

Abstract
Brain metastases are the most lethal cancer lesions; 10–30% of all cancers metastasize to the brain, with a median survival of only ~5–20 months, depending on the cancer type. To reduce the brain metastatic tumor burden, gaps in basic and translational knowledge need to be addressed. Major challenges include a paucity of reproducible preclinical models and associated tools. Three-dimensional models of brain metastasis can yield the relevant molecular and phenotypic data used to address these needs when combined with dedicated analysis tools. Moreover, compared to murine models, organ-on-a-chip models of patient tumor cells traversing the blood brain barrier into the brain microenvironment generate results rapidly and are more interpretable with quantitative methods, thus amenable to high throughput testing. Here we describe and demonstrate the use of a novel 3D microfluidic blood brain niche (μmBBN) platform where multiple elements of the niche can be cultured for an extended period (several days), fluorescently imaged by confocal microscopy, and the images reconstructed using an innovative confocal tomography technique; all aimed to understand the development of micro-metastasis and changes to the tumor micro-environment (TME) in a repeatable and quantitative manner. We demonstrate how to fabricate, seed, image, and analyze the cancer cells and TME cellular and humoral components, using this platform. Moreover, we show how artificial intelligence (AI) is used to identify the intrinsic phenotypic differences of cancer cells that are capable of transit through a model μmBBN and to assign them an objective index of brain metastatic potential. The data sets generated by this method can be used to answer basic and translational questions about metastasis, the efficacy of therapeutic strategies, and the role of the TME in both.
Introduction
Brain metastases are the most lethal cancer lesions; 10–30% of all cancers metastasize to the brain, with a median survival of only ~5–20 months, depending on the cancer type1 , 2 . A principal question that arises when studying cancer metastasis is how sub clones migrate from the humoral environment of the bloodstream into an organ such as the brain3 , 4 . This question has led to many variations of migration, invasion, and extravasation assays. All these methods share the critical step of counting or measuring properties of cells that move from one location to another in response to a stimulus. Most migration assays readily available are used to study two-dimensional (2D) migration of cancer cells. These have elucidated a wealth of knowledge; however, they do not recapitulate the three-dimensional nature of the in vivo system that other methods can provide5 . Therefore, it is necessary to study the tumor micro-environment (TME) in three-dimensional (3D) systems, but the analysis approaches available for 3D structures are limited and often inconsistent.
One of the most popular 3D tools is a Boyden chamber that consists of a membrane suspended at the bottom of a well, separating two distinct regions. Boyden introduced the assay to study leukocyte chemotaxis4 . The bottom regions may be varied by chemistry or other means6 , 7 to induce cells in the upper region to migrate to the lower region. The most common approach to quantifying the number of cells that have migrated is to release the cells from the bottom of the membrane using a buffer solution, lyse them, and then count them based on the quantity of DNA content in the solution7 . This indirect approach is prone to operator error due to technique variability and the procedure destroys information about the cancer phenotype and the micro-environment.
Variations of the Boyden chamber assay involve fixation of migratory cells that remain on the membrane, but only provides a count of cells that are no longer viable for continued study6 , 8 , 9 .
Due to limitations of the Boyden chamber and the growth of innovations in the microfluidic community, migration assay chips have been developed which observe the motion of cells in response to a stimulus in one direction rather than three10 , 11 , 12 . These migration assays facilitate control over factors such as flow or single cell separation13 , 14 that enable better interpretation of the results; however, their 2D format inevitably loses some dynamic information. Recent studies have focused on extravasation (i.e., the movement of cells from circulation into a tissue, such as the blood brain barrier) in a 3D environment14 , 15 . The extravasation distance into tissue and probing behavior that occurs at the cellular barrier/membrane is more refined than measurements gleaned using either the Boyden chamber or a 2D microfluidic migration device16 . Thus, devices that enable appropriate imaging and analysis of 3D extravasation are critical to capture these sophisticated measurements but are lacking in the literature.
Independent of migration assays, robust imaging techniques have been developed for magnetic resonance imaging (MRI) and tomography that are able to identify and accurately reconstruct tissue in 3D space17 , 18 . These techniques acquire images in z-stacks and segment portions of the image based on the properties of the tissue and then convert the segmented images into three-dimensional meshes19 , 20 , 21 . This allows physicians to visualize in 3D individual organs, bones, and vessels to aid in surgical planning or aid in diagnosis of cancer or heart disease22 , 23 . Here, we will show that these approaches can be adapted for use on microscopic specimens and 3D extravasation devices.
To this end, we developed the innovative confocal tomography technique, presented herein, which affords flexibility to study the extravasation of tumor cells across a membrane by adapting existing tomography tools. This approach enables the study of the full gamut of cancer cell behaviors as they interact with a cellular barrier, such as an endothelial cell layer. Cancer cells exhibit probing behaviors; some may invade but remain close to the membrane, while others traverse the barrier readily. This technique is capable of yielding information about the phenotype of the cell in all dimensions24 . Using this approach to study the TME is both relatively inexpensive, easy to interpret, and reproducible, when compared to more complex in vivo murine models. The presented methodology should provide a strong basis for the study of many types of tumors and micro-environments by adapting the stromal region.
We describe and demonstrate the use of a 3D microfluidic blood brain niche (μmBBN) platform (Figure 1) where critical elements of the barrier and niche (brain microvascular endothelial cells and astrocytes) can be cultured for an extended period (approximately up to 9 days), fluorescently imaged by confocal microscopy, and the images reconstructed using our confocal tomography technique (Figure 2); all aimed to understand the development of micro-metastasis and changes to the tumor micro-environment in a repeatable and quantitative manner. The blood brain barrier interface with the brain niche is composed of brain microvascular endothelial cells that are strengthened by basement membrane, astrocyte feet, and pericytes25 . We selectively focused on the astrocyte and endothelial components given their importance in the formation and regulation of the blood brain barrier. We demonstrate how to fabricate, seed, image, and analyze the cancer cells and tumor micro-environment cellular and humoral components, using this platform. Finally, we show how machine learning can be used to identify the intrinsic phenotypic differences of cancer cells that are capable of transit through a model μmBBN and to assign them an objective index of brain metastatic potential24 . The data sets generated by this method can be used to answer basic and translational questions about metastasis, therapeutic strategies, and the role of the TME in both.
Figure 1: Experimental workflow.
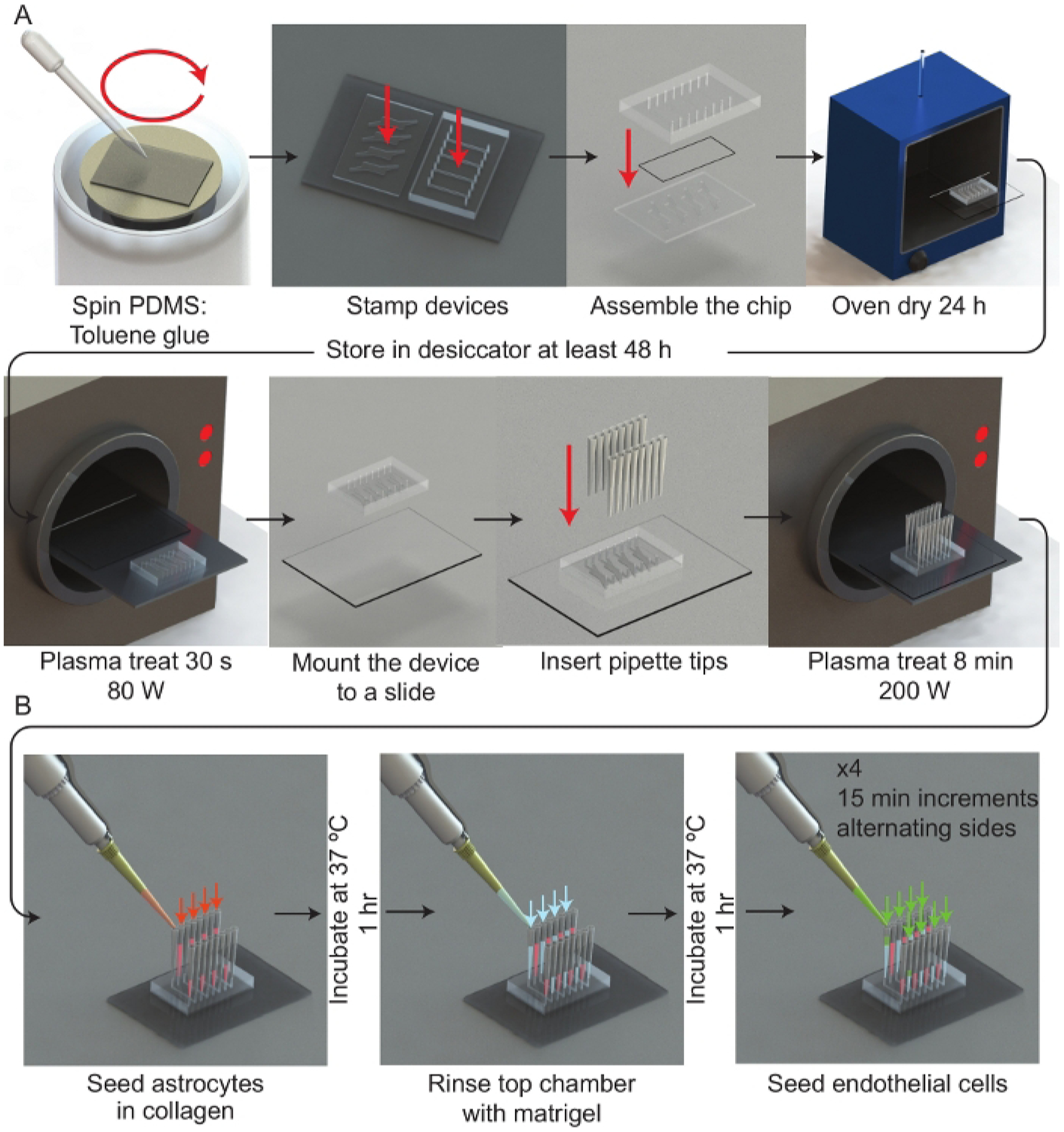
(A) Schematic representation of the microfluidic device assembly process. A spinner is used to deposit a thin film of PDMS:toluene glue onto a 50 mm × 75 mm glass slide. Each half of the μmBBN device is stamped channel side facing the glue and then assembled with a polycarbonate membrane (5 μm pores) between the μmBBN device parts. The μmBBN devices are placed in a 37 °C oven for 24 h to cure the glue. Devices are then dried in a vacuum desiccator for at least 48 h prior to experimental use. A μmBBN device and 50 mm × 75 mm glass slide are activated with a plasma treatment and bonded together. Standard P200 pipettes cut at the tips are inserted into all μmBBN device inlets and outlets. The completed μmBBN device is then sterilized by an 8 min (200 W) plasma treatment then transferred to a sterile secondary container. (B) Schematic overview of utilizing the microfluidic device scaffolding to create a cellular blood brain barrier and brain niche micro-environment. Inside a biosafety cabinet, a mixture of astrocytes in collagen are seeded into the bottom μmBBN device chamber and allowed to solidify for 1 h at 37 °C. Matrigel is used to coat the membrane through the top flow chamber for 1 h at 37 °C. Then endothelial cells are seeded into one tip of the top chamber and allowed to flow and settle for 15 min. This seeding is repeated x4, alternating sides of the flow chamber.
Figure 2: Confocal tomographic analysis overview.
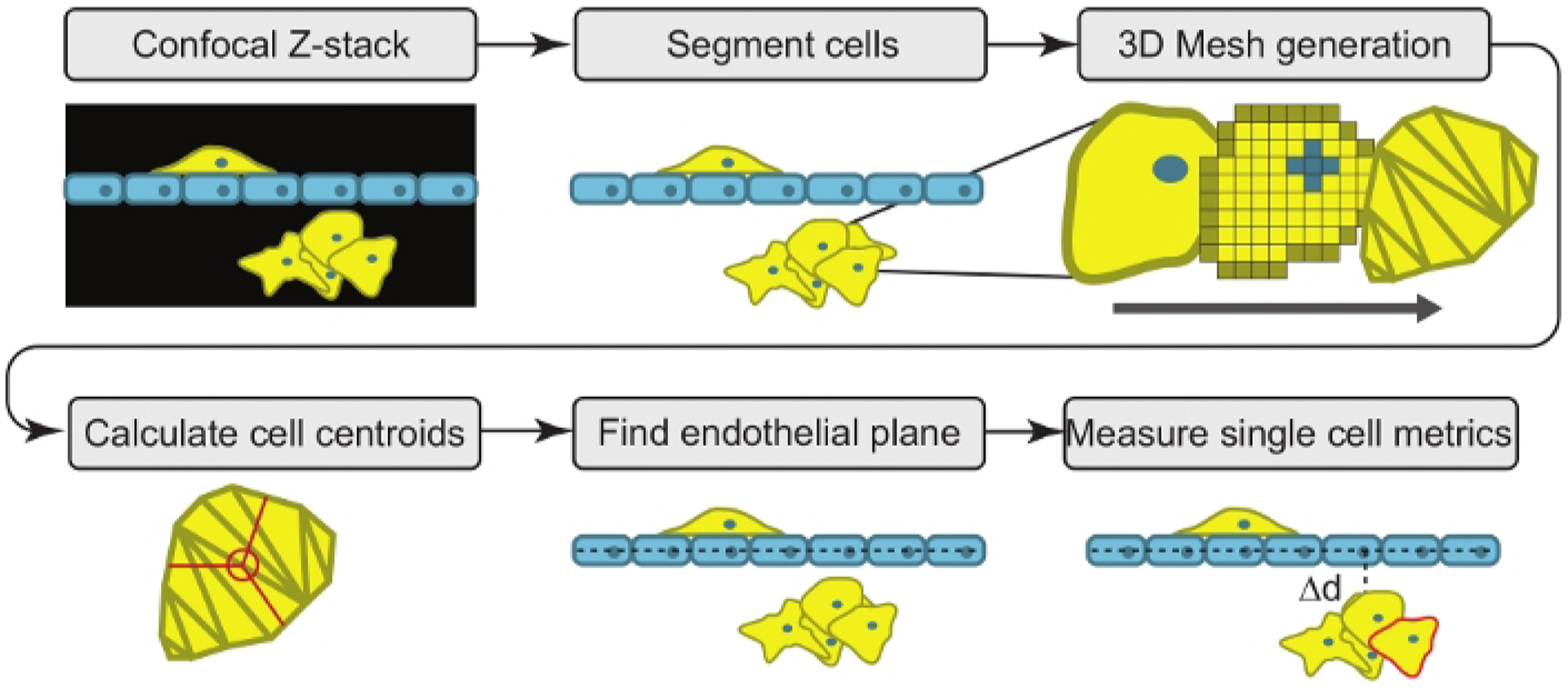
The software begins by converting a microscopic confocal Z-stack image into a 3-D model of the cells using segmentation and 3D meshes. The program then calculates the center position of each cell (centroid) and fits a plane to the endothelial barrier. Phenotypic measurements of each single cell are then tabulated.
Protocol
1. Prepare the blood brain barrier niche mold
NOTE: The culturing device used in this platform is a PDMS based scaffold that we build a cellular blood brain barrier niche upon. It is made of two parts separated by a porous membrane. To prepare the blood brain barrier niche two SU-8 molds made using photolithography are necessary26 , 27 . The protocol will be described for the 100 μm thick mold first and then notes will be given for the 200 μm thick mold.
- To prepare the mold, clean a 4” silicon wafer using acetone with a squeeze bottle and then dry it with a nitrogen gun.
- Bake the silicon wafer on a hotplate for 10 min at 200 °C to remove all residual solvent.
- In turn center the silicon wafer onto the chuck of a spin coater and dispense 1 mL of SU8-2075 for the top mold. Spin for 5 s at 500 rpm (acceleration 300 rpm/s) to disperse the photoresist and then 30 s at 2200 rpm (acceleration 300 rpm/s) to obtain a 100 μm thick SU-8 coating. Optimization may be necessary to achieve the specified thickness.
Soft bake the wafer on a hotplate at 65 °C for 2 min and then immediately at 98 °C for 20 min.
Place the wafer into a photolithography lamp and position the mask centered onto the wafer according to standard procedures. Expose the SU-8 coated wafers with a radiance of 230 mJ/cm2 of UVB (360 nm ± 10 nm). An exposure matrix experiment may be performed to determine the optimum dosage. Mask designs are available in Supplemental File 1.
Perform a post-exposure bake at 65 °C for 2 min and then immediately at 98 °C for 10 min to improve adhesion. Cool the wafer to 50 °C.
Remove the un-exposed resist using SU-8 photo-developer. Rinse the wafer in developer for 5 min in a bath and then use a spray bottle filled with SU-8 developer in a chemical hood to agitate and remove remaining uncured SU-8. A 4x microscope can be used to observe if all the uncured SU-8 has been removed. A white line on the edges of the photoresist features indicates the SU-8 has not been removed fully.
Perform a final hard bake in an oven at 110 °C for 60 min.
- Follow the same procedure for the 200 μm thick mold using SU-8 2075 but adjust the protocol to the following:
- Spin coater settings: Spin for 5 s at 500 rpm (acceleration 300 rpm/s) to disperse the photoresist and then 30 s at 1300 RPM (acceleration 300 rpm/s) to obtain a 200 μm thick coating.
- Soft bake at 65 °C for 2 min then at 98 °C for 40 min.
- Exposure time of 340 mJ/cm2 .
- Post exposure bake: 2 min at 65 °C, and then 98 °C for 15 min.
-
Finally, silanize each wafer by placing them in a vacuum chamber inside of a chemical hood with a plastic container in which 3 drops (~150 μL) of silanizing solution (Trichloro perfluoro octyl silane) have been placed. Pull a vacuum and leave overnight to allow the vapor to coat the wafer. This step reduces adhesion between the SU-8 and PDMS, increasing the life of the mold.
CAUTION: Trichloro perfluoroctyl silane should be always handled in the fume hood and kept away from water sources.
Place individual wafer molds into 150 mm Petri dishes using two strips of double-sided tape. Ensure the wafers are flat. An alternative is to fabricate an aluminum mold within which the wafer can be placed. Because the aluminum mold is enclosed it will produce casts with a uniform thickness, whereas the Petri dish method is sensitive to the tilt of the surface is it placed on. The improved flatness of the PDMS casts reduces downstream confocal imaging time.
2. Form and assemble the PDMS blood brain barrier (BBB) device
Mix 75 g of PDMS at a ratio of 1:10 (Crosslinker:Base) by weight in a plastic cup.
Pour the PDMS over the molds (1 mm thick for the 200 μm thick mold and 4 mm for the 100 μm thick mold) and degas in a vacuum desiccator for one hour or until all the bubbles have been removed. Place in a 65 °C oven overnight. The 1 mm thickness may be adjusted based on the working distance of the 10x objective in the confocal microscope.
After the PDMS has cured use a blade to gently cut against the wafer through the PDMS around the edges. Peel the PDMS off and use a blade to cut along the rectangular guides and a 1.5 mm biopsy punch to open the inlets and outlets on the device. Cover the PDMS device parts with 48 mm wide packing tape to keep it clean from dust and debris.
Next use dissection scissors to cut a 5 mm × 50 mm rectangle of polycarbonate membrane with 5 μm pores and store it inside of a Petri dish for later use.
Gather the following: 200 μL pipette tip, 2 mL of 1:10 PDMS mixed with toluene at a ratio of 2:3 by weight in a glass vial, a Pasteur pipette with a squeeze bulb, the prepared PDMS upper and lower parts, the membrane, three 50 mm × 75 mm glass slides and transport it all to a spin coater. The following steps for assembly and cell seeding of the device are depicted in Figure 1.
Use the Pasteur pipette to transfer 1 mL of PDMS:toluene glue solution into a 50 mm × 75 mm glass slide on the chuck of the spin coater. Spin for 5 seconds at 100 rpm (acceleration 300 rpm/s) and 30 seconds at 2000 rpm (acceleration 300 rpm/s).
Place the slide on a table and cover it with one PDMS upper chamber and one lower chamber so that the PDMS faces with features molded into it contact the slide and the glue is transferred to the PDMS face, outlining the perimeter of all features.
Flip the PDMS upper chamber onto another slide with the glue coating facing up and carefully place the membrane across the device between the inlets and outlets. Place the 200 μL tip in the PDMS:toluene glue solution until it has wicked some into the tip. Touch the tip between each inlet and outlet to place a small drop near the edge of the membrane where it contacts the PDMS.
Remove the other half of the device and place it on with the glue coating down while aligning the inlets and outlets on the two parts.
Place the assembled device into an oven at 37 °C overnight to cure the glue. Transfer to a vacuum bell jar with desiccant and let dehydrate for two days. This is a crucial step to allow the Toluene to evaporate and to improve the consistency of seeding the device by regulating the absorbed water vapor in the laboratory air.
3. Seed the brain micro-environment into the device
- Cell culture and reagents: Before beginning this protocol, obtain the following reagents and cells. Cultivate all cell lines in an incubator set at 37 °C in 5% CO2.
- Maintain human triple-negative breast cancer cell line MDA-MB-231 (ATCC HTB-26) and MDA-MB-231-BR-GFP cells (obtained from Patricia Steeg, PhD) in DMEM with 4.5 g/L glucose, supplemented with 2 mM L-glutamine, 10% FBS and 1x antibiotic-antimycotic. Create MDA-MB-231-GFP fluorescent cells by transducing MDA-MB-231 cell with empty vector pLL-EV-GFP lentivirus. Sort the transduced GFP+ population using fluorescence-activated cell sorting (FACS) prior to experimentation.
- Maintain human brain microvascular endothelial cells hCMEC/D3 in EGM-2 medium. Create hCMEC/D3-DsRed fluorescent cells by transducing hCMEC/D3 with empty vector pLL-3.7-dsRed lentivirus. Remove all non-transduced, non-fluorescent cells from the culture using FACS prior to experimental use.
- Maintain normal human astrocytes (NHA) in DMEM supplemented with 4.5 g/L glucose, 10% FBS, 2 mM Glutamax, 1 mM sodium pyruvate, 1x N-2 growth supplement, and 1x antibiotic-antimycotic. Immortalize the astrocytes by transducing pLOX-TERT-iresTK lentiviral vector (Addgene 12245). Create the vector using plasmid psPAX2 and envelope plasmid pMD2.G (Addgene 12260 and 12259).
Remove a μmBBN device from the vacuum desiccator and place onto a metal or paper (i.e., tape) surface with the inlets facing down and put it into a plasma chamber. Also place a 50 mm × 75 mm glass slide into the plasma chamber. Pull a vacuum and then treat with plasma at 80 W for 30 s.
Quickly remove the glass slide and device from the plasma chamber and place the device with the inlets facing up onto the glass slide aligned using a guide (Supplemental File 2). This will create a permanent bond between the PDMS and glass slide and cannot be re-positioned.
Next cut the tips off 16, 200 μL pipette tips, 2 mm from the tip. Insert the pipette tips into all the inlets and outlets. The device can be placed back into the vacuum desiccator at this point if not ready to seed the cells.
Place the device back in the plasma chamber and treat with plasma for 8 min at 200 W. After the device has cooled (5 min) from the plasma treatment place it inside a sterile secondary container, like a transparent pipette tip box. Perform the next step within ~15 min or the effectiveness of the plasma treatment may be reduced leading to clogging.
Several days before the experiment culture Petri dishes of 1 × 106 endothelial cells (hCMEC/D3-DsRed) and 1 × 106 astrocytes (NHA). While the device is undergoing the 8 min plasma treatment, prepare a collagen solution consisting of 0.5 mL of 3 mg/mL PureCol type I bovine collagen with 64 μL of 0.8 M NaHCO3 and 20 μL of 10x high-glucose (250 mM) MEM. Suspend 5.0 × 105 NHA cells in the collagen solution. Maintain the solution on ice while not in use.
Transfer 120 μL of the collagen/astrocyte/microglia solution into the device through the pipette tip for the bottom chamber (Figure 1 Red arrows). Allow the solution to wick across the chamber into the opposite pipette tip. After all four channels of the device have been filled place the chip in the CO2 incubator at 37 °C and 5% CO2 for 1 h or until the collagen has set.
After the collagen sets, fill all pipette tips feeding the bottom chamber with a mixture of complete media. For chips containing endothelial cells and astrocytes, a 50:50 mix of endothelial:astrocyte media is used.
Coat the upper chamber with 2% growth-factor reduced Matrigel in complete endothelial media using the upper chamber pipette tip (Figure 1 Blue arrows) and place in the incubator for 1 hr.
Rinse the upper chamber with the indicated media mixture and alternate which tip is seeded with endothelial cells (Figure 1 Green arrows). Suspend 1 × 106 endothelial cells in 1 mL of endothelial media and seed 30 μL every 15 min into alternating upper chamber tips for even coverage. Seed endothelial cells into each upper chamber tip twice, for a total of 4 times per chamber.
After the final seeding of endothelial cells, fill all the tips with the media mixture and place the device in the incubator at 37 °C and 5% CO2, changing media in both chambers every 12 h.
4. Monitor progression of the endothelial layer formation
- Complete coverage of the channel by the endothelial layer is observed after 3 days. Use one of two methods to monitor the coverage of the endothelial barrier: Fluorescence or TEER. the hCMEC/D3-DsRed fluorescence and according % coverage across the channel area can be quantified using ImageJ.
- Open the TIFF file in ImageJ representative of the hCMEC/D3-DsRed barrier. In the ImageJ software, click File > Open to select the file.
- Merge all the Z-layers of the image using the maximum intensity following these key commands and options: Image > Stacks > Z project > All Z slices, Maximum intensity.
- Perform a color threshold that is the same across all microfluidic chips assessed using this method. For the study presented we employed a threshold of 450. Use the threshold menu in ImageJ at: Image > Adjust > Threshold.
- Set the measurements to be recorded using the following commands and options: Analyze > Set Measurements > Limit to threshold, Area Fraction.
- Select a representative region of the microfluidic channel to measure by drawing a box. The box tool is located on the main menu of ImageJ. Measure 3 technical replicates positioned at the beginning, middle, and end of each microfluidic channel using the same box size.
- Analyze each channel and record the area fraction, representing the % endothelial coverage. Export these measurements as spreadsheet files to plot and visualize the data using the following commands: Analyze > Measure.
- As an alternative, use Impedance spectroscopy-based TEER to measure endothelial tight junctions per area. Quantification of the endothelial barrier using TEER is a proxy for the integrity of the endothelial layer as a barrier.
- Position two electrodes in the inlet and outlet of the upper and lower chambers.
5. Seed cancer cells into the device
After the endothelial layer has matured, seed cancer cells into the device. Prepare a 1 mL solution of 1 × 106 cancer cells in complete cancer cell media.
Exchange the media in the chip to replenish cell culture nutrients.
Seed each top chamber channel with 30 μL of cancer cells in suspension and then place the device back in the incubator for a 15 min. Always seed the cancer cells on the same side of all four top chamber channels within a single device.
Exchange the device with new media and the refill the tips every 12 hours until the device is imaged for metastatic behavior.
6. Image the tumor micro-environment by confocal imaging
At the desired experimental timepoint (1, 2, or 9-days), use confocal imaging to capture a 3D image of the channel. We perform this step on a Nikon A1 using the settings described here. This step is automated, and each channel requires 20–40 min to image depending on how many fluorescent channels are included and the Z-depth needed to cover the positions of all the cells.
Turn the microscope on, open the software and place the incubator cover onto the microscope.
Set the microscope stage heater to 37 °C and CO2 to 5% if available.
After the microscope incubator has stabilized, place the device into the microscope stage using the 50 mm × 75 mm mount.
Focus on one side of the device (left if to be used with the provided analysis software) with a 10x objective and set the Z height as zero. Under z-stack settings, include a range of 100 μm above and 200 μm below the focus plane. Then use the stitching setting to set the number of X and Y fields to 1 and 9 respectively with 15% overlap. Set the pinhole to its recommended minimum and the z-layer height to 9 μm. Adjust the bright field exposure so that the porous membrane is visible. Turn on the excitation lasers for the dsRed (561 nm) and GFP (488 nm) channels and adjust the fluorescent laser powers and cutoffs so that each channel is visible without overexposing the pixels.
Verify all fields are in focus when stepped over. If so, enter an output file name (001.nd2) for the image and start the experiment to automatically capture the 3D confocal image.
7. Measure the tumor micro-environment via confocal tomography
- Use confocal tomography an approach to estimate a set of metrics and measurements that describe the individual cells and the tumor micro-environment within the device. Confocal tomographic analysis (Figure 2) converts a confocal z-stack into a three-dimensional representation of the cells. Using a custom python script within the Jupyter notebook/lab environment, a plane is then matched to the layer of cells which form the blood brain barrier like membrane30 . Finally, make phenotypic measurements of the cancer cell populations (Table 1).
- Perform this analysis at the end of an experiment or over a time course. Install python and the appropriate libraries according to the referenced software guide, then open the software from Windows Command Prompt by running the command “conda activate”, followed by “jupyter lab”. The Jupyter environment will load within the default browser.
- From the Jupyter file explorer double click the Jupyter notebook “contom.ipynb” to open it. Run the cell below the title Import libraries, custom classes/functions and setup the notebook by clicking the notebook cell and then clicking the play button. All notebook cells below are executed using the same approach. Note that here notebook “cell” refers to a block of python code within the Jupyter notebook.
- Prepare the data. This algorithm uses the visualization toolkit (VTK) to manipulate and display the z-stack and three-dimensional data17 .
- Place the provided .XLSX file (“Experiment Tracker.xlsx”) in the same windows folder as the Jupyter notebook. The file tracks experiments and interfaces with the Jupyter notebook. Place the ND2 file from section 6 into a subfolder called “\Experiment_XXX\Confocal\” below the Jupyter notebook location. Additional experiment folders can be added within by adjusting the “XXX” to the numerical ID assigned to new folders.
-
Label the first experiment folder “Experiment_001” and the ND2 file “001.nd2”. First, convert the ND2 imaging file into a stitched multi-image TIFF file separated by color channel. Do this by executing the notebook cell below the title “Read the confocal z stack into memory” with the Save_tiff_from_ND2 () function uncommented30 . The ND2 file is a proprietary imaging format from Nikon, thus it is necessary to convert it to a format that open source software is compatible with.NOTE: The TIFF (Tag Image File Format) is used because it is ubiquitous, 16 bit compatible, easily imported into VTK, and multiple images can be stored in a single file, which is appropriate for z-stack images. Executing the notebook cell will read in an image from the ND2 file, extract information of the color and XYZ positions then store that image in a numpy array according to a pre-determined structure. It will then save the array as a TIFF file using the python library tifffile.
- Convert imaging data into 3D model
- Import the TIFF file into VTK (vtkTIFFReader) using a 3D rendering to visualize the cells (Figure 2). Select a threshold based on the color of the cells in the image. To clarify, the VTK object represents a block of pixels (X, Y, Z) in space (volume) but only certain pixels (green or red) represents cells, the rest are background or noise (black).
- Therefore, set an opacity filter on the volume which removes the background confirm that what fluorescence remains is only the cells. Do this using the Jupyter notebook cell titled, Change opacity values for each microscope channel by adjusting the Channel_alpha value variables (i.e., GFP_alpha). Visualize the effect using the notebook cell titled View a 3D rendering to verify the threshold are set correctly.
- Save the opacity values in the spreadsheet to use in the next step. Convert the volume data into individual 3D objects, each representing a cell in the image using a technique called marching cubes31 . This algorithm extracts a polygonal mesh of an isosurface from three-dimensional discrete scalar fields of voxels.
- Use the opacity value in the marching cubes algorithm to separate each cell from the background. Complete this step for all fluorescent cells identified in each microscope channel by running Convert voxel image into a triangular mesh and save as a VTK file in the notebook.
- Fitting a plane to the membrane
- Fit a plane to the endothelial barrier by first locating the cell centroids (notebook cell: Analyze the RFP channel (endothelial barrier)). Iterate through the list meshes in the volume and extract the regions that are not connected using a PolyDataConnectivityFilter from VTK. Calculate the centroid of each mesh and add the measurement to a list of centroids filtering for meshes that are too large or too small (<50, >1000000 voxels).
- Fit a plane to the list of centroids for the endothelial cells using a minimization of error method (notebook cell: Fit a plane to the RFP centroids (endothelial barrier)) (Figure 2, Figure 3)32 . Inspect the fit of the plane by plotting the plane and centroids and adjust manually if necessary (using theta, beta and z) by running the notebook cell titled Visualize RFP centroids and plane fit.
- After the plane is properly fitted, save the normal of the plane to the Experiment Tracker File .XLSX file for future use.
-
Analyze the descriptive features of individual cancer cells for the following descriptors (Table 1).
NOTE: If the computer performing the analysis is lagging, high throughput analysis via high performance computing is an option. This algorithm is useful on standard laptops for the analysis of small numbers of cells, however VTK is not well suited to a large number of individual objects (>1000). Therefore, it is optional to use the adapted algorithm to function on a high-performance computing cluster. This enables rapid analysis of experiments with many cells (Figure 2). All of 7.5 is accomplished by running the notebook cells titled Analyze the remaining microscope channels for phenotypic descriptors and Read in the Experiment_tracker information and analyze the channels that exist.- Measure the extravasation of the cancer cells: After characterizing the endothelial layer with a plane, measure the volume of each cancer cell that has migrated through the membrane. Clip each cell (Boolean) so that the mesh below the membrane is kept and the portion above the membrane is removed33 . Then close the open mesh (vtkFillHoles).
- Recalculate the normal and the new centroid of the clipped mesh. Measure the volume and position of each clipped cancer cell for analysis. Volume is equivalent to the number of voxels the mesh fills of each single cell. Calculate the distance between the endothelial plane and position of each cancer cell.
- Measure the cellular phenotype: Calculate the morphology of each cancer cell by factoring its shape, volume and position.
Validate the measurements and save to a spreadsheet or plot. Run the notebook cell titled Check that centroids were measured accurately and a render will pop up showing the centroids identified on top of the imaged volume by cell type. After all the experiments are done export the complete data set as a single spreadsheet file by typing the experiments to be included by ID in the notebook cell titled Export the data as a single Data.XLSX file for the variables experiments replacing the given experiment IDs. If verify the completed data set, plot it using the notebook cell titled Distance extravasated strip plot. The plot will appear in the notebook environment and save to file.
Table 1: List of descriptors by feature type.
The phenotypic characterization of tumor cells is represented using a panel of descriptors. The red box indicates the descriptors that have been used to predict brain metastatic probability via machine learning.
| Feature | Descriptor |
|---|---|
| Tumor cell | % extravasated into the niche |
| Tumor cell | Volume |
| Tumor cell | Sphericity |
| Tumor cell | Distance extravasated |
| Tumor cell | Live cell 2D migration |
| Micro-metastasis | Porosity |
| Micro-metastasis | Stromal interaction |
| Micro-metastasis | Age |
| Micro-metastasis | Growth rate |
| Stromal cell | Volume |
| Stromal cell | Distance from nearby cancer cells |
| Stromal cell | Distance from endothelial barrier |
| Stromal cell | Shape |
Figure 3: Endothelial barrier coverage and plane fitting.
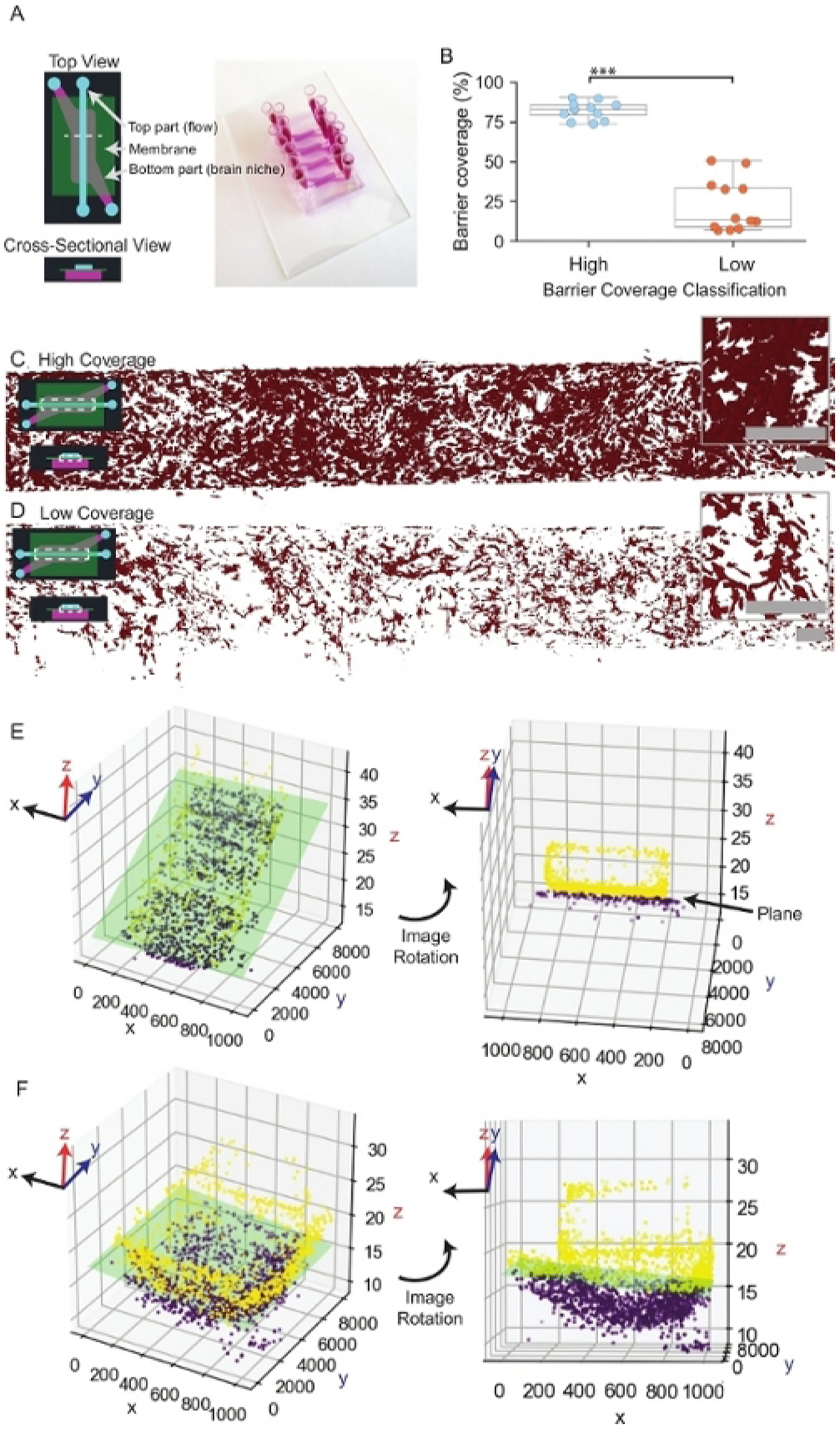
(A) Representative schematic and image of a μmBBN device. The dashed white line within the top view schematic indicates the area of the device represented in the cross-sectional view. (B) Comparison of high and low endothelial coverage of μmBBN devices prior to the application of cancer cells. Welch two-sample t-test, *** p < 0.1*10−4 . (C) Representative image of high endothelial coverage. The dashed white box within the inset schematic of a μmBBN device indicates the location of the endothelial cells within the device. Scale bars of overview and inset images = 200 μm. (D) Representative image of low endothelial coverage. Scale bars of overview and inset images = 200 μm. (E) Example plane fit of a flat endothelial barrier. The green rectangle represents the position of the endothelial plane. Dots represent single endothelial cells comprising the barrier. Yellow dots are endothelial cells above the plane, and purple dots are cells that fall below the plane. Endothelial cells above the plane (yellow dots) exhibit a tendency to grow up the sidewalls and top of the device to form a tube. (F) Example plane fit of a μmBBN device with a curved plane.
8. Analyze the related characteristics using Artificial Intelligence
NOTE: Identify metastatic phenotypic features using artificial intelligence algorithms.
Perform binary classification using Orange according to the scheme shown in Figure 5 and Supplemental File 3. Start Orange from a second Windows Command Prompt by typing “conda activate” followed by “python -m Orange.canvas” and click New from the prompt. Orange is a drag and drop based software, so arrange the functions by dragging each item from the left menu onto the canvas to match Supplemental File 3. After that is complete double click the File icon and select the Data.XLSX file.
- Filter the data to remove bad measurements, defined as those that failed a Boolean operation or giving parametric variable values outside of known bounds using the “Select Rows” icon. Double click the icon and set conditions that correspond to the filter such as “Sphericity is between 0 and 1”. Create conditions for 8.2.1, 8.2.2 and 8.2.3.
- Filter distance extravasated measurements to range from −100–200 μm.
- Filter sphericity measurements of cell shape to range from 0–1.
- Filter cancer cell volume measurements to range from 0–2000 voxels.
- Use all parametric variables (Table 1) to classify brain-metastatic MDA-MB-231-BR-GFP and non-brain metastatic MDA-MB-231-GFP. Double click Select Columns icon from the canvas. Using the > button to move Available Variables into either Features, Target Variables, or Meta Attributes. The only variable that should be a Target Variable is a Metastatic label which defines if cells in the data set are considered metastatic (1) or not (0). Experiment variables can be placed in the Meta Attributes section.
Sample the data into a training (80%) and test set (20%). Double click the Data sampler icon and select a Sampling type of Fixed proportion of data: set to 80% and select replicable and stratify check boxes. Stratify and cross-validate the training set using 10 folds against each model/classifier. Double click Test & Score and select Cross validation with Number of folds: set to 10 and Stratified checked. Set the target class to 1.
In this method, use Neural Networks and Random Forest learning algorithms as they are robust to the data. Within the Random Forest icon, choose the Number of Trees to be 50 and do not select any other options. Within the Neural Network icon choose Neurons per hidden layer: 100, Activation: ReLu, Solver: Adam, Alpha: 0.0001, and Max Iterations: 200. However, these settings will vary significantly by study and should be well understood before application.
After setting up the canvas double click each icon from File to Sample Data and either hit Apply or Send Data. Double click Test & Score and the training data will begin to be used to develop a model using the algorithms. After training the machine learning model, re-open Test & Score and select Test on Test Data and close the popup window to score the performance of the model by classifying the cells in the chip according to the probability they are brain metastatic from 0 to 1.
Save the machine learning model performance to a file (Table 2). Include the area under the curve (AUC) of the ROC, the accuracy and the F1 score. Save a second file that contains the individual metastatic indices and classification probabilities. Double click the Save icon and click Save As to write to file the classification probabilities. Similarly, the ROC curve can be viewed by double clicking ROC Analysis icon and the model performance can be calculated by double clicking the Confusion Matrix icon.
Figure 5: Representative results of machine learning classification of cancer cells.
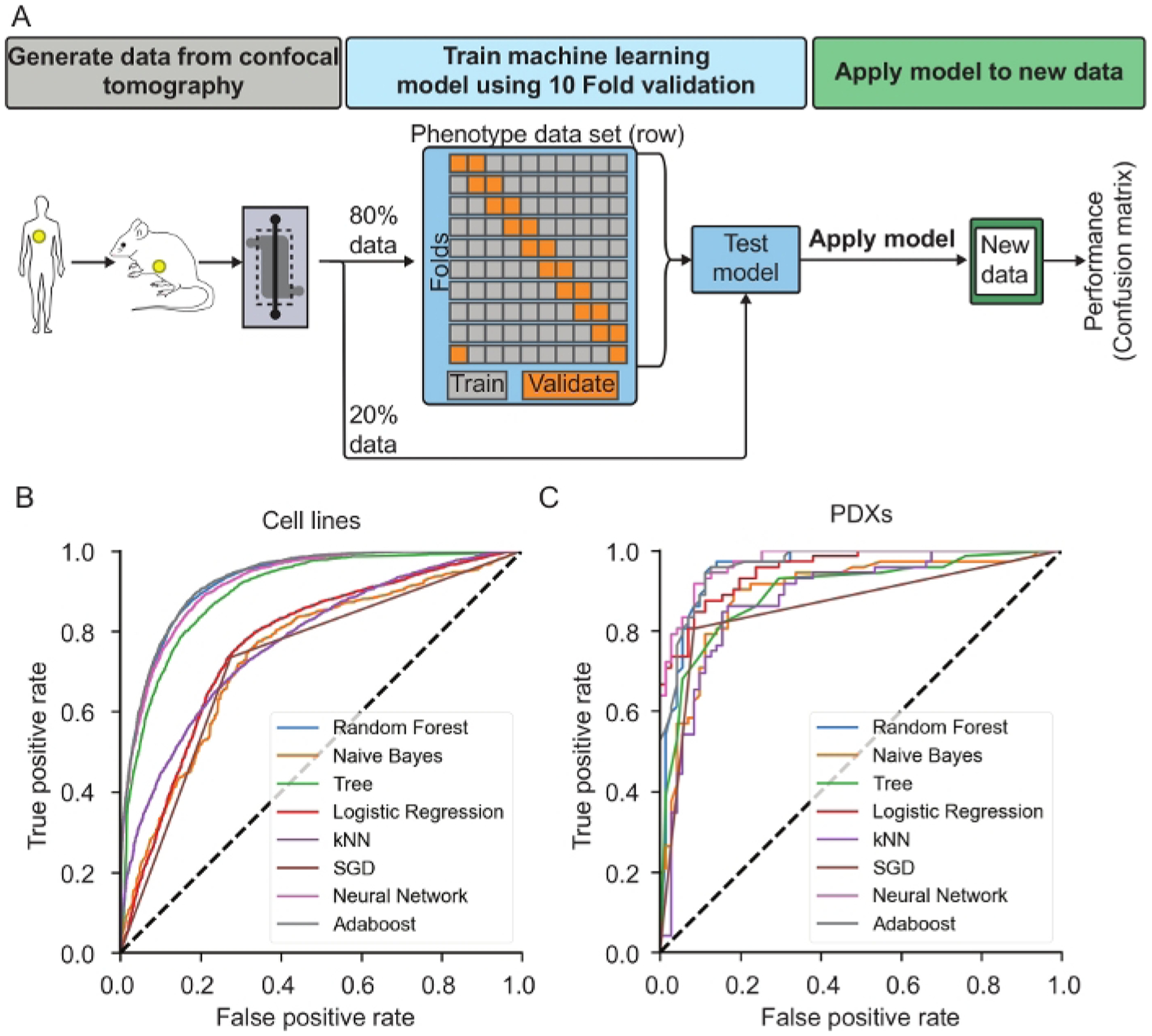
(A) Machine learning overview. Demonstrates process of splitting data collected from confocal tomography, filtering the data, training the machine learning algorithm using 10-fold validation and then testing the model against a random sample of 20% of the data the was reserved. The selected model can then be applied on new data to collect the metastatic index of individual cells. (B) ROC curves showing the performance of 8 different machine learning algorithms for MDA-231-BR-GFP and MDA-231-GFP cells culture for 1, 2 and 9-Days before imaging. This is representative of the type of curve to be analyzed to understand the performance of the trained model. (C) ROC curves for 8 different machine learning algorithms applied to patient derived xenograft (PDX) dissociated cells cultured for 2-Days. This is representative of the type of curve to be analyzed to understand the performance of the trained model. Reproduced from reference24 with permission from the Royal Society of Chemistry.
Table 2:
Comparison of machine learning methods to classify cancer cells and PDX cancer cells by brain metastatic potential.
| Cancer cells | |||
|---|---|---|---|
| Method | AUC | Accuracy | F1 |
| Neural Network | 0.925 | 0.84 | 0.847 |
| AdaBoost | 0.928 | 0.853 | 0.86 |
| Random Forest | 0.925 | 0.849 | 0.855 |
| Decision Tree | 0.898 | 0.817 | 0.827 |
| kNN | 0.775 | 0.702 | 0.718 |
| Logistic Regression | 0.769 | 0.735 | 0.751 |
| Naive Bayes | 0.745 | 0.715 | 0.73 |
| SGD | 0.73 | 0.73 | 0.737 |
| PDX Cancer cells | |||
| Method | AUC | CA | F1 |
| Neural Network | 0.972 | 0.881 | 0.878 |
| Random Forest | 0.964 | 0.888 | 0.887 |
| AdaBoost | 0.957 | 0.881 | 0.879 |
| Tree | 0.954 | 0.867 | 0.865 |
| Logistic Regression | 0.897 | 0.832 | 0.831 |
| Naive Bayes | 0.896 | 0.846 | 0.849 |
| kNN | 0.882 | 0.818 | 0.814 |
| SGD | 0.861 | 0.86 | 0.853 |
Representative Results
Using this technique, we analyzed cell types labeled with different fluorescent proteins or dyes. We demonstrate the use of this approach with a μmBBN chip formulated with hCMEC/D3-DsRed and non-fluorescent astrocytes. The brain microvascular endothelial cells were seeded onto a porous membrane (5 μm track etched pores) and placed in an incubator34 at 37 °C under 5% CO2. After three days the confluency of the endothelial layer was confirmed via microscopy and then cancer cells were placed on top of the endothelial layer. Two breast cancer cell lines, a brain-seeking clone termed MDA-MB-231-BR-GFP, and parental MDA-MB-231-GFP cells were input into the μmBBN chip containing an astrocytic brain niche35 , 36 , 37 , 38 , 39 , 40 , 41 . The μmBBN chips were imaged at 1, 2, and 9-Days using a confocal microscope with 3 channels (dsRed, GFP and Brightfield) using a 10x objective, with Z-slices every 9 μm and 1×9 XY stitching to cover the entire membrane area. This microscope maintained a temperature of 37 °C during the imaging process to minimize stress on the cells.
A representative image of a complete μmBBN chip (Figure 3A) and endothelial coverage (Figure 3B–D) is shown. Endothelial barriers with high and low coverage are quantified to ascertain μmBBN chips are suitable for the addition of cancer cells (Figure 3B). Representative images of a μmBBN chip with a confluent endothelial barrier that is acceptable for experimentation (Figure 3C) and a μmBBN chip with especially poor endothelial coverage that is not suitable for the addition of cancer cells are provided (Figure 3D). Long-term culturing of endothelial cells can result in coverage that spans beyond the membrane separating the top and bottom μmBBN chip chambers. Endothelial cells often grow both on top of and directly beneath the membrane, which does not affect cancer cell movement into the brain niche space but can make the plane fit difficult. Due to the variability of the μmBBN chip fabrication and endothelial coverage of the membrane, representative planes fit to a normal flat endothelial barrier (Figure 3E), and an atypical curved membrane are displayed for reference (Figure 3F). Drying the device in an oven at a temperature above 40 °C may cause a curved membrane as shown.
We observed phenotypic differences of the MDA-MB-231-BR-GFP and parental MDA-MB-231-GFP cell line when encountering astrocytic μmBBN that were quantified using the developed confocal tomographic analysis. The 4 phenotypic descriptors used for input into the machine learning algorithm (Table 1) are displayed in Figure 4.
Figure 4: Quantification of cellular phenotypes of brain-metastatic and parental cell phenotypes in astrocytic blood brain niche microfluidic chips.
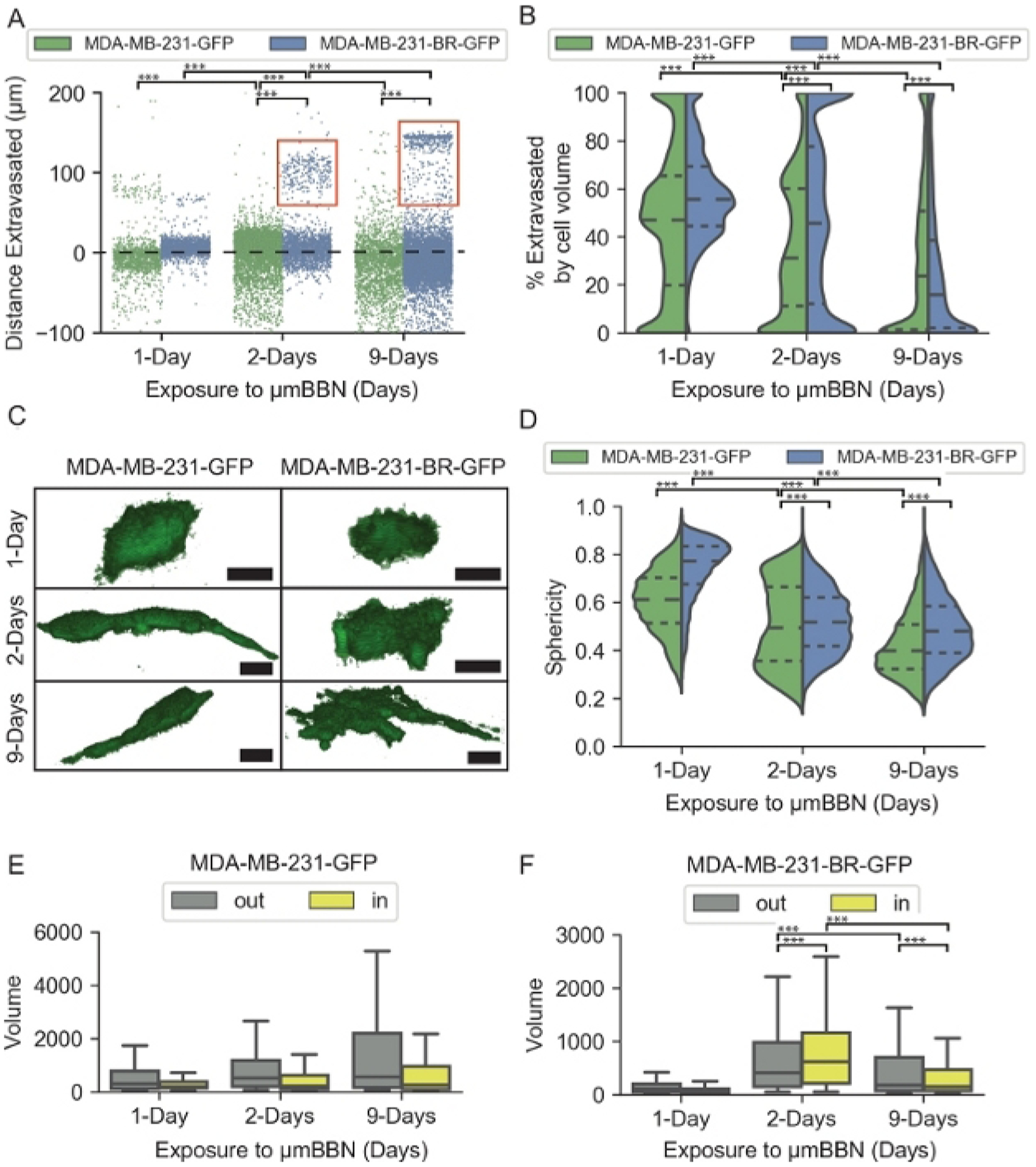
(A) Strip plot of distance in μm of cancer cells from the endothelial barrier at 1, 2, and 9-Days. The dashed black line at 0 μm represents the endothelial barrier. Red boxes indicate a subset of MDA-MB-231-BR-GFP cells that migrated far into the brain niche. (B) Violin plot of the percent total volume of cancer cells extravasated through the endothelial barrier at 1, 2, and 9-Days. Short dashed lines represent quartiles, longer dashed line represents the mean. (C) Representative images of the morphology of cancer cells in μmBBN device. Scale bar = 25 μm. (D) Violin plot of the sphericity of cancer cells in μmBBN device at 1, 2, and 9-Days. Sphericity ranges from 1: spherical to 0: not spherical. (E) Box plot of MDA-MB-231-GFP cell volume in the μmBBN device in voxels for cells resting outside the endothelial barrier (out) and cells that extravasated through the barrier (in). (F) Box plot of MDA-MB-231-BR-GFP cell volume in the μmBBN device in voxels for cells resting outside the endothelial barrier (out) and cells that extravasated through the barrier (in). The box displays the quartiles and whiskers extend to show the proportion of the interquartile range past the low and high quartiles. Pairwise Wilcoxon Rank Sum and Kruskal-Wallis with Dunn’s multiple comparisons, *** p < 0.1*10−4 . Reproduced from reference24 with permission from the Royal Society of Chemistry.
Distance extravasated represents the distance in μm between the endothelial barrier and each cancer cell position in the μmBBN chip (Figure 4A). The endothelial barrier is located at 0 μm. Distances <0 μm represent cancer cells that remained in the flow chamber. Distances >0 μm indicate cancer cells that have extravasated through the endothelial barrier and entered the brain niche space. After 1-day of exposure to the μmBBN both MDA-MB-231-BR-GFP and MDA-MB-231-GFP were positioned within the endothelial barrier. However, after 2 and 9-Days of interaction a subset of the MDA-MB-231-BR-GFP migrated >100 μm into the astrocytic brain niche, whereas the parental MDA-MB-231-GFP cells remained in proximity to the endothelial barrier. We resolved the cancer cell positions at the endothelial barrier/brain niche interface by calculating the volume of each cancer cell that has passed through the endothelial barrier. The resulting percentage represents cancer cell extravasated by volume (Figure 4B). A 0% extravasated by cell volume indicates cancer cells that remain on top of the endothelial barrier, ranging to 100% when a cancer cell has completely extravasated through the endothelial barrier and resides in the brain niche space. The parental MDA-MB-231-GFP cells initially interact with the endothelial barrier after 1-Day of exposure to the μmBBN chip that were <50% extravasated by volume. At 2, and 9-Days the MDA-MB-231-GFP cells maintain a substantial proportion of cells that remained on top of the barrier away from the brain niche space. The MDA-MB-231-BR-GFP cells maintained a proportion of cells that were >100% extravasated, especially at the 2-Day timepoint.
The cancer cell shape changed dramatically over the duration of exposure to the μmBBN. Representative images of the cancer cells at each timepoint are depicted in Figure 4C. Morphological quantification of the cancer cell shape was calculated using sphericity, that accounts for cell volume and surface area (Figure 4D). The sphericity metric ranges from 0–1, with 1 representing a perfect sphere. Both MDA-MB-231-BR-GFP and MDA-MB-231-GFP cells were highly spherical 1-day post seeding into the μmBBN chip. After 2- and 9-days of interaction in the μmBBN chip, both cancer cell lines trended to decrease their spherical in shape, albeit at different rates. In addition to cancer cell shape, the volume in voxels of each cancer cell is also quantified using the confocal tomographic analysis. Each cancer cell line was stratified into two groups in Figure 4E–F: “out”, representing the cancer cells that transited through the endothelial barrier (>90% extravasated through the barrier) and “in”, the population of cells that interact with the endothelial barrier but do not extravasate through (<90% extravasated). The cancer cell subpopulations that extravasated into the astrocytic niche were smaller in size compared to the cancer cells that remained in interaction with the endothelial barrier but did not fully extravasate through to the brain.
The brain-seeking MDA-MB-231-BR-GFP revealed a phenotypic pattern in the μmBBN chips distinct from the parental MDA-MB-231-GFP that can be exploited to differentiate between brain-metastatic and non-brain metastatic cancer cells using machine learning. The data was randomly separated into training and validation datasets to train the model and perform validation tests. To train the model, a total of 38,859 cells were used after filtering the data and the model was tested against 9,714 individual cells. The trained model was applied to the cancer cell lines and PDX-generated cancer cells that were analyzed in astrocytic μmBBN chips (4 isolated from patient brain metastases of a variety of primary tumor types, and 1 primary breast cancer tumor) to generate an index of brain metastatic probability (Figure 5). Eight different machine learning classification methods were tested: Naïve bayes, random forest, decision tree, k-nearest-neighbor (kNN), stochastic gradient descent, neural network, and Adaboost. The result of each method is shown in Table 2. Neural network and Adaboost were the 2 best performing classification methods that are recommended for use with data generated using the μmBBN platform with an AUC of 0.920 and 0.928, respectively. Moreover, they showed an accuracy of 0.833 and 0.853. The average of precision and recall (F1) for the Neural network and Adaboost methods were 0.847 and 0.860. From previous work where we applied this approach to PDX samples of non-metastatic breast tumor and known metastatic samples (breast, lung, ovarian, tongue) we found that the same approach applied to the PDX samples enabled accurate identification of the metastatic cells from the non-metastatic ones. Table 2 shows the results for each machine learning algorithm applied to the PDX data wherein the same methods proved to be the most robust (Neural network and Adaboost (Random Forest). Of the 143 cells used in the testing set for the PDX samples, 71 were non-metastatic, 46 were metastatic breast, 11 were metastatic tongue, 13 were metastatic lung and 2 were metastatic ovarian. Each cell type produced an overall accuracy of 0.88 but individual had the following approximate accuracies: non-metastatic breast: 0.96, metastatic breast: 0.80, metastatic tongue: 0.80, metastatic lung: 0.92, metastatic ovarian: 1.0
Discussion
We have developed and presented a new method that adapts tools often utilized in clinical imaging analyses for measurement of extravasation and migration of cancer cells through an endothelial barrier into brain tissue. We pose this approach can be useful for both in vivo and in vitro measurements; we have demonstrated its use on a 3D microfluidic system recapitulating brain vasculature. Cancer cell measurements including distance extravasated, percent extravasated by volume, sphericity, and volume are quantified using this technique. Distance extravasated and percent extravasated by volume permit the user to reconstruct the position of the cancer cells within the chip to assess extravasation across the barrier and migration within the tissue. Cell shape measurements such as sphericity and volume are related to the dynamic movements or function of the cell at each timepoint. Migratory MDA-MB-231-BR-GFP cells exhibit a high level of sphericity when initially introduced into the μmBBN device and become less spherical in shape as they decrease their movement and begin to colonize the niche. Overall cell volume of the MDA-MB-231-GFP and MDA-MB-231-BR-GFP differed due to the difference in shape of the cell lines. Cancer cells that cross the endothelial barrier are more rounded than the cells that do not traverse through the barrier, thus smaller round cells may be able to extravasate across the endothelial layer more efficiently.
Two critical steps exist within the protocol that facilitate success. The first occurs during the assembly of the μmBBN device upper and lower parts that are separated by the porous membrane. Upper and lower device parts must be mated so that the inlets and outlets overlap but are not occluded by the membrane in between to promote proper flow. All μmBBN devices with poor mating of the upper and lower device parts or membrane alignment are discarded to minimize fluctuations in quality. Subsequent baking to cure the PDMS:toluene glue to assemble the device is critical to perform at 37 °C to produce μmBBN devices with membranes that are flat. Baking temperatures that exceed 37 °C tend to produce devices with a curved membrane that makes image analysis difficult, especially when fitting a plane to the endothelial barrier. The second critical step happens during the seeding of the endothelial barrier. Seedings should occur at a minimum of fifteen minute intervals between the two inlets feeding the top part of the device to ensure random distribution of the endothelial cells onto the membrane of the device. Seeding of the collagen mixture containing astrocytes into the chip may require troubleshooting and modification to the lab-specific protocol. If the membrane surface of the device is not hydrophobic, the collagen will fill both the bottom and top parts of the device and set so that it clogs all flow through the top part of the device. The purpose of the final plasma gas treatment is to make the device surface hydrophobic. In this case, we recommend adjustment of the plasma gas treatment of the device to increase hydrophobicity.
We have observed some limitations with this approach. For example, the approach used to induce cell fluorescence can impact the imaging quality. When using live cell tracking dyes, the fluorescent pattern is made of small spots, while transfected or transduced expression of fluorophores produces a uniform pattern. The spotty pattern requires additional and sometimes error prone clustering of pixels. Additionally, the sensitivity of the measurement is dependent on the care taken during imaging. Higher resolution images and more z slices improves resolution, but also takes more time to image and analyze. Cells that are touching can also be incorrectly analyzed as a large single cell. This is a problem for many automated imaging systems but can be addressed in two ways. The first is that the endothelial layer has many cells touching, but their combination has negligible impact on the final location of the cutting plane. The second is the number of cancer cells is low enough that it is rare they are touching. Errors introduced when fitting the plane can occur for several reasons. The first is that some endothelial cells may have migrated away from the central membrane during cell culture. This can result in endothelial cells coating the entire channel or invading the collagen filled space, thereby skewing the measurement. Another source of error is, paradoxically, the manual adjusting of the plane to fix the previous error. However, repeatability and reproducibility studies have found this to have minimal impact. Finally, we observed that under certain circumstances the Boolean cutting of the cell mesh may fail or the algorithm to close the cut mesh may also fail. The techniques used here are the current “state of the art”, and these problems are currently being addressed by algorithmic scientists.
The results from training the machine learning algorithms (AI) against data collected by confocal tomography of the μmBBN demonstrate that the considerable number of individual cells analyzed by this approach may help address issues of capturing heterogeneity found in cancer cells. An AUC greater than 0.9 is considered a high performing classifier. Here we demonstrated an AUC of 0.928. We expect that as the method is improved upon performance will continue to increase. As with all AI methods, care must be taken to select the training data set carefully so that it represents broadly the type of data expected to be tested against. For this reason, we may expect that the performance would degrade, if the model were applied directly to patient samples for example without first exposing the model to a robust collection of patient samples. We demonstrate this here to some extent by including 1-Day, 2-Day, and 9-Day measurements for the cancer cells in the model compared to the 2-Day day used for the previous work. We observe that the broad sampling reduces the performance of the model slightly and shows how sensitive some of the poorer performing methods may be, thus suggesting that users may want to test several models on their data. Table 2 describing PDX results shows an overall good performance. However, individual PDX types demonstrate that the proportion of cells measured from each sample differ and may influence the performance for each site of origin. For example, the ovarian sample only produced two cells for the testing set. In contrast, metastatic breast cancer which had a larger population. This data set was intended to demonstrate that the cells survive in the niche and can be analyzed, but also highlights interpretive care is needed. Alterations to the target variable may also guide researchers in identifying sub-clones with other traits such as interactions with stromal cells or quiescent behavior.
This approach is important as more labs adopt membrane on-a-chip systems such as blood brain barrier on a chip, lung on a chip and gut on a chip11 , 42 . The majority of these chips are assembled so that the membrane is parallel with the imaging system, which until now it has meant that it would be difficult to measure when and how many cells have moved from one side of the membrane to the other15 , 28 . Moreover, when compared to horizontally oriented chips, vertically oriented chips provide a much larger membrane area increasing the dynamic range of the experiment. In addition, because orientation of the membrane is not critical for this analysis technique, confocal tomography could potentially enable new in vitro experiments. For instance, imaging the extravasation of breast cancer cells from the murine fat pad into the blood stream would be approachable by these methods. This could help researchers identify how the cancer cells probe the interface between the breast ECM and vessel.
In conclusion, we presented a methodology to construct a 3D microfluidic device that recapitulates the blood brain niche and demonstrate how to use confocal tomography and machine learning for analysis. Using this platform, we identified brain-metastatic cancer cell characteristics that differentiate between brain metastatic and non-metastatic PDX cancer cells based on their behavior within the μmBBN device. Future work will improve the clinical applicability of this platform as a diagnostic towards predicting brain metastases. We believe that having presented this platform will be useful and interesting to labs which need to measure cells of any type migrating or extravasating across a membrane. This is of importance as pre-clinical models of the tumor micro-environment become increasingly sophisticated so to must the engineering of the models and supporting software. To aid in robust analysis of the data we have packaged this tool as a simple to use, shared python notebook with installation instructions30 .
Supplementary Material
Acknowledgments
We thank the Steeg Lab, at the National Cancer Institute for the generous donation of MDA-MB-231-BR-GFP cells. Confocal microscopy was performed at the University of Michigan Biointerfaces Institute (BI). Flow cytometry was performed at the University of Michigan Flow Cytometry Core. Viral vectors were created by the University of Michigan Vector Core. We also thank Kelley Kidwell for guidance in statistical analysis of these data.
FUNDING:
C.R.O. was partially supported by an NIH T-32 Training Fellowship (T32CA009676) and 1R21CA245597-01. T.M.W. was partially supported by 1R21CA245597-01 and the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR002240. Funding for materials and characterization was provided by National Cancer Institute of the National Institutes of Health under award number 1R21CA245597-01, P30CA046592, 5T32CA009676-23, CA196018, AI116482, METAvivor Foundation, and the Breast Cancer Research Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
Footnotes
Disclosures
There are no disclosures to declare.
References
- 1.Rastogi K et al. Palliation of Brain Metastases: Analysis of Prognostic Factors Affecting Overall Survival. Indian Journal of Palliative Care. 24 (3), 308–312 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang R et al. The Clinicopathological features and survival outcomes of patients with different metastatic sites in stage IV breast cancer. BMC Cancer. 19 (1), 1091 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valster A et al. Cell migration and invasion assays. Methods. 37 (2), 208–215 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Eccles SA, Box C, Court W Cell migration/invasion assays and their application in cancer drug discovery. Biotechnology Annual Review. 11, 391–421 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Brekhman V, Neufeld G A novel asymmetric 3D in-vitro assay for the study of tumor cell invasion. BMC Cancer. 9 415 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen HC Boyden chamber assay. Methods in Molecular Biology. 294, 15–22 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Poissonnier A, Legembre P Boyden Chamber Assay to Study of Cell Migration Induced by Metalloprotease Cleaved-CD95L. Methods in Molecular Biology. 1557, 117–123 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Li X, Yang H, Huang H, Zhu T CELLCOUNTER: novel open-source software for counting cell migration and invasion in vitro. BioMed Research International. 2014, 863564 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Little AC et al. IL-4/IL-13 Stimulated Macrophages Enhance Breast Cancer Invasion Via Rho-GTPase Regulation of Synergistic VEGF/CCL-18 Signaling. Frontiers in Oncology. 9, 456 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen SG et al. Macrophages Enhance Migration in Inflammatory Breast Cancer Cells via RhoC GTPase Signaling. Scientific Reports. 6, 39190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhatia SN, Ingber DE Microfluidic organs-on-chips. Nature Biotechnology. 32 (8), 760–772 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Huh D et al. Reconstituting organ-level lung functions on a chip. Science. 328 (5986), 1662–1668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen YC et al. Single-cell Migration Chip for Chemotaxis-based Microfluidic Selection of Heterogeneous Cell Populations. Scientific Reports. 5, 9980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi YP et al. Cancer-associated fibroblast promote transmigration through endothelial brain cells in three-dimensional in vitro models. International Journal of Cancer. 135 (9), 2024–2033 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Takeshita Y et al. An in vitro blood-brain barrier model combining shear stress and endothelial cell/astrocyte co-culture. Journal of Neuroscience Methods. 232, 165–172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kienast Y et al. Real-time imaging reveals the single steps of brain metastasis formation. Nature Medicine. 16 (1), 116–122 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Schroeder W, Martin K, Lorenson B The Visualization Toolkit. 4th edn Kitware, (2006). [Google Scholar]
- 18.Wells WM, Grimson WL, Kikinis R, Jolesz FA Adaptive segmentation of MRI data. IEEE Transactions on Medical Imaging. 15 (4), 429–442 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Agarwal SK, Singh S, Ghuman SS, Sharma S, Lahiri AK Radiological assessment of the Indian children with congenital sensorineural hearing loss. International Journal of Otolaryngology. 2014, 808759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mansfield P, Maudsley AA Medical imaging by NMR. The British Journal of Radiology. 50 (591), 188–194 (1977). [DOI] [PubMed] [Google Scholar]
- 21.Plewes DB, Kucharczyk W Physics of MRI: a primer. Journal of Magnetic Resonance Imaging. 35 (5), 1038–1054 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Batteux C, Haidar MA, Bonnet D 3D-Printed Models for Surgical Planning in Complex Congenital Heart Diseases: A Systematic Review. Frontiers in Pediatrics. 7, 23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thibault F et al. MRI for surgical planning in patients with breast cancer who undergo preoperative chemotherapy. American Journal of Roentgenology. 183 (4), 1159–1168 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Oliver CR et al. A platform for artificial intelligence based identification of the extravasation potential of cancer cells into the brain metastatic niche. Lab on a Chip. 19 (7), 1162–1173 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gril B et al. Reactive astrocytic S1P3 signaling modulates the blood-tumor barrier in brain metastases. Nature Communications. 9 (1), 2705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Badilescu S, Packirisamy M BioMEMS: Science And Engineering Perspectives. Taylor & Francis/CRC Press, (2011). [Google Scholar]
- 27.Liu C Foundations of MEMS. 2nd edn Prentice Hall, (2012). [Google Scholar]
- 28.Booth R, Kim H Characterization of a microfluidic in vitro model of the blood-brain barrier (muBBB). Lab on a Chip. 12 (10), 1784–1792 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Lockman PR et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clinical Cancer Research. 16 (23), 5664–5678 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan Oliver C, Con-tom TMW: A Jupyter Notebook for Analyzing the tumor micro-environment in confocal images. Zenodo. (Version 0.8), 3825719 (2020). [Google Scholar]
- 31.Lorensen WE, Johnson C, Kasik D, Whitton MC History of the Marching Cubes Algorithm. IEEE Computer Graphics and Applications. 40 (2), 8–15 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Kleinbaum DG, Kupper LL Applied regression analysis and other multivariable methods. Duxbury Press, (1978). [Google Scholar]
- 33.Berg M. d. Computational geometry : algorithms and applications. 2nd, rev. edn Springer, (2000). [Google Scholar]
- 34.Esch MB, Post DJ, Shuler ML, Stokol T Characterization of in vitro endothelial linings grown within microfluidic channels. Tissue Engineering Part A. 17 (23–24), 2965–2971 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brinkley BR et al. Variations in cell form and cytoskeleton in human breast carcinoma cells in vitro. Cancer Research. 40 (9), 3118–3129 (1980). [PubMed] [Google Scholar]
- 36.Eigenmann DE et al. Comparative study of four immortalized human brain capillary endothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimization of culture conditions, for an in vitro blood-brain barrier model for drug permeability studies. Fluids and Barriers of the Central Nervous System. 10 (1), 33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griep LM et al. BBB on chip: microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomedical Microdevices. 15 (1), 145–150 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Weksler B, Romero IA, Couraud PO The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids and Barriers of the Central Nervous System. 10 (1), 16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dello Russo C et al. The human microglial HMC3 cell line: where do we stand? A systematic literature review. Journal of Neuroinflammation. 15 (1), 259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dun MD et al. Proteotranscriptomic Profiling of 231-BR Breast Cancer Cells: Identification of Potential Biomarkers and Therapeutic Targets for Brain Metastasis. Molecular & Cellular Proteomics. 14 (9), 2316–2330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xing F et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Molecular Medicine. 5 (3), 384–396 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang B, Radisic M Organ-on-a-chip devices advance to market. Lab on a Chip. 17 (14), 2395–2420 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.