

Abstract
The identification of biomarkers for patient stratification is fundamental to precision medicine efforts in oncology. Here, we identified two baseline, circulating immune cell subsets associated with overall survival in patients with metastatic pancreatic cancer who were enrolled in two phase II randomized studies of GVAX pancreas and CRS-207 immunotherapy. Single-cell mass cytometry was used to simultaneously measure 38 cell surface or intracellular markers in peripheral blood mononuclear cells obtained from a phase IIa patient subcohort (N = 38). CITRUS, an algorithm for identification of stratifying subpopulations in multidimensional cytometry datasets, was used to identify single-cell signatures associated with clinical outcome. Patients with a higher abundance of CD8+CD45RO−CCR7−CD57+ cells and a lower abundance of CD14+CD33+CD85j+ cells had improved overall survival [median overall survival, range (days) 271, 43–1,247] compared with patients with a lower abundance of CD8+CD45RO−CCR7−CD57+ cells and higher abundance of CD14+CD33+CD85j+ cells (77, 24–1,247 days; P = 0.0442). The results from this prospective–retrospective biomarker analysis were validated by flow cytometry in 200 patients with pancreatic cancer enrolled in a phase IIb study (P = 0.0047). The identified immune correlates provide potential prognostic or predictive signatures that could be employed for patient stratification.
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related mortality worldwide. Pancreatic ductal adenocarcinoma (PDAC) comprises over 90% of all pancreatic cancers, with less than a 5% 5-year survival rate (1). Because pancreatic cancer is usually diagnosed at an advanced stage due to lack of specific symptoms early in the disease, surgery is suitable only for a minority of patients. Unresectable and metastatic diseases have a median survival of 10 to 12 months when treated and 3 to 6 months when untreated, respectively (2).
GVAX and CRS-207 are cancer vaccines that are designed to target overexpressed tumor antigens and have been evaluated in clinical trials for PDAC. GVAX Pancreas is composed of two irradiated, GM-CSF–secreting allogeneic PDAC cell lines administered 1 day after treatment with low-dose cyclophosphamide (Cy) to inhibit regulatory T cells (Treg; ref. 3). GVAX induces T cells against a broad array of PDAC antigens, including mesothelin-specific T-cell responses that have been shown to correlate with survival (4). Mesothelin is a tumor-associated antigen overexpressed in most PDACs. CRS-207 is a recombinant live-attenuated, double-deleted Listeria monocytogenes (LADD-Lm) engineered to secrete mesothelin into the cytosol of infected antigen-presenting cells, which subsequently gets processed and presented in the context of MHC molecules (5, 6).
One of the major goals of immunotherapy research is to determine why some patients exhibit responses to therapy, whereas others gain only short-term benefits, if at all. Because most immunotherapies function by stimulating the immune system to target tumors, understanding the immune competence of patients is critical to determining which patients are most likely to benefit from such therapies. Cellular correlates of immune competence can be assessed using flow cytometry, which permits the reliable measurement of up to 12 to 16 parameters per cell via fluorescently labeled antibodies. Mass cytometry facilitates immune phenotyping of at least 40 cell surface or intracellular parameters per individual cell (7) via metal-labeled antibodies, thereby generating a more comprehensive view of immune cell phenotypes and functions. Biaxial manual gating is traditionally used to analyze cytometry data. However, this approach often relies on biases and a priori knowledge, while at times overlooking undefined cell populations. Biaxial manual gating is not scalable to single-cell, multiparametric mass cytometry data derived from large-scale experiments comprising multiple subjects and timepoints (8). Traditional statistical methods, such as principal component analysis, and novel computational methods have been applied to the analysis of high-dimensional mass cytometry datasets, such as SPADE, viSNE, Wanderlust, and ACCENSE (8). CITRUS (cluster identification, characterization, and regression) is another such data-driven approach for the identification of stratifying subpopulations in multidimensional mass cytometry datasets (9) and can be used to identify single-cell signatures that might be associated with clinical outcome. This approach has been used to identify signaling responses in subsets of circulating CD14+ monocytes that correlate with clinical parameters of surgical recovery, including functional impairment and pain (10).
In this study, mass cytometry and CITRUS were used to identify pretreatment single-cell signatures that were associated with overall survival (OS) in a subcohort of 38 patients with PDAC enrolled in a phase IIa randomized study of Cy/GVAX in combination with CRS-207 and Cy/GVAX alone (11). The candidate cellular biomarkers were further verified by prospective–retrospective flow cytometry analysis in a subcohort of 200 patients with pancreatic cancer enrolled in a phase IIb randomized study of Cy/GVAX in combination with CRS-207 and Cy/GVAX, CRS-207 monotherapy, and single-agent chemotherapy (ClinicalTrials.gov ID: NCT02004262). These results demonstrated that baseline cellular signatures of immune competency could be used to identify patients with pancreatic cancer most likely to respond to therapy and demonstrated the utility of mass cytometry as a platform for the identification of cellular biomarkers in a clinically relevant context.
Materials and Methods
Study design
A subcohort of 38 patients from a previously enrolled phase IIa trial of 90 patients (second line) was included in this analysis (11). The subcohort was selected based on clinical outcome to achieve an equal number of patients with best, poor, and worst responses. For prospective analysis, a subcohort of 200 patients (second and third lines) from a phase IIb trial of 303 patients was included in this analysis (ClinicalTrials.gov ID: NCT02004262). Briefly, previously treated patients with stage IV metastatic PDAC were enrolled. Inclusion criteria were Eastern Cooperative Oncology Group (ECOG) between 0 and 1, no radiographic ascites or clinically significant pleural effusions, and no implants or prosthetic devices that were not easily removed. Both studies were reviewed by the local Institutional Review Boards, biosafety committees, the FDA, and the National Institutes of Health Recombinant DNA Advisory Committee. These studies were conducted in accordance with Good Clinical Practice, the ethical principles of the Declaration of Helsinki, and the International Conference on Harmonisation E6 (Guideline for Good Clinical Practice). Informed written consent was obtained from the subjects for participation in the studies.
Phase IIa
Patients were randomly assigned at a ratio of 2:1 to two doses of Cy (Baxter)/GVAX (Johns Hopkins University, Baltimore, MD) followed by four doses of CRS-207 (Aduro BioTech; Arm A) or six doses of Cy/GVAX (Arm B) every 3 weeks. Stable patients were offered additional courses. Administered doses were as follows: Cy at 200 mg/m2, GVAX at 2.5 × 108 cells, each of Panc 6.03 and Panc 10.05, and CRS-207 at 1 × 109 colony-forming units. The primary endpoint was OS between arms. Secondary end points were safety and clinical response as defined by RECIST (version 1.1).
Phase IIb
Patients were randomly assigned at a ratio of 1:1:1 to two doses of Cy/GVAX followed by four doses of CRS-207 (Arm A) or six doses of CRS-207 (Arm B) at the dosages defined above, every 3 weeks or physician’s choice of single-agent chemotherapy (Arm C). Standard chemotherapy options for Arm C included capecitabine (1,000 mg/m2 orally twice a day, days 1–14 on a 21-day cycle), infusional 5-fluorouracil (2,400 mg/m2 continuous i.v. infusion over 46 hours on days 1 and 15 on a 28-day cycle), gemcitabine (1,000 mg/m2 i.v. on days 1, 8, and 15 on a 28-day cycle), irinotecan (150 mg/m2 i.v. on days 1 and 15 on a 28-day cycle), and erlotinib (100 mg orally once daily on a 21-day cycle). Stable patients were offered additional courses. The primary endpoint was OS between Arm A and Arm C in the primary cohort, which comprised third-line patients. OS was calculated based on start of treatment until time of death. OS, namely, best (median, 579; range, 247–1,247 days), poor (median, 92; range, 58–115 days), and worst (median, 38; range, 24–54 days) responders, and number of treatments received (≥6, 3–5, 1–2, respectively) were used to generate clinical response categories, for the phase IIa subcohort training set.
Patient sample processing
Peripheral blood mononuclear cells (PBMC) were isolated within 6 hours of blood draw (prior to treatment, and at 7 and 10 weeks after treatment) by qualified laboratory specialists and stored at −180°C until analysis. PBMCs were available for analysis from 38 patients from the phase IIa study and 200 patients from the phase IIb study. Briefly, whole blood was diluted 1:1 with 1X PBS (Mediatech), overlaid onto Ficoll-Paque Plus (GE Healthcare) in Leucosep tubes (Greiner Bio One). The tubes were centrifuged for 20 minutes at 800 × g at room temperature with the brake of the centrifuge not engaged. Following centrifugation, plasma was removed and frozen, and the remaining contents of the Leucosep tubes containing PBMCs were washed twice with 1X PBS. Cells were counted, and 10 million viable PBMCs per cryovial were frozen in 10% dimethylsulfoxide (Sigma) in FBS (Valley Biomedical) in a Mr. Frosty container (Nalgene) at −80°C for at least 16 hours but no longer than 24 hours. The PBMCs were transferred to liquid nitrogen for storage.
Mass cytometry
Mass cytometry was performed in the Human Immune Monitoring Center at Stanford University. PBMCs were thawed in warm complete Roswell Park Memorial Institute (RPMI) media (RPMI supplemented with 10% BSA, penicillin/streptomycin; Thermo Fisher Scientific), washed twice, resuspended in FACS buffer (PBS supplemented with 2% BSA, 2 mmol/L EDTA, and 0.1% sodium azide), and viable cells were counted by Vi-Cell (Beckman Coulter). Only samples with a cell viability ≥80% were included in subsequent analyses. Cells were added to a V-bottom microtiter plate (Corning) at 2 million viable cells per well and washed once by pelleting and resuspension in fresh FACS buffer. The cells were stained for 60 minutes on ice with 50 μL of the following surface antibody–polymer conjugate cocktail listed in Supplementary Table S1A. Antibodies were chosen to facilitate the identification of major immune cell types in peripheral blood. All antibodies were from purified unconjugated, carrier-protein-free stocks from BD Biosciences, Biolegend, Fluidigm, Southern Biotech, Abcam, or R&D Systems. The polymer and metal isotopes were from Fluidigm. The cells were washed twice by pelleting and resuspension with 250 μL FACS buffer. The cells were resuspended in 100 μL PBS buffer containing 2 μg/mL DOTA-maleimide (Macrocyclics) containing natural-abundance indium. The cells were washed twice by pelleting and resuspension with 250 μL PBS. The cells were resuspended in 100 μL 2% paraformaldehyde in PBS and placed at 4°C overnight. The next day, the cells were pelleted and washed by resuspension in fresh PBS. The cells were resuspended in 100 μL Permeabilization Buffer (1x in PBS; eBiosciences) and placed on ice for 45 minutes before washing twice with 250 μL PBS. For intracellular staining, the cells were resuspended in 50 μL intracellular antibody cocktail (Supplementary Table S1A) in FACS buffer for 1 hour on ice before washing twice in FACS buffer. The cells were resuspended in 100 μL iridium-containing DNA intercalator (1:2,000 dilution in PBS; Fluidigm) and incubated at room temperature for 20 minutes. The cells were washed twice in 250 μL MilliQ water. The cells were diluted in a total volume of 700 μL in MilliQ water before injection into a CyTOF2 (Fluidigm). At least 200,000 events were acquired per sample. Data were normalized using Normalizer v0.1 MCR (Mathworks). Gating was performed in Cytobank (Beckman Coulter). Dead cells, doublets, and debris were removed from FCS files prior to analysis in Cytobank.
Flow cytometry
Multicolor flow cytometry panels were developed to enumerate the cell subsets identified by mass cytometry (Supplementary Table S1B). Each panel was optimized to ensure minimal spectral overlap among fluorochromes and minimal antibody interference. Inter- and intraassay precision and repeatability were determined to be <10% CV. Only samples with a cell viability ≥80% as determined by Guava Viacount (Millipore) were included in the analysis. One million live PBMCs per patient were stained for each panel. Three deidentified healthy donor PBMCs isolated and cryopreserved according to the procedures above were included per run to ensure that interrun variability was <10%. PBMCs were washed with 1x PBS and incubated with LIVE/DEAD Fixable Aqua Dead Cell Stain kit (Thermo Fisher) for 20 minutes at room temperature. PBMCs were washed and stained with a cocktail of fluorescently labeled antibodies (Supplementary Table S1B) for 30 minutes at 4°C. Cells were washed 3 times and analyzed on the X-20 LSRFortessa (BD Biosciences). Application settings for each panel were used for all data acquisition. At least 200,000 events were acquired per sample. Gating was performed in Cytobank. Dead cells, doublets, and debris were removed from FCS files prior to analysis in Cytobank. Major immune cell lineages were identified using manual gating analysis (Supplementary Fig. S1).
Data and statistical analysis
For CITRUS analysis of mass cytometry data, patients were allocated into best, poor, and worst responder groups as defined above (9). Note that 10,000 events per patient were clustered with a minimum cluster size of 1%. All cell surface markers were used for clustering. Clusters were characterized in terms of abundance features or the proportion of a sample’s cells that belong to a cluster. Predictive and correlative models were used to detect the associations between cluster abundance and clinical outcome categorization using significance analysis of microarrays (sam) and nearest shrunken centroid (pamr; ref. 9). All clusters identified by CITRUS were verified by manual gating. One-way ANOVA was used to analyze differences between multiple groups. The unpaired t test was used to analyze differences between two groups. P values <0.05 were considered significant. Analysis was performed using GraphPad Prism v6.0.
Results
Mass cytometry identification of candidate immune cell subsets associated with OS
The training set for this study comprised patients with stage IV PDAC from a previously enrolled phase IIa clinical trial of 90 patients who had received one prior chemotherapy treatment (second-line patients; ref. 11). Patients were randomized and enrolled into one of two treatment arms. Arm A patients received Cy/GVAX in combination with CRS-207, whereas Arm B patients received Cy/GVAX only. Treatments were administered at 3-week intervals. A subcohort comprising 38 of these patients was selected as the training set for immune analysis on the basis of OS. The patients in the subcohort were selected to ensure equal distribution of best (n = 14), poor (n = 12), and worst (n = 12) responders based on their OS [median, 579 (247–1,247), 92 (58–115), 38 (24–54) days, respectively] and the number of treatments they received (≥6, 3–5, 1–2, respectively). Baseline clinical and demographic characteristics among the subcohort did not differ among patients in each response category or among treatment arms (Table 1). PBMCs obtained at baseline and at 7 and 10 weeks after treatment were analyzed by mass cytometry using a panel of 38 metal-tagged antibodies (Supplementary Table S1A). Major immune cell subsets including CD8+ T cells, CD4+ T cells, B cells, natural killer (NK) cells, myeloid cells, and γδ T cells were identified by manual gating analysis at baseline and over the course of treatment (Fig. 1A; Supplementary Fig. S1).
Table 1.
Clinical and demographic characteristics of the phase IIa patients used in the training set.
| Number of subjects (% of treated) | Arm A N = 18 | Arm B N = 20 | All arms N = 38 | P value (Arm A vs. Arm B) |
|---|---|---|---|---|
| Age (yr) | ||||
| Median | 63 | 64 | 63.5 | 0.7478 |
| Range | 52–83 | 47–80 | 47–83 | |
| Sex, N (%) | ||||
| Male | 78 | 65 | 71 | 0.3858 |
| Female | 22 | 35 | 29 | |
| ECOG, N (%) | ||||
| 0 | 67 | 60 | 63 | 0.6706 |
| 1 | 33 | 40 | 37 | |
| Sites of metastases, N (%) | ||||
| Liver | 78 | 75 | 76 | 0.6267 |
| Lung | 61 | 35 | 47 | |
| Other | 33 | 35 | 34 | |
| Disease status at study entry, N (%) | ||||
| Stable disease | 22 | 25 | 24 | 0.8406 |
| Progressive disease | 78 | 75 | 76 | |
| Prior chemotherapy for metastatic disease, N (%) | ||||
| 0 | 33 | 15 | 24 | 0.1378 |
| 1 | 17 | 45 | 32 | |
| 2+ | 50 | 40 | 45 | |
| Previous surgical resection, N (%) | ||||
| Yes | 72 | 55 | 63 | 0.2718 |
| No | 28 | 45 | 37 | |
Note: P values calculated for continuous variables using t test and for categorical variables using χ2 test.
Figure 1.
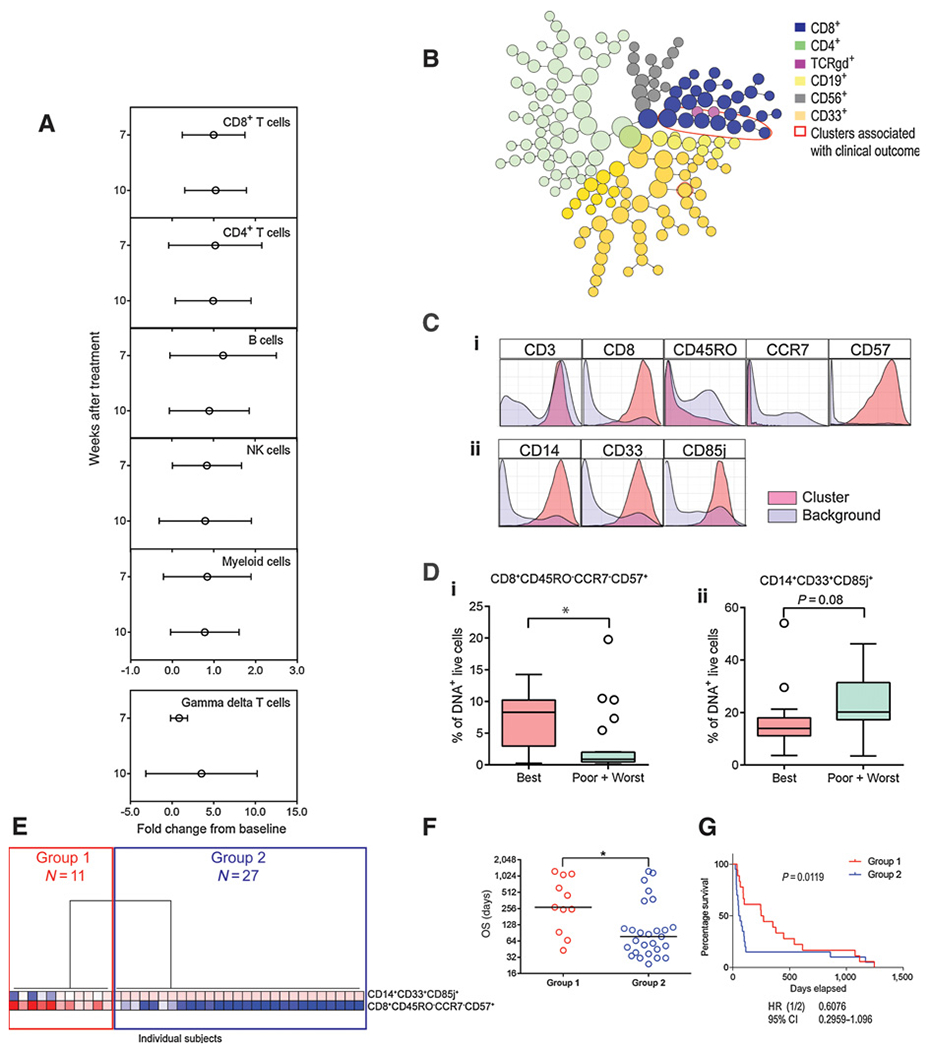
The abundance of two candidate immune cell subsets is correlated with clinical outcome at baseline in patients with pancreatic cancer. Unstimulated PBMCs from the 38-patient phase IIa subcohort were stained with a panel of 38 metal-tagged antibodies specific to cell surface and intracellular markers and analyzed by mass cytometry. Live, DNA+ events were used for analysis. A, Manual gating analysis of major immune cell subsets and their kinetics over the course of treatment. Shown is the mean fold change (±SD) from baseline at weeks 7 and 10 after treatment for CD8+ T cells, CD4+ T cells, B cells, NK cells, myeloid cells, and γδ T cells. B, CITRUS plot showing clusters comprised of phenotypically similar cells at baseline. Major immune cell lineages are indicated by color. Cell subsets associated with clinical outcome are outlined in red. C, Histogram representation of cell surface marker expression in the two immune cell subsets associated with clinical outcome (pink) compared with total, live DNA+ cells (purple). D, Boxplot representation of the abundance of the two immune cell subsets (i. CD8+CD45RO−CCR7−CD57+ and ii. CD14+CD33+CD85j+) at baseline determined by manual gating in patients with different clinical outcome categorization. P values determined by one-way ANOVA. *, P < 0.05. Line represents the median, and whiskers represent Tukey. E, Hierarchical clustering of the abundance of manually gated cell subsets associated with clinical outcome in the 38 patients; the two major groups of patients are indicated. F, OS in the two groups identified in E. Dots represent individual patient values; lines represent median values. P values were determined by unpaired t test. *, P < 0.05. G, Kaplan–Meier plots of groups identified in E. P value determined using the log-rank test. Repeat experiments were not performed due to limited patient material. CI, confidence interval.
The single-cell data were also analyzed by CITRUS to determine if any cell subsets were associated with OS. In CITRUS analysis, clusters of phenotypically similar cells that corresponded to major immune cell subsets, based on the expression of canonical lineage markers, were identified. At baseline, the abundance of eight cell clusters that belonged to CD8+ T-cell and myeloid cell lineages was associated with OS (Fig. 1B). The phenotypes of the clusters correlated with OS across both treatment arms and were characterized by CD8+CD45RO−CCR7−CD57+ and CD14+CD33+CD85j+ surface expression (Fig. 1C). The CD8+ clusters were also CD45RA+CD127−HLA-DR1o/−XD381o/−PD-1+, whereas the CD14+ clusters were also HLA-DR+CD11b+CD15−PD-L1+. Other than the markers listed above, the clusters were heterogeneous in terms of their expression of the additional markers included in the mass cytometry panel (Supplementary Fig. S2).
Manual gating analysis of the CD8+CD45RO−CCR7−CD57+ and CD14+CD33+CD85j+ subsets was used to verify the CITRUS results (Supplementary Fig. S3). The baseline median frequency of manually gated CD8+CD45RO−CCR7−CD57+ cells was higher in best responders compared with poor or worst responders (P = 0.0100). The median frequency of CD14+CD33+CD85j+ cells was higher in poor or worst responders compared with best responders, albeit the latter was not significant (P = 0.0785; Fig. 1D; Table 2). Unsupervised hierarchical clustering of manually gated cell subset abundance associated with clinical response in the 38 patients revealed two major groups of patients, denoted Group 1 (N = 11) and Group 2 (N = 27). These groups mainly differed based on the frequencies of the CD8 cluster. Patients in Group 1 had an elevated abundance of CD8+CD45RO−CCR7−CD57+ cells and a lower abundance of the CD14+CD33+CD85j+ cells, whereas patients in Group 2 had lower abundance of CD8+CD45RO−CCR7−CD57+ cells and higher abundance of the CD14+CD33+CD85j+ cells (Fig. 1E). Patients in Group 1 demonstrated longer OS compared with patients in Group 2 [median OS, range (days), Group 1 = 271, 43–1,247; Group 2 = 77, 24–1,247; P = 0.0442; Fig. 1F and G]. Group 1 overlapped with the best/poor responder classification, whereas Group 2 overlapped with the poor/worst response classifications above in terms of OS. Group 1 patients had higher OS compared with Group 2 patients in both arms of the study, thus suggesting to be a prognostic factor for survival [median OS, range (days) in Group 1 vs. Group 2; Arm A, 350,43–1,247 vs. 77, 24–1,247; Arm B, 271, 247–615 vs. 75, 31–380]. However, this analysis was limited due to a low number of patients in each group when the groups were substratified by treatment. The frequencies of CD8+CD45RO−CCR7−CD57+ and CD14+CD33+CD85j+ cells were not different between Arm A and Arm B (Arm A vs. Arm B for CD8+CD45RO−CCR7−CD57+, P = 0.5530; CD14+CD33+CD85j+, P = 0.7170) patients at baseline, determined by manual gating analysis, suggesting that the treatment arms were balanced (Supplementary Fig. S4). To further verify the mass cytometry results, the frequencies of CD8+CD45RO−CCR7−CD57+ and CD14+CD33+CD85j+ cells were determined in the same set of patients by both flow and mass cytometry (N = 38; Supplementary Table S1B). The frequencies of both subsets measured using the two independent methods were correlated (Supplementary Fig. S5).
Table 2.
Frequencies of immune cell subsets associated with OS at baseline and at 7 and 10 weeks after treatment determined by manual gating analysis of mass cytometry data.
| CD8+CD45RO−CCR7−CD57+ | ||||||
|---|---|---|---|---|---|---|
| Best |
Poor/Worst |
|||||
| Time point | Median (%) | Range (%) | Median (%) | Range (%) | P value | N |
| Baseline | 8.3 | 0.3–14.3 | 0.9 | 0.1–19.8 | 0.0100 | 38 |
| Week 7 | 5.0 | 0.2–8.3 | 0.0 | 0.2–7.6 | 0.0286 | 25 |
| Week 10 | 4.7 | 0.2–9.0 | 0.6 | 0.1–3.3 | 0.0707 | 15 |
| CD14+CD33+CD85j+ | ||||||
| Best |
Poor/Worst |
|||||
| Time point | Median (%) | Range (%) | Median (%) | Range (%) | P value | N |
| Baseline | 14.0 | 3.6–54.1 | 20.2 | 3.5–46.1 | 0.0785 | 38 |
| Week 7 | 8.1 | 2.9–25.3 | 12.5 | 5.5–40.5 | 0.0355 | 25 |
| Week 10 | 12.0 | 3.0–20.4 | 24.9 | 18.7–33.0 | 0.0023 | 15 |
Note: P values calculated using unpaired t test.
Baseline immune subsets associate with OS at 7 and 10 weeks after treatment
The identified baseline cellular biomarkers in the training set were measured longitudinally to determine if they remained associated with OS during treatment. Of the initial 38 patient subcohort, 25 patients survived to week 7, and 15 patients survived to week 10, independent of treatment arm. At week 7, all patients received two doses of Cy/GVAX at 3-week intervals and at week 10. Arm A patients also received two doses of Cy/GVAX followed by a single dose of CRS-207, and Arm B patients received three doses of Cy/GVAX at 3-week intervals. PBMCs were obtained 3 weeks following the final vaccination. Results from CITRUS analysis of patients at weeks 7 and 10 after treatment were concordant with the pretreatment analysis results (Fig. 2A). Manual gating verification of the CITRUS results revealed that the median frequency of CD8+CD45RO−CCR7−CD57+ cells was higher in best responders compared with poor responders at week 7 (P = 0.0286) and at week 10 (P = 0.0707). In contrast, the median frequency of CD14+CD33+CD85j+ cells was higher in poor responders compared with best responders at week 7 (P = 0.0355) and at week 10 (P = 0.0023; Fig. 2B and C; Supplementary Fig. S3; Table 2).
Figure 2.
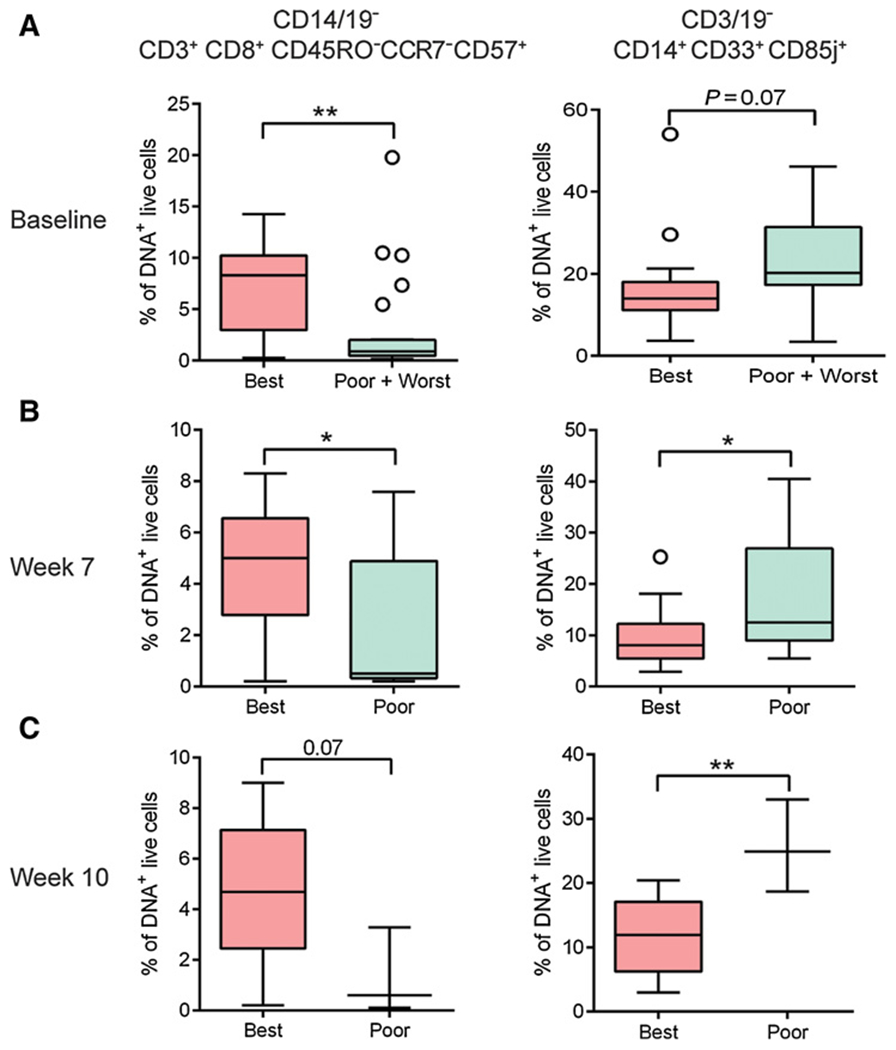
Candidate baseline prognostic immune cell biomarkers remain associated with clinical response at baseline and at 7 and 10 weeks after treatment. Unstimulated PBMCs from the same patients in Fig. 1 were analyzed by mass cytometry. Live, DNA+ events were gated and analyzed by manual gating. A–C, Boxplot representation of the abundance of the two immune cell subsets associated with survival in patients with differential clinical outcome categorization at baseline and at 7 and 10 weeks after treatment. Fourteen best, 12 poor, and 12 worst patients were included in baseline analysis; 13 best and 12 poor patients were included in week 7 analysis; and 13 best and 3 poor patients were included in week 10 analysis. Line represents the median, and whiskers represent Tukey. P values were determined by unpaired t test. *, P < 0.05; **, P < 0.005. Repeat experiments were not performed due to limited patient material.
Prospective–retrospective analysis of subsets associated with OS in a phase IIb PDAC trial
We next verified the results from our training set in a prospective–retrospective analysis of 200 patients enrolled in a phase IIb study. The validation cohort was derived from a study of 303 patients with metastatic pancreatic cancer (second- and third-line patients; ClinicalTrials.gov ID: NCT02004262) in which patients were randomized and enrolled into one of three treatment arms. Arm A patients received Cy/GVAX in combination with CRS-207, Arm B patients received CRS-207 monotherapy, and Arm C patients received single-agent chemotherapy. Treatments were administered at 3-week intervals. Baseline clinical and demographic characteristics in the subcohort of patients included in this analysis did not differ between treatment arms (Table 3). The baseline abundance of CD8+CD45RO−CCR7−CD57+ and CD14+CD33+CD85j+ cell subsets was measured by multicolor flow cytometry (Supplementary Table S1B). Unsupervised hierarchical clustering of the abundance of the two cell subsets revealed two major groups of patients independent of treatment assignment, denoted Group 1 (N = 45) and Group 2 (N = 155; Fig. 3A). These groups mainly differed in the frequencies of the CD8 cluster. Patients in Group 1 had elevated abundance of CD8+CD45RO−CCR7−CD57+ cells compared with patients in Group 2 (P < 0.0001). Patients in Group 2 had a higher abundance of CD14+CD33+CD85j+ cells compared with patients in Group 1 (P < 0.0001). The ratio of CD8+CD45RO−CCR7−CD57+ to CD14+CD33+CD85j+ cells was higher in Group 1 patients compared with Group 2 patients (P < 0.0001; Fig. 3B; Table 4).
Table 3.
Clinical and demographic characteristics of the phase IIb patients used in the validation set.
| Number of subjects (% of treated) | Arm A N = 81 | Arm B N = 79 | Arm C N = 40 | All arms N = 200 | P value |
|---|---|---|---|---|---|
| Age (yr) | |||||
| Median | 64 | 65 | 65 | 64.5 | 0.602 |
| Range | 41–91 | 36–82 | 45–84 | 36–91 | |
| Sex, N (%) | |||||
| Male | 53 | 53 | 45 | 52 | 0.6551 |
| Female | 47 | 47 | 55 | 49 | |
| ECOG, N (%) | |||||
| 0 | 40 | 34 | 45 | 39 | 0.4456 |
| 1 | 60 | 66 | 53 | 61 | |
| 2 | 3 | 1 | |||
| Sites of metastases, N (%) | |||||
| Liver | 73 | 68 | 78 | 72 | 0.8365 |
| Lung | 44 | 42 | 43 | 43 | |
| Other | 22 | 16 | 13 | 18 | |
| Disease status at study entry, N (%) | |||||
| Stable disease | 12 | 9 | 8 | 10 | 0.6418 |
| Progressive disease | 71 | 91 | 93 | 90 | |
| Prior chemotherapy for metastatic disease, N (%) | |||||
| 1 | 28 | 34 | 18 | 29 | 0.1632 |
| 2+ | 72 | 66 | 83 | 72 | |
| Previous surgical resection, N (%) | |||||
| Yes | 46 | 47 | 38 | 50 | 0.6024 |
| No | 54 | 53 | 63 | 51 | |
Note: P values calculated for continuous variables using one-way ANOVA and for categorical variables using χ2 test.
Figure 3.
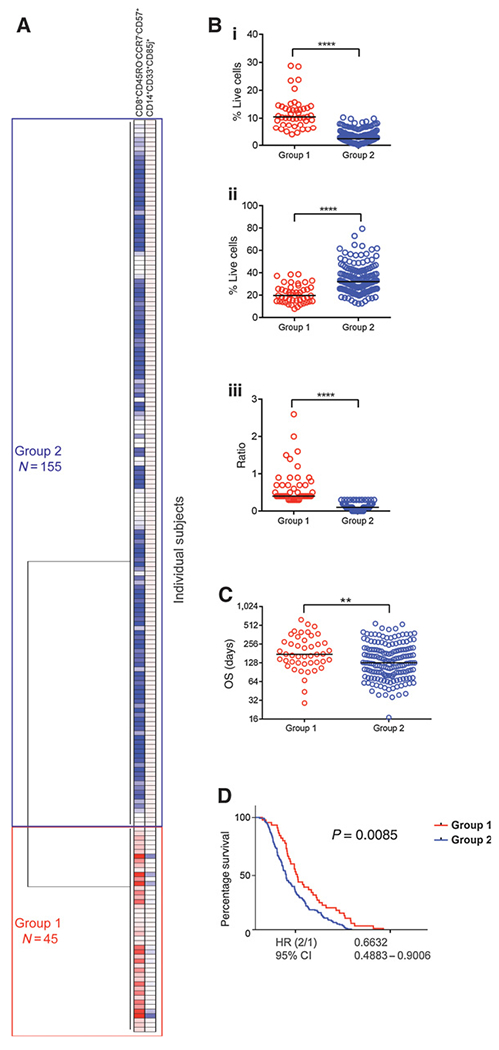
Immune subsets correlated with OS in a phase IIb trial of pancreatic cancer. Unstimulated PBMCs from a subcohort of 200 patients enrolled in a phase IIb study of pancreatic cancer were stained with flow cytometry antibody panels developed to enumerate candidate biomarker cell subsets identified in the phase IIa training set. A, Hierarchical clustering of the abundance of CD8+CD45RO−CCR7−CD57+ and CD14+CD33+CD85j+ cells in 200 patients; the two major groups of patients identified are indicated. The number of patients in each group is shown. B, The abundance of (i) CD8+CD45RO−CCR7−CD57+ and (ii) CD14+CD33+CD85j+ cells, and (iii) the ratio of CD8+CD45RO−CCR7−CD57+ to CD14+CD33+CD85j+ between patients in Group 1 and Group 2. C, OS for the two groups identified in A. Dots represent individual patient values; lines represent median values. P values were determined by unpaired t test. **, P < 0.005; ****, P < 0.00005. D, Kaplan-Meier plots of groups identified in A. P value determined using the log-rank test. Repeat experiments were not performed due to limited patient material. CI, confidence interval.
Table 4.
Frequencies of immune cell subsets associated with OS in groups identified by hierarchical clustering of flow cytometry data.
| Group 1 |
Group 2 |
||||
|---|---|---|---|---|---|
| Median (%) | Range (%) | Median (%) | Range (%) | P value | |
| CD8+CD45RO−CCR7−CD57+ | 10.5 | 4.0–28.8 | 2.4 | 0.1–10.3 | <0.0001 |
| CD14+CD33+CD85j+ | 19.8 | 7.9–38.7 | 32.0 | 12.4–79.5 | <0.0001 |
| Ratio | 0.4 | 0.3–2.6 | 0.1 | 0.0–0.3 | <0.0001 |
Note: P values calculated using unpaired t test.
Group 1 patients had a higher OS compared with Group 2 patients [median OS, range (days), Group 1 = 175, 29–631; Group 2 = 128, 71–547; P = 0.0047; Fig. 3C and D]. The two patient groups’ designations were independent of treatment (Supplementary Fig. S6), meaning Group 1 patients had higher OS compared with Group 2 patients in all three arms of the study [median OS, range (days) in Group 1 vs. Group 2; Arm A, 166,29–631 (N = 19) vs. 116,39–532 (N = 67); ArmB, 175, 96–492 (N = 17) vs. 128, 17–547 (N = 63); Arm C, 233, 94–374 (N = 9) vs. 140, 50–455 (N = 33)]. Although this analysis was limited due to the few patients in each group when substratified by treatment, the difference between Group 1 versus Group 2 was greater in Arms A and B compared with Arm C of the study (Group 1 vs. Group 2; Arm A, P = 0.0499; Arm B, P = 0.0695; Arm C, P = 0.3657). Subjects enrolled in the three treatments arms were balanced in terms of the ratio of CD8+CD45RO−CCR7−CD57+ to CD14+CD33+CD85j+ at baseline (median CD8+CD45RO−CCR7−CD57+ to CD14+CD33+CD85j+ ratio in Group 1, Group 2 in Arm A 0.4, 0.1; Arm B 0.5, 0.1; Arm C 0.4, 0.1; Supplementary Fig. S7). Taken together, these results recapitulated the findings from the training set, namely that the abundance of CD8+CD45RO−CCR7−CD57+ and CD14+CD33+CD85j+ cell subsets at baseline was associated with OS in patients with PDAC receiving Cy/GVAX and CRS-207, CRS-207 monotherapy, or standard-of-care chemotherapy.
Discussion
In this study, high-dimensional profiling of immune cells by mass cytometry was used to identify two circulating cell subsets (CD8+CD45RO−CCR7−CD57+ and CD14+CD33+CD85j+) that correlated with OS in patients with PDAC receiving immunotherapy. We confirmed the association of both biomarkers with OS in prospective–retrospective flow cytometry analysis of 200 patients with pancreatic cancer enrolled in a phase IIb immunotherapy trial. Further follow-up to verify these findings across multiple independent studies is required to determine whether these cellular biomarkers might be used in prognostic or predictive capacity for patient stratification. Based on the significant overlap observed in OS between the stratified groups identified in this study, additional biomarkers may be required to improve patient stratification.
One of the limitations of this work is the lack of functional characterization of the cellular biomarkers. Although potential prognostic and/or predictive values of biomarkers are independent of their biological function, future studies are required to further characterize the biological role of these subsets in clinical response. The CD8+CD45RO−CCR7−CD57+ cell subset associated with improved OS was a terminally differentiated effector phenotype, based on the downregulation of canonical memory markers, including CCR7, CD45RO, CD27, and CD127, and concomitant upregulation of CD45RA (12, 13). Activation markers, including HLA-DR and CD38, were not upregulated by these cells at steady state. The expression of CD57 on CD8+ T cells has been associated with conflicting functional attributes (14, 15). Some reports have associated CD8+CD57+ T cells with replicative senescence, shortened telomere lengths, and chronic activation due to persistent antigenic exposure (15), resulting in clonal expansion and an “exhausted T-cell phenotype” (16). In the current study, the CD8+CD45RO−CCR7−CD57+ cells associated with OS also expressed programmed cell death protein 1 (PD-1). However, due to PD-1’s sensitivity to cryopreservation (17), live monitoring in fresh blood will be required to verify this observation. Despite their association with lack of proliferation and propensity to undergo apoptosis upon T-cell activation, CD8+CD57+ T cells retain the ability to secrete cytokines upon encountering cognate antigen (14). It has been shown that CD8+CD57+ T cells can transiently upregulate telomerase activity and proliferate under certain stimulation conditions and that this population is heterogeneous in terms of its apoptotic potential (15). Elevated numbers of CD8+CD57+ T cells have been associated with chronic immune activation in the context of stress, aging, HIV-1, and other persistent viral and bacterial infections and various malignancies, including solid tumors and hemato-oncological diseases compared with healthy controls (15). In oncology, CD8+CD57+ cells have conflictingly been associated both with poor and improved OS (14,15). However, these studies have generally been limited to a few patients and low-resolution phenotypic analysis using flow cytometry. Therefore, it is likely that CD8+CD57+ T cells are heterogeneous and composed of various, functionally competing (cytotoxic and immunosuppressive) subsets, and their overall contribution to cell-mediated immunity in cancer or chronic viral infections likely depends on the prevalence of particular subsets. Their oligoclonal expansion and overcrowding of the immunological niche may be related to the increased occurrence of opportunistic infections and cancers (15, 18). On the other hand, the expanded oligoclonal T-cell population may contribute to protective responses, represented by high expression of perforin and secretion of cytokines such as IFNγ and/or TNFα (19).
In this study, CD14+CD33+CD85j+cells were elevated in poor and worst responders compared with best responders at baseline. CD85j is a product of the leukocyte Ig-like receptor/immunoglobulin-like transcript (LIR/ILT) molecular family of immunoreceptor tyrosine-based inhibitory motif (ITIM)–bearing inhibitory receptors. ITIM-bearing receptors negatively regulate cell functions when colligated with stimulating receptors that signal through ITAM. CD85j/LIR-1/ILT2 is detected on the surface of a proportion of NK cells and T cells, and on all B cells, monocytes, and myeloid dendritic cells (DC) but not plasmacytoid DCs (20). Ligands for CD85j include the nonclassical class I HLA-G protein, some alleles of HLA-A and -B loci, and the human CMV UL18 gene product, a viral homolog of HLA class I. It has been shown that these receptors function as negative regulators of monocyte and DC activation, presumably through recruitment of SHP-1 (21).
The concept of baseline immune competency as an effective tool for predicting response to treatment or overall clinical responses has been exemplified in the implementation of the “immunoscore,” where the frequency of CD4+ and CD8+ tumor-infiltrating T cells is enumerated by IHC (22). Previous studies have reported a prognostic combination model using baseline peripheral blood biomarkers (23–25). Standard clinical measurements, including low baseline lactacte dehydrogenase (LDA), high absolute eosinophil counts, and relative lymphocyte counts, are associated with improved survival in patients with melanoma treated with anti–CTLA-4 (25). Low baseline Lin−CD14+HLA-DR−/low myeloid-derived suppressor cell (MDSC) and CD4+CD25+FoxP3+ Treg frequencies are associated with improved survival in the same indication and treatment context (23, 25). The CD14+CD33+CD85j+ subset identified here to be associated with OS at baseline expressed HLA-DR+CD11b+CD15−, a phenotype that is not concordant with published descriptions of human MDSCs that are typically HLA-DR− (26).
In conclusion, here we identified and validated two circulating cellular biomarkers that correlated with OS in patients with PDAC enrolled in phase II immunotherapy trials. In future studies, we aim to determine if these markers are prognostic or predictive, and if they can be enhanced by integration with other parameters of immune competence, measured in circulation or in the tumor microenvironment. These data suggest a potential role of the identified biomarkers as predictive to select patients more likely to respond to CRS-207. Although clinical development of CRS-207 has been discontinued, it remains to be determined whether these cellular subsets are specific to cancer vaccines or may serve as general markers of immune competency and, thus, might also be relevant for other immuno-oncology therapies in PDAC or other cancers. To this end, more detailed biomarker analyses of CRS-207 clinical studies in other cancer types (such as mesothelioma, ovarian, peritoneal, and gastric cancer) and in other drug combinations (such as pembrolizumab, pemetrexed, and/or chemotherapy), along with preclinical in vivo models, are considered. The functional attributes of the immune cell subsets identified here and their relationship, if at all, to tumor-infiltrating lymphocytes in patients with PDAC warrant further investigation.
Supplementary Material
Acknowledgments
The authors thank Holden Maecker (Stanford University) for providing support for mass cytometry studies and John Frye, Jason Lo, and Kelly Chu (Aduro Medical Affairs) for their assistance in preparing this article for submission. This study was sponsored and funded by Aduro BioTech.
Disclosure of Potential Conflicts of Interest
N. Nair is Senior Director, Translational Medicine at Aduro BioTech. D.T. Le reports receiving other commercial research support from Aduro BioTech. E.M. Jaffee reports receiving commercial research grants from Aduro BioTech and Bristol-Myer Squibb; has ownership interest (including patents) in Aduro BioTech; and is a consultant/advisory board member for Genocea, DragonFly, CSTONE, Adaptive BioTech, Achilles, Parker Institute, National Cancer Advisory Board, and Lustgarten Foundation. C. Whiting is a director at Tempest Therapeutics, Inc.. T. Müller is Vice President, Translational & Predictive Medicine at Aduro BioTech. D.G. Brockstedt has ownership interest (including patents) in Aduro BioTech. No potential conflicts of interest were disclosed by the other authors.
Footnotes
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
References
- 1.International Agency for Research on Cancer. World cancer report 2008. Cancer Control 2008;199:512. [Google Scholar]
- 2.Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13. [DOI] [PubMed] [Google Scholar]
- 3.Laheru D, Lutz E, Burke J, Biedrzycki B, Solt S, Onners B, et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res 2008;14:1455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas AM. Mesothelin-specific CD8+ T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med 2004;200:297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brockstedt DG, Giedlin MA, Leong ML, Bahjat KS, Gao Y, Luckett W, et al. Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc Natl Acad Sci U S A 2004;101:13832–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, Nemunaitis J, et al. A live-attenuated listeria vaccine (ANZ-100) and a live-attenuated listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res 2012;18:858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011;332:687–96.21551058 [Google Scholar]
- 8.Chester C, Maecker HT. Algorithmic tools for mining high-dimensional cytometry data. J Immunol 2015;195:773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruggner RV, Bodenmiller B, Dill DL, Tibshirani RJ, Nolan GP. Automated identification of stratifying signatures in cellular subpopulations. Proc Natl Acad Sci U S A 2014;111:E2770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaudillière B, Fragiadakis GK, Bruggner RV, Nicolau M, Finck R, Tingle M, et al. Clinical recovery from surgery correlates with single-cell immune signatures. Sci Transl Med 2014;6:255ra131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol 2015;33:1325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med 1997;186:1407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sallusto F, Lenig D, Förster R. Pillars article : two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 2014;192:840–4. [PubMed] [Google Scholar]
- 14.Strioga M, Pasukoniene V. CD8+ CD28− and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011;134:17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Focosi D, Bestagno M, Burrone O, Petrini M. CD57+ T lymphocytes and functional immune deficiency. J Leukoc Biol 2010;87:107–16. [DOI] [PubMed] [Google Scholar]
- 16.Wood KL, Twigg HL, Doseff AI. Dysregulation of CD8+ lymphocyte apoptosis, chronic disease, and immune regulation. Front Biosci 2009;14:3771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell DE, Tustin NB, Riedel E, Tustin R, Taylor J, Murray J, et al. Cryopreservation decreases receptor PD-1 and ligand PD-L1 coinhibitory expression on peripheral blood mononuclear cell-derived T cells and monocytes. Clin Vaccine Immunol 2009;16:1648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinberger B, Welzl K, Herndler-Brandstetter D, Parson W, Grubeck-Loebenstein B. CD28-CD8+ T cells do not contain unique clonotypes and are therefore dispensable. Immunol Lett 2009;127:27–32. [DOI] [PubMed] [Google Scholar]
- 19.Eylar EH, Lefranc CE, Yamamura Y, Báez I, Colón-Martinez SL, Rodriguez N, et al. HIV infection and aging: enhanced interferon- and tumor necrosis factor-alpha production by the CD8+ CD28− T subset. BMC Immunol 2001;2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tenca C, Merlo A, Merck E, Elizabeth E, Bates M, Saverino D, et al. CD85j (leukocyte Ig-like receptor-1/Ig-like transcript 2) inhibits human osteoclast-associated receptor-mediated activation of human dendritic cells. J Immunol 2005;174:6757–63. [DOI] [PubMed] [Google Scholar]
- 21.Colonna M, Samaridis J, Cella M, Angman L, Allen RL, O’Callaghan CA, et al. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol 1998;160:3096–100. [PubMed] [Google Scholar]
- 22.Galon J, Franck P, Marincola FM, Angell HK, Thurin M, Lugli A, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med 2012;10:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother 2014;63:247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS One 2014;9:e87705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martens A, Wistuba-Hamprecht K, Geukes Foppen MH, Yuan J, Postow MA, Wong P, et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res 2016;22:2908–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greten TF. Myeloid-derived suppressor cells in pancreatic cancer: more than a hidden barrier for antitumour immunity? Gut 2014;63:1690–1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.