

Abstract
There is compelling evidence implicating intestinal permeability in the pathogenesis of nonalcoholic steatohepatitis (NASH), but the underlying mechanisms remain poorly understood. Here we examined the role of bile acids (BA) in western diet (WD)-induced loss of colonic epithelial barrier (CEB) function in mice with a genetic impairment in intestinal epithelial barrier function, junctional adhesion molecule A knockout mice, F11r−/−. WD-fed knockout mice developed severe NASH, which was associated with increased BA concentration in the cecum and loss of CEB function. Analysis of cecal BA composition revealed selective increases in primary unconjugated BAs in the WD-fed mice, which correlated with increased abundance of microbial taxa linked to BA metabolism. In vitro permeability assays revealed that chenodeoxycholic acid (CDCA), which was elevated in the cecum of WD-fed mice, increased paracellular permeability, while the BA-binding resin sevelamer hydrochloride protected against CDCA-induced loss of barrier function. Sequestration of intestinal BAs by in vivo delivery of sevelamer to WD-fed knockout mice attenuated colonic mucosal inflammation and improved CEB. Sevelamer also reduced hepatic inflammation and fibrosis, and improved metabolic derangements associated with NASH. Collectively, these findings highlight a hitherto unappreciated role for BAs in WD-induced impairment of the intestinal epithelial barrier in NASH.
Keywords: bile acids, intestinal permeability, microbiome, NAFLD, NASH, tight junction
1 |. INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) can be considered the hepatic manifestation of the metabolic syndrome (MetS), which is associated with chronic diseases including the type 2 diabetes mellitus (DM), essential hypertension, dyslipidemia, obesity, and hypothyroidism.1–3 An estimated 30%−40% of Americans have increased nonalcohol-related free fatty acid (FFA) and triglyceride (TGs) deposition in the hepatocytes, termed bland steatosis, or nonalcoholic fatty liver (NAFL).4,5 In approximately one-fifth of NAFL patients, disease progression can result in nonalcoholic steatohepatitis (NASH), which is characterized by increased inflammation and mild to moderate fibrosis. A subset of NASH patients is at significantly higher risk of developing cirrhosis and ultimately hepatocellular carcinoma (HCC).1,2 Despite ongoing research, mechanisms leading to NAFLD progression remain poorly understood and no effective treatment for NASH exists nor do we have a therapy to prevent disease progression.
Recent advances in our understanding of NASH pathogenesis underscore the contribution of the gut-liver axis in NAFLD progression. The concept of gut-liver-axis in NAFLD progression emerged from human and animal studies demonstrating an association between increased intestinal epithelial permeability and serum endotoxin level, a potent driver of hepatic inflammation.6–8 We have recently demonstrated that western diet (WD) feeding of mice with a compromised intestinal epithelial barrier (junctional adhesion molecule A knockout mice, F11r−/−), results in the rapid development of steatohepatitis with significant fibrosis.9 NAFLD progression in this model of compromised intestinal epithelial barrier correlated with further increase in intestinal epithelial permeability to gut bacterial products as well as perturbations in gut microbial composition.9 While this study confirmed the role of intestinal epithelial permeability and associated microbial dysbiosis in NAFLD progression, the mechanisms by which WD compromises intestinal epithelial barrier remain unclear.
Bile acids (BA) are amphipathic sterols synthesized in the liver from cholesterol, stored in the gall bladder, and released in the small intestine postprandially to facilitate the absorption of dietary fat, cholesterol, and fat-soluble vitamins.10,11 BAs secreted into the intestine are reabsorbed (~90%−95%) and recirculated to the liver via the entero-hepatic circulation. While the majority of BA reuptake occurs in the ileum, a small percentage of BAs is also reabsorbed in the colon. Colonic absorption of BAs is facilitated by gut microbiota, which converts the conjugated BAs into more hydrophobic molecules to enable their reabsorption by passive diffusion across the cell membrane.12–14 The entero-hepatic cycling of BAs is a highly efficient and tightly orchestrated process that not only provides an effective recycling mechanism to conserve and maintain a constant BA pool, but also serves as a mechanism to restrict BAs to the intestinal and hepatobiliary compartments.15 Disruption of this process, primarily due to BA malabsorption or excessive biotransformation by gut microbiota, can increase the passage of BAs to the colon.12,16–18 Colonic epithelial cells are susceptible to BA-induced cytotoxicity, and both in vitro and in vivo studies demonstrate that micromolar concentrations of BA can potentially compromise the integrity of the intestinal epithelial barrier.19–25 The role of BAs in WD-induced loss of intestinal epithelial barrier function in NASH is poorly defined, but a recent study reporting increased fecal BA concentrations in NASH patients,26 suggests a role for BAs in promoting colonic epithelial permeability in NASH.
The present study was designed to examine the role of BA in WD-induced impairment of intestinal epithelial barrier function in WD-fed F11r−/− mice, which develop severe steatohepatitis within 8 weeks of feeding a WD.9 Our results provide compelling evidence for a novel mechanism whereby WD-induced increases in the passage of primary, hydrophobic BA, CDCA to the colon compromises colonic epithelial barrier. We demonstrate that the BA-binding resin sevelamer hydrochloride attenuates NAFLD progression by protecting mice from BA-induced loss of colonic epithelial barrier function. In light of recent reports implicating gut-derived antigens in promoting the progression of NAFL to NASH, our findings have important clinical implications for the treatment of NASH.
2 |. MATERIALS AND METHODS
2.1 |. Mice
Junctional adhesion molecule A (JAM-A) knock out mice (F11r−/−) were generated as previously described.9 Mice were bred and maintained at Emory University and the University of Pittsburgh Divisions of Animal Resources. All animal studies were approved by the Institutional Animal Care and Use Committees.
2.2 |. BA feeding experiment
C57BL/6 mice were fed CDCA (3mg/gm body weight, Millipore Sigma, St. Louis, MO) mixed with FITC-conjugated dextran (4 kDa) (0.6 mg/gm body weight, Millipore Sigma, St. Louis, MO) solution by oral gavage following a 6 hours fast. After 3 hours, blood was collected and fluorescence intensity was measured using Fluorescence Spectrophotometer (Synergy 2, BioTek, Winooski, VT) as described previously.9
2.3 |. Diet and feeding studies
Five to six-week-old male F11r−/− mice were fed a ND or WD ad libitum for 12 weeks as described previously.9 After 12 weeks of feeding, mice were fed either ND or WD alone (control) or ND or WD mixed with sevelamer hydrochloride for 4 weeks. The western diet (WD) consisted of 0.2% cholesterol, 20% protein, 43% CHO, 23% fat (6.6% trans-fat), and 2.31% fructose (TD.130885; Harlan Laboratories).9 The normal diet (ND) is the standard mouse chow that contains 16% protein, 61% carbohydrate, and 7.2% fat.
2.4 |. Histopathology
Hematoxylin and eosin (H&E) and Sirius Red staining were performed on formalin-fixed liver and colon tissue sections, as previously reported [4]. A Zeiss Light Microscope (Zeiss, Jena, Germany) was employed to obtain the photomicrographs of the histologic sections.
2.5 |. Bile acid analysis
Bile acids extracted from liver and cecal content were quantified using liquid chromatography-mass spectrometry (LC-MS) analysis. Briefly, the liver samples were homogenized in PBS and cecal samples were sonicated in PBS. Samples were deproteinized with acetonitrile and centrifuged; subsequently, the supernatants were lyophilized under nitrogen and solvated in a 1:1 (v/v) mixture of methanol and water. LC-MS analysis was carried out on the SCIEX 5500 QTRAP LC-MS system (SCIX, Framingham, MA). Analyses of all samples were performed using a C18 reverse-phase HPLC column (Thermo Fisher Scientific). MultiQuant 3.0.2 software was used for the data processing and quantification.
2.6 |. Cell culture and in vitro permeability assay
Caco2 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in DMEM supplemented with 10% FBS, 1% penicillinstreptomycin, and 1 mmol/L of sodium pyruvate as described previously.27
For the in vitro permeability assay, Caco2 cells (passage number 23–35) were cultured in 6- or 12-well trans-well chambers (Corning, NY, USA) at a density of 4 × 104 cells/cm2 for 21 days as described previously.27 Fully differentiated Caco-2 cell monolayers were treated with 20 μM individual BAs or BAs premixed with 0.2 M sevelamer. FITC-conjugated dextran (4 kDa) (Millipore Sigma, St. Louis, MO) dissolved in Hanks’ balanced salt solution (HBSS) was added to the apical compartment and incubated for 2 hours in a tissue culture incubator. CA, CDCA, TCA, and TDCA were dissolved in HBSS, while LCA was dissolved in methanol and then diluted with HBSS. At the end of the study, transepithelial electrical resistance (TEER) was measured using a Millicell-ERS voltohm meter (Millipore, Temecula, CA, USA). Media from the basal compartments was collected and fluorescent intensity was measured using Fluorescence Spectrophotometer (Synergy 2, BioTek, Winooski, VT). FITC-dextran concentrations were determined from a standard curve generated by serial dilutions of 4 kDa FITC-dextran.
2.7 |. Immunofluorescence microscopy
Liver and intestinal cryosections were stained for immunofluorescence (IF) microscopy as described previously [4]. Cryosections were visualized using an Axioskop 2 plus microscope (Zeiss, Jena, Germany). The antibodies used for IF staining were obtained from Thermo Fisher Scientific (Rockford, IL).
2.8 |. Serological analysis
AST and ALT Activity Assay Kits (Sigma-Aldrich, St. Louis, MO) were used to estimate serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentrations.
2.9 |. Glucose tolerance test and insulin tolerance test
Glucose tolerance test (GTT) and insulin tolerance test (ITT) were conducted at 16 weeks of WD feeding, as previously reported [4]. A hand-held glucometer (Freestyle Flash, Abbott Laboratories, Abbott Park, IL) was used to determine the blood glucose concentration.
2.10 |. Quantitative real-time PCR
Total RNA was isolated from the liver, followed by cDNA synthesis and qRT-PCR, based on the previously described methods [4]. All data were normalized against 18S rRNA and were reported as fold change in gene expression as compared to those in ND-fed control mice.
2.11 |. Statistical analysis
ANOVA, in conjunction with post hoc analysis for multiple group comparisons, was used to analyze statistical differences. The data reported were from three independent experiments. A p value < .05 was considered statistically significant.
3 |. RESULTS
3.1 |. NASH development in WD-fed mice is associated with increased BAs in the cecum and alterations in the composition of the cecal BA pool
To induce NASH, F11r−/− mice were fed a WD for 16 weeks, F11r−/− mice fed a ND served as controls. WD-fed F11r−/− mice developed key histological features of NASH including hepatic steatosis with increased accumulation of triglycerides in hepatocytes, increased immune cell infiltration, and increased fibrosis in the perivascular and perisinusoidal regions (Figure 1A,B). Increased collagen deposition in the liver of WD-fed F11r−/− mice correlated with a significant increase in α smooth muscle actin (SMA) mRNA transcripts, a key hepatic stellate cell (HSC) activation marker,28 as well as mRNA transcripts for tissue inhibitors of metalloproteinase (TIMP)-1 and collagen I, [αI and αII] (Figure 1B,C–F). Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were also significantly higher in WD-fed mice compared to levels in the ND-fed mice demonstrating severe hepatic injury in these mice (Figure 1G–H).
FIGURE 1.
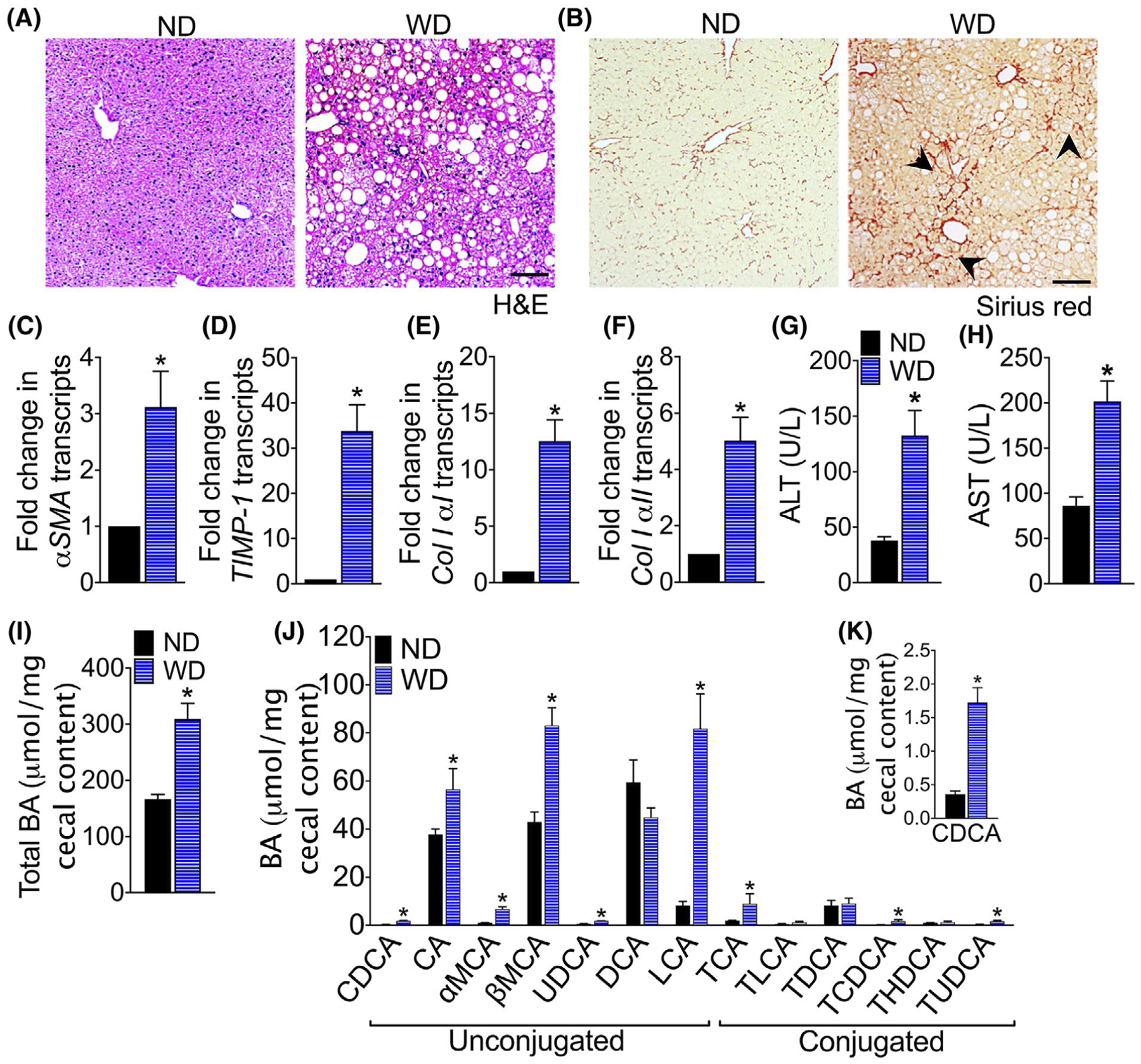
NASH development in western diet-fed mice is associated with increased colonic bile acids. Photomicrographs of A, Hematoxylin and Eosin (H&E) and B, Sirius Red stained liver tissue sections of F11r−/− mice fed a normal chow (ND) or western diet (WD) for 16 weeks (n = 5). Scale bar 20 μm. Black arrowhead, collagen deposition in the liver. C-F, Quantitative reverse transcription PCR analysis of hepatic C, α smooth muscle actin (αSMA), D, tissue inhibitor of metalloproteinases (TIMP)-1, E, collagen I αI, and F, collagen I αII transcripts. Serum G, ALT and H, AST levels. I, Total bile acid (BA) in the cecal content and J-K, composition of cecal BAs. CDCA, chenodeoxycholic acid; CA, cholic acid; MCA, muricholic acid; UDCA, ursodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; TCA, taurocholic acid; TLCA, taurolithocholic acid; TDCA, taurodeoxycholic acid; TCDCA, taurochenodeoxycholic acid; THDCA, taurohyodeoxycholic acid and TUDCA, tauroursodeoxycholic acid. Data are presented as means ± SEM. * P < .05 ND vs WD-fed mice
To determine whether NASH development in WD-fed F11r−/− mice was associated with increased passage of BAs to the cecum, we quantified total BAs in the cecal content. As seen in Figure 1I, the total cecal BA concentration in WD-fed mice was twofold higher relative to the ND-fed mice. Analysis of the BA composition by liquid chromatography-mass spectrometry (LC-MS) revealed that WD consumption increased BAs with high affinity for farnesoid X receptor (FXR): chenodeoxycholic acid (CDCA), cholic acid (CA), lithocholic acid (LCA), as well as BAs, that are FXR antagonists: α and β muricholic acid (MCA), and urodeoxycholic acid (UDCA) (Figure 1J–K). The concentration of conjugated BAs taurocholic acid (TCA), taurochenodesoxycholic acid (TCDCA), and tauroursodeoxycholic acid (TUDCA) were also significantly higher in the WD-fed mice. The colonic BAs in ND-fed mice consisted mainly of primary-unconjugated BAs except for the secondary BA deoxycholic acid (DCA). Together, these data demonstrate that WD consumption not only increases the passage of BAs to the cecum, but also alters the composition of cecal BA.
3.2 |. Sevelamer protects the epithelium from BA-induced loss of epithelial barrier
To determine whether the BAs that were increased in the cecum of WD-fed mice promoted epithelial permeability, we examined the effects of BAs on permeability across monolayers of Caco2 cells in vitro, a well-established model of human intestinal epithelial barrier function.29 Caco2 cell monolayers grown on transwell inserts were treated with specific BAs that were differentially represented in the cecal content of WD-fed F11r−/− mice and change in permeability was determined by the paracellular flux of 4kDa FITC-Dextran. As shown in Figure 2A,B, CDCA, but not CA, TCA, TDCA or LCA, significantly increased the paracellular flux of FITC-dextran. The CDCA-induced increase in paracellular permeability correlated with a significant decrease in TEER, consistent with CDCA induced the impairment of barrier function in Caco2 cells (Figure 2C,D). To determine whether sevelamer, which binds and sequesters BAs,30 protects the epithelial barrier from CDCA-induced loss of barrier function, we treated Caco2 monolayers with CDCA premixed with sevelamer. As shown in Figure 2A,B, CDCA premixed with sevelamer did not increase the paracellular flux of FITC-dextran across the Caco2 cell monolayers and did not reduce TEER, suggesting that sevelamer neutralizes the effect of CDCA on Caco2 monolayers. To determine whether CDCA-induced gut permeability in vivo, we measured intestinal permeability in CDCA-fed mice using the FITC-dextran permeability assay. As shown in Figure 2E, oral gavage of CDCA induced a ninefold increase in the paracellular flux of 4kDa FITC-Dextran. Together, these data suggest that WD-induced increase in the passage of CDCA to the cecum of F11r−/− mice impaired colonic epithelial barrier.
FIGURE 2.
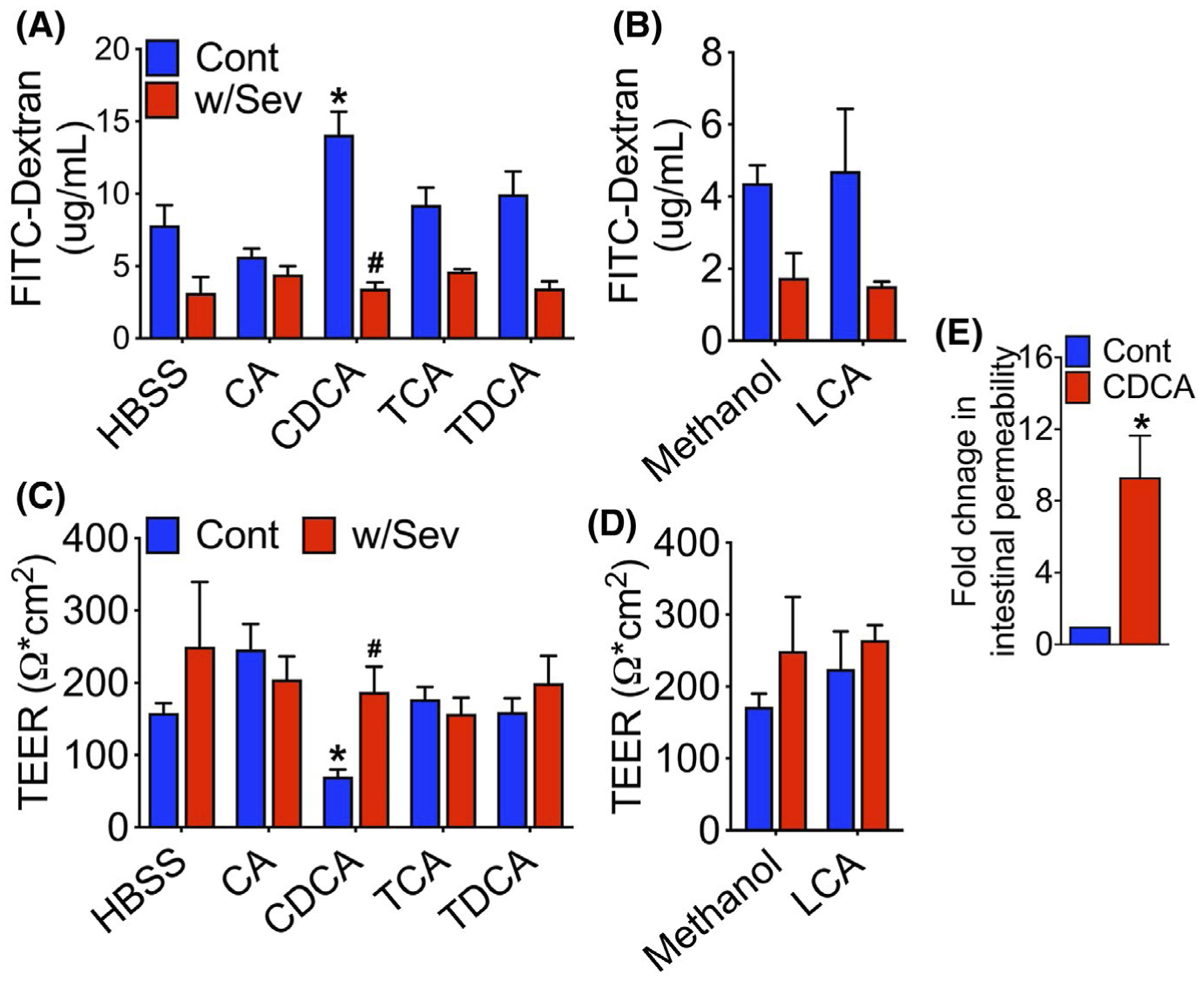
Sevelamer protects epithelial barrier from bile acid-induced increase in paracellular permeability. A,B, Paracellularflux of 4 kDa FITC-Dextran across a Caco2 monolayer treated with individual bile acid (BAs) or BAs mixed with sevelamer (Sev). Fully differentiated Caco-2 cell monolayers were treated with individual BAs or BAs premixed with sevelamer for 2 hours in the presence of 4 kDa FITC-Dextran. FITC-dextran flux was determined by measuring the fluorescence intensity of media in the basal chamber. C-D, Transepithelial electrical resistance (TEER) of Caco2 monolayers treated with individual BAs or BAs mixed with sevelamer. TEERwas measured before adding BAs and again after 2-hours incubation. TEER readings are reported as the normalized values after 2-hours incubation. E, Fold change in the intestinal permeability to FITC-dextran in control and CDCA-fed mice (n = 3–4 mice per cohort). CA, cholic acid; CDCA, chenodeoxycholic acid; TCA, taurocholic acid; TDCA, taurodeoxycholic acid; and LCA, lithocholic acid. Data are representative means ± SEM of at least four individual monolayer inserts. *P < .05 HBSS or methanol vs BA-treated wells. #P < .05 BA vs BA plus sevelamer-treated wells
3.3 |. Sequestration of BAs improves WD-induced disruption of the colonic epithelial barrier
Next, we sought to determine whether the sequestration of BAs would protect F11r−/− mice from WD-induced loss of colonic epithelial barrier. To address this question, a cohort of F11r−/− mice fed a ND or WD for 12-weeks were administered sevelamer mixed with diet for an additional four weeks (Figure 3A). At the end of the study, we analyzed the respective BA composition of the cecal contents, which revealed a significant increase in BAs in the mice fed a WD mixed with sevelamer compared to the cohort of mice fed the WD alone; however, no differences in the concentration of total BAs were observed between mice fed a ND or ND mixed with sevelamer (Figure 3B). Analysis of the composition of the BA in the cecal content revealed a significant increase in the concentration of CDCA, CA, LCA, TCA TLCA, and TCDCA in sevelamer-treated ND and WD fed mice demonstrating that sevelamer sequestered both hydrophobic and hydrophilic BAs (Figure 3C).
FIGURE 3.
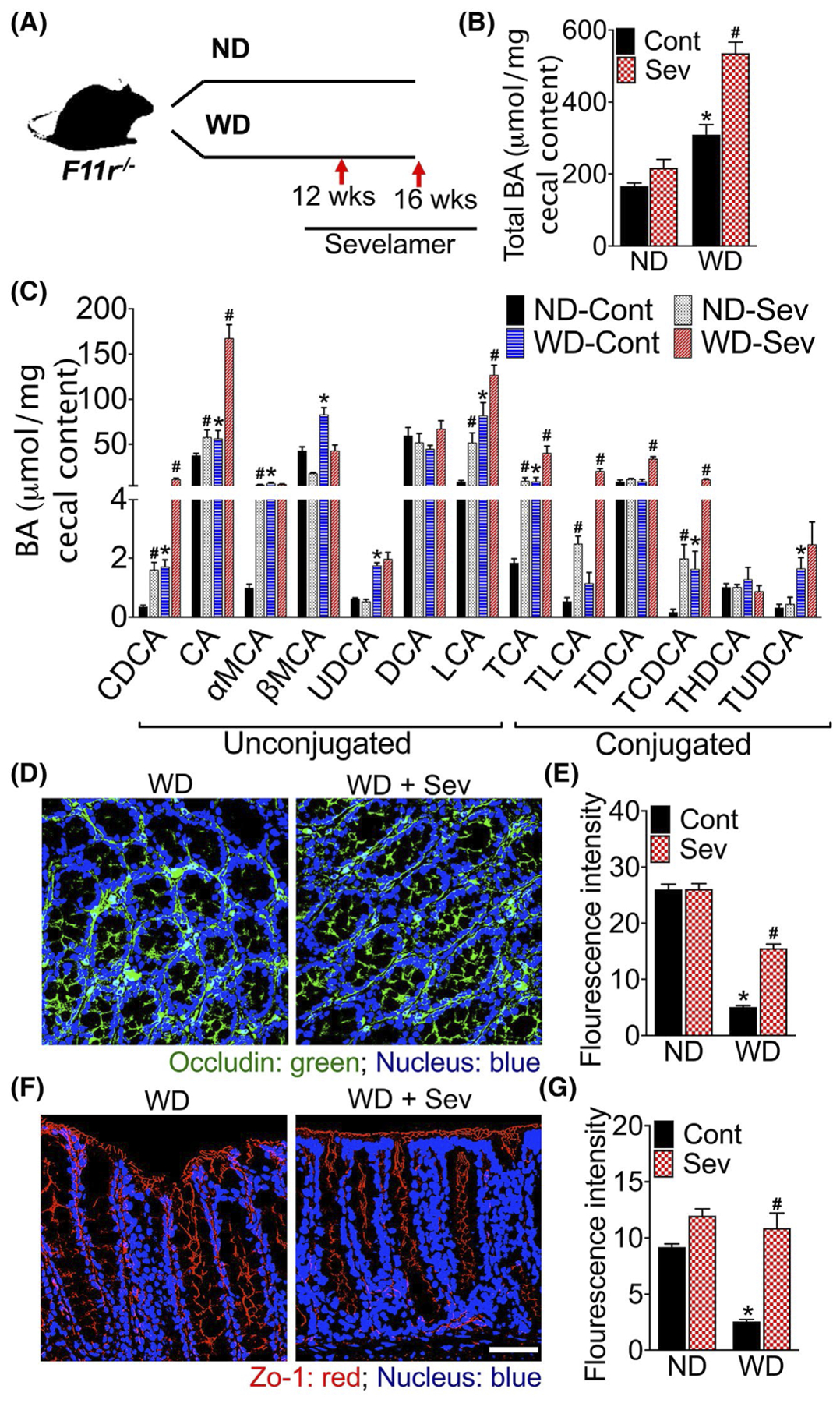
Sequestration of intestinal bile acids improves epithelial barrier in western diet-fed mice. A, Study design. Mice were fed a normal chow(ND) or western diet (WD) for 12 weeks followed by either the ND or WD with or without sevelamer (Sev) for an additional 4 weeks (n = 5 mice per cohort). B, Total bile acid (BA) in the cecal content, andC, composition of cecal BAs. CDCA, chenodeoxycholic acid; CA, cholicacid; MCA, muricholic acid; UDCA, ursodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; TCA, taurocholic acid; TLCA, taurolithocholic acid; TDCA, taurodeoxycholic acid; TCDCA, taurochenodeoxycholic acid; THDCA, taurohyodeoxycholic acid and TUDCA, tauroursodeoxycholic acid. Data are presented as means ± SEM. *P < .05 ND vs WD fed mice. #P < .05 between control and sevelamer-treated mice. D-G, Confocal microscopic images with quantification of D-E, occludin and F-G, ZO-1 immunofluorescence in the colonic tissue. Scale bar 20 μm
To determine whether the sequestration of intestinal BAs improves colonic epithelial barrier, we examined colonic expression and distribution of the tight junction (TJ) proteins, occludin and zonula occludens (ZO)-1 using confocal laser scanning microscopy. As evident in the photomicrograph (Figure 3D–G and Figure S1A,B), compared with ND fed mice, occludin and ZO-1 expression were markedly reduced in the colonic mucosa of WD-fed mice. Sevelamer treatment restored WD-induced reduction and redistribution of occludin and ZO-1 in the colonic mucosa of mice fed a WD. No significant differences in occludin and ZO-1 expression were observed between mice fed a ND or ND mixed with sevelamer (Figure S1A,B). Together, these data suggest that the sequestration of BAs improves WD-induced loss of colonic epithelial barrier by restoring the structural integrity of epithelial TJs.
3.4 |. Bile acid sequestration attenuates WD-induced mucosal inflammation
As seen in Figure 4A and Figure S2A, WD consumption resulted in severe colonic mucosal inflammation indicated by increased infiltration of immune cells in the mucosa. Further analysis revealed a marked increase in the infiltration of Ly6G + neutrophils and monocytes as well as myeloperoxidase (MPO) expressing cells in the colonic mucosa of WD-fed F11r−/− mice relative to ND-fed mice (Figure 4B,C and Figure S2B–C). The transcript levels of proinflammatory cytokines, TNFα, and IL1β were significantly higher in the colonic mucosa of WD-fed mice relative to the ND-fed mice demonstrating that WD-consumption resulted in severe mucosal inflammation in these mice (Figure 4D,E). The transcript levels of monocyte chemoattractant protein (MCP)-1, C-C chemokine receptor (CCR)-2, and F4/80 remained unchanged (Figure S2D,F). Sevelamer administration attenuated WD-induced mucosal inflammation as evident by a marked reduction in immune cell infiltration in the colonic mucosa and a significant decrease in transcript levels of TNFα (Figure 4A–D). Levels of IL1β, however, remained unchanged (Figure 4E).
FIGURE 4.
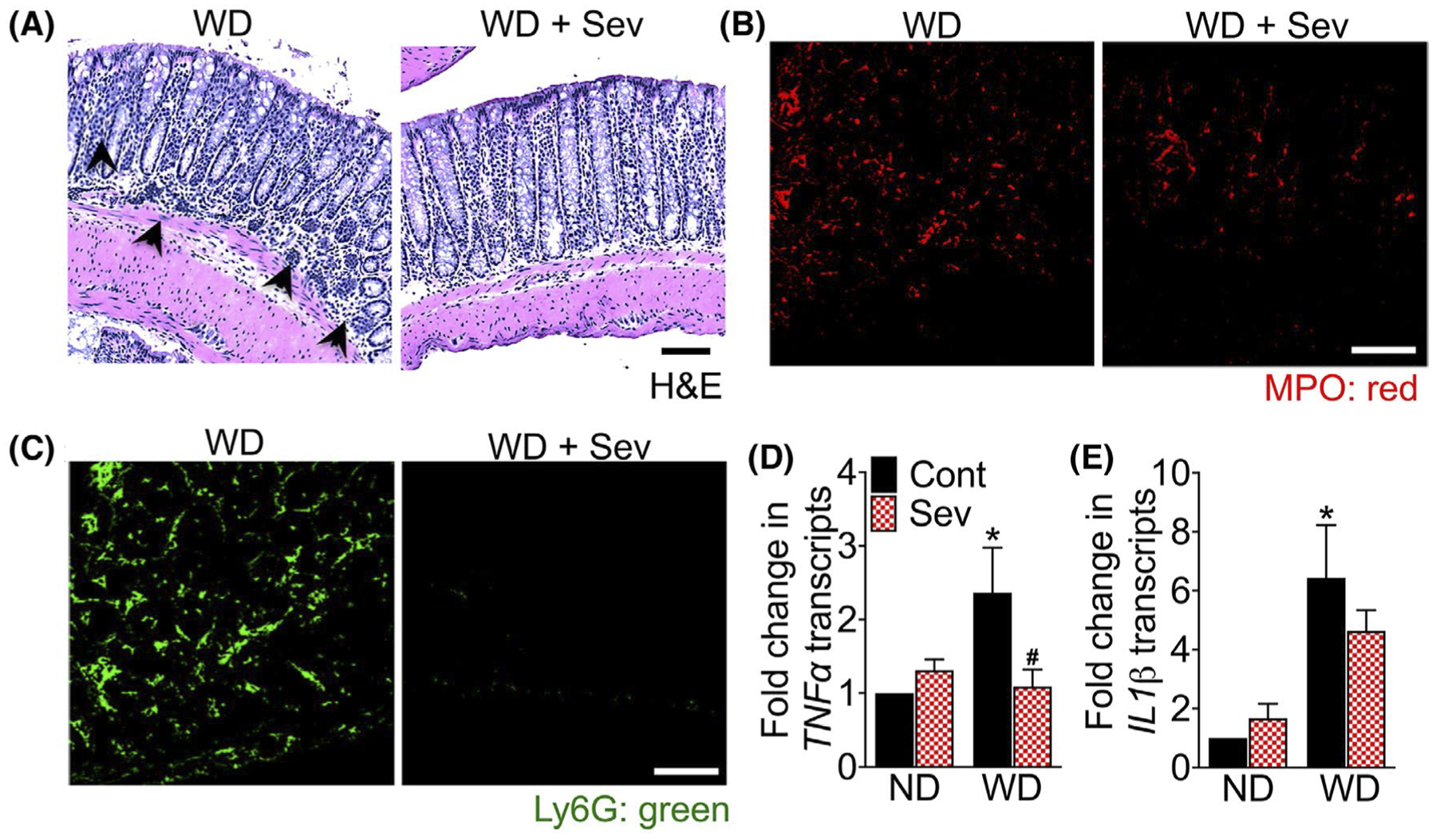
Intestinal bile acid sequestration attenuates western diet-induced mucosal inflammation. A, Photomicrographs of Hematoxylin and Eosin (H&E) stained colonic mucosal tissue sections of F11r−/− mice fed a normal chow (ND) or western diet (WD) for 12 weeks followed by either the ND or WD with or without sevelamer (Sev) for an additional 4 weeks (n = 5 mice per cohort). Scale bar 20 μm. Black arrowhead, immune cell infiltration. B-C, Confocal microscopic images of B, myeloperoxidase (MPO) and C, Ly6G expression in the colonic tissue. Scale bar 20 μm. D-E, Quantitative reverse transcription PCR analysis (RT-PCR) of colonic D, TNFα and E, IL1β transcripts. Data are presented as means ± SEM. *P < .05 ND vs WD fed mice. #P < .05 control vs sevelamer-treated mice
3.5 |. Sequestration of intestinal BAs attenuates hepatic inflammation and fibrosis in WD-fed mice
To determine whether the administration of sevelamer improved NASH, we examined histological and biochemical features of NASH in sevelamer-treated WD-fed F11r−/− mice. In agreement with a recent study,31 sevelamer markedly reduced hepatic steatosis, inflammation, and fibrosis in the WD-fed mice (Figure 5A–P and Figure S3A–C). Improvement in hepatic inflammation in sevelamer-treated WD-fed mice was confirmed by a significant reduction in serum AST and ALT levels (Figure 5B,C), and reduction in hepatic TNFα, IL1β, and IL6 levels (Figure 5D–F). Administration of sevelamer significantly decreased mRNA transcripts of αSMA, TIMP-1, and collagen I [αI and αII] (Figure 5H–K) demonstrating a significant reduction in HSC activation and collagen deposition in WD-fed mice. Sevelamer treatment also reduced hepatic macrophage infiltration (Figure 5L and Figure S3C), which correlated with a significant decrease in F4/80, MCP-1, CD68, and CCR2 transcripts. Together, these data demonstrate that the administration of sevelamer not only attenuated hepatic steatosis and inflammation, but also ameliorated hepatic fibrosis in WD-fed mice.
FIGURE 5.
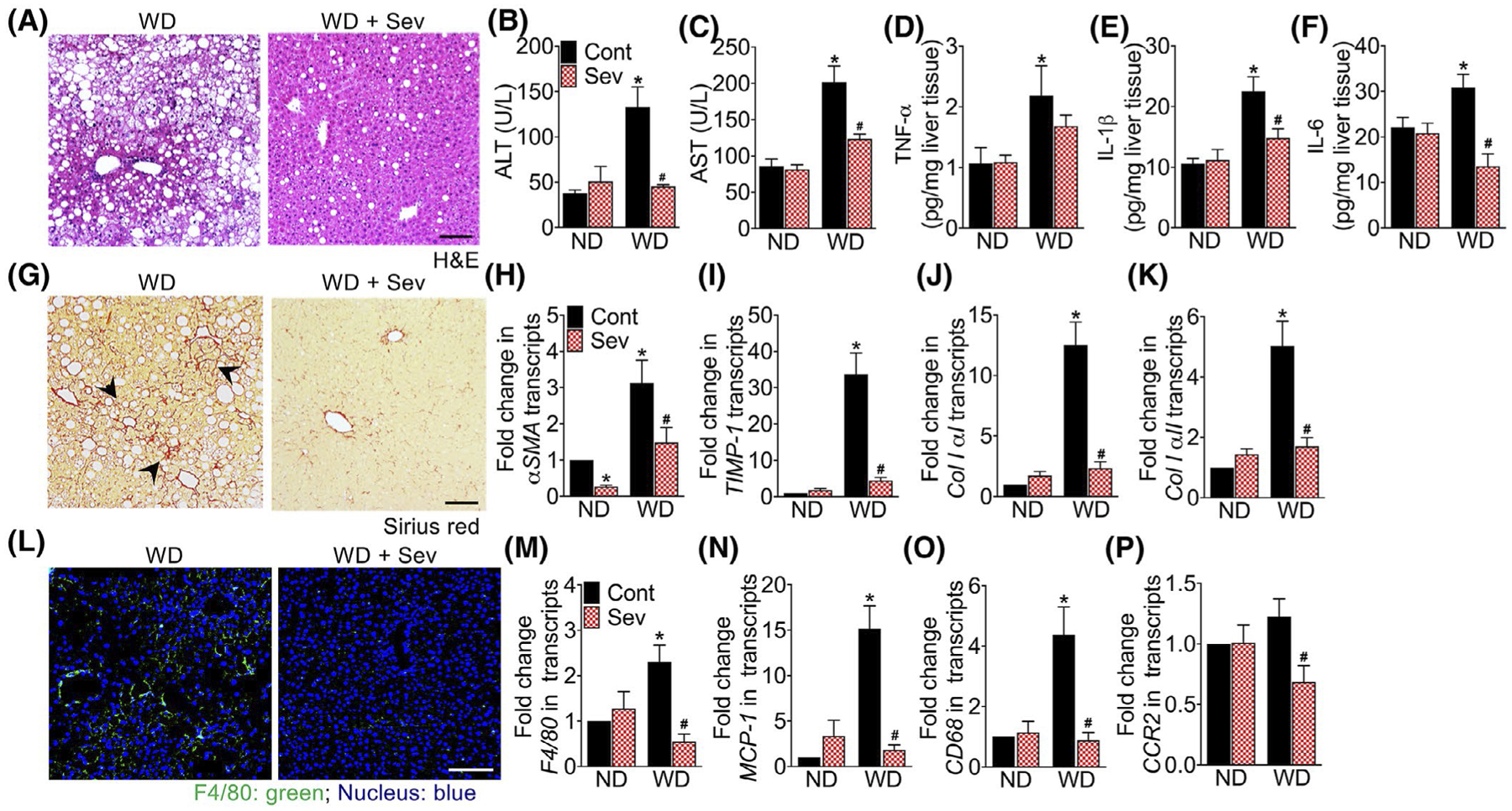
Sequestration of intestinal bile acids protects mice from western diet-induced hepatic inflammation and fibrosis. Photomicrographs of A, Hematoxylin and Eosin (H&E) stained liver tissue sections of F11r−/− mice fed a normal chow (ND) or western diet (WD) for 12 weeks followed by either the ND or WD with or without sevelamer (Sev) for an additional 4 weeks (n = 5 mice per cohort). Scale bar 20 μm. B, C, Serum B, ALT and C, AST levels. D-F, Quantification of hepatic proinflammatory cytokines D, TNFα, E, IL-1β, and F, IL-6. Photomicrographs of G, Sirius Red stained liver tissue sections. Scale bar 20 μm. Black arrowhead, collagen deposition in the liver. H-K, Quantitative reverse transcription PCR (RT-PCR) analysis of hepatic H, α smooth muscle actin (αSMA), I, tissue inhibitor of metalloproteinase (TIMP)-1, J, collagen IαI and K, collagen IαII transcripts. L, Confocal microscopic images of tissue-resident macrophage marker F4/80 expression in the liver. Scale bar 20 μm. M-P, RT-PCR analysis of hepatic M, F4/80, N, monocyte chemoattractant protein (MCP)-1, O, CD68, and P, C-C chemokine receptor (CCR)-2 transcripts. Data are presented as means ± SEM. *P < .05 ND vs WD fed mice. #P < .05 control vs sevelamer-treated mice
3.6 |. Sequestration of intestinal BAs improves metabolic parameters in WD-fed mice
Examination of metabolic parameters in sevelamer-treated mice revealed a significant reduction in body weight in WD-fed F11r−/− mice, but not in ND-fed mice (Figure 6A). Reduction in body weight was most significant in the first 2 weeks of treatment, after which body weight of sevelamer-treated WD-fed mice remained stable (Figure 6A). Sevelamer treatment significantly decreased liver weight/body weight and visceral fat weight/body weight ratios, respectively (Figure 6B,C). While sevelamer treatment reduced the liver weights of WD-fed mice compared to the ND-fed mice, visceral fat weight remained higher (Figure 6B,C) in the former. Even after 4 weeks of sevelamer treatment, the visceral fat weight of WD-fed mice remained twofold higher than the ND-fed mice (Figure 6C).
FIGURE 6.
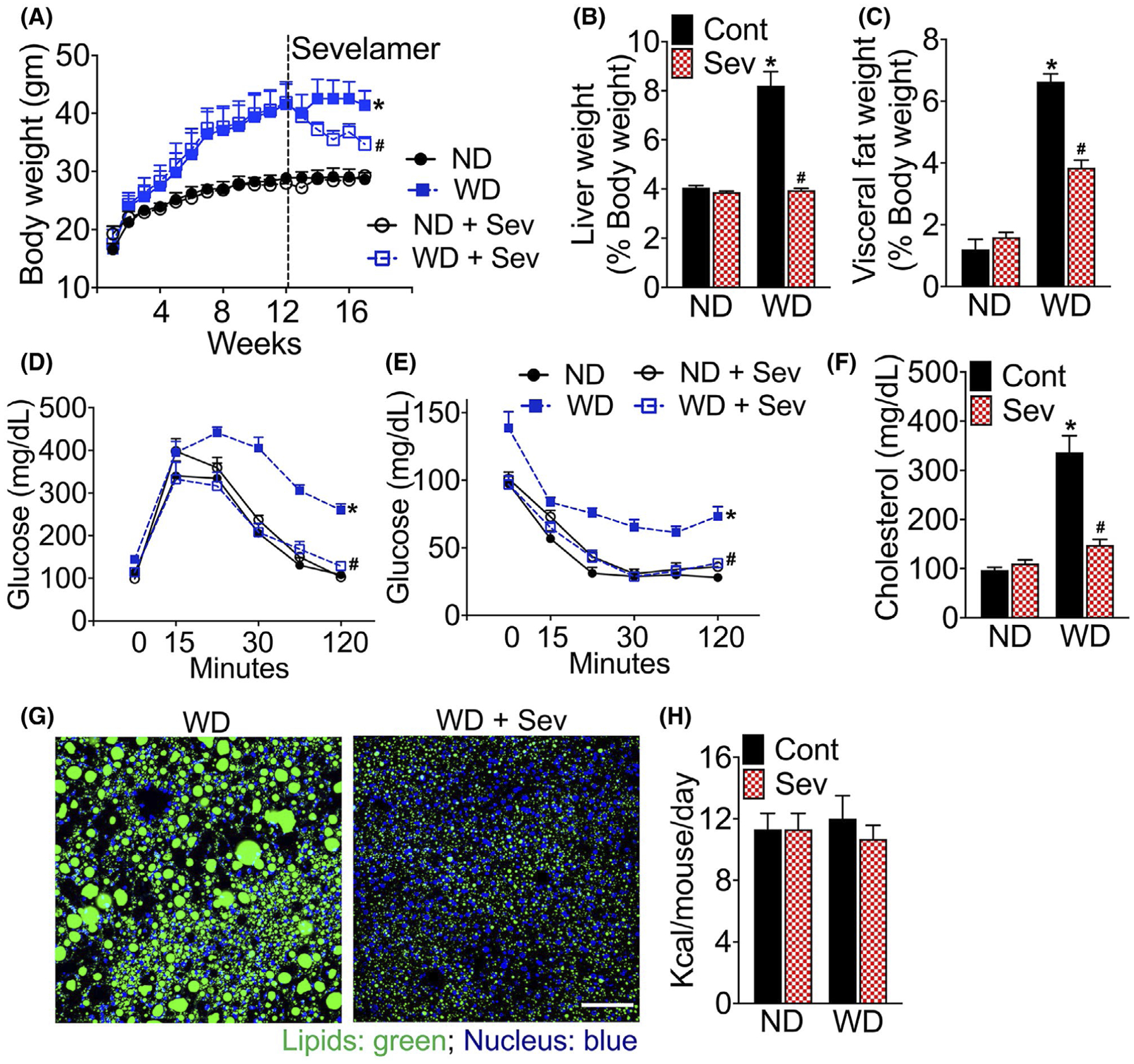
Intestinal bile acid sequestration improves metabolic parameters and glucose homeostasis. A, Changes in the body, B, liver, and C, visceral fat weight in F11r−/− mice fed a normal chow (ND) or western diet (WD) for 12 weeks followed by either the ND or WD withor without sevelamer (Sev) for an additional four weeks (n = 5 mice per cohort). Changes in the liver and visceral fat weights are reported as the percent of body weight. D, Glucose, and E, insulin tolerance after 16 weeks of total feeding. F, Changes in the serum cholesterol levels. Confocal microscopic images of G, BODIPY (lipids) stained liver tissue sections. Scale bar 20 μm. H, Average daily calorie intake. Data are presented as means ± SEM. *P < .05 ND vs WD fed mice. #P < .05 control vs sevelamer-treated mice
We observed improved metabolic parameters in sevelamer-treated WD-fed mice including improved glycemic control and insulin sensitivity (Figure 6A–E). We observed no significant differences in metabolic parameters between mice fed a ND or ND and sevelamer (Figure 6A–E). Sevelamer treatment significantly reduced serum cholesterol levels to levels comparable to those recorded in the ND-fed mouse cohort (Figure 6F), suggesting that the sequestration of intestinal BA improves whole body cholesterol metabolism in WD-fed mice. Sevelamer treatment also reduced hepatic fat deposition in the WD-fed mice (Figure 6G). No differences in average calorie intake were observed between control and sevelamer-treated WD-fed mice (Figure 6H).
3.7 |. Intestinal BA sequestration reverses WD-induced alteration in hepatic BA composition and increases hepatic BA synthesis
To understand the mechanism of sevelamer-mediated improvement in metabolic parameters, we examined hepatic BA composition in sevelamer-treated WD-fed F11r−/− mice. As shown in Figure 7A–B, WD consumption did not increase total BA concentrations in the liver; rather, it altered hepatic BA composition. WD consumption significantly increased TCDCA, but decreased the TDCA in the liver (Figure 7B).
FIGURE 7.
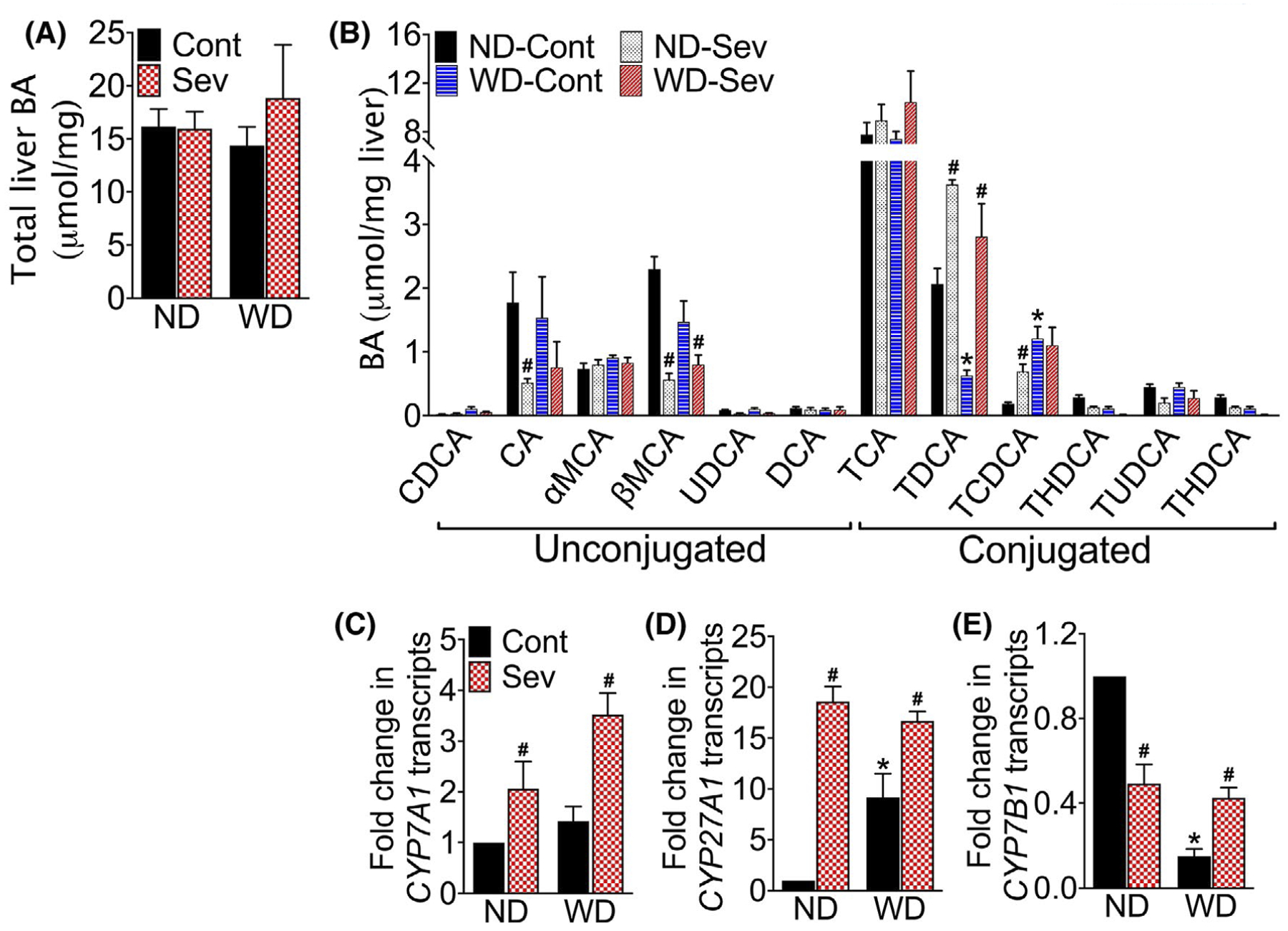
Intestinal bile acid sequestration restores western diet-induced alterations in the hepatic BA pool. A, Total hepatic BA content and B, composition in mice fed a normal chow (ND) or western diet (WD) for 12 weeks followed by either the ND or WD with or without sevelamer (Sev) for an additional four weeks (n = 5 mice per cohort). CDCA, chenodeoxycholic acid; CA, cholic acid; MCA, muricholic acid; UDCA, ursodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; TCA, taurocholic acid; TLCA, taurolithocholic acid; TDCA, taurodeoxycholic acid; TCDCA, taurochenodeoxycholic acid; THDCA, taurohyodeoxycholic acid and TUDCA, tauroursodeoxycholic acid. C-E, RT-PCR analysis of C, CYP7A1, D, CYP27A1, and E, CYP7B1 transcripts in the liver. Data are presented as means ± SEM. *P < .05 ND vs WD fed mice. #P < .05 control vs sevelamer-treated mice
Sevelamer treatment did not change the total BA concentration in the liver, but decreased βMCA concentration and restored TCDCA concentrations to that seen in the livers of ND-fed mice (Figure 7A,B). To understand the effect of sevelamer on hepatic BA synthesis, we analyzed key enzymes that regulate BA biosynthesis in the liver. As shown in Figure 7C–E, sevelamer treatment significantly increased the expression of key enzymes involved in the classical and alternative BA synthesis pathways, cholesterol 7α-hydroxylase (CYP7A1), sterol 27-hydroxylase (CYP27A1), and oxysterol 7α-hydroxylase (CYP7B1) transcripts. Together, these data suggest that the sequestration of BAs activates both classic and alternative BA synthesis pathways in the liver without altering hepatic BA pool size.
3.8 |. WD increases the abundance of microbial taxa associated with BA metabolism
Since microbiota play a significant role in metabolism, and the absorption and excretion of BAs,32 we analyzed cecal mucosa-associated microbiota by 16S rRNA sequencing followed by phylogenetic analyses and a comparison of the microbial community structure using the unweighted UniFrac algorithm as described previously.9 As seen in Figure 8A,B, WD consumption decreased diversity and altered composition of colonic mucosa-associated microbial composition. At the order level, WD consumption resulted in the expansion of Bacteroidales and Clostridiales, major gut microbial taxa with bile salt hydrolase activity33,34; as well as Desulfovibrionales, a bacterial taxa associated with colitis35; and Deferribacterales (Figure 8C). At the family level, WD increased the abundance of Bacteroidaceae, Odoribacteraceae, and Desulfovibrionaceae, but decreased S24–7 (Figure 8D). Sevelamer treatment did not significantly alter the microbial composition in the ND or WD fed mice, except for a significant reduction in the abundance of Deferribacteraceae in the WD-fed mice. Together, these data suggest a link between increased BA metabolizing bacterial taxa in the cecum and increased unconjugated BAs in the cecal content of WD-fed mice.
FIGURE 8.
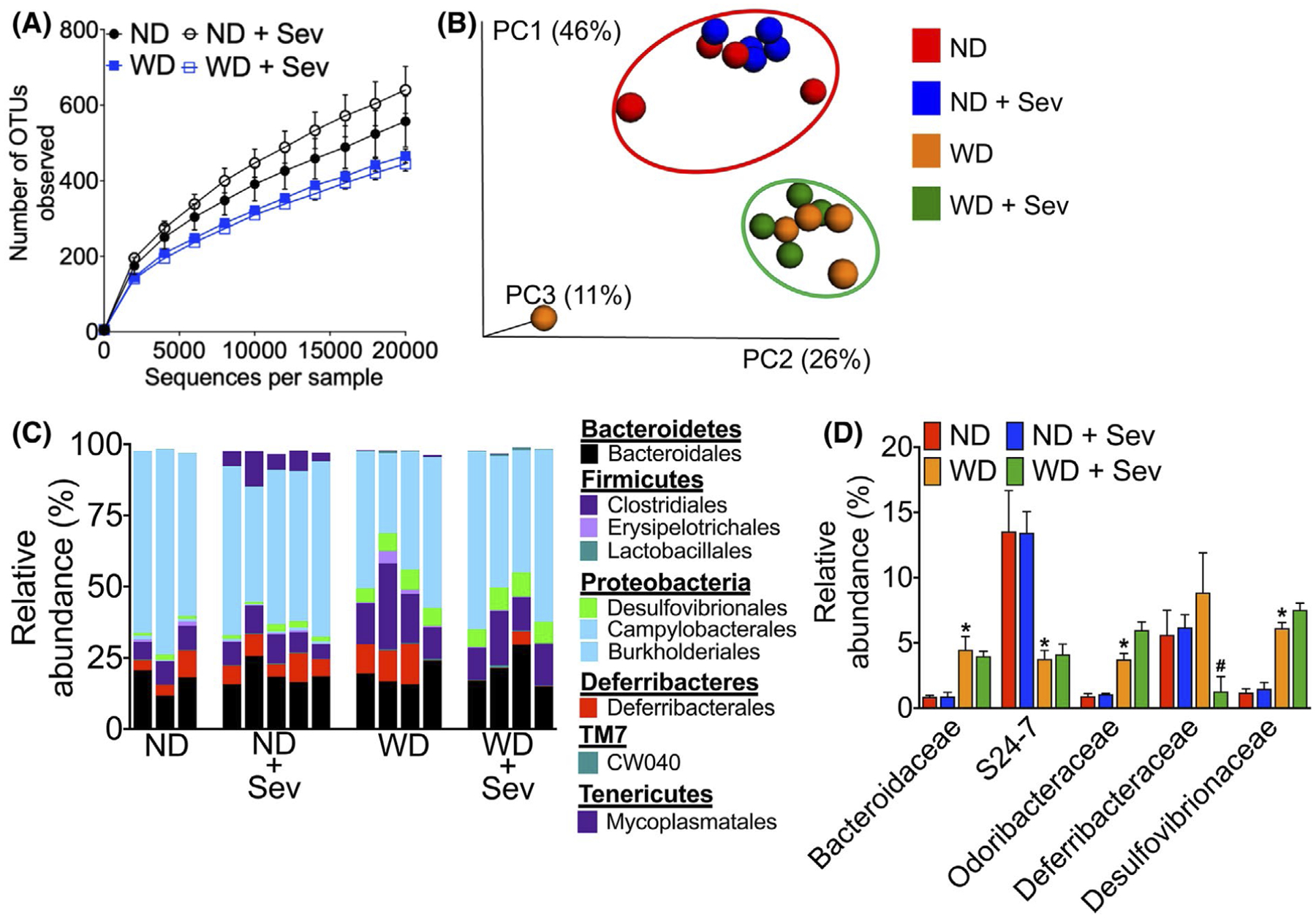
Western diet increases the abundance of gut microbial taxa associated with bile acid metabolism. A, Microbiota richness and diversity in the colonic mucosa in mice fed a normal chow (ND) or western diet (WD) for 12 weeks followed by either the ND or WD with or without sevelamer (Sev) for an additional four weeks (n = 3–5 mice per cohort). B, Jackknifed principal coordinate analysis (PCoA) of the unweighted UniFrac distance matrix of the colonic mucosa-associated microbiota. Symbols represent data from individual mice color-coded by the indicated metadata. The ovals represent clustering by treatment groups. C, Relative abundance of bacterial orders in the colonic mucosa. Bars represent individual mice. Labels indicate families with average relative abundances >1% in at least one treatment group. D, Relative abundance of mucosa-associated bacterial families significantly altered in WD or WD plus sevelamer-treated mice. Data are presented as means ± SEM. *P < .05 ND vs WD fed mice. #P < .05 control vs sevelamer-treated mice
4 |. DISCUSSION
In the present study, we provide a potential mechanistic explanation underpinning the loss of intestinal epithelial barrier function in NASH by demonstrating that WD-induced increase in the primary BA, CDCA in the cecum disrupts the colonic epithelial barrier in mice that ultimately develop severe NASH. We show that the effect of CDCA on colonic mucosa was attenuated by the oral administration of the BA-binding resin sevelamer, secondary to a marked decrease in hepatic inflammation and fibrosis. Collectively our data implicate CDCA as a contributing factor in WD-induced loss of colonic epithelial barrier in NASH and provide evidence that BA-binding resins may represent a promising treatment for NASH. Furthermore, hepatic injury in F11r−/− mice following 16 weeks of WD consumption recapitulates key physiological, metabolic, and histological features of human NASH with fibrosis, demonstrating the utility of this accelerated rodent model of progressive NASH for preclinical studies.
Under normal physiological conditions, only a small amount of BA reaches the colon, where it is metabolized by resident microbiota to facilitate reabsorption and excretion in the stool. Higher concentrations of BAs in the colon, however, disrupt colonic epithelial barrier integrity ultimately resulting in loss of barrier function.19–22 The extent of BA-induced cytotoxicity and subsequent tissue injury depends on the conjugation status, concentration, and the duration of exposure.36 At abnormally high concentrations, BAs disrupt cell membranes, cause oxidative/nitrosative stress, and apoptosis.23,36,37 In contrast, prolonged exposure to high concentrations of BAs results in genomic instability and apoptosis resistance, which can lead to the development of cancer.38,39 Our results demonstrate an association between elevated cecal BA levels, including the highly cytotoxic primary BA, CDCA, and colonic epithelial injury in WD-fed mice. As demonstrated by other investigators, CDCA is a potent inducer of epithelial permeability.21,24,25 This together with our data from in vitro permeability assay and in vivo BA-feeding study implicate CDCA as a major player in driving intestinal epithelial permeability, and ultimately NAFLD progression. While CA did not induce epithelial permeability in our in vitro permeability assay, CA is well known to be highly cytotoxic to intestinal epithelial cells,23 and conceivably chronic exposure to a high concentration of CA may contribute to colonic tissue injury. Suggesting that similar mechanisms may be at play in human NASH, a recent study reported increased fecal BA concentrations in NASH patients, and interestingly NASH patients also present with significantly higher levels of CA and CDCA in the stool.26 Collectively, these results suggest that diet-induced changes in the composition of the cecal BA pool could contribute to increased intestinal epithelial permeability that would promote NAFLD progression to NASH with fibrosis.9
Sevelamer hydrochloride has profound BA-binding abilities due to its unique cooperative binding property that enables it to bind more hydrophobic BAs, thereby acting as a sink for more BA binding, ultimately leading to net excretion of BAs in the stool.30 A significant sevenfold increase in CDCA and threefold increase in CA in the cecum content of sevelamer-treated WD-fed mice is a testament to the effectiveness of sevelamer in sequestering these two cytotoxic BAs from the colon. This correlated with a marked reduction in colonic mucosal inflammation and restoration of the colonic epithelial barrier in sevelamer-treated WD-fed mice, suggesting that the ability of sevelamer to bind and neutralize excess BAs in the colon played a central role in protecting the colonic mucosa from the harmful effect of cytotoxic BAs. Collectively these data further establish the role of BAs in diet-induced impairment of colonic epithelial barrier in NASH.
Microbial metabolism of BAs not only increases the diversity and hydrophobicity of the BA pool, but also facilitates passive absorption and excretion of BAs.12 Most gut microbial taxa including Lactobacilli, Bifidobacteria, Clostridium, and Bacteroides have bile salt hydrolase activity and can deconjugate BAs.33,40,41 BA deconjugation, however, prevents the active reuptake of BAs via the ASBT transporter responsible for the majority of BA reuptake in the terminal ileum resulting in the increased passage of BAs to the colon.15 Accordingly, in the absence of bacteria in the germ-free or antibiotic-treated mice, BA pool consists of primary-conjugated BAs, and fecal excretion of BAs is decreased in germ-free mice.32,40 In agreement with these findings, an increase in unconjugated BAs in the colon of WD-fed mice was associated with increased abundance of Clostridium and Bacteroides implicating their potential role in increasing unconjugated BAs in the colon of WD-fed mice. Likewise, an increase in cecal LCA also correlated with higher abundance of Clostridium, which are among a small number of intestinal anaerobes with 7-dehydratase activity, the enzyme required for the production of DCA and LCA.12,33,42 Despite marked improvement in colonic mucosal injury in sevelamer-treated mice, sevelamer treatment did not significantly alter the abundance of taxa belonging to either Clostridium or Bacteroides. An increase in unconjugated BAs in the cecum of sevelamer-treated WD-fed mice suggests that the ability of sevelamer to sequester and neutralize the cytotoxic effect of BAs protected mucosa from BA-induced injury.
Improvement in metabolic derangements associated with NAFLD and NASH in the sevelamer-treated WD-fed mice may be explained in part by increased hepatic BA biosynthesis, which is the major pathway for catabolizing dietary cholesterol and maintaining whole body-cholesterol homeostasis.10,11 Increased BA biosynthesis in transgenic mice overexpressing CYP7A1, the rate-limiting enzyme in hepatic BA biosynthesis, improves metabolic homeostasis, high-fat diet-induced obesity, and insulin resistance.43,44 In agreement with these reports, our data demonstrate a significant increase in hepatic CYP7A1 expression, which was also associated with a significant decrease in serum cholesterol in sevelamer-treated WD-fed mice. Sevelamer treatment also increased hepatic CYP27A1 expression in the WD-fed mice, suggesting that intestinal BA sequestration activates both classic and alternative BA synthesis pathways.45,46 A similar improvement in metabolic parameters was also observed in apical sodium-dependent bile acid transporter (ASBT) inhibitor-treated WD-fed mice where the inhibition of intestinal BA reuptake was associated with increased BA biosynthesis.47 These data suggest that the inhibition of intestinal BA reuptake improves metabolic parameters by increasing hepatic BA biosynthesis. Alternatively, it is possible that the ability of sevelamer to bind lipids or alter intestinal lipid absorption via the neutralization of intestinal BAs may also contribute to the improvement in metabolic derangements in sevelamer-treated WD-fed mice. Since intestinal BA critically regulates hepatic BA synthesis and whole-body metabolism,48–50 further studies are needed to delineate how alterations in the composition of the colonic BA pool or sequestration of intestinal BAs modulates lipid absorption and intestinal BA receptor expression, and to determine their effects on hepatic BA synthesis.
The modest outcomes of recent early phase clinical trials for NASH with fibrosis that are examining the efficacy of various nuclear hormone receptor modulators, fatty acid synthesis inhibitors, anti-apoptotic drugs, anti-fibrotics, and immunomodulators, as well as drugs used for T2DM and autoimmune diseases, demonstrate the complexity to finding effective long-term treatments for NASH.51 There is a significant resurgence in interest in BAs as crucial players mediating the metabolism of carbohydrates and lipids as well as in hepatic inflammation and fibrosis. Therefore, further studies are needed to fully understand the respective mechanistic contributions of BAs not-only with respect to regulation of lipid and carbohydrate metabolism, but also with respect to intestinal epithelial permeability, cellular injury, and repair, as well as hepatic inflammation and fibrosis.
Supplementary Material
ACKNOWLEDGMENTS
This research project was supported in part by the University of Pittsburgh Biospecimen Processing and Repository Core and Advanced Cell and Tissue Imaging Centre of the Pittsburgh Liver Research Centre supported by the NIH/NIDDK Digestive Disease Research Core Center grant P30DK120531; Emory University Integrated Cellular Imaging Microscopy Core of the Emory and Children’s Pediatric Research Center, and the Emory Integrated Lipidomics Core.
FUNDING INFORMATION
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award number K01DK110264 to RR and DK072564 and DK061379 to CP, and by the National Institute on Alcohol Abuse and Alcoholism award number F31AA024960 to DMC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the US Food and Drug Administration, the US Department of Health and Human Services, or the US Government.
Abbreviations:
- CEB
colonic epithelial barrier
- MetS
Metabolic Syndrome
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
Footnotes
CONFLICT OF INTEREST
All authors declare no conflicting interests.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol. 2013;28(Suppl 1):68–76. [DOI] [PubMed] [Google Scholar]
- 2.Kanwal F, Kramer JR, Mapakshi S, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology. 2018;155(1828–1837):e1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams LA, Lymp JF, St Sauver J, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. [DOI] [PubMed] [Google Scholar]
- 5.O’Grady MJ, Capretta JC. Assessing the economics of obesity and obesity interventions. Washington, DC: Campaign to End Obesity:1–40. [Google Scholar]
- 6.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. [DOI] [PubMed] [Google Scholar]
- 8.Luther J, Garber JJ, Khalili H, et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol Gastroenterol Hepatol. 2015;1:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman K, Desai C, Iyer SS, et al. Loss of junctional adhesion molecule a promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology. 2016;151(4):733–746.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiang JY. Recent advances in understanding bile acid homeostasis. F1000Research. 2017;6:2029 10.12688/f1000research.12449.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramirez-Perez O, Cruz-Ramon V, Chinchilla-Lopez P, Mendez-Sanchez N. The role of the gut microbiota in bile acid metabolism. Ann Hepatol. 2017;16:s15–s20. [DOI] [PubMed] [Google Scholar]
- 14.Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41–50. [DOI] [PubMed] [Google Scholar]
- 15.Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56:1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camilleri M Bile acid diarrhea: prevalence, pathogenesis, and therapy. Gut Liv. 2015;9:332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oduyebo I, Camilleri M. Bile acid disease: the emerging epidemic. Curr Opin Gastroenterol. 2017;33:189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Staley C, Weingarden AR, Khoruts A, Sadowsky MJ. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl Microbiol Biotechnol. 2017;101:47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrasa JI, Olmo N, Lizarbe MA, Turnay J. Bile acids in the colon, from healthy to cytotoxic molecules. Toxicol In Vitro. 2013;27:964–977. [DOI] [PubMed] [Google Scholar]
- 20.Stenman LK, Holma R, Eggert A, Korpela R. A novel mechanism for gut barrier dysfunction by dietary fat: epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol. 2013;304:G227–234. [DOI] [PubMed] [Google Scholar]
- 21.Sarathy J, Detloff SJ, Ao M, et al. The Yin and Yang of bile acid action on tight junctions in a model colonic epithelium. Physiol Rep. 2017;5:e13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raimondi F, Santoro P, Barone MV, et al. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G906–913. [DOI] [PubMed] [Google Scholar]
- 23.Araki Y, Katoh T, Ogawa A, et al. Bile acid modulates transepithelial permeability via the generation of reactive oxygen species in the Caco-2 cell line. Free Radic Biol Med. 2005;39:769–780. [DOI] [PubMed] [Google Scholar]
- 24.Munch A, Soderholm JD, Ost A, Carlsson AH, Magnusson KE, Strom M. Low levels of bile acids increase bacterial uptake in colonic biopsies from patients with collagenous colitis in remission. Aliment Pharmacol Ther. 2011;33:954–960. [DOI] [PubMed] [Google Scholar]
- 25.Munch A, Strom M, Soderholm JD. Dihydroxy bile acids increase mucosal permeability and bacterial uptake in human colon biopsies. Scand J Gastroenterol. 2007;42:1167–1174. [DOI] [PubMed] [Google Scholar]
- 26.Mouzaki M, Wang AY, Bandsma R, et al. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS ONE. 2016;11:e0151829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chopyk DM, Kumar P, Raeman R, Liu Y, Smith T, Anania FA. Dysregulation of junctional adhesion molecule-A contributes to ethanol-induced barrier disruption in intestinal epithelial cell monolayers. Physiol Rep. 2017;5(23):e13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96:736–749. [PubMed] [Google Scholar]
- 30.Braunlin W, Zhorov E, Guo A, et al. Bile acid binding to sevelamer HCl. Kidney Int. 2002;62:611–619. [DOI] [PubMed] [Google Scholar]
- 31.McGettigan BM, McMahan RH, Luo Y, et al. Sevelamer improves steatohepatitis, inhibits liver and intestinal farnesoid X receptor (FXR), and reverses innate immune dysregulation in a mouse model of nonalcoholic fatty liver disease. J Biol Chem. 2016;291:23058–23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sayin SI, Wahlstrom A, Felin J, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–235. [DOI] [PubMed] [Google Scholar]
- 33.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. [DOI] [PubMed] [Google Scholar]
- 34.Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci U S A. 2008;105:13580–13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devkota S, Wang Y, Musch MW, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hegyi P, Maleth J, Walters JR, Hofmann AF, Keely SJ. Guts and gall: bile acids in regulation of intestinal epithelial function in health and disease. Physiol Rev. 2018;98:1983–2023. [DOI] [PubMed] [Google Scholar]
- 37.Ignacio Barrasa J, Olmo N, Perez-Ramos P, et al. Deoxycholic and chenodeoxycholic bile acids induce apoptosis via oxidative stress in human colon adenocarcinoma cells. Apoptosis. 2011;16:1054–1067. [DOI] [PubMed] [Google Scholar]
- 38.Debruyne PR, Bruyneel EA, Li X, Zimber A, Gespach C, Mareel MM. The role of bile acids in carcinogenesis. Mutat Res. 2001;480–481:359–369. [DOI] [PubMed] [Google Scholar]
- 39.Bernstein C, Holubec H, Bhattacharyya AK, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol. 2011;85:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narushima S, Ito K, Kuruma K, Uchida K. Composition of cecal bile acids in ex-germfree mice inoculated with human intestinal bacteria. Lipids. 2000;35:639–644. [DOI] [PubMed] [Google Scholar]
- 41.Narushima S, Itoha K, Miyamoto Y, et al. Deoxycholic acid formation in gnotobiotic mice associated with human intestinal bacteria. Lipids. 2006;41:835–843. [DOI] [PubMed] [Google Scholar]
- 42.Kitahara M, Takamine F, Imamura T, Benno Y. Assignment of Eubacterium sp. VPI 12708 and related strains with high bile acid 7alpha-dehydroxylating activity to Clostridium scindens and proposal of Clostridium hylemonae sp. nov., isolated from human faeces. Int J Syst Evol Microbiol 50 Pt. 2000;3:971–978. [DOI] [PubMed] [Google Scholar]
- 43.Li T, Francl JM, Boehme S, Chiang JY. Regulation of cholesterol and bile acid homeostasis by the cholesterol 7alpha-hydroxylase/steroid response element-binding protein 2/microRNA-33a axis in mice. Hepatology. 2013;58:1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li T, Owsley E, Matozel M, Hsu P, Novak CM, Chiang JY. Transgenic expression of cholesterol 7alpha-hydroxylase in the liver prevents high-fat diet-induced obesity and insulin resistance in mice. Hepatology. 2010;52:678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–693. [DOI] [PubMed] [Google Scholar]
- 46.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. [DOI] [PubMed] [Google Scholar]
- 47.Rao A, Kosters A, Mells JE, et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci Transl Med. 2016;8:357ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang S, Suh JM, Reilly SM, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med. 2015;21:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li F, Jiang C, Krausz KW, et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun. 2013;4:2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drucker DJ. Minireview: the glucagon-like peptides. Endocrinology. 2001;142:521–527. [DOI] [PubMed] [Google Scholar]
- 51.Lemoinne S, Friedman SL. New and emerging anti-fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr Opin Pharmacol. 2019;49:60–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.