

Abstract
Solid tumors elicit a detectable immune response including the infiltration of tumor-associated macrophages (TAMs). Unfortunately, this immune response is co-opted into contributing toward tumor growth instead of preventing its progression. We seek to reestablish an antitumor immune response by selectively targeting surface receptors and endogenous signaling processes of the macrophage subtypes driving cancer progression. RP-182 is a synthetic 10-mer amphipathic analog of host defense peptides that selectively induces a conformational switch of the mannose receptor CD206 expressed on TAMs displaying an M2-like phenotype. RP-182-mediated activation of this receptor in human and murine M2-like macrophages elicits a program of endocytosis, phagosome-lysosome formation, and autophagy and reprograms M2-like TAMs to an antitumor M1-like phenotype. In syngeneic and autochthonous murine cancer models, RP-182 suppressed tumor growth, extended survival, and was an effective combination partner with chemo- or immune checkpoint therapy. Antitumor activity of RP-182 was also observed in CD206high patient-derived xenotransplantation models. Mechanistically, via selective reduction of immunosuppressive M2-like TAMs, RP-182 improved adaptive and innate antitumor immune responses, including increased cancer cell phagocytosis by reprogrammed TAMs.
INTRODUCTION
Recent advances in immunotherapy have transformed the care of many patients with cancer. Immunotherapy approaches in the form of checkpoint inhibitor monoclonal antibody (CIMA) or chimeric antigen receptor T cell therapy have become first- or second-line treatment options and afford some patients sustained, durable treatment responses generally not observed with standard systemic chemotherapy (1, 2). To date, these positive findings are limited to immunologically “hot” cancers, whereas in the greater majority of solid organ cancers, which are classified as immunologically “cold,” the promise of immunotherapy via T cell activation has so far largely bypassed patients. These tumors create an immune milieu, which excludes cytotoxic T cells or induces an exhausted T cell phenotype through an abundance of immune-evasive cues frequently involving innate immune cells (3). Strategies that reinvigorate innate immune cells are underrepresented within current immuno-oncology therapies (4).
Tumor cells attract and reprogram innate immune cells including tumor-associated macrophages (TAMs) to support tumor growth and metastatic spread (5). Although the dichotomous M1-versus-M2 classification fails to capture the ontogeny and tissue-specific cues and stress responses of TAMs, in general terms, M1-like TAMs are proposed to be the more common phenotype in early tumor stages, whereas M2-like TAMs are more prominent in more evolved cancers (6). There is both intra- and intertumoral heterogeneity in the intratumoral macrophage population, and M1- and M2-like TAMs coexist within tumors (7). M2-like TAMs may promote tumor growth directly via the excretion of cancer-promoting factors or indirectly via angiogenesis, the nurturing of cancer stem cells, or the generation of an immune-evasive microenvironment (8). A number of challenges in the development of anti-TAM therapies have emerged: systemic toxicities associated with targeting the systemic macrophage population, the sometimes rapid adaptation of the macrophage pool resulting in escape from treatment, and a lack of selectivity for macrophage subtypes and disease sites (9).
Innate defense regulators (IDRs) are an emerging class of immunomodulators, which are fundamentally different from the better-known immune checkpoint inhibitors or immunocytokines. IDRs are synthetic peptide analogs of naturally occurring antimicrobial peptides (AMPs), more often called host defense peptides (HDPs) (10, 11). HDPs are an ancient aspect of the innate immune system. They are ubiquitously expressed in complex organisms and form the first-line defense and “immediate” response to injury and infection, which is often mediated by their lytic and direct antimicrobial functions (12, 13). HDPs are highly sequence divergent, but specific structural properties are conserved to retain activity (13). For those structures that adopt an α helix, these properties are chain length, exhibition of amphipathy, and enhanced hydrophobic moment. In particular, the antimicrobial activity of these commonly 10- to 40-amino acid peptides is intimately linked to the amphipathic spatial distribution of cationic charges, usually via clustering of amino acids lysine and arginine, versus oppositely directed hydrophobic amino acids. In some cases, synthetic designs based on these natural peptides have entered clinical testing for infectious disease indications (14). HDPs are also encoded by internal sequences of collagens and complement and virulence factors, and more recently, the role of HDPs in modulating cell signaling pathways and especially their immune effector functions are gaining a greater appreciation (15, 16). For example, HDPs mediate monocyte and leukocyte attraction to sites of tissue injury, regulate the local equilibrium of pro- and anti-inflammatory cues via Toll-like (TLR) or C-type lectin receptors, and are involved in the regulation of adaptive immune responses (17–19). Classification tools such as the heuristic Molly font have recently been developed to aid the identification and design of biophysically similar motifs (13). Biophysically conserved motifs may hint at structure-function relations that may allow the design of synthetic IDRs with therapeutic applications. We suspected that certain IDRs might specifically target and be capable of reprogramming TAMs.
Here, we identified the IDR RP-182 as an immunotherapy agent derived from in silico biophysical homology screening. RP-182 triggers a conformational switch of the mannose receptor CD206 expressed on M2-like macrophages, induces endocytosis, phagosome-lysosome formation, and apoptosis in these cells, and shifts the population of TAMs from M2-like macrophages toward an M1 phenotype, increasing innate and adaptive antitumor immune responses and improving outcomes in a variety of human and murine cancer models.
RESULTS
Biophysical homology screening of HDPs identifies synthetic IDR RP-182
We screened a database of 431 α-helical AMPs and HDPs (http://aps.unmc.edu/AP/main.php) using Molly font under the hypothesis that phylogenetically conserved structural domains within naturally occurring HDPs harbor important innate immune functions and that such select structure/function paradigm domains can be isolated and optimized for the design of effective therapeutics. Instead of homology comparisons using primary amino acid alignments, Molly font (Molly Hydrophobicity Wheel) assesses the three key biophysical features: hydrophobicity, electrostatic charge of amino acids, and steric amino acid volume to detect structural homologies via their unique conserved biophysical nature (fig. S1A) (13). Of the screened 431 peptides, we identified 129 peptides, or 30%, that have a preserved 10-amino acid domain consistent with the structural determinants of a secondary α-helical structure with amphiphilic surface topology (data file S1). To our surprise, biophysically similar sequences were also identified in human collagens and various microbial virulence factors (www.ncbi.nlm.nih.gov/ipg/docs/about/), possibly indicating peptide structure involved in conserved, shared innate immune functionalities (fig. S1B and table S1). The synthetic design RP-182 was optimized for maximum amphipathy of the original conserved 10-mer sequence by increasing hydrophobicity (hydrophobic moment vector <μ>) and positive charge density as visualized in Molly font (Fig. 1A). RP-426 was designed as a control to test the impact of hydrophobicity on activity. Next, to identify possible leads of putative binding partners of RP-182 and to examine whether RP-182 and the original 10-mer homology sequences share common IDR functions, we conducted in silico docking studies on human C-type lectin receptors, which are target receptors of HDPs and major regulators of innate immune processes in higher organisms (20). The animal lectin database (www.imperial.ac.uk/research/animallectins/ctld/mammals/humanvmousedata.html) contained 86 membrane-associated human C-type lectin-like domain (CTLD)-containing proteins, 24 of which had crystal structures available. Using the ClusPro server (https://cluspro.org), the crystal structures were interrogated for binding to RP-182 and biophysically similar 10-mer peptide fragments from 23 representative HDPs, virulence factors, and internal collagen sequences (table S2) (21). Figure 1B shows the 10 CTLD-containing proteins with the highest predicted binding affinity to RP-182, and fig. S2 shows binding affinities of the top receptor/ligand combinations for other 10-mer homology motifs, identifying mannose receptor 1 (CD206) as the target with the highest in silico affinity.
Fig. 1. RP-182 induces the closed conformation of CD206.
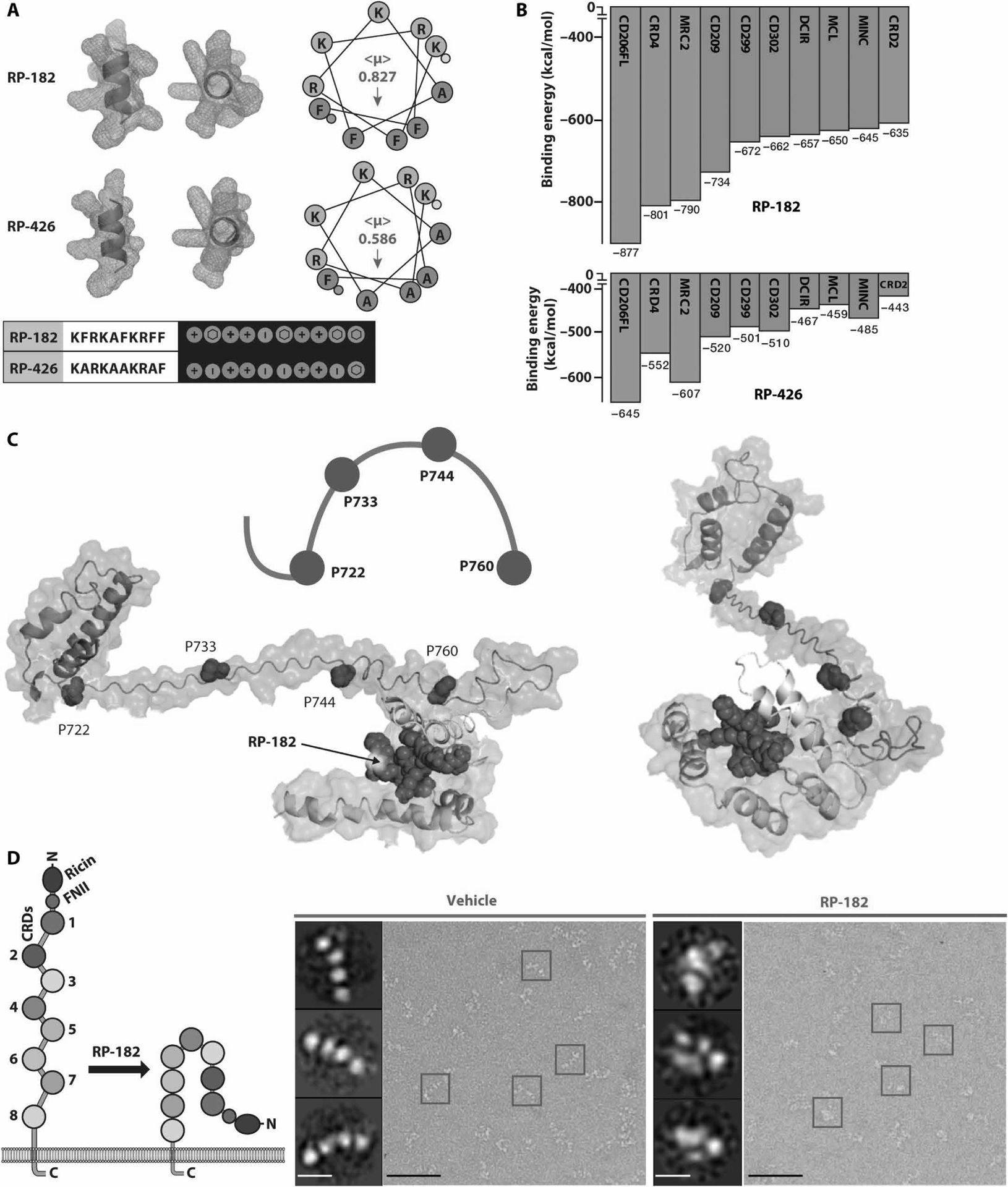
(A) Secondary α-helical structures, helical hydrophobicity wheel projections, and Molly font alignments (bottom) of RP-182 and RP-426. Amphiphilic surface topology shown in cyan (hydrophobic) and magenta (hydrophilic) colors. (<μ>), hydrophobic moment vectors; red arrows indicate strength and direction; yellow and green dots represent N and C termini. (B) Relative binding energies for the top 10 C-type lectin receptors using ClusPro. Green bars, C-type lectin class VI receptors, blue class II receptors. CRD4, carbohydrate recognition domain 4 of CD206. (C) Model of conformational bend in CRD4 and CRD5 of CD206 induced by RP-182. Hydrophobic plane of RP-182 bound to CRD5 (cyan color). (D) Negatively stained electron micrographs of CD206 proteins incubated with vehicle (blue squares) and RP-182 (red squares) and corresponding 2D single-particle images (insets); schematic of open “elongated” and “closed” conformations on left. Scale bars, 50 nm; insets, 5 nm.
CD206 is a member of the group 6, C-type lectin receptor family and undergoes a conformational change from open, “elongated” to closed state upon ligand binding or as pH in the surrounding environment decreases (fig. S3A) (22, 23). We next created in silico models of full-length human CD206, which we aligned with small-angle x-ray scattering (SAXS) data (fig. S3, B to D). The SAXS scattering profiles of CD206 and structure parameters extracted from SAXS data can be found at www.sasbdb.org/data/SASDG54/jdbt3at6ao/. The molecular weight estimated by SAXS data revealed that CD206 forms a dimer in solution (fig. S3B). The dimer of CD206, based on Model 1 as monomer, achieved the best fit (minimum χ2) to SAXS experimental data (fig. S3, C and D). Therefore, Model 1 was selected and used to repeat docking studies, which confirmed CD206 being the top binding partner of RP-182 and all except two of the 10-mer peptide sequences with biophysical homology (table S2). On the basis of Model 1, RP-182 was predicted by ClusPro to nestle into a cavity of CRD5 and to engage via three equidistantly spaced prolines (P722, P733, and P744) within CRD4. P760 acts as a fulcrum enabling bending of the CRD4 “handle” and rolling in of the receptor, inducing the closed, “globular” state of the receptor (Fig. 1C). To confirm the above binding studies, we first determined ratios of open versus closed CD206 particles incubated with RP-182 and controls, visualized by electron microscopy. Upon incubation with RP-182, the open, elongated conformation of CD206 switched to the closed, globular conformation (Fig. 1D and fig. S4A). The half-maximal effective concentration (EC50) of RP-182 to induce the closed conformation of CD206 measured ~11 μM (fig. S4B). Homology motifs (10-mer) from peptides LL37F1 or AVP1, predicted to bind less to CD206, had lower ratios of closed-to-open conformations compared to peptides RP-832C and RP-182, which were predicted to bind with higher affinity (−1146 and −877 kcal/mol, respectively) (fig. S4, A and C). Next, using microscale thermophoresis (MST), we measured binding of RP-182 to human CD206 and determined a dissociation constant (KD) of ~8 μM; the binding affinity of RP-426 to CD206 was about 10 times lower (KD = 85 μM) (fig. S4D). A KD of ~19 μM was measured for binding of RP-182 to murine CD206. Binding of RP-182, but not control peptide RP-426, to endogenous CD206 in human and murine macrophages was confirmed by cellular thermal shift assay (CETSA) (fig. S5). CETSA assesses target engagement of ligands via thermostability shifting of target protein(s) in a cell-based context. Human and murine M2-polarized macrophages incubated with RP-182, but not with RP-426, showed changed thermostability (>4°) of CD206 compared to vehicle control, indicating interaction of RP-182 and the CD206 receptor in the natural environment (fig. S5). To further map the binding region of RP-182, we performed mass spectrum studies of recombinant CD206 cross-linked to RP-182 derivative NCGC-00510434. NCGC-00510434, which displays similar KD binding to recombinant CD206 as wild-type RP-182, contained a diazirine-substituted phenylalanine and a C-terminally attached biotin (fig. S6). Fragment analysis of trypsin-digested CD206 pulled down with NCGC-00510434 identified the CRD5 sequence NFGDLVSIQSESEKK, which aligned with peptide analysis of CD206 covalently cross-linked to NCGC-00510434, followed by digestion, pulldown, and sequencing of peptide fragments, as well as the CRD5 motif previously predicted by in silico studies using the CD206 SAXS structure to be the binding region of RP-182 (fig. S7).
In summary, RP-182 is a synthetic HDP derived from a conserved homology sequence found across a diverse range of peptide and protein regulators involved in innate immune processes. It selectively induces a conformational switch from the open to the closed state in the mannose receptor CD206, which is different from the conformational change of CRD3 associated with lower pH or the binding of collagen to the fibronectin II domain (23).
RP-182 induces a program of phagocytosis, autophagy, and apoptosis in human and murine M2 macrophages
To investigate the impact of RP-182 binding and the induced conformational change of CD206 on cell function, we first examined the effects of RP-182 by global RNA-sequencing (RNA-seq) analysis. Volcano plot analysis of gene expression changes between vehicle- and RP-182-treated M2 bone marrow-derived macrophages (BMDMs) showed differentially expressed genes (DEGs) skewed toward up-regulation. Seven among the eight top DEGs were cytokines or regulators of the classical proinflammatory M1 phenotype showing ≥10- to 100-fold increased expression after 2 hours of treatment (Fig. 2A). Transcriptomic changes after RP-182 treatment occurred selectively in M2-polarized macrophages with no genes differentially expressed in M1 macrophages after 2 hours, six DEGs after 6 hours, and eight DEGs after 24 hours of treatment with RP-182 (fig. S8). Cytoscape Functional Gene Ontology (GO) Enrichment and Network Analyses identified pathways of inflammation and macrophage activation up-regulated in RP-182-treated M2 BMDMs, including C-type lectin receptor, nuclear factor κB (NF-κB), tumor necrosis factor (TNF), and TLR signaling (Fig. 2B). The most commonly represented genes identified by Leading-Edge Analysis after Gene Set Enrichment Analysis were imputed into Pathway Studio, which identified processes of endocytosis, phagocytosis, autophagy, and apoptosis as top biological pathways affected by RP-182 in M2 macrophages (Fig. 2C and fig. S9, A and B). Proteomic analysis of binding partners in CD206 complexes pulled down after 10 min of treatment with RP-182 compared to bead-only control showed enrichment of proteins involved in similar cell processes (Fig. 2D; data file S2; and fig. S9, C and D). To confirm results of above analyses, we next evaluated BMDMs polarized into M1 and M2 by electron microscopy. RP-182 induced phagosomes in M2- but not M1-polarized BMDMs (Fig. 2E). M2-selective induction of phagocytosis was confirmed by up-regulation of the early and late endosomal markers RAB5a and RAB7 and the lysosomal-associated membrane protein 1 (LAMP1) (Fig. 2F). Upon treatment with RP-182, CD206 was increasingly detected in the cytoplasm and induced RAB7-positive phagosomes costained with CD206, findings in line with the known internalization of the mannose receptor (Fig. 2G) (22, 24). Selective induction of phagosomes in the M2 phenotype upon treatment with RP-182 was equally observed in M2-polarized CD14+ peripheral monocytes isolated from healthy volunteers (fig. S10) and in BMDMs polarized into an “in vitro TAM-like” phenotype after coculture with conditioned medium from cancer cells (fig. S11). Control peptide RP-426 did not induce phagocytosis (fig. S12). RP-182 activated NF-κB signaling (Fig. 2H and fig. S13A). Next, we extended treatment time to 24 hours and measured induction of autophagy and apoptosis across several time points. RP-182 sequentially induced phagocytosis, autophagy, and apoptosis in M2 macrophages with no effect on M1 cells (Fig. 2I and figs. S13B and S14). RP-182 also induced cleaved caspase 3 and 7, known downstream substrates of activated caspase 8 (Fig. 2J). Upon 48 hours of exposure to increasing concentrations of RP-182, RP-182 reduced M2- but not M1-polarized macrophages detected by a double-staining cell viability assay with calculated half-maximal inhibitory concentration (IC50) of 1.1 μM for human M2 and 3.4 μM for murine M2 (Fig. 2K and fig. S15A). Control peptide RP-426 did not show any activity (fig. S15B), and RP-182 did not affect growth of mesenchymal stem cells, murine and human cancer cells, fibroblasts, endothelial cells, or DC2.4 dendritic cells (fig. S16).
Fig. 2. RP-182 selectively induces phagocytosis, autophagy, and apoptosis in M2-polarized macrophages.

(A) Volcano plot of RNA-seq analysis of vehicle-versus RP-182-treated M2 macrophages. Red color indicates transcripts with false discovery rate (FDR) q < 0.05, orange transcripts with −1 < log2[FC] >1, transcripts with both in blue. FC, fold change. (B) Cytoscape Functional GO Enrichment and Network Analyses of DEGs of vehicle versus RP-182-treated M2 BMDMs. (C) Pathway Studio graph of GO Cell Processes of the most common genes across enriched gene sets in RP-182-treated M2 BMDMs. Dashed arrows indicate positive regulation, and blue arrows indicate positive expression. (D) Protein network and related cellular processes of CD206 interactome induced by RP-182 in M2 macrophages. (E) Electron microscopy images of M1- and M2-polarized BMDMs. Scale bars, 2 μm; insets, 0.5 μm. (F) Immunofluorescence images of BMDMs polarized into M2 stained with anti-RAB5a, RAB7, LAMP1, and CD206 antibodies and nuclei stained with DAPI (4′,6-diamidino-2-phenylindole). Quantification of induced fluorescence on bottom. For all figures, data shown are representative of three independent experiments and normalized to corresponding vehicle treatment unless indicated otherwise. a.u., arbitrary units. (G) Immunofluorescent images of M2 BMDMs stained with anti-NF-κBp65. (H) Immunofluorescence images of M2 BMDMs costained with anti-CD206 (green) and anti-RAB7 (red). White arrows indicate costaining for both markers. (I) Quantification of activation of phagocytosis (RAB7), autophagy (LC3), and apoptosis (cleaved caspase 8) in M1 and M2 macrophages over time. (J) Quantification of cleaved caspase 3 and 7 after 24 hours of treatment. (K) Cell viability of human and murine M1 (blue curves) and M2 (red curves) macrophages after 48 hours of treatment with RP-182 relative to vehicle treatment.
RP-182 reprograms M2 macrophages toward an M1-like phenotype
The observation that viable cell fractions after 48 hours of treatment with RP-182 at the highest concentrations were greater than the initial fraction of CD206-negative cells (31% viable cells after maximum response versus 6.8% CD206-negative cells in human M2 macrophages; 17.2% viable cells versus 12.7% CD206-negative cells in M2 BMDMs) led us to examine a possible second mechanism of action of RP-182. We speculated that M2 macrophages reprogrammed by RP-182 toward an M1-like phenotype may lose CD206 expression and might not be subject to the cell killing function of RP-182. Flow cytometry experiments with CD11b+F4/80+Gr1− macrophages gated on alive cells using the M1 marker CD86 and M2 marker CD206 showed rapid induction of CD86 expression with an increase in the CD86+CD206+ double-positive macrophage fraction (87.8% versus 10.3% in vehicle-treated control) within 30 min of starting treatment with RP-182 (Fig. 3A and fig. S17). Induction of CD86 expression was associated with loss of CD206 expression, resulting in a CD86+CD206− M1-like fraction not expressing the M2 marker CD206 of 10.6% after 24 hours of treatment (Fig. 3A). Increased CD86 expression in M2 BMDMs treated with RP-182 was accompanied by up-regulation of M1 cytokines and markers, including interleukin-1β (IL-1β), IL-12β, TNF-α, and inducible nitric oxide synthase (iNOS) expressed by M1 macrophages (Fig. 3B and fig. S18A). Induction of M1 and loss of M2 markers were also observed in M2 BMDMs treated with RP-182 isolated by fluorescence-activated cell sorting (FACS) (fig. S18B). The increase in M1 cytokine expression was selective for CD86+ macrophages and not observed in CD206+CD86− cells (Fig. 3B). The induced M1-like CD86+CD206− and double-positive CD206+CD86+ macrophage cell populations showed decreased numbers of cells staining positive for the programmed cell death protein 1 (PD-1) (8.52 and 18.6% versus 66.2%) and inhibitory regulatory membrane glycoprotein signal regulatory protein α (SIRP-α) (2.81 and 7.84% versus 16.9%) immune checkpoint compared to vehicle-treated CD86−CD206+ M2 cells (Fig. 3C and fig. S19). Macrophage fractions staining positive for the M1 cytokines TNF-α and IL-12β were increased (19.4 and 16.0% versus 4.97% and 17.1 and 12.7% versus 5.74%, respectively) compared to vehicle control. The phenotypic switch induced by RP-182 toward M1 was accompanied by increased bacterial phagocytosis, a function more commonly associated with the M1 phenotype (Fig. 3D). The rate of apoptosis in the reprogrammed M1-like CD86+CD206− macrophages after treatment with RP-182 was lower than in the CD206+CD86− and CD206+CD86+ double-positive cells (Fig. 3E). Both pharmacological blockade of RP-182-induced NF-κB signaling and autophagy, previously shown to be induced by RP-182 in Fig. 2 (G and I), suppressed reprogramming effect of RP-182 toward the M1 phenotype (Fig. 3F). To examine whether gene expression changes induced by RP-182 support reprogramming of M2 BMDMs toward an M1-like phenotype, we analyzed gene expression matrices from RNA-seq data. Pearson’s correlation analysis of gene expression matrices derived from global RNA-seq data of RNA isolated from M1, M2, and M2 BMDMs treated with RP-182 showed a high degree of similarity between the three datasets. Using an M1 M2 marker set previously described for characterization of macrophage phenotypes in BMDMs, RP-182-treated M2 macrophages displayed greater similarity to untreated M1 than to untreated M2 cells (Fig. 3G) (25). In summary, in addition to induction of phagocytosis, autophagy, and apoptosis in M2 macrophages, the synthetic HDP RP-182 induces a shift toward an M1-like phenotype.
Fig. 3. RP-182 reprograms M2 macrophages toward an M1-like phenotype.
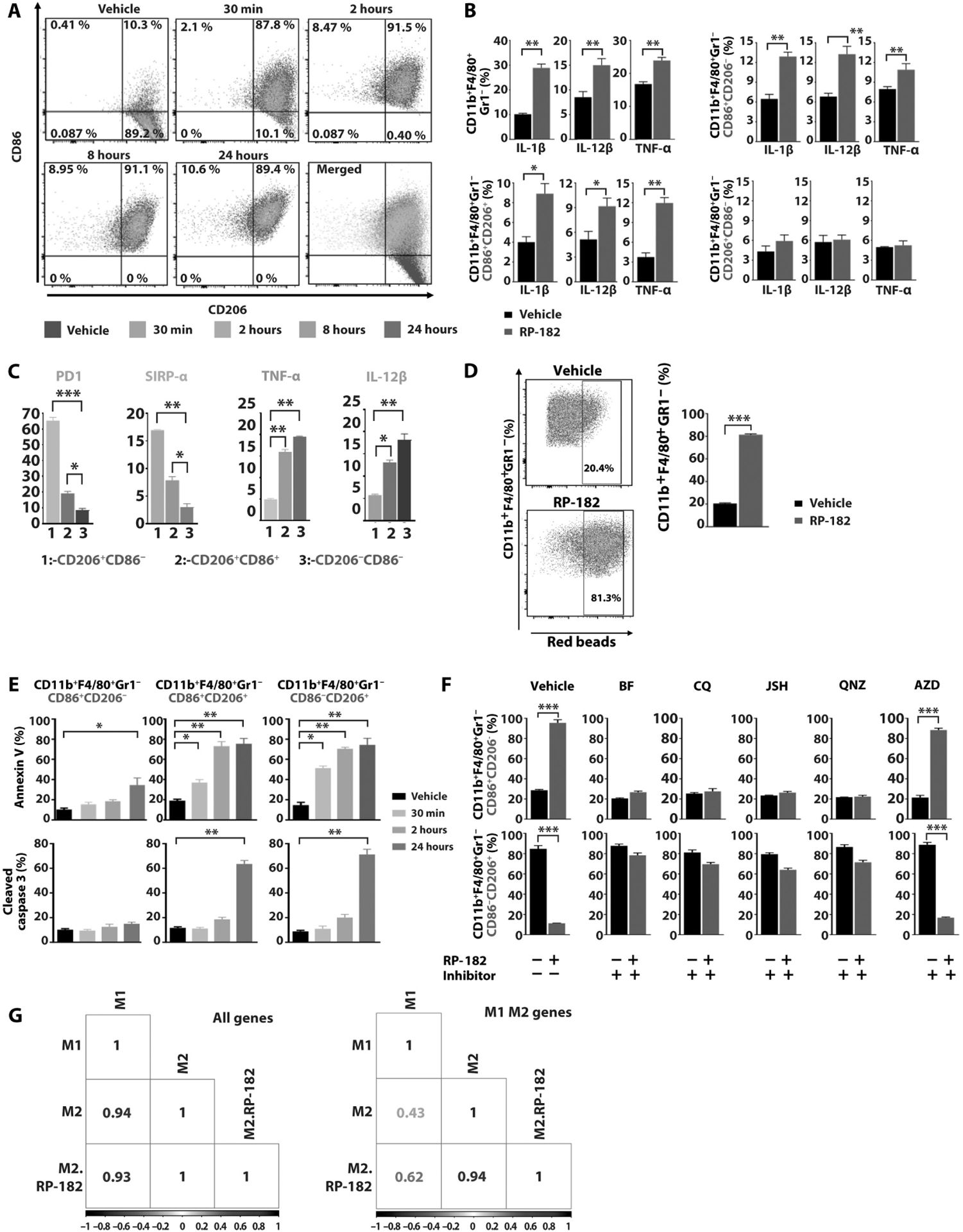
(A) Flow cytometry plots of CD86 and CD206-positive CD11b+F4/80+Gr1− macrophage fractions of M2 BMDMs after treatment with vehicle or RP-182 at indicated time points. (B) Quantification of fractions of IL-1β, IL-12β, and TNF-α-positive CD11b+F4/80+Gr1− cells, and of CD86+CD206−, CD86+CD206+ double-positive, and CD86−CD206+ subpopulations of M2 BMDMs by flow cytometry. Data shown are representative of three independent experiments conducted in triplicate. (C) Immune checkpoint and M1 cytokine-positive macrophage populations in vehicle-treated CD86−CD206+ M2 BMDMs, and RP-182-treated CD86+CD206− and CD86+CD206+ subpopulations. Univariate histograms with percentage of positive cell fractions, quantifications of n = 3 independent experiments are shown. (D) Flow cytometry analysis and quantification of CD11b+F4/80+Gr1− cell fractions with phagocytosed Escherichia coli-covered latex beads (red beads), quantification of n = 3 independent experiments. (E) Quantification of annexin V-positive (top) and cleaved caspase 3-positive cell fractions (bottom) of CD86+CD206−, CD206+CD86+, and CD86−CD206+ M2 BMDMs treated with RP-182 at indicated time points. (F) Quantification of vehicle- and RP-182-treated CD86+ (top) and CD206+ (bottom) CD11b+F4/80+Gr1− BMDM-M2 cells coincubated with NF-κB inhibitors JSH-23 and QNZ (EVP4593), autophagy inhibitors bafilomycin (BF) and chloroquine (CQ), and MEK inhibitor AZD6244 (selumetinib; AZD). (G) Pearson’s correlation analysis of gene expression matrices between samples using global RNA-seq data (left) and M1 M2 marker set derived from BMDMs (right). Values range from 0 to 1, where a high value indicates high degree of correlation between two sample sets.
RP-182 mechanism of action depends on CD206 and initiates RAC1/CDC42 activation and IQGAP1 recruitment
When pharmacodynamic readouts of RP-182 treatment were overlaid, the close EC50 and IC50 activities of RP-182 suggested a shared mechanism of action mediated by a common CD206 target (Fig. 4A). To show that the M2-selective action of RP-182 is indeed CD206 dependent, we isolated BMDMs from B6.129P2-Mrc1tm1Mnz/J mice, which are deficient in CD206 (24). We first confirmed that with the exception of CD206, there was no difference in the expression profile of M1 and M2 markers between CD206wt and CD206−/− BMDMs upon polarization into M1 and M2 populations (fig. S20). In contrast to M2-polarized macrophages from wild-type mice, M2-polarized macrophages isolated from B6.129P2-Mrc1tm1Mnz/J mice failed to show induction of phagocytosis, autophagy, or apoptosis (Fig. 4B) and showed no change in viability (Fig. 4C) and no induction of M1 cytokines upon treatment with RP-182 (Fig. 4D). To better understand the downstream CD206 signaling mechanisms induced by RP-182, we revisited the proteomic analysis of CD206 complexes from M2-polarized BMDMs treated with vehicle or biotinylated RP-182 (Fig. 2D and data file S2). Previous studies identified growth factor receptor-bound protein 2 (GRB2) as an intracellular signaling adaptor molecule of mannose CD206 receptor activation, and GRB2 was highly enriched in the CD206 pulldown from RP-182-treated BMDMs (26). Coimmunoprecipitation and immunoblotting studies in M2 macrophages showed that binding of RP-182 to CD206 recruits GRB2 and activates Rac family small guanosine triphosphatase (GTPase) 1 (RAC1)/cell division cycle 42 (CDC42)/p21 (RAC1)-activated kinase 1 (PAK1) signaling (Fig. 4, E and F). IQ motif-containing GTPase activating proteins 1 and 2 (IQGAP1 and IQGAP2), which were 9- and 76-fold enriched in CD206 complexes pulled down from RP-182-treated cells compared to control, are effectors of small GTPases RAC1/CDC42 in cytoskeletal dynamics stabilizing the GTP-bound, active state of RAC1/CDC42 and are involved in endocytosis/phagocytosis (27, 28). In M2 BMDMs, RP-182 increased the binding of IQGAP1 to the CD206 complex and induced membranous recruitment within 10 min (fig. S21, A and B). Blockade of RAC1/CDC42 signaling abrogated RP-182-induced IQGAP1 membrane translocation and induction of phagocytosis (Fig. 4G and fig. S21C). Treatment with autophagy inhibitors prevented RP-182-induced LC3 expression but did not affect induction of cleaved caspase 8, suggesting that caspase 8 activation, which did not occur in the presence of NF-κB inhibitors, is not part of an autophagolysosomal cascade but driven by RAC1/CDC42/PAK1-mediated NF-κB signaling (Fig. 4,H and I, and fig. S21D). Induction of apoptosis was mediated by RP-182-induced autocrine TNF-α signaling triggered by NF-κB activation (fig. S22). Blockade of TNF-α signaling abrogated induction of cleaved caspase 8 and 3, whereas conditioned medium from RP-182-treated M2 BMDMs activated apoptosis, which was not observed in the presence of anti-TNF-α antibodies. These data suggest that RP-182 binding to CD206 recruits GRB2 and the RAC1/CDC42 effector IQGAP1 and activates RAC1/CDC42/PAK1 signaling promoting phagocytosis and autophagy and costimulates NF-κB signaling, which is associated with induction of apoptosis via autocrine TNF-α signaling (fig. S22).
Fig. 4. Biological activity of RP-182 is CD206 dependent and initiates activation of RAC1/CDC42 signaling.

(A) RP-182 dose-response curves of binding to recombinant CD206 (MST assay; red curve), induction of the closed conformation of CD206 [electron microscopy (EM); green curve], induction of phagocytosis (anti-RAB7; purple), and M2 cell viability (blue) in M2 BMDMs. (B) Immunofluorescence images of BMDMs derived from wild-type and CD206−/− mice polarized into M2 stained with anti-RAB7, LC3, and cleaved caspase 8, quantification of three independent experiments at the bottom. (C) Cell viability of M2 BMDMs derived from CD206wt (red curve) and CD206−/− (brown curve) mice. (D) Flow cytometry plots of CD86+ and CD206+ fractions (top) and quantification of M1 marker-positive cells in CD11b+F4/80+Gr1− M2-BMDMs isolated from CD206−/− mice. Mean percent positive cells of n = 2 independent experiments in triplicate are shown. (E) Immunoblot (IB) of lysates immunoprecipitated (IP) with biotinylated peptides (left) or with anti-GRB2 antibody (right). Proteins visualized using anti-GRB2 and anti-CD206 antibodies. Preloading control superoxide dismutase 1 (SOD1) shown at the bottom. (F) Immunoblot analysis of lysates from M2 BMDMs immunoprecipitated for active GTP-bound form of RAC1/CDC42 and visualized with anti-CDC42 or anti-RAC1 antibodies (left). Immunoblots of M2 lysates with anti-phospho-PAK1 and anti-phospho-AKT on the right. Quantification of band intensities summarizes n = 3 independent experiments. (G) Quantification of RAB7 immunofluorescence in M2 BMDMs treated with RP-182 and preincubated with ZCL278 (left) or NSC23766 (right). Data shown are representative of two independent experiments and normalized to corresponding vehicle treatment unless indicated otherwise. (H) LC3 and cleaved caspase 8 quantification in M2 macrophages preincubated with chloroquine and bafilomycin; n = 2 in triplicate. (I) Quantification of phagocytosis (RAB7) and cleaved caspase 8 in M2 BMDMs in the presence of NF-κB inhibitors JSH23 and QNZ; n = 2 in triplicate.
CD206high expression status is associated with decreased intratumoral immunity in human and murine pancreatic cancers
CD206 expression status as a surrogate for M2 macrophage population varied considerably across clinical pancreatic cancer resection specimens (Fig. 5A and fig. S23A). CD206 was overexpressed in two of three available independent gene expression sets of clinical pancreatic cancer specimens compared to matched uninvolved normal pancreas, with no significant difference in gene set GSE28735 (gene sets GSE15471, GSE16515, and GSE28735; fig. S23B). Overall survival (OS) of patients with pancreatic cancers was lower in the CD206high clinical cases [hazard ratio (HR) 1.87, 95% confidence interval (CI) 1.165 to 2.813; log-rank test; P = 0.003] (Fig. 5B). Infiltrating CD8+ T cells measured by CD8 transcript expression further separated outcomes of clinical cases with high M2-like populations (HR 6.09, 95% CI, 1.338 to 10.16; log-rank test; P = 0.0006) (Fig. 5C). To examine whether the adverse disease outcome in CD206high cases is supported by immune subpopulation correlations in human pancreatic cancers or across solid organ cancers, we investigated correlations between intratumoral macrophage subsets and surrogates of intratumoral CD8+ T cell function in The Cancer Genome Atlas (TCGA) pan-cancer and pancreatic adenocarcinoma datasets. After selecting tumors with M2-high fractions and low abundance of M1-like macrophages, we found a negative correlation with CD8 transcripts, as well as measures of CD8+ T cell function, including low expression of two previously described T cell activation response signatures (Fig. 5, D and E) (29). To study the association of CD206 and clinical outcome further, we generated murine pancreatic cancers in CD206-deficient B6.129P2-Mrc1tm1Mnz/J mice. There was a discernable difference in survival of murine pancreatic KPC tumors grown in CD206−/− versus CD206 wild-type mice, with CD206-deficient tumors showing prolonged OS (median OS of KPC CD206−/− versus CD206 wild type, 32 days versus 25 days; P = 0.0278; Fig. 5F). KPC tumors in CD206−/− mice showed absent CD206 expression and, in line with the negative correlation of CD206high and CD8 T cell function in the human cancer specimens, significantly increased intratumoral CD8+ T cell numbers (P = 0.004) compared to KPC tumors generated in CD206-proficient C57B/L6 wild-type mice (Fig. 5G). KPC tumors in CD206−/− mice attracted an equal number of TAMs compared to KPC tumors grown in wild-type mice. The TAM population of CD206−/− mice was skewed toward an M1-like phenotype compared to TAMs of KPC tumors grown in wild-type mice (Fig. 5H).
Fig. 5. Infiltration of CD206high TAMs is associated with clinical outcome and intratumoral immunity.
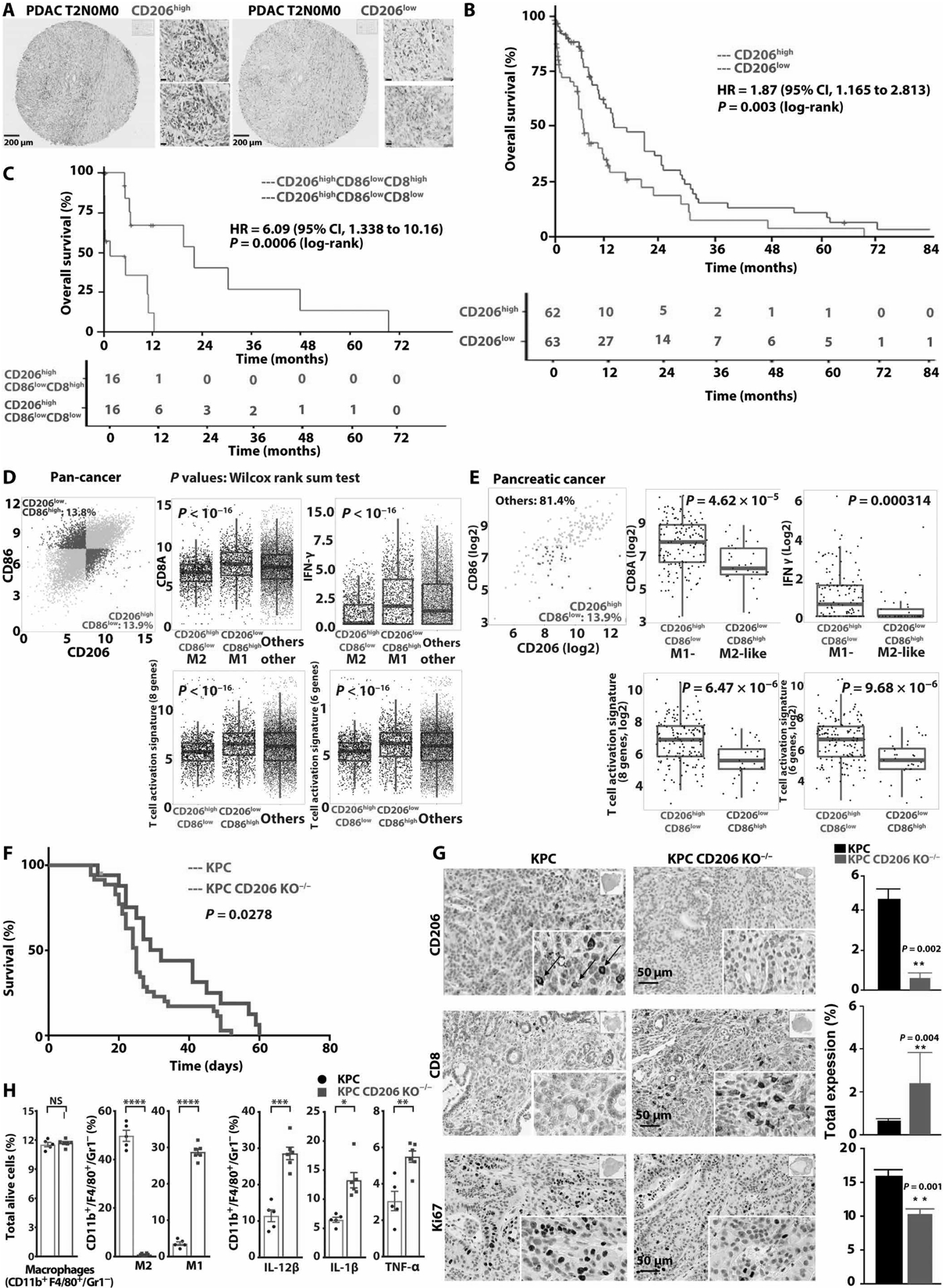
(A) Immunohistochemical staining of CD206 in CD206high and CD206low pancreatic ductal adenocarcinoma (PDAC) [scale bars, 10 μm (insets)]. (B) Kaplan-Meier plots of overall survival of 125 patients with PDAC stratified by CD206 expression. Log-rank test. (C) Kaplan-Meier analysis of patients with CD206high TAMs stratified by CD8high versus CD8low. (D) Correlation of CD8A, IFNG expression, eight and six gene CD8 T cell activation signatures (bottom), and M2 marker expression in clinical specimens from TCGA cancer dataset. Samples across all cancers were divided into CD206high and CD86low using median of all specimens. (E) Correlation of macrophage subtypes and CD8+ T cell function in pancreatic cancer TCGA dataset. (F) Kaplan-Meier analysis of KPC tumors allografted in CD206−/− B6.129P2-Mrc1tm1Mnz/J mice (blue curve) and in C57B/L wild-type mice (red curve). Log-rank test, two tailed. (G) Immunohistochemical staining of KPC wild-type (KPC) and KPC tumors generated in CD206−/− B6.129P2-Mrc1tm1Mnz/J mice (KPC CD206 KO−/−), quantification on right (n ≥ 4 per group). (H) Quantification of flow cytometry of TAM subpopulations in KPC tumors grown in C57B/L6 wild-type and CD206−/− mice (n ≥ 5 per group).
RP-182 mediates antitumor activity and reprograms the tumor microenvironment
Next, we tested RP-182 in the autochthonous genetically engineered Pdx1-Cre, LSL-KrasG12D/+, Trp53R172H/+ (KPC) and Ink4a(p16)/Arf(p19) (KP16) mice. Both models spontaneously develop pancreatic tumors and recapitulate the biology of human pancreatic cancer. Kaplan-Meier analysis and tumor growth measurements showed extension of survival and antitumor activity of RP-182 monotherapy yielding similar gains in survival and tumor suppression as gemcitabine (median OS of 20.5 days versus 32 days in vehicle versus RP-182-treated KPC animals; P = 0.0125; 27 days versus 31.5 days in KP16 animals; P = 0.0241) (Fig. 6, A to D). Animals treated with the combination of RP-182 and gemcitabine were afforded the greatest extension of survival in both models, with outcome in the combination cohort improved compared to single-agent treatment (34 days versus 44 days in gemcitabine versus combination group in KP16 mice; P = 0.0006 and 24.5 days versus 42.6 days in KPC; P = 0.0002, respectively) (Fig. 6, A and C). Tumor tissues harvested at study endpoint showed reduced stromal CD206-positive macrophages and decreased nuclear Ki67 expression (Fig. 6E). RP-182 induced E-cadherin expression and reduced expression of the epithelial-to-mesenchymal transition (EMT) marker vimentin (Fig. 6F). In vitro, the expression of the EMT markers vimentin and SNAIL in murine pancreatic cancer cells was induced upon coculture with M2 BMDMs but reduced when macrophages were pretreated with RP-182 (fig. S24). Flow cytometry studies of tumor digests from KP16 mice treated for 7 days with RP-182 alone or RP-182 in combination with gemcitabine confirmed reduced M2-like TAM fractions in RP-182 and RP-182 in combination with gemcitabine-treated mice (10.3% versus 4.61%, P = 0.001; 10.3% versus 3.91%, P = 0.0003, respectively) (Fig. 6G). RP-182 also decreased immunosuppressive CD4-positive regulatory T (Treg) cells, and in combination with gemcitabine, reduced myeloid-derived suppressor cells (MDSCs) (8.75% versus 4.99%, P = 0.015). Either alone or in combination with gemcitabine, RP-182 increased intratumoral CD8+ T cells (1.74% versus 3.40%, P = 0.032, and 1.74% versus 4.99%, P = 0.020, respectively) (Fig. 6G and fig. S25). The reduction in the MDSC population occurred nearly exclusively in the CD206high monocytic MDSC subset, whereas CD206low polymorphonuclear MDSCs did not show any change (fig. S26).
Fig. 6. RP-182 exerts antitumor activity and improves tumor immunogenicity.
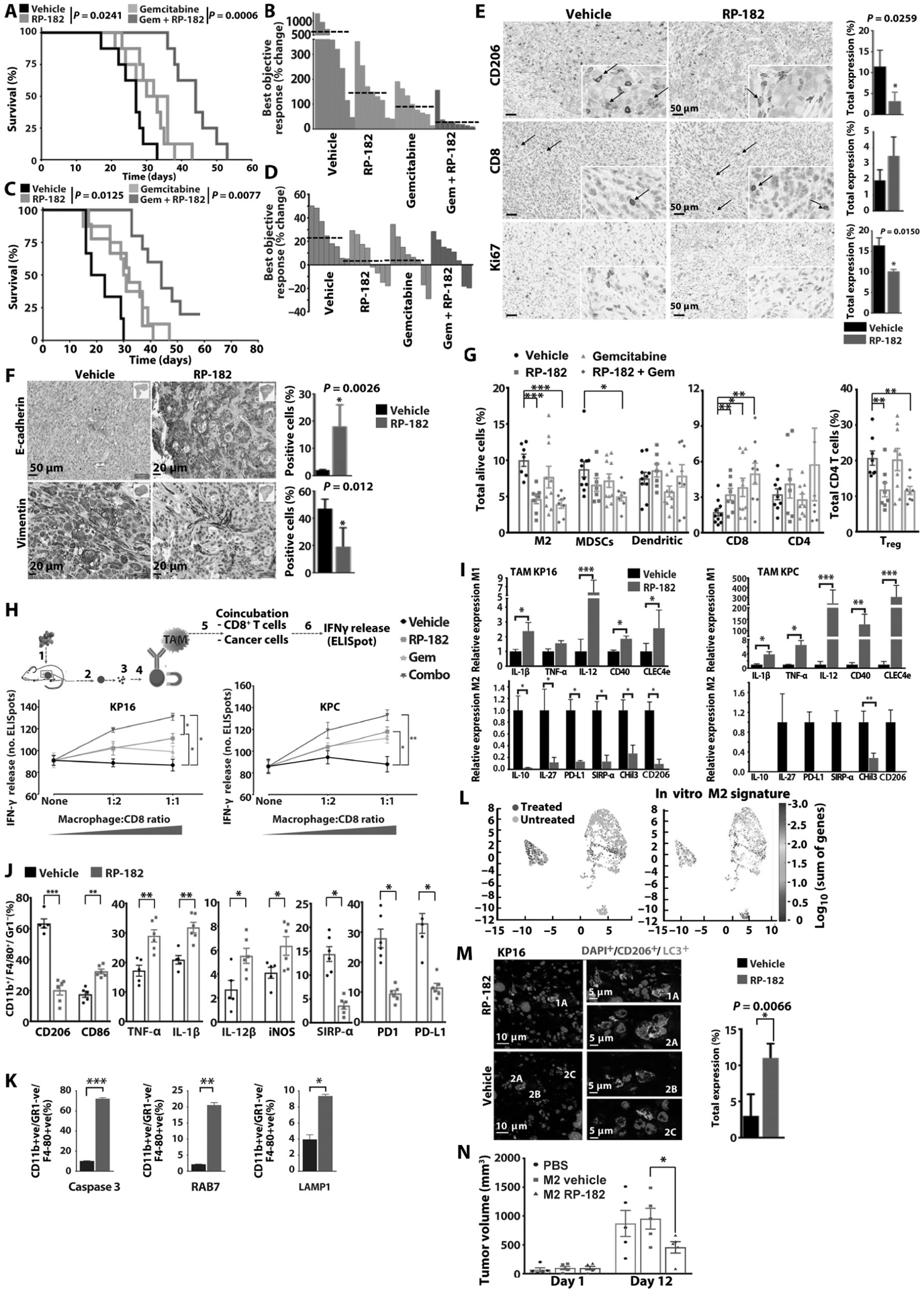
(A) Kaplan Meier analysis of KP16 mice. Log-rank test, two-tailed. (B) Waterfall plot of best objective response in KP16 mice. (C) Kaplan-Meier analysis of KPC mice. (D) Waterfall plot of best objective response in KPC mice. (E) Images of immunohistochemical stains of tumors from KP16 mice treated with vehicle or RP1–82. Quantification depicts mean percent positive cells by computer-based tissue analysis of n = 4 per group. Arrows indicate membranous staining of CD206-positive cells or CD8+ T cells. (F) Immunohistochemical staining of E-cadherin and vimentin, quantification of n ≥ 4 per group on right. (G) Percentages of CD206-positive M2 macrophage, MDSC, dendritic, CD8-, and CD4-positive cell fractions of total cells in KP16 tumors treated for 7 days. Percentages of FoxP3-positive Tregs of CD4+ T cells shown on the right. (H) IFN-γ-positive T cells after addition of TAMs isolated from KP16 and KPC tumors to cocultured cancer and splenic CD8+ T cells from tumor-bearing mice. (I) Quantification of quantitative reverse transcription PCR analysis of M1 and M2 gene expression of TAMs isolated from KPC and KP16 tumors (n ≥ 3 per group in triplicate). (J) Quantification of CD11b+F4/80+/Gr1− TAM fractions expressing M1 cytokines and immune checkpoints is shown. (K) Quantification of cleaved caspase 3, RAB7, and LAMP1-positive CD11b+F4/80+Gr1− TAM fractions by flow cytometry in KPC tumors of vehicle and RP-182-treated mice (n ≥ 5 mice per group). (L) t-distributed stochastic neighbor embedding (t-SNE) plots after tumor single-cell sequencing of CD11b+and filtering out KRT19+, CD11c+, Ly6G+ cells (n = 4 per group) of vehicle- (light blue) versus RP-182-treated (dark blue) tumors; color bar indicates log10[molecules per cell]. Increased expression of RP-182-induced DEGs identified in M2 BMDMs in vitro (“in vitro gene M2 signature”) was projected onto CD11b+KRT19−CD11c−Ly6G− cluster from RP-182-treated tumors (right), color bar indicates log10(sum of in vitro gene expression changes). (M) Images of immunofluorescence of KP16 tumors treated with RP-182 (top) and vehicle (bottom) costained with anti-CD206 (red) and anti-LC3 (green), computer-based quantification of costaining cells (of total cells, n = 4 per group). Insets show representative images of cells scored as costaining for CD206 and LC3 (1A, 2A), and cells scored as LC3 (2B) and CD206 (2C) positive. (N) Tumor growth after intratumoral cell transfer of M2 BMDMs into KPC allografts (n = 5).
Next, we isolated equal numbers of TAMs from treated murine KPC and KP16 pancreatic tumors and evaluated their impact on T cell function. Whereas TAMs isolated from vehicle-treated animals did not induce increments of interferon-γ (IFN-γ) release, TAMs isolated from animals treated with RP-182, or RP-182 in combination with gemcitabine, showed activation of T cell function (Fig. 6H), suggesting a switch of the TAM population toward an antitumor, proinflammatory M1-like phenotype. Gene expression analysis of TAMs isolated from tumors of RP-182-treated animals and flow cytometry analysis of the TAM population confirmed a switch from decreased M2 to an increased M1 fraction in RP-182-treated animals (Fig. 6, I and J, and fig. S27). Increased fractions of macrophages staining positive for the M1 cytokines IL-1β, IL-12β, TNF-α, and M1 marker iNOS were observed in RP-182-induced double-positive CD86+CD206+ and the CD86+CD206− M1-like cells but not in the CD86−CD206+ M2-like TAMs (fig. S28), in line with the reprogramming effect of RP-182 observed in M2 BMDMs in vitro. Consistent with RP-182’s mechanism of action in vitro, caspase 3-, RAB7-, and LAMP1-positive TAM fractions were increased in RP-182 versus vehicle-treated tumors (10.9% versus 72.1%, 2.7% versus 19.8%, and 3.9% versus 9.2%, respectively) (Fig. 6K and fig. S29A). Induction of apoptosis and phagocytosis was selective for CD11b+F4/80+Gr1− macrophages, whereas CD11b−CK19–9+ cancer cells showed only minimal or no changes (fig. S29B). Furthermore, when applying the previous DEG set obtained from RP-182-treated versus untreated M2 BMDMs onto whole-transcriptome analysis of single cells from KPC tumor digests, there was a significant enrichment of genes altered by RP-182 in vitro (adjusted P values of top markers displayed on a log10 fold-change color scale) in the TAM cell cluster formed by the treated cohort (Fig. 6L). Dual staining of RP-182-treated tumors with the markers LC3 and CD206 showed that RP-182 induced autophagosome formation in CD206-positive TAMs, phenocopying the induced LC3 expression in human and murine M2-like macrophages in vitro (Fig. 6M). RP-182-induced changes in M2 macrophages were associated with a tumor growth restricting impact upon intratumoral in vivo transfer of M2 BMDMs pretreated with RP-182 (Fig. 6N).
RP-182 increases antitumor immunogenicity, cooperates with immune checkpoint inhibition, and improves disease outcome in models of cancer and inflammation
Next, we examined the effect of RP-182 on intratumoral T cell function. Intratumoral T cells from animals treated with RP-182 (P < 0.05), or RP-182 in combination with gemcitabine (P < 0.01), showed significantly stronger activation upon coculture with cancer cells compared to T cells isolated from animals treated with vehicle control, suggesting improved tumor antigen recognition after RP-182 treatment (Fig. 7A). Improved T cell function was selective for T cells isolated from tumors and not observed in T cells isolated from spleens. To link the above increased tumor cell recognition to the observed antitumor activity in vivo, we repeated the efficacy studies with RP-182 and gemcitabine treatment in mice depleted of CD8+ T cells. Mice void of CD8+ T cells and treated with RP-182 and gemcitabine displayed a reduced extension of survival when compared to mice treated with isotype control, indicating the involvement of CD8+ T cells in RP-182’s mechanism of action (Fig. 7B). Previous studies on the tumor microenvironment of solid organ cancers have identified high numbers of TAMs as a major impediment to the efficacy of immune checkpoint therapy, and activation of cytotoxic T cells in tumors can be followed by up-regulation of immune checkpoints blunting effective antitumor responses (30). Murine pancreatic cancers showed increased programmed death ligand 1 (PD-L1) expression on CK19-positive cancer cells upon treatment with RP-182 (Fig. 7C). To test whether this increased checkpoint expression can be exploited for combination therapies and whether anti-TAM therapy via RP-182 might cooperate with PD-L1 immune checkpoint inhibition in pancreatic cancer not known to respond to single agent anti-PD-1/PD-L1 therapy, we combined RP-182 with anti-PD-L1 treatment. Antitumor activity of the combination was enhanced compared to single agent therapy (P = 0.0215) (Fig. 7D).
Fig. 7. RP-182 increases antitumor activity of CD8+ T cells and inhibits tumor growth in CD206high patient-derived pancreatic cancer xenografts.
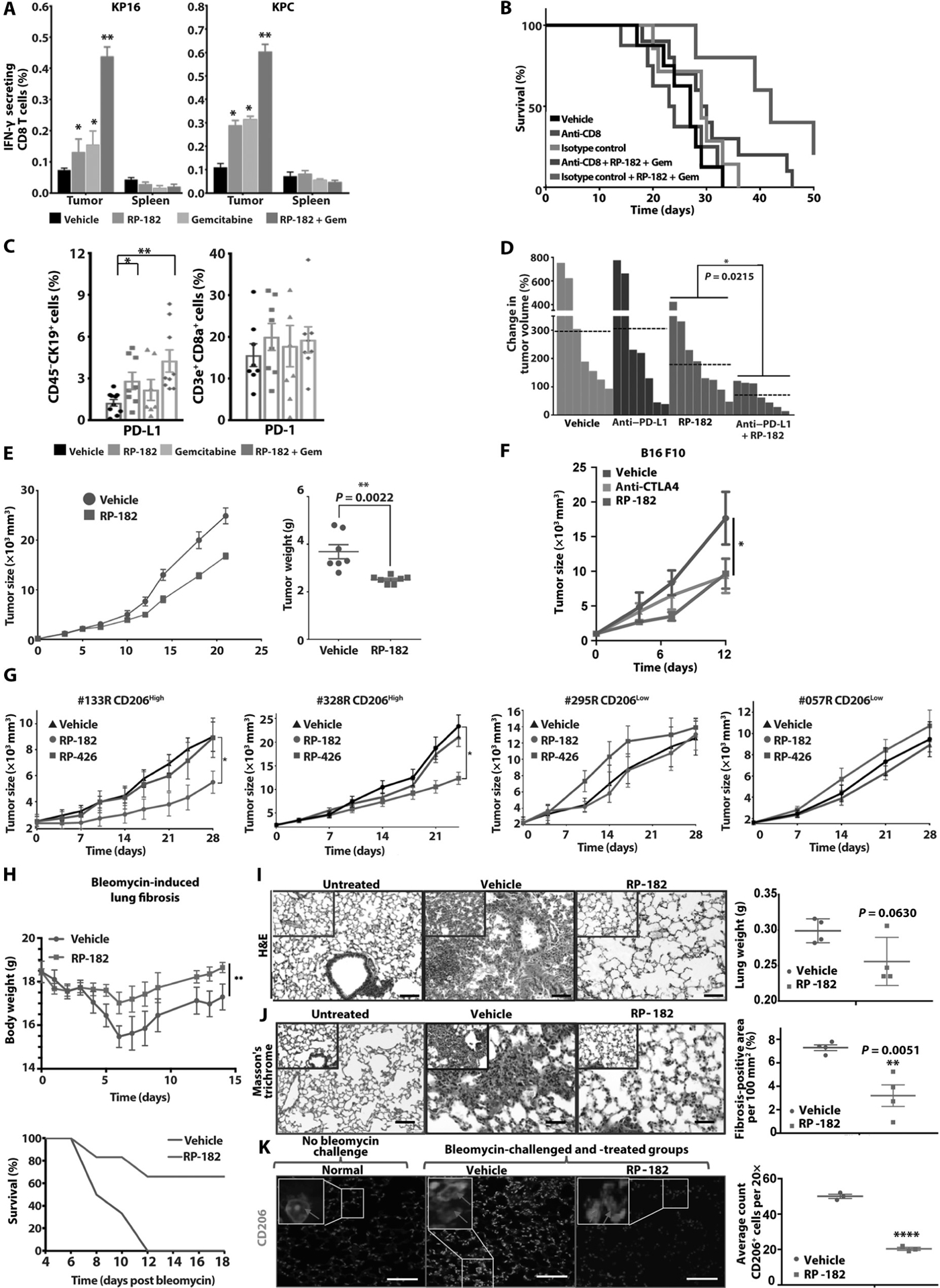
(A) Quantification of enzyme-linked immune absorbent spots (ELISpots) (IFN-γ-secreting CD8+ T cells of added total CD8+ T cells) after coculture of KP16 (left) and KPC cancer cells (right) and CD8+ T cells isolated from tumors and spleens. (B) Kaplan-Meier analysis of KP16 mice treated with anti-CD8 neutralizing antibody, anti-IgG2 isotype control, and indicated combinations. (C) Quantification of flow cytometry analysis of CD45−CK19+ cancer cells expressing PD-L1 (left) and PD-1 expression on CD45+CD3e+CD8+ T cells (right) in KP16 tumors. (D) Best objective response (BOR) of KP16 tumors treated with vehicle (gray bars), anti-PD-L1 injections (purple), RP-182 (red), and anti-PD-L1 in combination with RP-182; n ≥ 7 animals per group. E. Tumor growth of CT-26 allografts, tumor weights at study endpoint shown on right. (F) Tumor growth of murine B16 melanoma. (G) Pancreatic cancer patient-derived xenotransplantation models that are CD206high (#133R, #328R) or CD206low (#295R, #057R) treated with RP-182, RP-426, or vehicle control; n ≥ 7 per group. (H) RP-182 administration rescues mice with chemically induced pulmonary fibrosis. Quantification of total body weights (in grams; top) and Kaplan-Meier survival analysis, error bars indicate SDs. (I) Pulmonary inflammatory infiltrates after bleomycin instillation. Images of H&E stains, quantification of measured weights of excised lungs on right; n = 4 per group. Scale bars, 50 μm. (J) Images of Masson’s trichrome staining, quantification of percent fibrosis positive area of 100 mm2 examined lung surface on right. Scale bars, 50 μm. (K) Reduction of CD206-positive alveolar cell infiltrate in RP-182-treated mice. Immunofluorescence lung field images of noninstilled and bleomycin-instilled mice treated with vehicle or RP-182 stained with DAPI and anti-CD206 (green). Scale bars, 100 μm. Red arrows in insets indicate CD206-positive cells. Quantification of number of CD206-positive cells shown on right, n = 4 per group.
Next, we examined whether the above antitumor activity extends onto additional cancer models, including patient-derived xenotransplantation models. RP-182 reduced growth of CT-26 colon tumors and murine B16 melanomas, where it showed equal efficacy to cytotoxic T lymphocyte-associated protein 4 (CTLA4) checkpoint inhibition (Fig. 7, E and F). Using previously genotyped human pancreatic cancer tissues from the National Cancer Institute (NCI) Patient-Derived Models Repository (https://pdmr.cancer.gov/), patient-derived xenografts (PDX) with CD206high and CD206low expression were generated and treated with vehicle, control peptide RP-426, or RP-182. Whereas RP-182 reduced tumor growth in the CD206high PDX models compared to vehicle and RP-426 control, there was no effect in the CD206low models (Fig. 7G). Considering that CD206-positive, alternatively activated macrophages are involved in other disease processes, we tested RP-182 in a bleomycin lung fibrosis model. Treatment with RP-182 resulted in increased animal weight, improved OS, and diminished pulmonary fibrosis (Fig. 7, H to K). Correlative lung tissue studies showed reduction of M2-like macrophages measured by expression of CD206 (Fig. 7K). These findings indicate that RP-182 can modulate macrophage activity across several murine and human cancer models and possibly also noncancerous disease models driven by CD206-positive macrophages, suggesting a wide applicability. CD206 expression status may aid future selection of tumors most likely to respond.
RP-182 induces cancer cell phagocytosis by M1-like macrophages
To confirm that RP-182 can efficiently engage CD206-positive target cells in pancreatic tumors after systemic administration, we dosed KPC mice with RP-182 (20 mg/kg) carrying biotin (NCGC-00510434; fig. S6B). Multicolor confocal microscopy measuring staining intensities across linear sectional distances (in micrometers) showed notable colocalization of RP-182 with CD206-positive cells in the microenvironment of pancreatic KPC tumors, suggesting that RP-182 is binding its target (Fig. 8A). Photon xenogene quantification of Alexa Fluor 480-RP-182 in organs showed enrichment in tumor and kidney compared to other organs (fig. S30A). In line with its selectivity for its target CD206 and CD206-expressing M2 macrophages, treatment with RP-182 mediated a survival gain C57BL/6 wild-type mice allografted with KPC tumors but not in CD206-deficient B6.129P2-Mrc1tm1Mnz/J mice with KPC tumors lacking the target receptor of RP-182 (Fig. 8B). The smaller impact of RP-182 on OS in allografts compared to autochthonous KPC tumors shown in Fig. 6C might be due to differences between spontaneous KPC tumors and KPC tumors generated from allografted cells. RP-182 did not induce detectable hematological changes in the blood of dosed animals or any change in total body or selected organ weights upon preliminary toxicity testing (fig. S30, B and C).
Fig. 8. RP-182 increases cancer cell phagocytosis.

(A) Immunofluorescence images of KPC tumors treated with biotinylated RP-182 (bio-RP-182) and costained with anti-CD206 (green), streptavidin phycoerythrin (PE) (red), and DAPI (blue). Laser intensity profiles (upper right of linear scanning of random tissue section measuring intensity (fluorescence intensity, y axis) versus distance (in micrometers; x axis). Colocalization of CD206-expressing cells (green) and bio-RP-182 (red channel) (lower right) generating yellow/purple emission. (B) Kaplan-Meier analysis of KPC wild-type (left) and KPC CD206 KO−/− allografts (right) treated with vehicle (black) or RP-182 (red). (C) Phase-contrast images of M2 BMDMs incubated with carboxyfluorescein succinimidyl ester (CFSE)-labeled cancer cells (green). Inset shows a representative macrophage with engulfed green-labeled cancer cell and induced cytoplasmatic vacuoles and vesicles. (D) Quantification of the number of CFSE-positive macrophages of total number of M2 BMDMs (in %, phagocytic index). (E) CFSE-positive fractions in CD11b+F4/80+Gr1−CD86+ and CD206+ BMDMs by flow cytometry after coculture of CFSE-labeled KPC cancer cells and treatment with vehicle or RP-182. (F) Tumor growth of KPC xenografts (n ≥ 7 per group). (G) H&E images of KP16 tumors treated with vehicle (top) and RP-182 (bottom). Arrows indicate ballooned, vesicle-filled macrophages with peripheral nuclei, and asterisks indicate ingested cellular debris. Scale bar, 10 μm. (H) Transmission electron microscopy of KP16 tumors treated with vehicle or RP-182. TAMs with intracellular vesicles are indicated by arrows, and clasping of cancer cell and partial or complete cancer cell phagocytosis events are highlighted by solid red lines. Inset (dashed line) shows complete engulfment of cancer cell by TAM. Scale bars, 1 μm.
The loss of the SIRP-α receptor involved in the “do-not-eat me” signaling of innate immune cells on macrophages isolated from murine pancreatic tumors treated with RP-182 (Fig. 6J) prompted us to explore whether T cell-independent innate mechanisms of RP-182 such as cancer cell phagocytosis might contribute to the antitumor activity of RP-182 (8). RP-182 increased cancer cells phagocytosis [measured by engulfed carboxyfluorescein succinimidyl ester (CFSE)-positive cells; phagocytotic index after 2 hours of treatment with RP-182] of several murine and human cancer cell lines by 28.2 to 46.6% (Fig. 8, C and D, movies S1 and S2). At baseline, cancer cell phagocytosis was primarily observed in the CD86-positive M1 population and not in CD206-positive M2 macrophages (21.7% versus 1.08%, respectively) and increased after exposure to RP-182 in the M1 population (Fig. 8E and fig. S31). Although the increase in cancer cell phagocytosis by M1 macrophages is similar to the fraction of reprogrammed CD86-positive M1 cells, possibly suggesting that newly reprogrammed M1-like macrophages by RP-182 account for the increase, it cannot be ruled out that improved M1 function due to the reduction of suppressive M2 cues is, in part, responsible for the observed increased cancer cell phagocytosis effect of RP-182. To show that innate mechanisms of action are involved in RP-182’s antitumor activity, we established KPC, MDA-MB231 breast, and C4–2 prostate tumors in homozygous nu/J mice, which are deficient in mature T lymphocytes and unable to mount cell-mediated antitumor immune responses but retain B cell, natural killer, and myeloid cell functions. RP-182 monotherapy reduced tumor growth across these tumor models and improved antitumor activity of the standard gemcitabine and reduced metastatic dissemination in the MDA-MB231 model (Fig. 8F and fig. S32). Hematoxylin and eosin (H&E) staining of RP-182-treated tumor sections revealed features of macrophage activation and cancer cell phagocytosis (Fig. 8G). Tissue sections interrogated by electron microscopy showed multiple complete inclusions of cancer cells in TAMs, partial phagocytosis of cancer cells, or clasping of activated macrophages onto cancer cells compared to vehicle-treated tumors (Fig. 8H). Thus, RP-182 works through CD206 to enhance adaptive and innate immune cell functions in tumors that are not considered responsive to immune checkpoint blockade.
DISCUSSION
TAMs, which recognize and infiltrate affected tissues, are well positioned to initiate a profound antitumor immune response. Unfortunately, many tumors are able to alter the behavior of these cells and coax them into supporting vascularization, tumor growth, invasion, and metastasis (5, 7). Either these cells need to be selectively killed or we need to develop approaches to shift their behavior toward a more antitumor response. Initially, we considered that RP-182 was selectively killing these problematic macrophages, initiating an apoptotic process that then depletes them at tumor sites. However, upon further evaluation of RP-182’s biological actions within M2 macrophages and other immune cells, we showed that this peptide also alters the function of these macrophages, shifting them from an immunosuppressive state to a proinflammatory, phagocytosing phenotype able to mediate antitumor immune activity.
The cell surface mannose receptor is one of several types of pattern recognition receptors that have evolved to exploit some of the essential surface structural features of related, common pathogens, such as the mannose-containing proteins or internal collagen sequences, to mediate its scavenger function during tissue repair (22). We believe that the discovery of biophysically conserved sequences in many HDPs from different species (~30% of AMP/HDP database) and in diverse proteins such as microbial outer membrane virulence factors of different microbial organisms or in conserved sequences across different collagens, which show top-ranking binding to the mannose receptor, indicates the conservation of important structural features as part of the pattern recognition function of the receptor (13, 31). Several recent discoveries confirm that collagens and outer membrane microbicidal virulence factors contain internal peptide sequences involved in innate immune regulation (19, 31). In addition, successful pathogens have evolved by developing counter-measures to usurp pattern recognition and exploit entry into macrophages via the CD206 receptor (26). Thus, the biophysical similarity studies that we performed during the drug discovery campaign seem to suggest that the CD206 receptor is also a pattern recognition receptor for protein-based pathogen-associated molecular pattern recognition, and pattern recognition is not limited to outer membrane glycoproteins. We propose that future biophysical homology screening campaigns should include related protein databases beyond the canonical function of the interrogated class or family of proteins, which may yield additional biosimilar hits, increasing the confidence of the motif probe.
RP-182 activates phagocytosis and autophagy in M2-like macrophages via the mannose receptor CD206, reverting these cells into an antitumor M1-like phenotype with increased M1 cytokine production, as well as the ability to phagocytose cancer cells and to aid tumor antigen recognition of intratumoral CD8+ T cells. These altered functions were associated with select changes in a relatively small gene set involved in M1 and M2 function. Both activation of autophagy and NF-κB signaling, together with metabolic reprogramming of M2-like macrophages, have previously been shown to transform M2 macrophages toward an M1 phenotype (32, 33). In addition, RP-182 induces apoptosis via cleaved caspase 8 and an autocrine-positive feedforward loop involving TNF-α signaling, promoting the depletion of this population and further shifting the balance toward the proinflammatory, antitumor M1 phenotype. In addition, phagocytic activation of M2 macrophages in the tumor environment reduces collagen deposition and fibrosis, promoting tissue recovery (34). We tested RP-182 in a lung fibrosis model characterized by the extravasation of CD206-positive alveolar macrophages. The observed decreased deposition of collagen and reduced fibrosis associated with RP-182 treatment in this inflammatory model appear consistent with the known antifibrotic activity of activated M2 macrophages. Induction of phagocytosis and autophagy by RP-182 in CD206-positive M2-like macrophages was followed by induction of apoptosis, reduction of M2-like macrophages in the TAM population, and increased CD8 cytotoxic T cell infiltration and function. The altered TAM phenotype was also associated with a less EMT-like cancer phenotype in RP-182-treated tumors, which might explain the observed cooperativity with chemo- and CIMA therapy (35). The increased M1 population transformed from M2-like cells improved innate antitumor immunity via increased cancer cell phagocytosis both in vitro and in vivo. Results from the studies in human CD206high versus CD206low PDX models and in CD206−/− knockout allografts suggest that CD206 expression status might be used as a possible biomarker for this approach.
Peptide-based immuno-oncology therapeutics might confer major advantages compared to current monoclonal antibody therapy in difficult-to-penetrate, desmoplastic solid organ cancers: The reduced molecular size of a drug facilitates the extravasation from the endothelial system and the diffusion within the tumor (36). In addition, peptides have been reported to be more flexible than antibodies because they can acquire conformations that minimize the total polar surface area of the molecule, facilitating the partition toward the lipid bilayer and aqueous solution states (37). It can be speculated that increased trans- and paracellular diffusion of amphipathic peptide designs might yield important improvements in intratumoral drug distribution (38). On the other hand, the same flexibility makes them more vulnerable to hydrolysis by peptidases and results in a low half-life in comparison to antibodies. PR-182 binding to CD206-positive cells in the tumor induced phagocytosis and autophagy, initiating a distinct signaling cascade in M2-like TAMs, and therefore, from a pharmacodynamics point of view, exposure of the peptide seems to be sufficient for a robust biological response.
Although peptide-based therapeutics have seen accelerated translation and early clinical development as antimicrobial and antifungal biopharmaceuticals, there is a lack of peptide-based immune modulators targeting dysfunctions of innate and adaptive immune responses in cancer, despite the recognition that HDPs could be a fertile source of such immunomodulators (15, 16). There have been reports on synthetic peptides developed as vehicles to aid intratumoral drug delivery. These include the HDP LyP-1 targeting p32 expressed on activated TAMs and other cells, the anti-M2 peptide M2pep coupled to a toxin with unknown mechanism or molecular target, or recently the peptide sequence CSPGAKVRC (named “UNO”), able to carry drug-loaded nanoparticles targeting CD206 into xenotransplanted tumors (39–41). Overall, although they also aimed to reduce M2-like cues of TAMs, these peptide designs differ from the IDR described in this study, and comparisons to RP-182 are limited in part because their function is primarily restricted to molecular probe designs. Previously described IDRs in monocyte and macrophage in vitro models with unknown target(s) developed primarily for infectious disease indications were reported to also affect NF-κB signaling, albeit in an inhibitory function compared to RP-182 (42, 43). Targeting tumor-promoting TAM subpopulations within a precision medicine approach has recently been shown to be a promising antitumor strategy by monoclonal antibody therapy against the macrophage surface marker MARCO (macrophage receptor with collagenous structure), and we believe that selective depletion and reprogramming of CD206-positive M2-like TAMs by RP-182 described in this study might be advantageous to the more global reprogramming of TAMs using, for example, colony-stimulating factor 1 receptor blockade (44, 45).
The study also has some limitations. The single-cell sequencing results in KPC tumors treated with RP-182 show a heterogeneous response to the IDR. Although differences in response to RP-182 might be due to inherent heterogeneity of TAM populations, we cannot exclude that additional factors in CD206high TAMs co-regulate the response to RP-182. Inherent differences in TAM phenotypes might also determine whether RP-182 predominantly induces reprogramming toward an M1-like phenotype versus M2 killing. In this regard, differences in intratumoral exposure might have contributed to the variabilities in CD206high M2 reprogramming and cell killing. Although costaining of biotin-labeled RP-182 and CD206-positive cells in KPC tumors confirms that RP-182 can reach its cellular target cells in vivo, formal pharmacokinetic studies in tumor-bearing mice coupled with dose-response testing have yet to be carried out. Similarly, the modest effect of RP-182 in KPC tumors grown in CD206 wild-type mice but not CD206−/− knockout mice might be, in part, due to limited intratumoral exposure of RP-182, and differences between CD206 wild-type and CD206−/− mice treated with RP-182 might have been larger if studies had been performed in combination with gemcitabine.
In summary, this report shows that biophysical similarities beyond primary amino acid sequence alignments can detect homologies between HDPs and regulators of the innate immune system and that these motifs can be used to design effective therapeutics. The IDR RP-182 is a 10-mer synthetic HDP derived from screening for biophysical homologies across HDPs and mediators involving innate immune processes. A conformational switch of the mannose receptor CD206 by RP-182 reprograms M2-like TAMs in the tumor stroma and improves intratumoral innate and adaptive antitumor immunity and tumor control. RP-182 should be evaluated in cancer types with large TAM populations and possibly noncancerous diseases driven by CD206high population.
MATERIALS AND METHODS
Study design
Previous studies from our laboratory have identified phylogenetically conserved structural motifs within HDPs or other classes of immune regulators. However, their functional implications and whether these conserved biophysical homology domains can be used for the design of effective immune modulators are unknown. Therefore, our primary objective was to rigorously evaluate the mechanism of action and efficacy of RP-182, an α-helical synthetic HDP, derived from a biophysical homology screen across HDPs and other IDRs. The relevance of RP-182 for targeting the mannose receptor CD206 on CD206high M2-like macrophages, including CD206high TAMs, was tested in human and murine macrophage models and in preclinical animal models of pancreas, colon, breast, and prostate cancer, and melanoma. RP-182 was evaluated in four PDX of pancreatic cancer with different expression of CD206. RP-182 was also evaluated in a nonmalignant bleomycin lung fibrosis model. All efficacy studies in the genetically engineered murine models of pancreatic cancer, KPC [Pdx1-CRE/Lox-stop-Lox(LSL)-KrasG12D/+;Tp53R172H/+] and KP16 [Pdx1-CRE/Lox-stop-Lox(LSL)-KrasG12D/+;Ink4a/Arfflox/flox] mice, enrolled a minimum of 8 to 10 tumor-bearing animals per group and were repeated at least once. All mice were weighed before the beginning of the experiments, and mice were randomized to different treatment groups after transabdominal ultrasound imaging before administration of the first dose. Efficacy studies in pancreatic cancer PDX models #133R, #328R, #295#, and #057R enrolled 8 to 10 mice per treatment group (with the exception of PDX #295, which had n = 7 in the vehicle group), and studies in colon, breast, and prostate cancer models enrolled 7 to 10 mice per treatment group. Animals were randomized to treatment groups. Quantifications of tissue staining were carried out via computer-based algorithm, and quantification of CD206+LC3+ costaining shown in Fig. 6M was done manually without blinding to treatment groups.
Animal protocols
Colonies of transgenic mice were established at NCI in Bethesda, MD, and all animal experiments were conducted according to protocols and policies approved by the Animal Care and Use Committees (ACUC) of the National Institutes of Health (NIH). All animal studies were conducted under ACUC-approved protocols SB-210 and SB-211. Mice with individual genes for Pdx1-CRE, LSL-KrasG12D/+, LSL-Tp53R172H/+, and LSL-Ink4a/Arfflox/flox were obtained from NCI’s Mouse Repository, Frederick National Laboratory of Cancer Research (https://frederick.cancer.gov/science/technology/mouserepository) and crossed to create animals with the triple genotype of Pdx1-CRE; LSL-KrasG12D/+; Ink4a/Arfflox/flox (KP16) or Pdx1-CRE; LSL-KrasG12D/+; LSL-Tp53R172H/+ (KPC). B6.129P2-Mrc1tm1Mnz/J mice were obtained from The Jackson Laboratory (JAX stock no. 007620). Genotypes were verified using polymerase chain reaction (PCR) methods performed by Transnetyx Inc.
Human pancreatic cancer tissues for xenotransplantation were obtained from the NCI Patient-Derived Models Repository (https://pdmr.cancer.gov/) initiative and subcutaneously implanted into nonobese diabetic severe combined immunodeficient IL2Rγnull immunodeficient mice (F0 generation). After tumors reached 2 cm, tumors were explanted, cut into equal pieces, and regenerated in another generation (F1 generation). Treatment experiments were carried out in F2 mice.
Syngeneic murine models of cancer included the murine CT-26 colon cancer and B16 melanoma models. About 1 × 106 CT-26 cells/100 μl medium were implanted subcutaneously into 6- to 8-week-old BALB/c mice. When tumors reached ~50-mm3 volume, mice were started on treatment as outlined below. Tumor volume (mm3) was calculated as (L × W2)/2, with L for length (mm) and W for width (mm) during two-dimensional caliper measurements, and total body weights were recorded twice per week. Two hours after the last injections, mice were euthanized, and tumors were excised and weighed. Similarly, 0.5 × 106 murine B16 melanoma cells were subcutaneously injected into the flank of BALB/c animals, and treatment was started upon tumors reaching ~50 to 100 mm3 volume. Human breast MDA-MB23, prostate C4–2, or KPC cells (0.5 × 106) were subcutaneously injected into the flank of homozygous female athymic (nu/J) nude mice. KPC tumors were treated for 3 weeks after tumors reached 250 mm3, C4–2 tumors were treated for 4 weeks, and MDA-MB231 tumors were treated for 6 weeks after tumors had reached 100 mm3 volume, at which point the draining lymph node basin was removed for H&E staining determination of locoregional metastatic index (number of lymph nodes involved by cancer divided by the total number of excised and examined lymph nodes in the draining basin).
For the bleomycin lung fibrosis studies, to facilitate intratracheal bleomycin instillation, animals were anesthetized via isoflurane inhalation vaporized at concentrations of up to 4% for a short period of time. A single dose of bleomycin (0.5 mg/kg; 1 to 4 U/mg) in sterile isotonic saline (total volume, 50 μl) was intratracheally administered via a 22-gauge plastic cannula to a total of n = 12 BALB/c mice, and the same volume of sterile saline was administered to a control group of mice (n = 4 mice). Mice instilled with bleomycin (catalog no. S1214, Selleckchem Inc.) were randomized on day 1 to receive RP-182 (20 mg/kg) or vehicle control via daily intraperitoneal injection. Mice underwent daily weight measurements, and animal survival was measured from the first day of treatment until death. Animals in control and RP-182 treatment cohorts were allowed to progress under continuous treatment conditions until they reached the study endpoint (determined as 20% weight loss, recognizable signs of morbidity, general lack of reflexes, abnormal posture, loss of ability to ambulate, labored respiration, inability to drink or feed, or being determined as moribund by study veterinarian), upon which, to minimize animal suffering, animals were euthanized in accordance with the ACUC animal care guidelines.
Animal imaging
Mice with the KP16 and KPC genotypes were imaged with ultrasound weekly, starting at 6 weeks of life. Ultrasound imaging was performed using a 40-mHz transducer and a Vevo700 ultrasound machine (Visualsonics). Mice were anesthetized with isoflurane (Baxter), shaved, and injected intraperitoneally with 1.5 ml of normal saline. B-mode images were recorded to obtain tumor measurements.
Treatment protocols
KP16 and KPC mice were treated for 7 days before harvest of tumors used in flow cytometry, pulldown of immune cells, or immune assays, or until a predefined study endpoint. All animal treatments started after ultrasound confirmed a pancreatic tumor measuring ≥4 to 5 mm and randomization of individual animals to treatment groups. Animal survival was measured from the first day of treatment until death. Animals in control and treatment cohorts were allowed to progress under continuous treatment administration until they reached study endpoint. For experiments in KP16 and KPC mice, normal saline as vehicle, RP-182 (20 mg/kg; PolyPeptide), gemcitabine (50 mg/kg; Fresenius Kabi), or RP-182 in combination with gemcitabine was injected intraperitoneally at a final administered volume of 200 μl. RP-182 was injected every other day, and gemcitabine was injected two times a week. Anti-PD-L1 (BioLegend, catalog no. 124329) was administered three times weekly at 150 μg per mouse by intraperitoneal injection. Anti-CTLA4 antibody (Bio X Cell; 9D9) was administered at 100 μg twice a week via intraperitoneal injection. For CD8 depletion, two doses of 100 μg of anti-mCD8 (Bio X Cell, catalog no. BE0061) per mouse were administered on days 1 and 5. Rat isotype control immunoglobulin G1 (IgG1) (Bio X Cell, catalog no. BE0090) was given at equivalent doses at the same schedule. Mice with CT-26, MDA-MB231, C4–2, and B16 tumors received RP-182 (10 mg/kg) via intraperitoneal injection daily for tumor growth studies, gemcitabine was given at 50 mg/kg twice a week, and the docetaxel dose delivered to the C4–2 model was 2.5 mg/kg daily for a total of 7 days. For intratumoral injections, 50,000 BMDMs pretreated for 2 hours with vehicle or 20 μM RP-182 were injected on days 2, 5, 7, and 9 into KPC tumors ≥500 mm3 grown in C57B/L wild-type mice. Before injection, M2 BMDMs grown and polarized on T75 flasks were washed twice, lifted and counted, and resuspended in <50 μl of Hanks’ balanced salt solution for injection.
Bleomycin lung fibrosis model
To facilitate intratracheal bleomycin instillation, BALB/c mice obtained from Charles River Laboratories were anesthetized for a short period of time. A single dose of bleomycin (0.5 mg/kg; 1 to 4 U/mg) in sterile isotonic saline (total volume, 50 μl) was intratracheally administered via a 22-gauge plastic cannula to a total of n = 12 mice, and the same volume of sterile saline was administered to a control group of mice (n = 6). Mice instilled with bleomycin were randomized on day 1 to receive RP-182 (20 mg/kg) by daily intraperitoneal injection or vehicle control. Mice underwent daily weight measures, and animal survival was measured from the first day of treatment until death. Animals in control and RP-182 treatment cohorts were allowed to progress under continuous treatment conditions until they reached study endpoint. Only “warm” necropsy specimens (lungs) were used for tissue analysis. Lungs were weighed before fixation in formalin and embedding in paraffin and stained with H&E, Masson’s trichrome, and anti-CD206. ImageJ was used to compare the extent of fibrosis between the vehicle and RP-182-treated groups.
Murine and human macrophages
Murine monocyte precursor cells were obtained by flushing out the bone marrow from the femurs of 6- to 8-week-old healthy C57B/L mice. Experiments were conducted according to protocols and policies approved by the ACUC of the NIH (ACUC protocol SB-210–3) and NIH’s policy on humane care and use of laboratory animals (https://olaw.nih.gov/home.htm). After 1 week of incubation at 37°C with 5% CO2, bone marrow progenitors were polarized into M1 and M2 macrophages with the use of cytokines. Human macrophages were obtained from peripheral blood mononuclear cells (PBMCs) acquired from deidentified healthy human donors from the NIH, Department of Transfusion Medicine (under Institutional Review Board-approved NIH protocol 99-C-0168) using Classical Monocyte Isolation Kit, human (catalog no. 130-117-337, Miltenyi Biotec) and were depleted of CD14-positive cells and polarized into M1 and M2 macrophages with the use of cytokines(46).
Statistical methods
Data were statistically analyzed using SPSS software version 16 (IBM). Tumor volumes were compared between all four groups using best objective response (BOR, best response recorded from the start of the study treatment compared to any of the follow-up measurements) or absolute measurements (in mm3). Continuous data, including tumor volumes, gene expression, or immune cell population percentages, were compared using Student’s t test in GraphPad Prism. The log-rank test was used to compare Kaplan-Meier curves. Error bars indicate SEM unless otherwise indicated. Calculated P values are indicated directly and with asterisks (*P < 0.05, **P < 0.01, and ***P < 0.001). Original data are available in data file S3.
Supplementary Material
Fig. S1. Biophysical homology screening using Molly font identifies conservation of 10-mer structural motif across HDPs, outer surface virulence factors, and collagens.
Fig. S2. Docking of biophysical 10-mer homology motifs onto C-type lectin receptors.
Fig. S3. Full-length CD206 model.
Fig. S4. RP-182 binds to CD206.
Fig. S5. RP-182 but not RP-426 induces a cellular thermostability shift of CD206 in human and murine M2 macrophages.
Fig. S6. Abbreviated synthesis scheme of RP-182 analogs with diazirine-containing phenylalanine and biotin.
Fig. S7. RP-182 binds to carbohydrate recognition domain 5 of the CD206 receptor.
Fig. S8. Gene expression changes induced by RP-182 in M1- and M2-polarized BMDMs.
Fig. S9. RNA-seq analysis of vehicle-versus RP-182-treated M2 macrophages and enrichment analysis of binding partners to CD206.
Fig. S10. RP-182 activates phagocytosis and phagolysosome formation in M2- but not M1-polarized human macrophages derived from PBMCs of healthy volunteers.
Fig. S11. RP-182 activates phagocytosis, autophagy, and apoptosis in BMDMs cocultured with conditioned medium from PANC1 cancer cells.
Fig. S12. RP-182 but not control peptide RP-426 induces phagocytosis in M2-polarized BMDMs.
Fig. S13. RP-182 reduces IKK-α and activates autophagy and caspase 8 in M2-polarized BMDMs.
Fig. S14. RP-182 activates autophagy and apoptosis in human M2 macrophages.
Fig. S15. RP-182 but not RP-426 induces cell death in M2-polarized macrophages.
Fig. S16. RP-182 does not inhibit growth of pluripotent progenitor cells, cancer cells, fibroblasts, or endothelial or dendritic cells.
Fig. S17. Gating strategy for the determination of CD86+ and CD206-positive CD11b+F4/80+Gr1− macrophage fractions.
Fig. S18. Gating strategy for the determination of M1 cytokine-positive CD11b+F/40+Gr1− macrophages and M1 and M2 marker expression profiles of M2 BMDMs isolated by FACS after treatment with RP-182.
Fig. S19. Gating strategy for the determination of cytokine- and checkpoint-positive cell fractions of CD86- and CD206-positive macrophage populations.
Fig. S20. M1 and M2 gene expression profiles of BMDMs isolated from B6.129P2-Mrc1tm1Mnz/J (CD206−/−) and wild-type C57BL/6 (CD206+/+) mice after polarization into M1 and M2.
Fig. S21. RP-182 induces binding of IQGAP1 to CD206 and recruitment of IQGAP1 to the cell membrane of M2-polarized macrophages.
Fig. S22. Blockade of RP-182-induced TNF signaling abrogates autocrine activation of apoptosis in M2-polarized macrophages.
Fig. S23. CD206 expression in human pancreatic cancer.
Fig. S24. Pretreatment of M2-polarized macrophages with RP-182 reduces induction of expression of EMT markers in KPC cancer cells upon coculture.
Fig. S25. Gating strategy for the identification of tumor-infiltrating CD8+ T cell and CD206high M2-like TAM fractions.
Fig. S26. RP-182 in combination with gemcitabine reduces CD206high monocytic MDSCs in autochthonous KPC tumors.
Fig. S27. Gating strategy for determination of PD-1, SIRP-α, and TNF-α-positive TAM populations in KPC mice.
Fig. S28. RP-182-induced CD86+ TAM populations in KPC mice have increased M1 cytokine and M1 marker expression.
Fig. S29. RP-182 induces phagocytosis, phagolysosome formation, and apoptosis in TAMs but not in CD11b−CK19+ cancer cells.
Fig. S30. Tissue distribution and toxicity measures of RP-182 in mice.
Fig. S31. RP-182 induces cancer cell phagocytosis in CD86- but not CD206-positive M2 BMDMs.
Fig. S32. RP-182 restricts tumor growth of MDA-MB231 breast cancer and C4–2 prostate cancer xenografts grown in T cell-deficient mice.
Table S1. Origins of representative 10-mer biophysical homology peptide sequences within HDPs, virulence factors, and collagens.
Table S2. Relative binding affinities (in kcal/mol) of individual 10-mer homology sequence ligand-lectin receptor combinations by ClusPro web portal identifies CD206 as top binding target.
Table S3. List of primary and secondary antibodies used in immunocytochemistry, tissue immunohistochemistry, Western blots, immunoprecipitation, and flow cytometry experiments with manufacturers and catalog numbers.
Data file S1. Biophysical homology screening of AMPs and HDPs using Molly font.
Data file S2. Characterization of binding partners of CD206 induced by RP-182 in M2 macrophages.
Data file S3. Source data.
Movie S1. RP-182 increases phagocytosis of cancer cells.
Movie S2. Engulfment and internalization of cancer cells by RP-182-treated macrophages.
Acknowledgments:
We thank L. Roy for help with the medical illustrations and visual artwork. We thank the NCI Patient-Derived Models Repository (PDMR), NCI-Frederick, Frederick National Laboratory for Cancer Research, Frederick, MD (pdmr.cancer.gov) for PDX models 193399-133-R, 381576-328-R, 165739-295-R, and 466636-057-R and S. A. Rosenberg for the provision of melanoma cell line 2183.
Funding: This research was supported, in part, by the Intramural Research Program of the NIH, NCI, Center for Cancer Research (ZIA BC 011267; to U.R.), the Intramural Research Program of the NIH, National Center for Advancing Translational Sciences (NCATS) (ZIA TR000302-02 to J.M. and A. Simeonov), donations from “Running for Rachel” and the Pomerenk family via the Rachel Guss and Bob Pomerenk Pancreas Cancer Research Fellowship, as well as funding from U54-MD007585-26 NIH/NIMHD (to C.C.Y.) and U54 CA118623 (NIH/NCI) (to C.C.Y.) and via Department of Defense Grant (PC170315P1, W81XWH-18-1-0589), UNCF/Merck Postdoctoral Science Research Fellowship (to J.C.Z.), and Prostate Cancer Foundation Young Investigator Award (to J.C.Z.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research used 12-ID-B beamline of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. The mass spectroscopy experiments and data analysis were performed by Poochon Scientific, LLC (Frederick, MD).
Footnotes
Competing interests: J.M.J., H.L., and C.C.Y. have equity interest in Riptide Bioscience Inc., which develops RP-182, RP-185, and RP-832c. J.M.J., H.L., C.C.Y., and U.R. hold a patent on RP-182 (“Peptide-Based Methods for Treating Pancreatic Cancer”; U.S. patent no. 10,016,480). All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. The data for the SAXS model of CD206 have been deposited in the Small-Angle Scattering Biological Data Bank (www.sasbdb.org/data/SASDG54/jdbt3at6ao/). Pancreatic cancer PDX models 193399–133-R, 381576–328-R, 165739–295-R, and 466636–057-R can be requested at www.pdmr.nci.gov and RP-182, RP-185, and RP-832c from J.M.J., C.C.Y., or U.R. under material transfer agreement with waived cost recovery.
SUPPLEMENTARY MATERIALS
http://stm.sciencemag.org/cgi/content/full/12/530/eaax6337/DC1
Materials and Methods
Reference (46)
View/request a protocol for this paper from Bio-protocol.
REFERENCES AND NOTES
- 1.Rosenberg SA, Decade in review—Cancer immunotherapy: Entering the mainstream of cancer treatment. Nat. Rev. Clin. Oncol 11, 630–632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribas A, Wolchok JD, Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gajewski TF, Schreiber H, Fu Y-X, Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moynihan KD, Irvine DJ, Roles for innate immunity in combination immunotherapies. Cancer Res. 77, 5215–5221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, Coussens LM, Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Qian B, Pollard JW, Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang M, McKay D, Pollard JW, Lewis CE, Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 78, 5492–5503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P, Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol 14, 399–416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cassetta L, Pollard JW, Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov 17, 887–904 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Scott MG, Dullaghan E, Mookherjee N, Glavas N, Waldbrook M, Thompson A, Wang A, Lee K, Doria S, Hamill P, Yu JJ, Li Y, Donini O, Guarna MM, Finlay BB, North JR, Hancock REW, An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol 25, 465–472 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Achtman AH, Pilat S, Law CW, Lynn DJ, Janot L, Mayer ML, Ma S, Kindrachuk J, Finlay BB, Brinkman FSL, Smyth GK, Hancock REW, Schofield L, Effective adjunctive therapy by an innate defense regulatory peptide in a preclinical model of severe malaria. Sci. Transl. Med 4, 135ra64 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Gallo RL, Hooper LV, Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol 12, 503–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaynes JM, Bernard GC, Structure/function link between cytokine domains and natural and designed lytic peptides: Medical promise, in Small Wonders: Peptides for Disease Control, Rajasekaran K, Cary JW, Jaynes JM, Montesinos E, Eds. (American Chemical Society, 2012), chap. 2, pp. 21–45. [Google Scholar]
- 14.Mishra B, Reiling S, Zarena D, Wang G, Host defense antimicrobial peptides as antibiotics: Design and application strategies. Curr. Opin. Chem. Biol 38, 87–96 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mansour SC, Pena OM, Hancock RE, Host defense peptides: Front-line immunomodulators. Trends Immunol. 35, 443–450 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Hancock REW, Haney EF, Gill EE, The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol 16, 321–334 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schröder JM, Wang JM, Howard OMZ, Oppenheim JJ, β-defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 286, 525–528 (1999). [DOI] [PubMed] [Google Scholar]
- 18.Mookherjee N, Brown KL, Bowdish DME, Doria S, Falsafi R, Hokamp K, Roche FM, Mu R, Doho GH, Pistolic J, Powers J-P, Bryan J, Brinkman FSL, Hancock REW, Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J. Immunol 176, 2455–2464 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Abdillahi SM, Maaß T, Kasetty G, Strömstedt AA, Baumgarten M, Tati R, Nordin SL, Walse B, Wagener R, Schmidtchen A, Mörgelin M, Collagen VI contains multiple host defense peptides with potent in vivo activity. J. Immunol 201, 1007–1020 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Drickamer K, Taylor ME, Recent insights into structures and functions of C-type lectins in the immune system. Curr. Opin. Struct. Biol 34, 26–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S, The ClusPro web server for protein-protein docking. Nat. Protoc 12, 255–278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Pomares L, The mannose receptor. J. Leukoc. Biol 92, 1177–1186 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Hu Z, Shi X, Yu B, Li N, Huang Y, He Y, Structural insights into the pH-dependent conformational change and collagen recognition of the human mannose receptor. Structure 26, 60–71.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Lee SJ, Evers S, Roeder D, Parlow AF, Risteli J, Risteli L, Lee YC, Feizi T, Langen H, Nussenzweig MC, Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science 295, 1898–1901 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Gensel JC, Kopper TJ, Zhang B, Orr MB, Bailey WM, Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment. Sci. Rep 7, 40144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajaram MVS, Arnett E, Azad AK, Guirado E, Ni B, Gerberick AD, He L-Z, Keler T, Thomas LJ, Lafuse WP, Schlesinger LS, tuberculosis-initiated M human mannose receptor signaling regulates macrophage recognition and vesicle trafficking by FcRγ-chain, Grb2, and SHP-1. Cell Rep. 21, 126–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandt DT, Marion S, Griffiths G, Watanabe T, Kaibuchi K, Grosse R, Dia1 and IQGAP1 interact in cell migration and phagocytic cup formation. J. Cell Biol 178, 193–200 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owen D, Campbell LJ, Littlefield K, Evetts KA, Li Z, Sacks DB, Lowe PN, Mott HR, The IQGAP1-Rac1 and IQGAP1-Cdc42 interactions: Interfaces differ between the complexes. J. Biol. Chem 283, 1692–1704 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Ock C-Y, Keam B, Kim S, Lee J-S, Kim M, Kim TM, Jeon YK, Kim D-W, Chung DH, Heo DS, Pan-cancer immunogenomic perspective on the tumor microenvironment based on PD-L1 and CD8 T-cell infiltration. Clin. Cancer Res 22, 2261–2270 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A, Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiss G, Schaible UE, Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev 264, 182–203 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan H-Y, Wang N, Man K, Tsao S-W, Che C-M, Feng Y, Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis. 6, e1942 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu M, O’Connor RS, Trefely S, Graham K, Snyder NW, Beatty GL, Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated ‘don’t-eat-me’ signal. Nat. Immunol 20, 265–275 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madsen DH, Jürgensen HJ, Siersbæk MS, Kuczek DE, Grey Cloud L, Liu S, Behrendt N, Grøntved L, Weigert R, Bugge TH, Tumor-associated macrophages derived from circulating inflammatory monocytes degrade collagen through cellular uptake. Cell Rep. 21, 3662–3671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song W, Mazzieri R, Yang T, Gobe GC, Translational significance for tumor metastasis of tumor-associated macrophages and epithelial-mesenchymal transition. Front. Immunol 8, 1106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dewhirst MW, Secomb TW, Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 17, 738–750 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Firer MA, Gellerman G, Targeted drug delivery for cancer therapy: The other side of antibodies. J. Hematol. Oncol 5, 70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raucher D, Ryu JS, Cell-penetrating peptides: Strategies for anticancer treatment. Trends Mol. Med 21, 560–570 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Cieslewicz M, Tang J, Yu JL, Cao H, Zavaljevski M, Motoyama K, Lieber A, Raines EW, Pun SH, Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl. Acad. Sci. U.S.A 110, 15919–15924 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scodeller P, Simón-Gracia L, Kopanchuk S, Tobi A, Kilk K, Säälik P, Kurm K, Squadrito ML, Kotamraju VR, Rinken A, de Palma M, Ruoslahti E, Teesalu T, Precision targeting of tumor macrophages with a CD206 binding peptide. Sci. Rep 7, 14655 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fogal V, Zhang L, Krajewski S, Ruoslahti E, Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 68, 7210–7218 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yanashima K, Chieosilapatham P, Yoshimoto E, Okumura K, Ogawa H, Niyonsaba F, Innate defense regulator IDR-1018 activates human mast cells through G protein-, phospholipase C-, MAPK- and NF-κB-sensitive pathways. Immunol. Res 65, 920–931 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Wu BC, Lee AH-Y, Hancock REW, Mechanisms of the innate defense regulator peptide-1002 anti-inflammatory activity in a sterile inflammation mouse model. J. Immunol 199, 3592–3603 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Georgoudaki A-M, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, Dahan R, Harris RA, Rantalainen M, Klevebring D, Sund M, Brage SE, Fuxe J, Rolny C, Li F, Ravetch JV, Karlsson MCI, Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 15, 2000–2011 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG, CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, DeNicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA, Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Biophysical homology screening using Molly font identifies conservation of 10-mer structural motif across HDPs, outer surface virulence factors, and collagens.
Fig. S2. Docking of biophysical 10-mer homology motifs onto C-type lectin receptors.
Fig. S3. Full-length CD206 model.
Fig. S4. RP-182 binds to CD206.
Fig. S5. RP-182 but not RP-426 induces a cellular thermostability shift of CD206 in human and murine M2 macrophages.
Fig. S6. Abbreviated synthesis scheme of RP-182 analogs with diazirine-containing phenylalanine and biotin.
Fig. S7. RP-182 binds to carbohydrate recognition domain 5 of the CD206 receptor.
Fig. S8. Gene expression changes induced by RP-182 in M1- and M2-polarized BMDMs.
Fig. S9. RNA-seq analysis of vehicle-versus RP-182-treated M2 macrophages and enrichment analysis of binding partners to CD206.
Fig. S10. RP-182 activates phagocytosis and phagolysosome formation in M2- but not M1-polarized human macrophages derived from PBMCs of healthy volunteers.
Fig. S11. RP-182 activates phagocytosis, autophagy, and apoptosis in BMDMs cocultured with conditioned medium from PANC1 cancer cells.
Fig. S12. RP-182 but not control peptide RP-426 induces phagocytosis in M2-polarized BMDMs.
Fig. S13. RP-182 reduces IKK-α and activates autophagy and caspase 8 in M2-polarized BMDMs.
Fig. S14. RP-182 activates autophagy and apoptosis in human M2 macrophages.
Fig. S15. RP-182 but not RP-426 induces cell death in M2-polarized macrophages.
Fig. S16. RP-182 does not inhibit growth of pluripotent progenitor cells, cancer cells, fibroblasts, or endothelial or dendritic cells.
Fig. S17. Gating strategy for the determination of CD86+ and CD206-positive CD11b+F4/80+Gr1− macrophage fractions.
Fig. S18. Gating strategy for the determination of M1 cytokine-positive CD11b+F/40+Gr1− macrophages and M1 and M2 marker expression profiles of M2 BMDMs isolated by FACS after treatment with RP-182.
Fig. S19. Gating strategy for the determination of cytokine- and checkpoint-positive cell fractions of CD86- and CD206-positive macrophage populations.
Fig. S20. M1 and M2 gene expression profiles of BMDMs isolated from B6.129P2-Mrc1tm1Mnz/J (CD206−/−) and wild-type C57BL/6 (CD206+/+) mice after polarization into M1 and M2.
Fig. S21. RP-182 induces binding of IQGAP1 to CD206 and recruitment of IQGAP1 to the cell membrane of M2-polarized macrophages.
Fig. S22. Blockade of RP-182-induced TNF signaling abrogates autocrine activation of apoptosis in M2-polarized macrophages.
Fig. S23. CD206 expression in human pancreatic cancer.
Fig. S24. Pretreatment of M2-polarized macrophages with RP-182 reduces induction of expression of EMT markers in KPC cancer cells upon coculture.
Fig. S25. Gating strategy for the identification of tumor-infiltrating CD8+ T cell and CD206high M2-like TAM fractions.
Fig. S26. RP-182 in combination with gemcitabine reduces CD206high monocytic MDSCs in autochthonous KPC tumors.
Fig. S27. Gating strategy for determination of PD-1, SIRP-α, and TNF-α-positive TAM populations in KPC mice.
Fig. S28. RP-182-induced CD86+ TAM populations in KPC mice have increased M1 cytokine and M1 marker expression.
Fig. S29. RP-182 induces phagocytosis, phagolysosome formation, and apoptosis in TAMs but not in CD11b−CK19+ cancer cells.
Fig. S30. Tissue distribution and toxicity measures of RP-182 in mice.
Fig. S31. RP-182 induces cancer cell phagocytosis in CD86- but not CD206-positive M2 BMDMs.
Fig. S32. RP-182 restricts tumor growth of MDA-MB231 breast cancer and C4–2 prostate cancer xenografts grown in T cell-deficient mice.
Table S1. Origins of representative 10-mer biophysical homology peptide sequences within HDPs, virulence factors, and collagens.
Table S2. Relative binding affinities (in kcal/mol) of individual 10-mer homology sequence ligand-lectin receptor combinations by ClusPro web portal identifies CD206 as top binding target.
Table S3. List of primary and secondary antibodies used in immunocytochemistry, tissue immunohistochemistry, Western blots, immunoprecipitation, and flow cytometry experiments with manufacturers and catalog numbers.
Data file S1. Biophysical homology screening of AMPs and HDPs using Molly font.
Data file S2. Characterization of binding partners of CD206 induced by RP-182 in M2 macrophages.
Data file S3. Source data.
Movie S1. RP-182 increases phagocytosis of cancer cells.
Movie S2. Engulfment and internalization of cancer cells by RP-182-treated macrophages.