

Abstract
Nucleobase radicals are the major family of reactive intermediates formed when nucleic acids are exposed to hydroxyl radical, which is produced by γ-radiolysis and Fe • EDTA. Significant advances have been made in understanding the role of nucleobase radicals in oxidative DNA damage by independently generating these species from photochemical precursors. However, this approach has been used much less frequently to study RNA molecules. Norrish type I photocleavage of the tert-butyl ketone (2b) enabled studying the reactivity of 5′-benzoyl-5,6-dihydrouridin-6-yl (1b). High mass balances were observed under aerobic or anaerobic conditions, and O2 did not affect the photochemical conversion of the ketone (2b) to 1b. Competition studies with O2 indicate that the radical abstracts hydrogen atoms from β-mercaptoethanol with a bimolecular rate constant = 2.6 ± 0.5 × 106 M−1s−1. The major product formed in the presence of O2 was 5′-benzoyl-6-hydroxy-5,6-dihydrouridine (6). In contrast, 5-benzoyl-ribonolactone (7), a hypothetical product resulting from C1′-hydrogen atom abstraction by the peroxyl radical, could not be detected. Overall, tert-butyl ketone 2b is a clean source of 5′-benzoyl-5,6-dihydrouridin-6-yl (1b) and should prove useful for studying the reactivity of the respective radical in RNA.
Graphical Abstract
Introduction
It is widely accepted that oxidative DNA damage is associated with a variety of diseases and aging. Because of its larger copy number and shorter lifetime in many cells, RNA damage was perceived to be less significant biologically. This perception has begun to change as RNA oxidation has been posited as playing a role in Alzheimer’s disease, atherosclerosis, and inflamation.1–3 Although the appreciation of oxidative RNA damage as a cause and/or effect of disease is growing, its greatest impact on biology thus far has come from its use as a tool for studying RNA structure, folding dynamics, and protein binding.4–8 In this regard RNA cleavage by hydroxyl radical (HO•) generated by Fe•EDTA or via pulse radiolysis is the most frequently employed oxidant.9–13 These investigations rely upon the accessibility of the biopolymer to the extremely reactive HO• to deduce 3-dimensional structure. Studies on the effects of ionizing radiation on nucleic acids that employed RNA as a substrate implicate a variety of possible pathways by which HO• may produce direct strand breaks. Because of the poor selectivity of this reactive oxygen species, it is difficult to unequivocally characterize any one pathway. We and other research groups have utilized organic chemistry to study nucleic acid oxidation by independently generating reactive intermediates in nucleosides and nucleotides and at defined sites in oligonucleotides. 14–19 This has been a fruitful approach that has uncovered novel reaction pathways and helped resolve mechanistic controversies while improving our overall understanding of biologically important chemistry.20–26 Although most of these studies have focused on DNA, there has been limited investigation of RNA radical chemistry.27–29 The growing acceptance of the importance of RNA oxidation has prompted us to turn our attention to this nucleic acid molecule.
The primary structural difference between RNA and DNA is the presence of the 2′-hydroxyl group in the former. The 2′-hydroxyl group may have a significant effect on the reactivity of RNA radicals in several ways. The C2′-H bond dissociation energy is significantly lower in RNA compared to that in DNA. Computational experiments show that the C2′-H bond is the weakest carbohydrate carbon-hydrogen bond in nucleic acids.30 In addition, the C2′-hydroxyl group may facilitate heterolytic β-fragmentation from the C2′-radical that is analogous to those observed in C4′-radicals in DNA.17,31 The electronic effects of the 2′-hydroxyl group may affect nucleic acid radical reactivity in other more subtle, indirect ways.27 For instance, the altered conformation of RNA (A-form) compared to DNA (B-form) may influence radical reactivity by affecting the proximity of potential reactive centers to one another. One similarity between RNA and DNA is that nucleobase radicals resulting from hydroxyl radical addition to the π-bonds of the Watson–Crick bases are believed to account for more than 80% of the reactive intermediates generated.11 Independent generation of 2′-deoxypyrimidine nucleobase radicals has revealed the formation of tandem lesions via reaction with adjacent nucleotides but few if any direct strand breaks.14,24,25,32,33 In contrast, analogous pyrimidine nucleobase radicals are believed to produce direct strand breaks via intranucleotidyl and internucleotidyl hydrogen atom abstraction from the ribose rings.11–13 Given the predominance and proposed distinctive reactivity of RNA pyrimidine nucleobase radicals, we set out to independently generate one of them in order to begin to gain insight into their chemical behavior. Characterization of nucleotide radical reactivity at specific sites in RNA could facilitate extracting additional structural information from HO• cleavage experiments and shed light on why RNA oxidation is deleterious in cells. However, product analysis of radical reactions in biopolymers is challenging, and it is imperative to demonstrate that the radical of interest is cleanly generated in solution at the monomeric level before carrying out such studies.
Results and Discussion
Synthesis of a Photochemical Precursor to 5,6-Dihydrouridin-6-yl (1) and Product Standards.
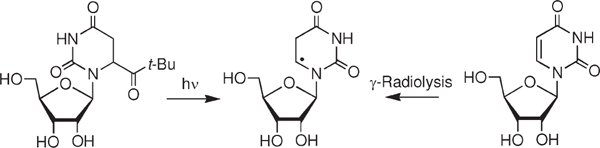
Photochemistry is a desirable method for independently generating nucleic acid radicals because this method provides temporal control, is often compatible with aqueous conditions, and can be carried out without exposing the biopolymer to conditions or reagents that may randomly damage it. Hydroxyl radical preferentially adds to the C5-position of pyrimidines (Scheme 1).11 We chose to synthesize a C6-radical precursor (2), which generates the analogous radical (1) resulting from the formal C5-hydrogen atom addition product instead of HO• addition (eq 1). Hydrogen atoms are formed in lower yields by γ-radiolysis but add to the pyrimidine double bonds and exhibit similar regioselectivity as hydroxyl radical.34 Ketone 2 was targeted for synthetic expediency. However, we cannot rule out that the absence of the C5-hydroxyl group (β-position of the radical) would make the radical less reactive. The Norrish type I photochemical cleavage reaction was previously used when studying the role of pyrimidine nucleobase radicals in DNA oxidation.24,25,32,35–38 Based upon this precedent and synthetic methods developed by Tanaka et al., the synthesis of 2 was straightforward (Scheme 2).39,40 The ribose protecting groups could be removed under fairly strong acidic conditions due to the greater stability of the glycosidic bond in the ribonucleoside compared to a 2′-deoxyribonucleoside.
Scheme 1.
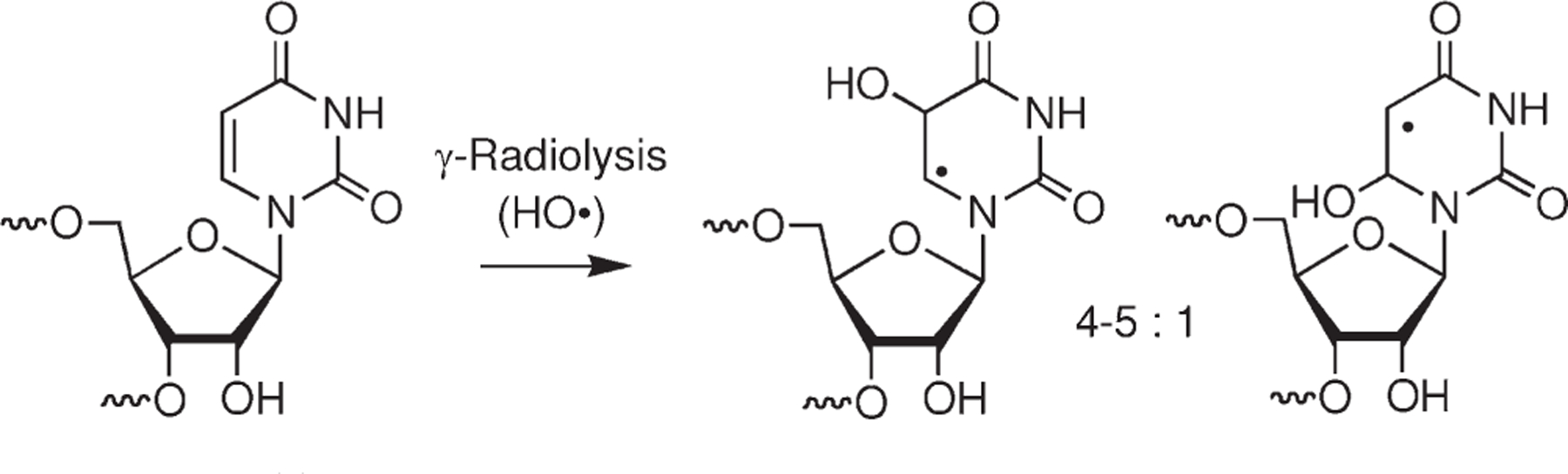
Generation of Nucleobase Radicals in RNA via Hydroxyl Radical Addition
Scheme 2.
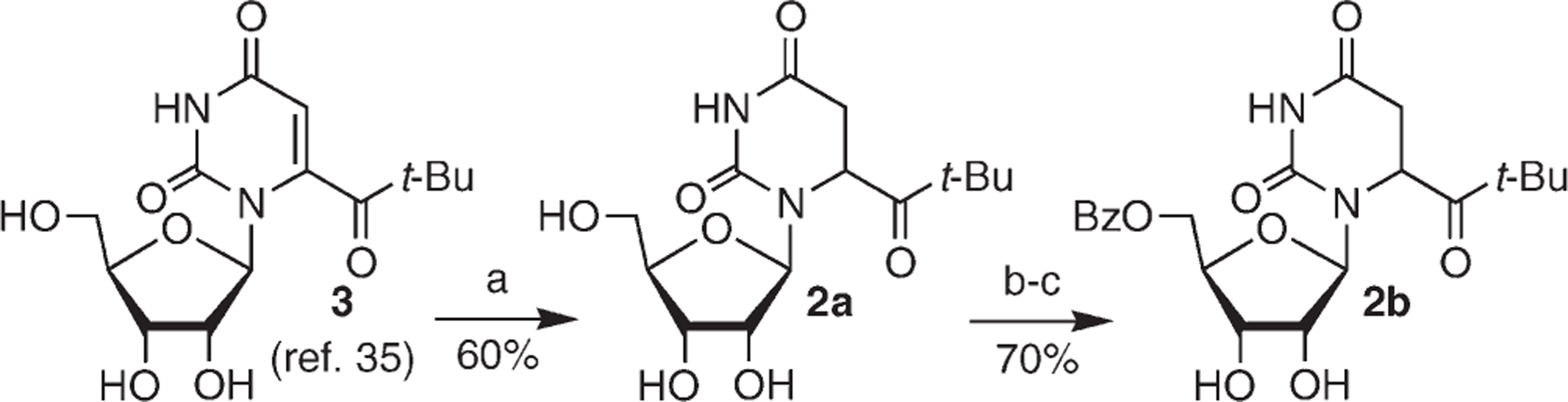
Synthesis of Photochemical Radical Precursors 2a and 2ba
aReagents and conditions: (a) 5% Rh/Al2O3, H2 (45 psi), MeOH; (b) (i) pTSA, dimethoxy propane, DMF, (ii) BzCl; (c) TFA/H2O (1:1).
 |
(1) |
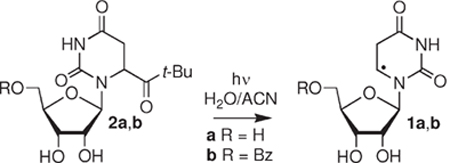
To enhance the detection of the ketone and the products (4–7) derived from 1 via HPLC using UV-detection, a benzoyl group was installed at the 5′-position (2b). The benzoyl derivatives of the radical precursor (2b), 5,6-dihydrouridine (4), and uridine (5) were prepared from the free ribonucleosides via a two-pot procedure that did not require purification of the intermediates.41 The C6-hydrate (6) was synthesized from 5 via the respective bromohydrins, which were prepared using a previously reported method.42,43 Only the major bromohydrin diastereomer was debrominated using zinc in acetic acid, because the C6-hydrate epimerizes in water. Finally, 5-benzoyl ribonolactone (7) was prepared via the reported procedure.44
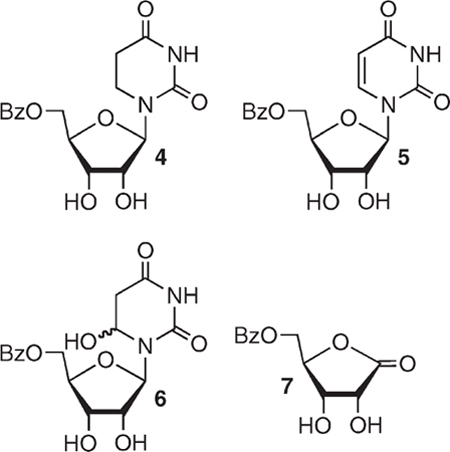
Photochemical Generation of 5,6-Dihydrouridin-6-yl Radical (1b).
As a prelude to using the tert-butyl ketone (2) to characterize the reactivity of 5,6-dihydrouridin-6-yl (1) and as a radical precursor in RNA, we needed to establish the fidelity of the monomer’s photochemistry. In initial experiments we characterized the mass balance of photolyses of 2b (≤50 μM) under aerobic and anaerobic conditions in the presence of a low (0.5 mM) but excess concentration of β-mercaptoethanol (Table 1). Photolyses were carried out to ∼60% conversion of the ketone. Two products, the C6-hydrate (6) and uridine (5), were detected under aerobic conditions in addition to starting ketone (2b). The average mass balance was greater than 85%. The mass balance is considerably lower under aerobic conditions in the absence of thiol. A more complex mixture of products is formed under these conditions, presumably as a result of reactions between alkyl and peroxyl radicals.
Table 1.
Product Yields and Mass Balance from Photolysis of 2ba
| % Yieldb |
|
||||
|---|---|---|---|---|---|
| conditions | 4 | 5 | 6 | 2b | mass balance (%) |
| aerobic | n.d. | 7.1 ± 0.3 | 37.6 + 0.1 | 41.5 ± 1.4 | 86.5 ± 1.6 |
| anaerobic | 50.8 ± 1.2 | n.d. | n.d. | 42.2 ± 1.1 | 93.6 + 2.1 |
Photolyses carried out for 90 mm in the presence of 0.5 mM β-mercaptoethanol.
Yields and mass balances are the avenge of 3 samples. n.d. = not detected.
The mass balance is even higher (>90%) under anaerobic conditions, where the only product detected is the dihydrouridine (4) formed by hydrogen atom abstraction by 1b. The requirement of O2 for the formation of the C6-hydrate (6) is consistent with the formation of this product via the peroxyl radical (8, Scheme 3) and not direct one-electron oxidation of 1b, followed by trapping by H2O.
Scheme 3.
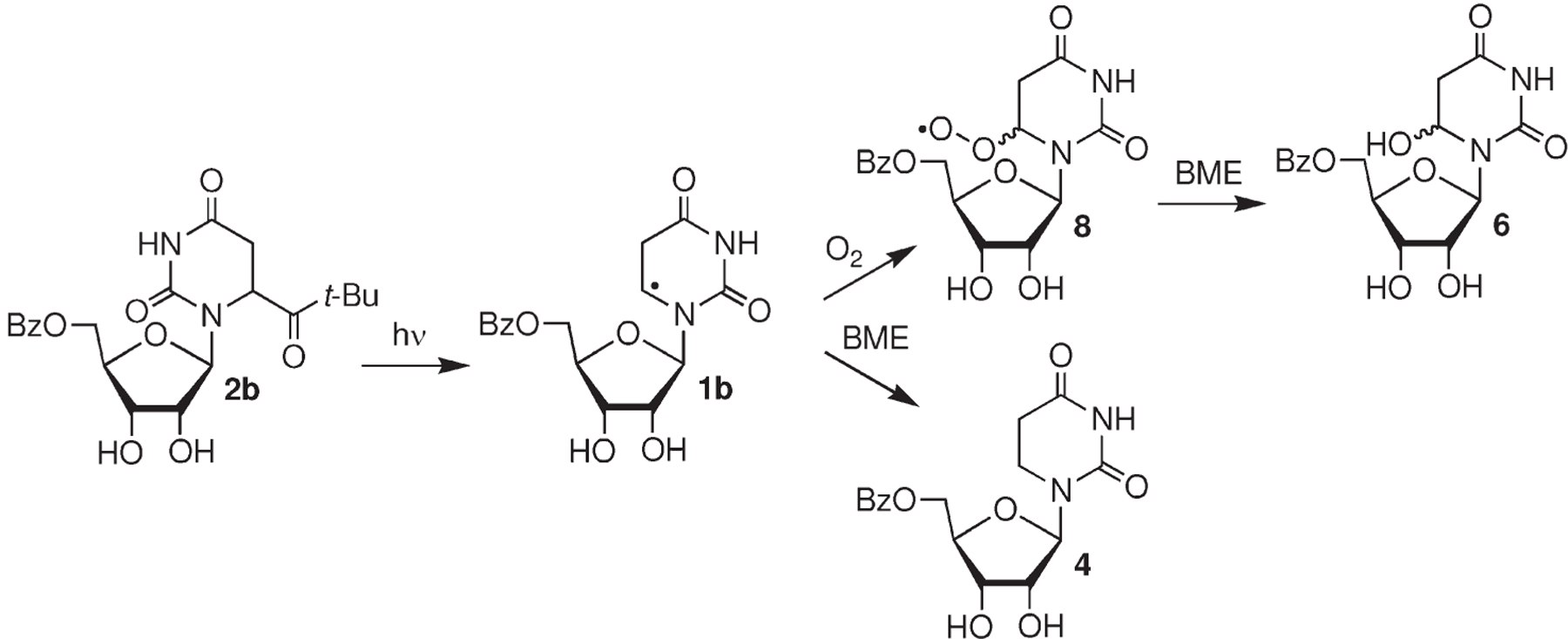
Competitive Trapping of 1b by O2 and β-Mercaptoethanol
In addition to determining the effect of O2 on the mass balance and product distribution, we examined its effect on the photochemical conversion of 2b (Figure 1) because dioxygen quenches the excited states of triplet ketones.45 However, we observed no qualitative difference in the photoconversion of 2b as a function of time in the presence or absence of O2. Furthermore, the ketone conversion was linear with respect to time, even at high conversion. This is consistent with a zero-order (nonchain) photochemical process.
Figure 1.
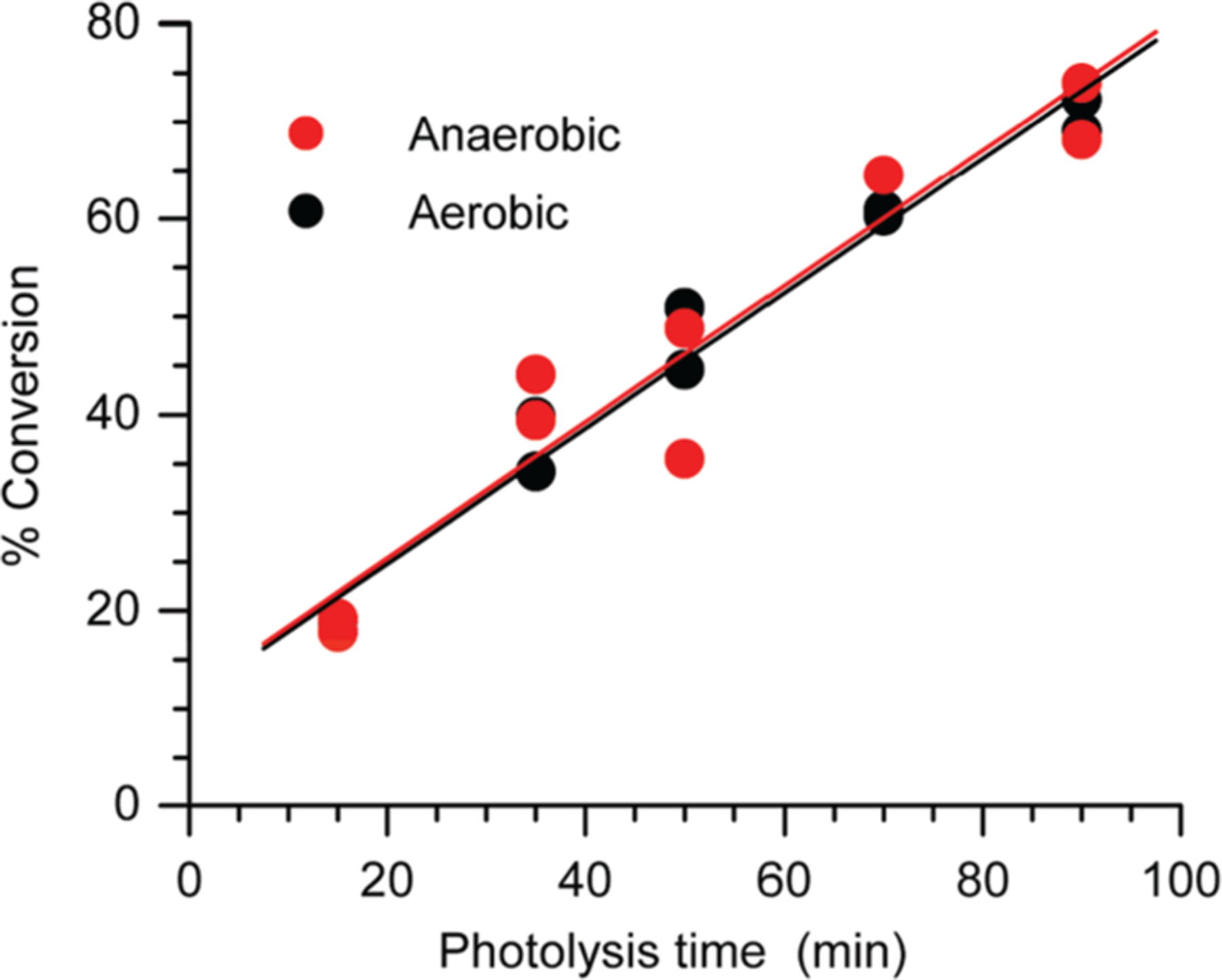
Photochemical conversion of 2b as a function of time under aerobic and anaerobic conditions.
Reactivity of 5′-Benzoyl-5,6-dihydrouridin-6-yl (1b) with β-Mercaptoethanol.
The competition between O2 and β-mercaptoethanol (BME) was explored as a function of thiol concentration (Figure 2). The average slope of this line (6.6 ± 1.3 × 10−3 M−1) is equal to the ratio of the rate constants for trapping 1b by BME and O2 times the reciprocal of the O2 concentration (0.2 mM, kRSH/(kO2[O2]), eq 2). The rate constant for the reaction of 1b with BME (kRSH = 2.6 ± 0.5 × 106 M−1 s−1) was estimated by assuming that the rate constant for O2 trapping of the nucleoside radical (kO2) is 2 × 109 M−1 s−1. This is more than 3-fold slower than the respective rate constant measured for the reaction of the 2′-deoxynucleoside analogue of 1 with BME, although it is not known why.35
| (2) |
Figure 2.
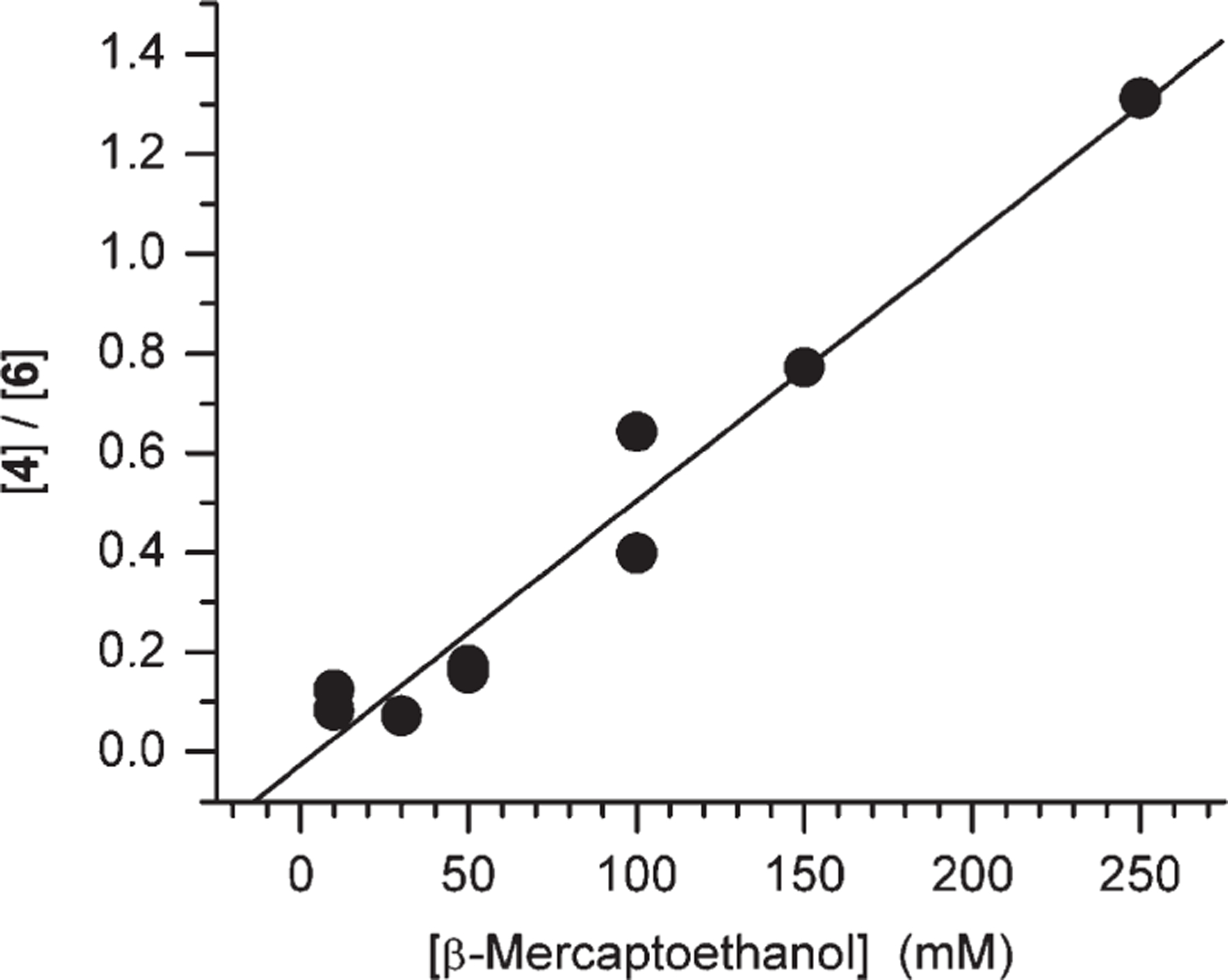
Ratio of 5,6-dihydrouridine (4) to 6-hydroxy-5,6-dihydrouridine (6) as a function of BME concentration.
Search for C1′-Hydrogen Atom Abstraction by the C6-Peroxyl Radical (8).
2-Deoxyribonolactone was produced in low yield by the 2′-deoxynucleoside analogue under aerobic conditions.35 The postulated mechanism involved C1′-hydrogen atom abstraction by the respective peroxyl radical and subsequent transformation of the C1′-radical via a known O2-dependent mechanism to the lactone.46,47 Analysis for ribonolactone (7) formation was carried out using aqueous (unbuffered) acetonitrile (10% acetonitrile) because it was unstable in aqueous buffer. We were unable to detect 5-benzoyl ribonolactone (7) upon photolysis of 2b (85 μM) under aerobic conditions. Using independently synthesized 7, we established the limit of detection of the lactone in these experiments to be 5%. We speculate that C1′-hydrogen atom abstraction by the electrophilic peroxyl radical (7) is even slower than the analogous reaction in the deoxyribonucleoside system, due to the inductive effect of the 2′-hydroxyl group (Scheme 4).
Scheme 4.
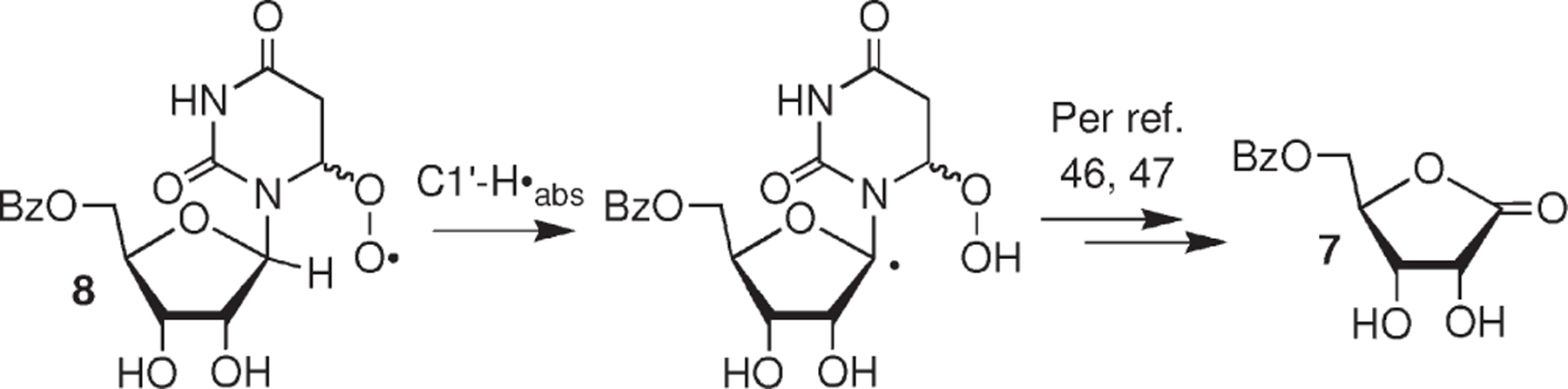
Hypothetical Formation of 5-Benzoyl Ribonolactone (7) from 8
Formation of 5′-Benzoyl-uridine (5) from 5,6-Dihydrouridin-6-yl (1b).
5′-Benzoyl-uridine (5) is observed under aerobic but not anaerobic conditions (Table 1), and its yield is as high as ∼20% at low concentrations of BME (<1 mM). Dehydration of the C6-hydrate was a possible source of this product; as such molecules are reported to undergo this reaction.11 However, examination of independently synthesized 6 showed that the hydrate is stable under the reaction conditions. Since benzoylated uridine (5) formation was observed only under aerobic conditions we focused our attention on the peroxyl radical (8) as its source. Formal elimination of the hydroperoxyl radical, which deprotonates to superoxide at pH 7.2, from 7 would yield 5′-benzoyl uridine. This process has been observed for other peroxyl radicals but is typically slow unless the incipient carbocation is especially stabilized.46–49 No evidence for superoxide elimination was detected using either epinephrine oxidation or cytochrome c reduction as a means for indirect detection (data not shown). In addition, uridine formation was efficiently quenched by β-mercaptoethanol at concentrations much lower than those used to compete with O2 for 1b. However, the yield varied nonlinearly with thiol concentration (Figure 3). This indicated that the thiol did not trap the peroxyl radical (7) in competition with a first-order process. The behavior is suggestive of quenching a radical-radical reaction, but we do not have any additional evidence for such a process. Overall, these experiments reveal that 5′-benzoyl uridine (5) results from a radical process but its yield is reduced to 1–2% at even very low concentrations of thiol.
Figure 3.
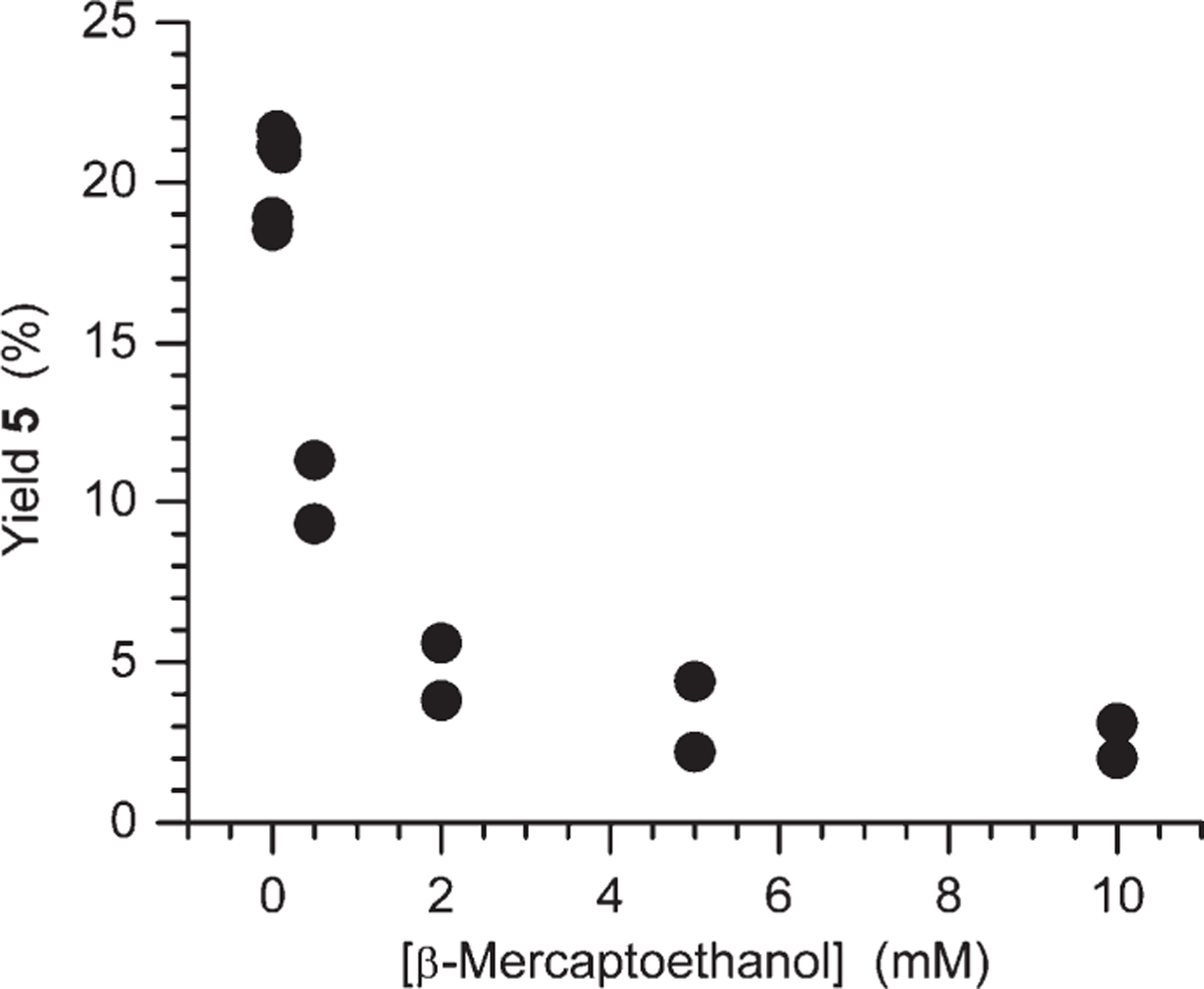
Dependence of uridine yield on β-mercaptoethanol concentration formed upon photolysis of 2b under aerobic conditions.
Conclusions
These studies establish that 5,6-dihydrouridin-6-yl can be generated cleanly via Norrish type I photocleavage. The mass balances are high, and under anaerobic conditions in the presence of mM thiol 1–2% of uridine is produced via an uncertain mechanism that involves the alkyl radical. The reactivity of 5,6-dihydrouridin-6-yl radical is similar to that of the 2′-deoxyribonucleoside analogue. However, it is slightly less reactive with thiol and intramolecular hydrogen atom abstraction from the C1′-position could not be detected. The C2′-position of RNA is expected to be more susceptible to hydrogen atom abstraction than it is in DNA, and it is possible some of the unaccounted for material (7–14%) in these experiments is consumed by this or other pathways involving intramolecular hydrogen atom abstraction.
Experimental Section
General Procedure for the Photolysis of 2b.
Solutions for all experiments were prepared in Eppendorf tubes to have the same concentration of 2b (50 μM) in a 10% CH3CN/phosphate buffer (10 mM, pH 7.2) mixture. The internal standard (dU) was added to yield a final concentration of 17 μM. The solutions were then vortexed and transferred into Pyrex tubes. Anaerobic photolyses were carried out in sealed tubes that were subjected to 3 freeze–pump–thaw degas cycles. All tubes were placed at a similar distance (ca. 5 cm) from the lamps and rotated during irradiation (350 nm). Experiments were carried out in multiple replicates (2 or 3). All photolysis samples were kept at room temperature.
Preparation of 2a.
To a high pressure flask equipped with a magnetic stir bar was added a solution of 339 (1.4 g, 4.3 mmol) in methanol (65 mL). Rh/Al2O3 (1.74 g, 0.85 mmol Rh) was then added to the flask. The flask was placed under 45 psi H2 atmosphere and vented to release the pressure. This was repeated 3, and the fourth time, the flask was kept at 45 psi H2. The reaction was allowed to stir at room temperature for 3 h. The mixture was filtered through Celite and washed ×3 with methanol (∼300 mL). The solvent was removed under reduced pressure, and purification was accomplished via flash column chromatography (5% methanol in dichloromethane) to give 0.84 g (60% yield) of a 3.4:1 mixture of diastereomers of 2a as a white foam. 1H NMR (CD3OD) δ 1.24 (s) and 1.26 (s) (9H), 2.80–2.85 (m, 1H), 3.14–3.20 (m, 1H), 3.66–3.87 (m, 3H), 3.97–4.00 (m, maj) and 4.10–4.14 (m, min) (2H) and 5.27–5.30 (m,1H), 5.53–5.54 (d, J = 4.0 Hz, min) and 5.68–5.69 (m, maj) (1H); 13C NMR (CDCl3) δ 26.4, 26.5, 33.2, 33.7, 42.9, 43.1, 52.8, 54.0, 60.9, 61.1, 69.6, 70.1, 71.4, 73.4, 83.5, 84.2, 89.0, 90.3, 153.8, 154.6, 168.35, 168.4, 211.4, 212.5. IR (neat) 1023.7 m, 1164.2 m, 1106.4 m, 1225.0 m, 1289.6 w, 1372.2, m, 1463.3 m, 1682.7 s, 1694.3 s, 1713.2 s, 2802.5 w, 2842.9 w, 2966.9 m, 3422.7 br s cm−1; HR-FABMS calculated for C14H23N2O7 (M + H+) 331.1517, found 331.1526.
General Procedure for Preparation of 5′-Benzoylated Compounds.
To a flame-dried round-bottom flask equipped with a magnetic stir bar were added the ribonucleoside (1.0 equiv), p-toluenesulfonic acid monohydrate (0.23 equiv), and 4 Å M.S. The contents were diluted with DMF (2.9 mL/mmol) and dimethoxy propane (3.9 equiv). The flask was heated to 45 °C for 2 h, at which time the reaction mixture was cooled to room temperature and Amberlyst A-21 resin (0.05 g/mmol) was added to neutralize the acid. The mixture was stirred for 20 min and then filtered through a pad of Celite. The Celite pad was washed with methanol, and then the combined solvents were removed under reduced pressure. The crude oil was used for the next step without purification.
Assuming 100% conversion in the first step, the crude acetonide was diluted with pyridine (9.8 mL/mmol), and then benzoyl chloride (1.5 equiv) was added dropwise over 5 min. The reaction mixture was stirred for 17 h at room temperature, at which time it was evaporated under reduced pressure. To ensure the pyridine was removed, toluene (1 mL/mmol) was added and removed under vacuum (×2). The material was then diluted with dichloromethane and transferred to a separatory funnel containing water. The layers were separated, and the dichloromethane was dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The benzoylated acetonide was used for the next step without further purification.
Again assuming 100% conversion to the product, a 1:1 mixture of trifluoroacetic acid/water (10.8 mL/mmol) was added to the crude benzoylated acetonide, and the mixture was stirred at room temperature for 21 h. The water and TFA were removed under reduced pressure. The final products were purified via flash column chromatography (8% MeOH in chloroform, unless specified otherwise) and isolated as white foams. When the reaction was carried out using 5.0 g (20.4 mmol) of uridine, 5.16 g (72% yield) of the desired, previously reported product 5′-benzoyl uridine (5) was obtained.41
Preparation of 4.
The reaction was carried out using 1.0 g (4.06 mmol) of dihydrouridine, and 0.487 g (34% yield) of the desired product was isolated. 1H NMR (CD3OD) δ 2.51–2.55 (m, 2H), 3.35–3.46 (m, 2H), 4.14–4.17 (m, 3H), 4.42–4.46 (dd, 1H, J = 4.8, 12 Hz), 4.60–4.64 (dd, 1H, J = 3.2, 12 Hz), 5.86–5.87 (d, 1H, J = 4.8 Hz), 7.49–7.53 (m, 2H), 7.61–7.66 (m, 1H), 8.04–8.05 (m, 2H); 13C NMR δ 30.5, 36.8, 64.2, 70.6, 71.2, 80.8, 89.0, 128.5, 129.3, 129.9, 133.2, 153.8, 166.4, 171.3; IR 599 w, 713 m, 762 w, 906 w, 1047 m, 1118 m, 1179 w, 1276 s, 1316 w, 1376 m, 1377 m, 1602 w, 1699 br s, 1713 br s, 2986 w, 2999 w, 3116 w, 3258 br m cm−1; HR-FABMS calculated for C16H19N2O7 (M + H+) 351.1204, found 351.1182.
Preparation of 2b.
The reaction was carried out using 100 mg (0.303 mmol) of 2a, and 62.2 mg (70% yield) of the desired product was isolated as a 5.9:1 mixture of diastereomers. Purification was carried out via flash chromatography using 4% MeOH in chloroform. 1H NMR (CD3OD) δ 1.10 (s, min) and 1.16 (s, maj) (1H), 2.60–2.67 (m, 1H), 3.05–3.15 (m, 1H), 4.02–4.21 (m, 3H), 4.39–4.45 (m, 1H), 4.55–4.58 (m, 1H), 4.91–4.93 (d, J = 7.6 Hz, min) and 4.98–5.00 (d, J = 7.6 Hz, maj) (1H), 5.36–5.37 (d, J = 4.0 Hz, maj) and 5.46 (s, min) (1H), 7.46–7.53 (m, 2H), 7.59–7.66 (m, 1H), 8.03–8.05 (d, 2H, J = 7.2 Hz); 13C NMR (CD3OD) δ 27.2, 27.7, 35.2, 44.7, 56.7, 65.9, 71.9, 72.9, 81.6, 94.9, 129.8, 129.9, 130.7, 130.8, 131.3, 134.6, 155.0, 167.9, 169.9, 170.1, 213.3; IR (neat) 584 w, 714 m, 763 w, 908 w, 969 w, 1026 m, 1069 m, 1109 m, 1179 w, 128 s, 1316 w, 12372 m, 1463 m, 1602 w, 1716 br s, 2971 m, 3247 br m cm−1; HR-FABMS calculated for C21H27N2O8 (M + H+) 435.1697, found 435.1759.
Preparation of 6.
To a round-bottom flask equipped with a magnetic stir bar was added the benzoylated uridine (1.0 g, 2.87 mmol) and a 1:1 mixture of THF/water (80 mL). The mixture was cooled to 0 °C, at which time NBS (0.612 g, 3.44 mmol) and calcium carbonate (0.43 g, 4.3 mmol) were added. The ice bath was removed, and the reaction was allowed to stir at room temperature for 4 h. The crude reaction mixture was then filtered through a pad of Celite, washed with methanol (∼200 mL), and concentrated under reduced pressure. Purification was done via flash column chromatography using a solvent gradient from 2% MeOH in chloroform to 5% MeOH in chloroform to give 578 mg (45% yield) of the less polar diastereomer and 155 mg (12% yield) of the more polar diastereomer. Less polar diastereomer: 1H NMR (CD3OD) δ 4.22–4.39 (m, 4H), 4.59–4.70 (m, 2H), 5.36 (s, 1H), 5.84 (s, 1H), 7.51–7.58 (m, 2H), 7.65–7.69 (m, 1H), 8.08–8.15 (m, 2H); 13C NMR (CD3OD) δ 40.3, 64.0, 70.3, 73.9, 77.0, 80.9, 89.8, 128.4, 129.2, 129.8, 133.2, 151.1, 166.45, 166.59; IR (neat) 567 w, 714 m, 763 w, 905 w, 1072 m, 1118 m, 1179 w, 1281 m, 1317 w, 1385 w, 1455 m, 1464 m, 1602 w, 1683 s, 1695 s, 1699 s, 1714 s, 1733 s, 2534 br m, 3440 br s cm−1; HR-FABMS calculated for C16H17BrN2O8 427.0168, found (M - OH) 427.0144. More polar diastereomer: 1H NMR (CD3OD) δ 4.20–4.36 (m, 3H), 4.40–4.42 (m, 1H), 4.54–4.58 (dd, 1H, J = 5.2, 12 Hz), 4.69–4.72 (dd, 2H, J = 2.8, 6.8 Hz), 5.33 (d, 1H, J = 2.4 Hz), 5.85–5.86 (d, 1H, J = 3.6 Hz), 7.52–7.56 (m, 2H), 7.64–7.69 (m, 1H), 8.10–8.12 (d, 1H, J = 7.2 Hz); 13C NMR(CD3OD) δ 41.3, 65.3, 71.4, 74.4, 78.4, 79.5, 81.6, 91.4, 129.7, 130.1, 130.69, 130.74, 130.8, 131.3, 134.6, 152.2, 167.9, 168.0; IR(neat) 554 w, 713 m, 762 m, 906 w, 1027 m, 1118 m, 1179 w, 1281 m, 1317 w, 1385 w, 1455 m, 1464 m, 1602 w, 1683 s, 1694 s, 1699 s, 1712 s, 1722 s, 2526 br m, 3409 br s cm−1.
To a round-bottom flask equipped with a magnetic stir bar were added the less polar diastereomer of the bromohydrin (359 mg, 0.81 mmol) and a 3:1 mixture of THF/water (8 mL). The solution was cooled to 0 °C, at which time Zn dust (75.8 mg, 1.16 mmol) and acetic acid (85 μL, 1.47 mmol) were added. The ice bath was removed, and the reaction was allowed to stir atroom temperature for 22 h. The mixture was then filtered through a pad of Celite and washed with methanol. After removing the solvent under reduced pressure 6 (81 mg, 28%) was purified via flash column chromatography (7% MeOH in chloroform). 1H NMR (CD3OD) δ 2.53–2.57 (d, 1H, J = 16.8 Hz), 2.85–2.90 (m, 1H), 4.21–4.29 (m, 3H), 4.53–4.69 (m, 2H), 5.33 (s, 1H), 8.05–8.07 (d, J = 7.6 Hz), 7.49–7.51 (t, 2H, J = 7.6 Hz), 7.62–7.66 (m, 1H), 8.05–8.07 (d, 2H, J = 7.8 Hz); 13C NMR (CD3OD) δ 39.9, 65.7, 71.7, 74.4, 75.3, 82.5, 92.1, 129.8, 130.7, 131.2, 134.7, 154.2, 168.0, 171.4; IR (neat) 712 m, 760 w, 892 w, 1026 m, 1061 m, 1120 m, 1276 s, 1316 w, 1384 m, 1474 m, 1601 w, 1701 br s, 3295 v br m cm−1; HR-FABMS calculated for C16H18N2O8 349.1063, found (M – OH+) 349.1029.
Supplementary Material
Acknowledgment.
We are grateful for support of this research from the National Institute of General Medical Sciences (GM-054996). J.T.S. thanks The Johns Hopkins University for a Sonneborn fellowship. M.J.E.R. thanks the NIGMS for a Research Supplement to Promote Diversity in Health-Related Research.
Footnotes
Supporting Information Available: General experimental methods, NMR spectra of previously unreported molecules and 7, and a table of HPLC response factors. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Martinet W; DeMeyer GRY; Herman AG; Kockz MM Eur. J. Clin. Invest 2004, 34, 323–327. [DOI] [PubMed] [Google Scholar]
- (2).Moreira PI; Nunomura A; Nakamura M; Takeda A; Shenk JC; Aliev G; Smith MA; Perry G Free Radical Biol. Med 2008, 44, 1493–1505. [DOI] [PubMed] [Google Scholar]
- (3).Dedon PC; DeMott MS; Elmquist CE; Prestwich EG; McFaline JL; Pang B Biomarkers Med 2007, 1, 293–312. [DOI] [PubMed] [Google Scholar]
- (4).Tullius TD; Greenbaum JA Curr. Opin. Chem. Biol 2005, 9, 127–134. [DOI] [PubMed] [Google Scholar]
- (5).Brenowitz M; Chance MR; Dhavan G; Takamoto K Curr. Opin. Struct. Biol 2002, 12, 648–653. [DOI] [PubMed] [Google Scholar]
- (6).Adilakshmi T; Bellur DL; Woodson SA Nature 2008, 455, 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chauhan S; Woodson SA J. Am. Chem. Soc 2008, 130, 1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Adilakshmi T; Lease RA; Woodson SA Nucleic Acids Res 2006, 34, e64/1–e64/7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ralston CY; Sclavi B; Sullivan M; Deras ML; Woodson SA; Chance MR; Brenowitz M Methods Enzymol 2000, 317, 353–368. [DOI] [PubMed] [Google Scholar]
- (10).Sclavi B; Sullivan M; Chance MR; Brenowitz M; Woodson SA Science 1998, 279, 1940–1943. [DOI] [PubMed] [Google Scholar]
- (11).von Sonntag C The Chemical Basis of Radiation Biology; Taylor & Francis: London, 1987. [Google Scholar]
- (12).Jones GDD; O’Neill P Int. J. Radiat. Biol 1990, 57, 1123–1139. [DOI] [PubMed] [Google Scholar]
- (13).Jones GDD; O’Neill P Int. J. Radiat. Biol 1991, 59, 1127–1145. [DOI] [PubMed] [Google Scholar]
- (14).Greenberg MM Org. Biomol. Chem 2007, 5, 18–30. [DOI] [PubMed] [Google Scholar]
- (15).Peng X; Pigli YZ; Rice PA; Greenberg MM J. Am. Chem. Soc 2008, 130, 12890–12891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Giese B; Beyrich-Graf X; Erdmann P; Giraud L; Imwindelried P; Müller SN; Schwitter U J. Am. Chem. Soc 1995, 117, 6146–6147. [Google Scholar]
- (17).Dussy A; Meggers E; Giese BJ Am. Chem. Soc 1998, 120, 7399–7403. [Google Scholar]
- (18).Zhang Q; Wang Y Chem. Res. Toxicol 2005, 18, 1897–1906. [DOI] [PubMed] [Google Scholar]
- (19).Romieu A; Bellon S; Gasparutto D; Cadet J Org. Lett 2000, 2, 1085–1088. [DOI] [PubMed] [Google Scholar]
- (20).Giese B Acc. Chem. Res 2000, 33, 631–636. [DOI] [PubMed] [Google Scholar]
- (21).Giese B Annu. Rev. Biochem 2002, 71, 51–70. [DOI] [PubMed] [Google Scholar]
- (22).Hong IS; Ding H; Greenberg MM J. Am. Chem. Soc 2006, 128, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hong IS; Greenberg MM J. Am. Chem. Soc 2005, 127, 3692–3693. [DOI] [PubMed] [Google Scholar]
- (24).Hong IS; Carter KN; Sato K; Greenberg MM J. Am. Chem. Soc 2007, 129, 4089–4098. [DOI] [PubMed] [Google Scholar]
- (25).Carter KN; Greenberg MM J. Am. Chem. Soc 2003, 125, 13376–13378. [DOI] [PubMed] [Google Scholar]
- (26).Bales BC; Pitié M; Meunier B; Greenberg MM J. Am. Chem. Soc 2002, 124, 9062–9063. [DOI] [PubMed] [Google Scholar]
- (27).Crich D; Mo X-SJ Am. Chem. Soc 1997, 119, 249–250. [Google Scholar]
- (28).Strittmatter H; Dussy A; Schwitter U; Giese B Angew. Chem., Int. Ed 1999, 38, 135–137. [Google Scholar]
- (29).Chatgilialoglu C; Duca M; Ferreri C; Guerra M; Ioele M; Mulazzani QG; Strittmatter H; Giese B Chem.;Eur. J 2004, 10, 1249–1255. [DOI] [PubMed] [Google Scholar]
- (30).Li M-J; Liu L; Wei K; Fu Y; Guo Q-XJ Phys. Chem. B 2006, 110, 13582–13589. [DOI] [PubMed] [Google Scholar]
- (31).Dizdaroglu M; Von Sonntag C; Schulte-Frohlinde DJ Am. Chem. Soc 1975, 97, 2277–2278. [DOI] [PubMed] [Google Scholar]
- (32).Greenberg MM; Barvian MR; Cook GP; Goodman BK; Matray TJ; Tronche C; Venkatesan HJ Am. Chem. Soc 1997, 119, 1828–1839. [Google Scholar]
- (33).Tallman KA; Greenberg MM J. Am. Chem. Soc 2001, 123, 5181–5187. [DOI] [PubMed] [Google Scholar]
- (34).von Sonntag C Free-Radical-Induced DNA Damage and Its Repair; Springer-Verlag: Berlin, 2006. [Google Scholar]
- (35).Carter KN; Greenberg MM J. Org. Chem 2003, 68, 4275–4280. [DOI] [PubMed] [Google Scholar]
- (36).Hong IS; Carter KN; Greenberg MM J. Org. Chem 2004, 69, 6974–6978. [DOI] [PubMed] [Google Scholar]
- (37).Barvian MR; Greenberg MM J. Am. Chem. Soc 1995, 117, 8291–8292. [Google Scholar]
- (38).Barvian MR; Greenberg MM J. Org. Chem 1995, 60, 1916–1917. [Google Scholar]
- (39).Tanaka H; Hayakawa H; Miyasaka T Tetrahedron 1982, 38, 2635–2642. [Google Scholar]
- (40).Tanaka H; Hayakawa H; Miyasaka T Chem. Pharm. Bull 1981, 29, 3565–3672. [DOI] [PubMed] [Google Scholar]
- (41).Kumar A; Walker RT Tetrahedron 1990, 46, 3101–3110. [Google Scholar]
- (42).Cadet J; Ducolomb R; Teoule R Tetrahedron 1977, 33, 1603–1615. [Google Scholar]
- (43).Carter KN; Greenberg MM Bioorg. Med. Chem 2001, 9, 2341–2346. [DOI] [PubMed] [Google Scholar]
- (44).Sa MM; Silveira GP; Castilho MS; Pavao F; Oliva G ARKIVOC 2002, 112–124.
- (45).Turro NJ Modern Molecular Photochemistry; Benjamin Cummings: Menlo Park, CA, 1978. [Google Scholar]
- (46).Tallman KA; Tronche C; Yoo DJ; Greenberg MM J. Am. Chem. Soc 1998, 120, 4903–4909. [Google Scholar]
- (47).Emanuel CJ; Newcomb M; Ferreri C; Chatgilialoglu CJ Am. Chem. Soc 1999, 121, 2927–2928. [Google Scholar]
- (48).von Sonntag C; Schuchmann H-P Angew. Chem., Int. Ed 1991, 30, 1229–1253. [Google Scholar]
- (49).Schuchmann MN; Von Sonntag CZ Naturforsch. B 1987, 42, 495–502. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.