(2):133–143. doi: 10.1016/j.cels.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 207.Chubukov V, Desmarais JJ, Wang G, Chan LJG, Baidoo EE, Petzold CJ, et al. Engineering glucose metabolism of Escherichia coli under nitrogen starvation. NPJ Syst Biol Appl. 2017;3(1):1–7. doi: 10.1038/npjsba.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Burg JM, Cooper CB, Ye Z, Reed BR, Moreb EA, Lynch MD. Large-scale bioprocess competitiveness: the potential of dynamic metabolic control in two-stage fermentations. Curr Opin Chem Eng. 2016;14:121–136. doi: 10.1016/j.coche.2016.09.008. [DOI] [Google Scholar]
- 209.Zhang B, Zhou N, Liu Y-M, Liu C, Lou C-B, Jiang C-Y, et al. Ribosome binding site libraries and pathway modules for shikimic acid synthesis with Corynebacterium glutamicum. Microb Cell Fact. 2015;14:71. doi: 10.1186/s12934-015-0254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rodríguez-García A, Combes P, Pérez-Redondo R, Smith MCA, Smith MCM. Natural and synthetic tetracycline-inducible promoters for use in the antibiotic-producing bacteria Streptomyces. Nucleic Acids Res. 2005;33(9):e87-e. doi: 10.1093/nar/gni086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Girbal L, Mortier-Barrière I, Raynaud F, Rouanet C, Croux C, Soucaille P. Development of a sensitive gene expression reporter system and an inducible promoter-repressor system for Clostridium acetobutylicum. Appl Environ Microbiol. 2003;69(8):4985–4988. doi: 10.1128/AEM.69.8.4985-4988.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ruegg TL, Pereira JH, Chen JC, DeGiovanni A, Novichkov P, Mutalik VK, et al. Jungle Express is a versatile repressor system for tight transcriptional control. Nat Commun. 2018;9(1):1–13. doi: 10.1038/s41467-017-02088-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mearls EB, Olson DG, Herring CD, Lynd LR. Development of a regulatable plasmid-based gene expression system for Clostridium thermocellum. Appl Microbiol Biotechnol. 2015;99(18):7589–7599. doi: 10.1007/s00253-015-6610-5. [DOI] [PubMed] [Google Scholar]
- 214.Nshogozabahizi JC, Aubrey KL, Ross JA, Thakor N. Applications and limitations of regulatory RNA elements in synthetic biology and biotechnology. J Appl Microbiol. 2019;127(4):968–984. doi: 10.1111/jam.14270. [DOI] [PubMed] [Google Scholar]
- 215.Ma AT, Schmidt CM, Golden JW. Regulation of gene expression in diverse cyanobacterial species by using theophylline-responsive riboswitches. Appl Environ Microbiol. 2014;80(21):6704–6713. doi: 10.1128/AEM.01697-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zhou LB, Zeng AP. Exploring lysine riboswitch for metabolic flux control and improvement of L-lysine synthesis in Corynebacterium glutamicum. ACS Synth Biol. 2015;4(6):729–734. doi: 10.1021/sb500332c. [DOI] [PubMed] [Google Scholar]
- 217.Jang S, Jang S, Xiu Y, Kang TJ, Lee S-H, Koffas MAG, et al. Development of artificial riboswitches for monitoring of naringenin in vivo. ACS Synth Biol. 2017;6(11):2077–2085. doi: 10.1021/acssynbio.7b00128. [DOI] [PubMed] [Google Scholar]
- 218.Marcano-Velazquez JG, Lo J, Nag A, Maness P-C, Chou KJ. Developing riboswitch-mediated gene regulatory controls in thermophilic bacteria. ACS Synth Biol. 2019;8(4):633–640. doi: 10.1021/acssynbio.8b00487. [DOI] [PubMed] [Google Scholar]
- 219.Jin DJ, Burgess RR, Richardson JP, Gross CA. Termination efficiency at rho-dependent terminators depends on kinetic coupling between RNA polymerase and rho. Proc Natl Acad Sci USA. 1992;89(4):1453–1457. doi: 10.1073/pnas.89.4.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nordeen SK, Green PP, Fowlkes DM. A rapid, sensitive, and inexpensive assay for chloramphenicol acetyltransferase. DNA. 1987;6(2):173–178. doi: 10.1089/dna.1987.6.173. [DOI] [PubMed] [Google Scholar]
- 221.Dürre P, Kuhn A, Gottwald M, Gottschalk G. Enzymatic investigations on butanol dehydrogenase and butyraldehyde dehydrogenase in extracts of Clostridium acetobutylicum. Appl Microbiol Biotechnol. 1987;26(3):268–272. doi: 10.1007/BF00286322. [DOI] [Google Scholar]
- 222.Brown SD, Guss AM, Karpinets TV, Parks JM, Smolin N, Yang S, et al. Mutant alcohol dehydrogenase leads to improved ethanol tolerance in Clostridium thermocellum. Proc Natl Acad Sci USA. 2011;108(33):13752. doi: 10.1073/pnas.1102444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Platteeuw C, Simons G, De Vos W. Use of the Escherichia coli beta-glucuronidase (gusA) gene as a reporter gene for analyzing promoters in lactic acid bacteria. Appl Environ Microbiol. 1994;60(2):587–593. doi: 10.1128/AEM.60.2.587-593.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Thorn K. Genetically encoded fluorescent tags. Mol Biol Cell. 2017;28(7):848–857. doi: 10.1091/mbc.e16-07-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, Shavit S, et al. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat Methods. 2006;3(8):623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
- 226.Su L, Jia W, Hou C, Lei Y. Microbial biosensors: a review. Biosens Bioelectron. 2011;26(5):1788–1799. doi: 10.1016/j.bios.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 227.Jha RK, Narayanan N, Pandey N, Bingen JM, Kern TL, Johnson CW, et al. Sensor-enabled alleviation of product inhibition in chorismate pyruvate-lyase. ACS Synth Biol. 2019;8(4):775–786. doi: 10.1021/acssynbio.8b00465. [DOI] [PubMed] [Google Scholar]
- 228.Alvarez-Gonzalez G, Dixon N. Genetically encoded biosensors for lignocellulose valorization. Biotechnol Biofuels. 2019;12(1):246. doi: 10.1186/s13068-019-1585-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Charubin K, Bennett RK, Fast AG, Papoutsakis ET. Engineering Clostridium organisms as microbial cell-factories: challenges & opportunities. Metab Eng. 2018;50:173–191. doi: 10.1016/j.ymben.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 230.Streett HE, Kalis KM, Papoutsakis ET. A strongly fluorescing anaerobic reporter and protein-tagging system for Clostridium organisms based on the fluorescence-activating and absorption-shifting tag protein (FAST) Appl Environ Microbiol. 2019;85(14):e00622–e719. doi: 10.1128/AEM.00622-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Monmeyran A, Thomen P, Jonquière H, Sureau F, Li C, Plamont M-A, et al. The inducible chemical-genetic fluorescent marker FAST outperforms classical fluorescent proteins in the quantitative reporting of bacterial biofilm dynamics. Sci Rep. 2018;8(1):10336. doi: 10.1038/s41598-018-28643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.