

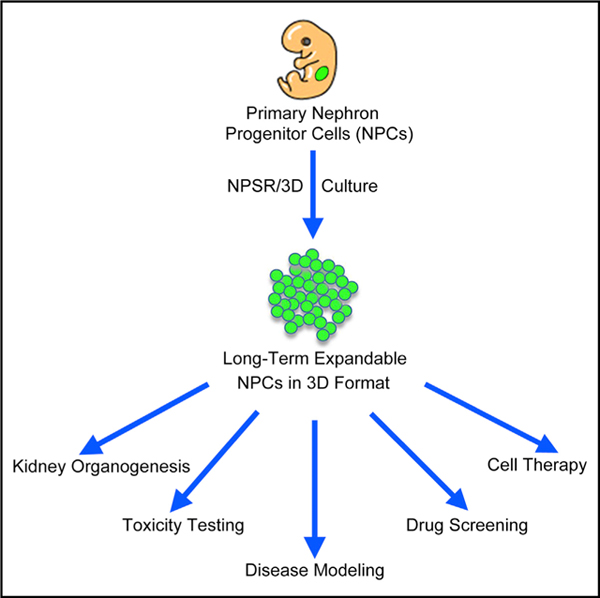
SUMMARY
Transit-amplifying nephron progenitor cells (NPCs) generate all of the nephrons of the mammalian kidney during development. Their limited numbers, poor in vitro expansion, and difficult accessibility in humans have slowed basic and translational research into renal development and diseases. Here, we show that with appropriate 3D culture conditions, it is possible to support long-term expansion of primary mouse and human fetal NPCs as well as NPCs derived from human induced pluripotent stem cells (iPSCs). Expanded NPCs maintain genomic stability, molecular homogeneity, and nephrogenic potential in vitro, ex vivo, and in vivo. Cultured NPCs are amenable to gene targeting and can form nephron organoids that engraft in vivo, functionally couple to the host’s circulatory system, and produce urine-like metabolites via filtration. Together, these findings provide a technological platform for studying human nephrogenesis, modeling and diagnosing renal diseases, and drug discovery.
In Brief
Li et al. report the derivation and long-term culture of mouse and human nephron progenitor cell lines under chemically defined conditions in 3D format. Expanded NPCs have nephrogenic potential in vitro and in vivo and allow the study of kidney organogenesis, gene editing, drug screening, and disease modeling.
Graphical Abstract
INTRODUCTION
Nephron progenitor cells (NPCs) give rise to all nephrons, the kidney functional units. Unlike some fish species where NPCs exist throughout their lifetime and can produce new nephrons upon kidney injury (Diep et al., 2011), mammalian NPCs are transient amplifying cells during development (Nishinakamura, 2016) and are not yet identified in the adult. Thus, alternative strategies to treat kidney failure are urgently needed, and one approach that holds great promise is the derivation of authentic nephron progenitor cell lines in vitro with intact generative capacity.
Pluripotent stem cells (PSCs) offer an attractive source toward the generation of NPCs. Recent studies have generated NPC-like cells from PSCs fulfilling several molecular and functional criteria such as NPC-specific marker gene expression and in vitro differentiation abilities (Araoka et al., 2014; Imberti et al., 2015; Lam et al., 2014; Mae et al., 2013; Morizane et al., 2015; Taguchi et al., 2014; Takasato et al., 2014, 2015; Toyohara et al., 2015). However, similar to embryogenesis, PSC differentiation only transiently passes through a nephron progenitor stage, and NPC-like cells do not persist long-term after the induction of nephron formation (Little, 2016). Therefore, development of culture conditions promoting long-term self-renewal of NPCs is warranted.
Kidney development has been extensively studied in mice, and various niche signals for the proper specification, proliferation, and differentiation of murine nephron progenitors have been identified (Costantini and Kopan, 2010; Dressler, 2006; Little and McMahon, 2012). This accumulated knowledge has greatly facilitated efforts in extending the lifespan of primary murine NPCs in culture. Bone morphogenetic protein (BMP)7 and FGF2 could maintain NPCs for 2–4 days in a monolayer culture (Dudley et al., 1999). Similarly, BMP7 together with FGF9/20 and heparin was able to maintain Six2+ NPCs for 5 days without losing nephrogenic potential (Barak et al., 2012). Based on these studies, further improvements significantly prolonged the survival of NPCs in culture: Brown et al. developed the nephron progenitor expansion medium (NPEM) that allowed for the in vitro expansion of murine NPCs up to ten passages (Brown et al., 2015), and Tanigawa et al. could culture mouse or rat NPCs for five passages using a different protocol (Tanigawa and Perantoni, 2016; Tanigawa et al., 2015, 2016). While these results have greatly advanced the field, there are still several notable limitations: (1) NPCs expanded in NPEM were unable to differentiate to glomeruli; (2) NPCs generated by both protocols suffered from molecular and functional heterogeneity and limited self-renewal in vitro; and (3) it remains unknown whether the cultured NPCs still retain an intact in vivo nephrogenic potential found in primary NPCs. With regard to humans, Brown et al. reported that human embryonic stem cell (hESC)-derived NPCs could be maintained in NPEM for no more than two passages and cultured human nephron progenitor cells (hNPCs) only gave rise to renal tubules, but not glomeruli, upon the second passage (Brown et al., 2015). Likewise, limited expansion (8 days) of human induced pluripotent stem cell (iPSC)-derived NPCs was reported by Tanigawa et al. Thus, culture conditions for long-term self-renewal of NPCs have yet to be achieved (Tanigawa et al., 2016).
Here, we describe a robust 3D culture system that enables the stable long-term in vitro propagation of functional murine and human nephron progenitors. Long-term cultured NPCs can be harnessed for rapid and efficient generation of nephron organoids, thereby providing an accessible system for modeling kidney development, renal toxicity testing, gene editing, and disease modeling (summarized in Tables S1 and S2).
RESULTS
Derivation and Long-Term Culture of Murine NPCs
Lineage tracing experiments established Six2 as a specific NPC marker (Kobayashi et al., 2008; Self et al., 2006). Therefore, we used an available mouse strain with a GFP cassette knocked in at the Six2 locus (Six2tm3(EGFP/cre/ERT2)Amc, Six2GCE for short hereafter) to facilitate purification of Six2-GFP+ NPCs from fetal or neonatal mouse kidneys by fluorescence activated cell sorting (FACS) and screened for conditions conducive to in vitro expansion of NPCs. We initially focused on isolated Six2-GFP+ cells from embryonic day 13.5 (E13.5) Six2GCE mice. Since in vivo NPCs are tightly packed together, we reasoned that cell-cell contact could play an important role in NPC survival and proliferation. Thus, to better preserve the native microenvironment, we cultured sorted Six2-GFP+ NPCs in 3D aggregates. Based on previous studies (Barak et al., 2012; Dudley et al., 1999), we tested the combination of BMP7, FGF9 (or FGF2), heparin, and Y27631 (BFHY) for culturing Six2-GFP+ cells in a 3D format. Interestingly, in BFHY medium, we observed robust proliferation of Six2-GFP+ NPCs cultured as aggregates for up to 7 days, after which the GFP signal dramatically decreased. Based on the BFHY/3D culture, next we sought to identify condition to support long-term NPC self-renewal in vitro by screening additional growth factors and chemical inhibitors (Table S3). Cell viability, growth rate, and GFP signal were used among other criteria. Through successive testing, we established a culture condition containing a cocktail of BMP7, FGF2, heparin, Y27632, LIF, and CHIR99021 that supports long-term in vitro expansion of Six2-GFP+ NPCs with high purity (we designated this culture medium as NPC self-renewal [NPSR] medium) (Figure 1A). Of note is that, to date, isolated Six2-GFP+ NPCs have been propagated in NPSR/3D culture for more than 17 months and 110 passages. NPCs grow fast as aggregates in NPSR medium. Starting with 3,000 cells, after 4–5 days, the aggregate can grow to ~1 mm in diameter containing ~100,000 cells (Figures 1B and 1C). Additionally, this growth rate was stably maintained after long-term passages with a consistent five or six doublings per passage (Figure 1D). Importantly, even after 60 passages, we found that 98.44% of cultured NPCs were still GFP+, with the GFP signal intensities similar to that of freshly isolated primary NPCs (Figure 1E). Moreover, we obtained similar results when the expressions of SIX2 and other NPC markers were quantified by immunostaining (SIX2+, ~96.2%; SALL1+, ~99.1%; and CITED1+, ~96.1%) (Figures 1F and 1G). These results show that highly pure NPCs with homogenous expression of NPC marker genes can be stably maintained in our NPSR/3D culture conditions. The expression of TWIST and VIMENTIN, but not E-CADHERIN (CDH1), in cultured NPCs further confirmed their mesenchymal identity (Figure 1H). By using the NPSR/3D culture system, we next tested the derivation of NPC lines from other developmental stages, including E11.5, E16.5, and postnatal day 1 (P1). Stable NPC lines could be easily derived from Six2-GFP+ cells isolated from all stages examined with a 100% success rate (Figure 1I). Similarly to E13.5-NPC lines, all E11.5-, E16.5-, and P1-derived NPC lines showed highly homogeneous GFP signal and NPC marker gene expression (Figures S1A and S1B).
Figure 1. NPSR/3D Culture Supports the Derivation and Long-Term Culture of Primary NPCs.

(A) Schematic of the experimental protocol.
(B) Time-lapse bright-field images showing the morphology and size of NPC aggregates within one passage cycle. Scale bar, 200 μm.
(C) Growth curve of NPCs within one passage cycle.
(D) Doubling times of NPCs from indicated passages.
(E) Flow cytometry analysis of Six2-GFP+ cells from E13.5 mouse fetal kidney (mEK) and E13.5-derived NPCs (P60).
(F) Immunofluorescence analysis of NPCs (P60). Scale bar, 100 μm.
(G) Quantification of data in (F). Data are presented as mean ± SD.
(H) Western blotting analysis in E13.5-derived NPCs (P60).
(I) Summary of NPC line derivation efficiency.
(J) 3D PCA plot of RNA-seq data. Different colors represent different developmental stages and different shapes indicate primary cells (squares) or cultured NPCs (triangles). Oval circles indicate three distinct clusters: cultured NPCs, early NPCs, and late NPCs.
See also Figure S1.
Subsequently, we performed RNA-sequencing (RNA-seq) analysis of cultured NPCs and their in vivo counterparts from different developmental stages. Six2-GFP– and Six2-GFP+ cells sorted from E12.5 Six2GCE mouse kidneys were used as controls. Comparative analysis of RNA-seq data identified 1,218 differentially expressed genes between Six2-GFP– and Six2-GFP+ cells and were subsequently used as NPC signature genes for further analysis (Table S4). Principal-component analysis (PCA) of the NPC signature genes revealed that primary NPCs are separated into two discrete groups according to their developmental stages: E11.5, E12.5, and E13.5 were clustered together, indicative of an early NPC identity, while E16.5 and P1 were found in the late NPC group. Interestingly, regardless of timing of derivation or passage number, all cultured NPCs are clustered together in a group much closer to the early NPC group than the late NPC group on both PC1 and PC3 axes (Figures 1J, S1C, and S1D). NPC lines expressed most of the known NPC marker genes (Six2, Cited1, Gdnf, and Hoxa11) and were expressed at levels similar to primary NPCs, while some other markers, including Eya1, Wt1, and Fgf9, showed varied gene expression among NPC lines (Figure S1E). Ureteric bud (UB) and stromal progenitor cell-specific genes were expressed at low levels in all NPC lines examined. Also, the same expression pattern of ITGA8+/PDGFRA–, two surface markers characteristic of primary murine NPCs (Taguchi et al., 2014), was also maintained in cultured NPCs (Figure S1E). Of note is that even after 80 passages, the global gene expression pattern of cultured NPCs was still very similar to that of early passage NPC lines, demonstrating the robustness of the NPSR/3D culture system for stable NPC self-renewal in vitro. Karyotyping analysis demonstrated genomic stability in long-term cultured NPCs (Figure S1F).
To gain mechanistic insights underlying NPC self-renewal in the NPSR/3D condition, we subtracted each of the six factors (FGF2, heparin, Y27632, LIF, BMP7, and CHIR99021) from the NPSR medium and evaluated the effects on cultured NPCs. We found that all six factors were indispensable for optimal propagation of NPCs in 3D culture. Removal of each component for as short as 4 days resulted in slower cell growth (FGF2, heparin, or Y27632), a decrease in Six2-GFP signal (LIF), or both (BMP7 or CHIR99021) (Figure S1G), and these effects became more pronounced when a longer time point was examined (Figure S1H). Of note is that NPCs cultured without FGF2 exhibited massive cell death after 2 days (Figures S1G and S1H), suggesting FGF2 has pro-survival role for NPSR/3D cultured NPCs. Consistently, global gene expression analysis supported the necessity of all six factors in maintaining self-renewal of cultured NPCs (Figure S1I).
Long-Term Cultured Murine NPCs Retain Nephrogenic Potential
To evaluate the nephrogenic potential of cultured NPCs in vitro, we first used spinal cord induction assay and co-cultured E13.5-derived NPCs (P60) with embryonic dorsal spinal cords in an air-liquid interface. We observed many tubular structures formed on day 3; between day 3 and day 7, their number increased (Figures 2A, 2B, and S2A). On day 7, by immunocytochemistry, we observed positive staining for glomerulus markers podocalyxin (PODXL), synaptopodin (SYNPO), and Wilms tumor 1 (WT1) (Figures 2C, 2D, and S2B); proximal tubule markers Lotus tetragonolobus lectin (LTL) and aquaporin 1 (AQP1) (Figures 2C, 2D, and S2C); and Henle’s loop/distal tubule markers E-cadherin (CDH1), distal tubule markers, PAX2, and Dolichos biflorus agglutinin (DBA) (Figures 2C, 2E, S2D, and S2E), suggesting proper differentiation of cultured NPCs to major segments of nephrons. Moreover, we found numerous PODXL+ glomeruli were adjacent to LTL+ proximal tubules, LTL+ proximal tubules were connected with CDH1+ Henle’s loop/distal tubules, and CDH1+/DBA– Henle’s loop were joined with CDH1+/DBA+ distal tubules (Figures 2C and S2E), indicating a proper glomerulus→proximal tubule→Henle’s loop→distal tubule organization. NPC lines derived from E11.5, E16.5, and P1 kidneys exhibited similar differentiation potential upon spinal cord induction (Figures S2F and S2G). Of note is that even after 100 passages (>15 months and 10139-fold expansion in culture), the nephrogenic potential of NPC lines obtained from different developmental stages was still intact (summarized in Figure 2F).
Figure 2. Cultured NPCs Retain Nephrogenic Potential In Vitro and Ex Vivo.
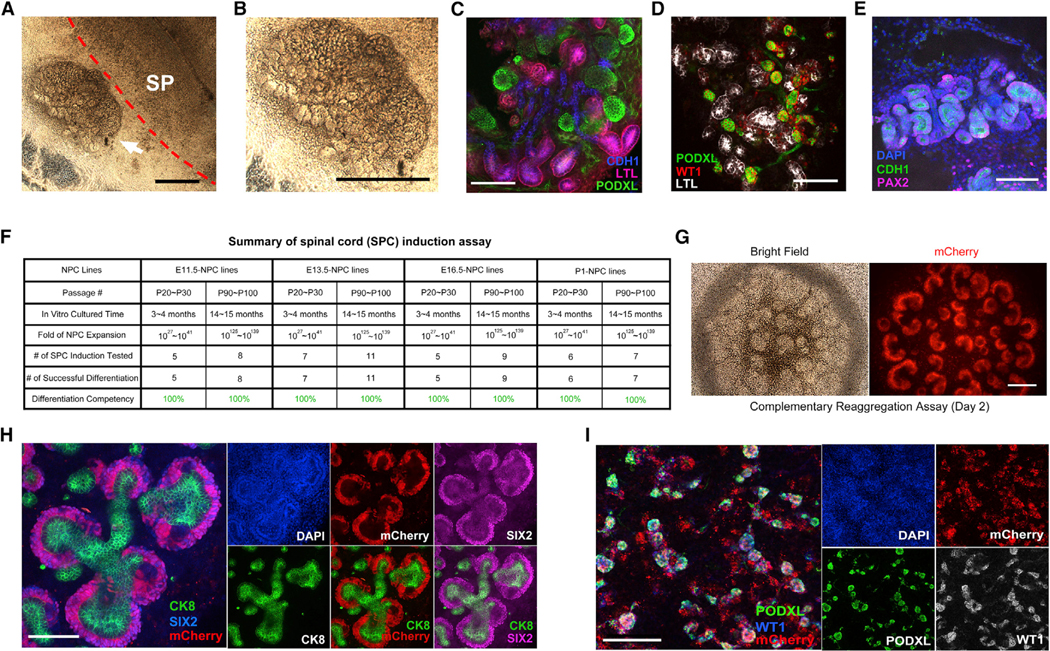
(A and B) Bright-field images of the spinal cord induction assay. E13.5-derived NPCs (P60) were co-cultured with E12.5 dorsal spinal cord for 7 days. The white arrow indicates the differentiated structures from cultured NPCs (a higher magnification image shown in B). Dashed red line indicates the boundary between spinal cord and differentiated NPCs. SP, spinal cord. Scale bar, 1mm.
(C–E) Whole-mount immunofluorescence analyses of E13.5-derived NPCs (P60) induced by dorsal spinal cord for 7 days. Scale bars, 100 μm.
(F) Summary of spinal cord induction assay results.
(G) Bright field and fluorescence images of reaggregates formed by mCherry-labeled NPCs plus Six2-GFP- cells in air-liquid interface cultured for 2 days. Scale bar, 200 μm.
(H) Whole-mount immunofluorescence analyses of the reaggregates showing in G. Scale bar, 100 μm.
(I) Whole-mount immunofluorescence analyses of reaggregates formed from mCherry-labeled NPCs plus Six2-GFP cells in air-liquid interface cultured for 7 days. Scale bar, 200 μm.
See also Figures S2.
During normal kidney development, reciprocal interactions between UB tips and NPCs underlie kidney morphogenesis. To test if cultured NPCs could interact with UB and commit to nephron formation, we devised a “complementary reaggregation assay.” To this end, we dissociated E11.5 kidneys from Six2GCE mice and depleted the Six2-GFP+ NPCs by FACS. The remaining Six2-GFP– population was mixed with mCherry-labeled NPCs to form aggregates and then transferred to an air-liquid interface to test if nephrogenesis could occur ex vivo (Figure S2H). Six2-GFP– cells reaggregated with primary Six2-GFP+ NPCs, or reaggregated unfractionated E11.5 kidney cells were used as positive controls. As shown in Figure S2I, time-lapse imaging revealed a dynamic nephrogenic process in reaggregates formed by Six2-GFP– cells and mCherry-labeled NPCs, which was indistinguishable from positive controls. In vivo, NPCs form condensed cap structure known as the cap mesenchyme surrounding each UB tip. Interestingly, in day 2 reaggregates, we found most if not all mCherry-labeled NPCs formed cap structures (Figure 2G). Immunofluorescence analysis confirmed SIX2 expression in mCherry+ cells in all cap structures examined (Figure 2H) and the expression of SOX9 in the ureteric epithelia tip cells adjacent to SIX2+ NPCs, suggesting normal interactions between UB and nephron progenitors were maintained (Reginensi et al., 2011) (Figure S2J). Furthermore, following a 7-day course of morphogenesis (Figure S2K), day 7 aggregates displayed formation of major nephron segments from mCherry+ cells (Figures 2I, S2L, and S2M).
To evaluate whether our cultured NPCs are functionally homogenous, we tested clonal derivation from single Six2-GFP+ cells. We observed that single Six2-GFP+ cells from either fetal kidneys or long-term cultured NPCs could not survive and died within 2 days under NPSR/3D condition, suggesting that cell-cell contact in an aggregate format and/or paracrine factors are essential for the survival and self-renewal of NPCs. To circumvent this problem, we developed a methodology to allow clonal expansion from single NPCs by using helper NPCs. For proof of concept, we first established an NPC line stably expressing mCherry and puromycin-resistance genes (mCherry/Puro-NPC). A single mCherry/Puro-NPC was mixed with 300–500 wild-type helper NPCs to form an aggregate, which allowed the single mCherry/Puro-NPC to proliferate (Figure S2N). Puromycin was then added to selectively kill the wild-type NPCs, leaving only mCherry/Puro-NPCs with homogeneous mCherry expression (Figure S2O). By using this method, we derived 50 single-cell clones from NPCs expressing puromycin-resistance gene (Puro-NPCs). Nine of the clones were randomly selected for further analysis. All the NPC marker genes examined were expressed in all nine clones; Six2, Osr1, and Hoxd11 were expressed at comparable levels, while expression levels of Eya1 and Wt1 varied among clones (Figures S2P and S2Q). More importantly, all the single clones (nine out of nine) examined efficiently differentiated to nephron structures upon spinal cord induction (Figure S2R). These results demonstrate an intact nephrogenic potential appear to be homogenously and stably retained in cultured murine NPCs.
Robust and Efficient Generation of Nephron Organoids from Cultured Murine NPCs
To relieve the spinal cord dependency, next we sought to establish a chemically defined method for nephron formation. Wnt signaling pathway is known to trigger the initiation of NPC differentiation (Carroll et al., 2005; Kispert et al., 1998; Kuure et al., 2007; Park et al., 2007). We thus started by treating cultured NPCs with a GSK3 inhibitor CHIR99021 (CH), which activates the canonical Wnt signaling pathway. CH treatment led to limited differentiation in cultured NPCs accompanied by cell death. Considering that FGF2 (F2) has a pro-survival effect on cultured NPCs, we next tested the combination of CH/F2 for differentiation and found that 2-day treatment is sufficient to faithfully reproduce the nephrogenesis induction conferred by the embryonic spinal cord (Figure 3A). We designated CH/F2-induced NPCs as fate-specified NPCs (FS-NPCs). Once induced, FS-NPCs could autonomously differentiate further in basal medium alone to form nephron organoids marked by numerous tubular formations (Figure 3B). Whole-mount staining of obtained nephron organoids identified major nephron segments (Figures 3C).
Figure 3. Robust and Efficient Nephron Organoid Formation from Cultured NPCs Facilitates Disease Modeling.
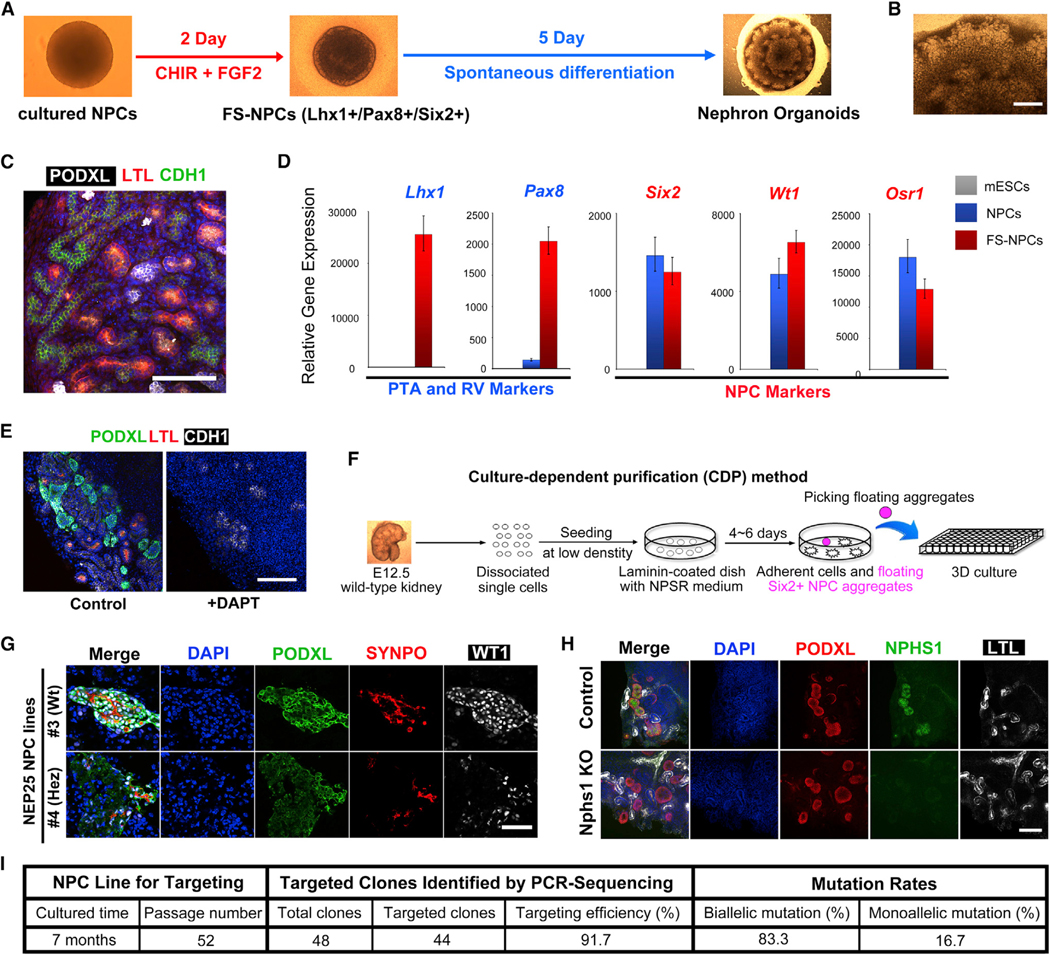
(A) Schematic of the differentiation from cultured NPCs to nephron organoids through fate-specified NPCs (FS-NPCs).
(B) Bright-field image showing part of the nephron organoid. Scale bar, 200 μm.
(C) Whole-mount immunofluorescence analyses of the nephron organoid. Scale bar, 100 μm.
(D) qRT-PCR analyses in mESCs (control), NPCs, and FS-NPCs. Data are presented as mean ± SD.
(E) Immunofluorescence analyses of nephron organoid treated with DAPT from day 2 to day 7. Scale bars, 100 μm.
(F) Schematic of the culture-dependent purification (CDP) method.
(G) Immunofluorescence analyses of nephron organoids generated from wild-type (#3) and transgenic (#4) NEP25 NPC lines treated with LMB2 (20 nM) for 4 days. Scale bars, 100 μm.
(H) Whole-mount immunofluorescence analyses of nephron organoids derived from control wild-type NPCs and Nphs1 knockout NPCs (Nphs1 KO). Scale bar, 50 μm.
(I) Efficiencies of CRISPR-Cas9 based gene targeting of nephrin in cultured NPCs.
See also Figure S3.
In vivo, NPCs first differentiate to pretubular aggregates (PTA) and renal vesicles (RV) before further maturing into segmented nephron structures. qPCR analysis indicated that PTA and RV markers Lhx1 and Pax8 were strongly upregulated, while NPC markers such as Six2, Wt1, and Osr1 were maintained at similar levels in FS-NPCs and cultured NPCs (Figure 3D). Immunofluorescence studies confirmed protein expression of both PTA/RV markers (LHX1 and PAX8) and NPC markers (SIX2 and SALL1) in FS-NPCs (Figures S3A and S3B). These results suggest that FS-NPCs resemble more closely the PTA than RV stage. To further investigate to what degree the in vitro nephron organoid formation mimics the normal nephrogenesis after transitioning to the PTA-like stage (FS-NPC), from day 2 to 7, we used DAPT to inhibit Notch signaling, which is indispensable for proximal tubule and glomerulus formation (Cheng et al., 2003). Upon DAPT treatment, both proximal tubule and glomerulus differentiation were inhibited in formed organoids (Figure 3E), providing additional evidence that nephron organoid formation from cultured NPCs might be a valid in vitro platform toward partially recapitulating in vivo nephrogenesis.
Next, we studied the utility of NPC-derived organoids for renal toxicity testing. To this end, we treated day 7 organoids with gentamicin, which selectively elicits proximal tubule injury (Morizane et al., 2015; Whiting and Brown, 1996). Massive cell death was observed in LTL+ proximal tubules as evidenced by active caspase-3 (CASP3) staining and severely disrupted LTL+ tubules. In contrast, PODXL+ glomeruli remained intact with virtually no CASP3 staining and a normal morphology (Figures S3C and S3D). These results indicate that the nephron organoids generated from long-term cultured NPCs are suitable for nephrotoxicity testing.
Disease Modeling and Gene Editing using Murine NPCs
To date, various transgenic mouse strains have been generated for studying kidney development and disease modeling. We envision that efficient NPC line derivation and organoid formation methodologies like the one here described could be further expanded into existing mouse models for a better understanding of kidney morphogenesis and disease progression. Toward achieving these goals, we first need to liberate NPC line derivation from relying on the Six2-GFP reporter. To this end, we developed a culture-dependent purification (CDP) method for the transgene-independent derivation of NPC lines (Figure 3F). We found that after plating cells dissociated from whole E12.5 Six2GCE kidney onto laminin-coated plates, but not gelatin or Matrigel-coated plates, some cells detach and form aggregates after 4 days of culture (Figure S3E). These floating aggregates contained most if not all GFP+ cells. Similar to Six2GCE, when an outbred ICR strain was used, floating aggregates were formed after 4-day culture on laminin-coated plates, and further passages of these aggregates led to successful NPC line derivation. ICR-NPCs homogenously expressed the NPC markers SIX2 and SALL1 (Figure S3F), and upon spinal cord induction, ICR-NPCs also generated nephron structures, demonstrating their nephrogenic potential (Figure S3G).
Toward demonstrating the utility of NPC line derived by the CDP method for disease modeling, next we chose a well-established transgenic mouse model (NEP25) that allows for controlled induction of podocyte-specific injury via immunotoxin anti-Tac (Fv)-PE38 (LMB2) (Matsusaka et al., 2005). We generated four NPC lines from NEP25 mice using the CDP method (Figure S3H). Nephron organoids were efficiently obtained from both lines. Immunofluorescence analysis showed that compared to control (line 3, wild-type), while proximal tubules were not affected, glomeruli structures in the transgenic line-4-derived organoids were morphologically disrupted and the expression levels of glomerulus marker genes were significantly decreased, mimicking the phenotypes observed in NEP25 transgenic mice upon LMB2 administration (Figures 3G and S3I).
The long-term stability and functional homogeneity of cultured NPCs is technically advantageous for gene targeting. For proof of concept, we used the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system to knockout nephrin (Nphs1) in cultured NPCs. Stable NPC aggregates were obtained after successful delivery of gene targeting constructs and drug selection. 48 clones from single cells were generated using wild-type helper NPCs described above. DNA sequencing confirmed on-target mutations generated in 44 out of 48 clones (91.3%). To further discriminate between biallelic and monoallelic mutations, we performed PCR/TA-cloning followed by sequencing. Our results showed that among on-target clones, 83.3% harbored biallelic mutations, while 16.7% were monoallelically mutated (Figure 3I). Nphs1−/− (biallelic frameshift) clones were used for the generation of nephron organoids. Consistently, NEPHRIN expression was not detected in these nephrons, confirming a successful Nphs1 knockout (Figure 3H).
In Vivo Developmental Potential
As a more rigorous functional test, authentic NPCs are expected to participate in nephrogenesis in vivo. In utero delivery of NPCs to their cognate niche in mouse fetal kidneys is technically challenging. Since the nephrogenic niches may still persist a few days after birth in the mouse, we tested whether cultured NPCs could contribute to new nephron formation in neonatal kidneys. We transplanted NPCs under the subcapsule and/or into the cortex of neonatal P0–P1 kidneys but failed to detect any contribution. We interpret this result as a failure of exposing NPCs to the proper niche signals normally provided by UB tips within the fetal nephrogenic zone. Nephrogenic zones in the neonatal kidney are occupied by endogenous SIX2+ cells, thereby preventing transplanted NPCs from accessing the UB tips. We reasoned that, unlike self-renewing NPCs, FS-NPCs have already initiated nephrogenesis and therefore might be relieved of niche signal dependency for differentiation in vivo. Thus, we transplanted FS-NPCs inside the neonatal kidney cortex at P0–P1 stages (Figure 4A). 7 days after transplantation, we observed that the mCherry-labeled FS-NPCs generated numerous tubular structures within the kidney cortex (Figure 4B). Immunofluorescence analysis indicated that transplanted FS-NPCs engrafted and formed chimeric proximal tubules, distal tubules, and vascularized glomeruli with host cells (Figures 4C–4E), and in some cases, we observed entire LTL+ proximal tubules and BRN1+/DBA– Henle’s loop structures contributed by FS-NPCs (Figures 4F and 4G).
Figure 4. In Vivo Developmental Potential of NPCs and FS-NPCs.
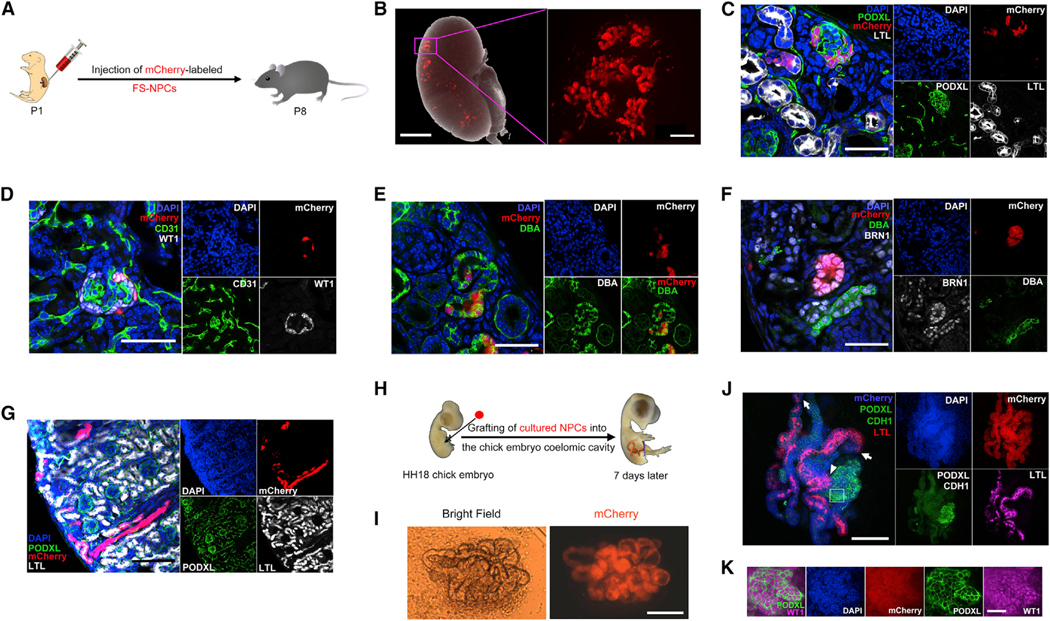
(A) Schematic of injecting mCherry-labeled mouse FS-NPCs into the kidney of neonatal mice (P1).
(B) A merged bright-field and fluorescence image showing the mCherry+ FS-NPCs contribution in a P8 mouse kidney after P1 injection (left), and a higher-magnification fluorescence image (right) (in the right panel, a black box was superimposed over the original software-generated scale bar and, as a replacement, a thicker white scale bar was added). Scale bars represent 1 mm (left) and 100 μm (right).
(C–G) Immunofluorescence analyses of P8 mouse kidney 7 days after injection of mCherry-labeled mouse FS-NPCs. Scale bars represent 50 μm (C–F) and 100 μm (G).
(H) Schematic of grafting mCherry-labeled mouse NPCs to the coelomic cavity of the HH18 stage chick embryo.
(I) Bright field and fluorescence images of dissected out mCherry+ tubular structures from the chick embryo 7 days after grafting. Scale bar, 100 μm.
(J) Whole-mount immunostaining of the dissected mCherry+ tubular structure from (I). The white arrows indicate the connecting sites of CDH1+ Henle’s loop/distal tubules and LTL+ proximal tubules. The white arrowhead indicates the connecting site of PODXL+ glomeruli and LTL+ proximal tubules. Scale bar, 100 μm.
(K) Higher-magnification immunofluorescence images for PODXL and WT1 in the region indicated by the white box in (J). Scale bars, 25 μm.
To test whether NPCs could generate nephrons in vivo if exposed to signals present earlier in development, we took advantage of the developing chick embryo, a well-established and more accessible system for testing progenitor developmental potentials. Small clumps of mCherry-labeled NPC aggregates were grafted into the lateral plate mesoderm of stage HH18 chick embryos (Figure 4H). 7 days after transplantation, we dissected the chick embryo and found an intact mCherry+ tubular structure (Figure 4I). Whole-mount staining further indicated that the mCherry-labeled NPC aggregate differentiated into tubular structures in vivo containing PODXL+/WT1+ glomerulus, LTL+ proximal tubules, and CDH1+ Henle’s loop/distal tubules (Figures 4J and 4K). Importantly, these structures were spatially organized in a pattern mimicking an in vivo nephron with glomerulus, proximal tubules, and Henle’s loop/distal tubules sequentially connected (Figures 4J and 4K). These results strongly support an intact in vivo nephrogenic potential of cultured NPCs.
The robust in vivo nephrogenic potential led us to test whether a functional ectopic organoid with urine excretion capability could be generated from cultured NPCs. We transplanted mCherry-labeled NPCs together with spinal cord into the omentum of immunodeficient NSG mice (Figures S4A). 2 weeks later, we observed the formation of numerous cysts filled with fluid surrounded by mCherry+ cells (Figures 5A and 5B). We also observed host blood vessels infiltrating the mCherry+ structures (Figures 5A, 5B, and S4B). Immunofluorescence analysis not only confirmed the lining of cysts by mCherry+ cells (Figure S4C) but also revealed that mCherry-labeled NPCs could give rise to numerous LTL+ proximal tubules and DBA+ distal tubules (Figures 5C, S4C–S4E, and S4G) as well as PODXL+ and WT1+ glomerulus structures (Figures 5C, 5D, S4C, S4F, and S4G). Importantly, PODXL+ glomeruli were found connected to LTL+ proximal tubules (Figure S4G), and many CD31+/mCherry– endothelial cells integrated into the WT1+/mCherry+ glomerulus (Figure 5D), suggesting blood vessels from the host tissue invaded the NPC-derived glomeruli. To investigate whether the cyst fluid originated from the host circulatory system, we injected fluorescence-conjugated low-molecular-weight dextran through the tail vein. 2 hr after injection, we observed clear dextran accumulation in the cyst (Figure 5E). Importantly, we confirmed the presence of dextran in glomeruli and proximal tubule, but not in distal tubule, similar to control adult mouse kidney (Figure S4H), suggesting a functional connection between glomeruli and proximal tubule within the nephron organoid. Typical urine metabolites such as creatinine were also found in the cyst fluid, further suggesting that the cultured NPCs might have generated functional nephron-like structures with urine filtration ability in vivo (Figure 5F).
Figure 5. Cultured NPCs Generate Ectopic Nephron Organoids In Vivo with Urine-like Excretion and Improve Kidney Function in an Acute Kidney Injury Model.

(A) Bright-field image showing cystic structures derived from mCherry-labeled NPCs (P60) after 2 weeks of omentum transplantation. Red arrowheads mark the border of the cysts. Scale bar, 500 μm.
(B) Bright-field image and fluorescence image of cystic structures. White arrows represent the infiltration of host blood vessels. Scale bar, 500 μm.
(C and D) Immunofluorescence analyses of the cystic structures in (B). Scale bars represent 50 μm (C) and 100 μm (D).
(E) Lucifer-yellow dextran accumulation in the cyst after 2 hr of tail vein injection. Dashed circles indicate the boundaries of cysts. Scale bar, 500 μm.
(F) Creatinine levels in serum, urine, and cyst fluid. Data are presented as mean ± SD.
(G) Mouse survival is presented using Kaplan-Meier survival curves.
(H) Blood urea nitrogen (BUN) and serum creatinine (S-Cre) levels in cisplatin-induced AKI mice receiving indicated treatments. Data are presented as mean ± SD. (I and J) H&E and PAS staining images (I) and counting of pathological features (J) of histological sections from kidneys in (H). Data are presented as mean ± SD. Scale bars, 50 μm.
(K) BUN and S-Cre levels in AKI mice receiving conditioned medium (CM) or unconditioned KR5 medium (DMEM/F12 with 5% knockout serum replacer [KSR]) 4 days after cisplatin administration. Data are presented as mean ± SD.
(L and M) H&E and PAS staining images (L) and counting of pathological features (M) of histological sections from kidneys in (K). Data are presented as mean ± SD. Scale bars, 50 μm.
See also Figure S4.
Paracrine Factors Produced by Cultured NPCs Improve Kidney Function in an Acute Kidney Injury Model
The robust in vivo nephrogenic potential also prompted us to test whether FS-NPCs could repair renal damage upon acute kidney injury (AKI). Because of massive bleeding, we failed to deliver FS-NPCs in situ into the kidney cortex. Instead, we transplanted FS-NPCs under the renal subcapsule following cisplatin treatment. Our observations revealed that mice receiving FS-NPC transplants survived longer than the control group (Figure 5G). All control mice (n = 5) receiving cisplatin died within 8 days, during which time only one mouse out of six receiving FS-NPC transplantation died. Mice transplanted with FS-NPCs showed improved renal function as measured by several biochemical parameters, including significantly reduced blood urea nitrogen (BUN) and serum creatinine (S-Cre) levels, compared with mice receiving mouse embryonic stem cells (mESCs) or PBS as control (Figure 5H). Histological analyses further revealed significantly lowered levels of tubular dilation and necrosis, reduced numbers of urinary cast, and reduced occurrence of loss of tubular borders in mice receiving FS-NPCs than in mice receiving mESCs or PBS (Figures 5I and 5J). Interestingly, although transplanted FS-NPCs properly differentiated into nephron structures surrounded by some CD31+ host endothelial cells (Figures S4M–S4O), they localized outside of the kidney cortex and did not integrate (Figures S4I–S4L). This suggests that paracrine factors may provide some beneficial effects. To test this hypothesis, we injected conditioned medium (CM) collected from FS-NPCs intraperitoneally to NSG mice receiving cisplatin treatment. In contrast to control mice injected with unconditioned medium, which did not show any improvement in kidney functions, CM injection significantly decreased the levels of BUN and creatinine, mimicking the effects of FS-NPC transplantation (Figures 5K–5M). Based on these results, we conclude that the rescue effects observed were probably due to paracrine effects.
Derivation and Long-Term Culture of Human NPCs with Nephrogenic Potential
Next, we tested if the mouse NPSR/3D culture could be applied for the derivation of hNPC lines. We first analyzed 11-week human fetal kidney sections by immunofluorescence and found that, similar to mouse, SIX2+ cells formed cap structures surrounding CK8+/CDH1+ structures at the periphery of the fetal kidney, suggesting that SIX2 and CK8/CDH1 specifically labeled in vivo human NPCs and ureteric epithelial cells, respectively (Figure 6B). We generated aggregates using unfractionated human fetal kidney cells from 9 to 14 weeks of gestation and cultured in NPSR medium. After 1 week, however, the aggregates showed massive tubulogenesis and no SIX2+ cells were detected (data not shown). We speculated that non-NPC cells present in the human fetal kidney cell mixture interfered with hNPC line derivation. To enrich the primary hNPCs, based on our RNA-seq analysis, we selected surface markers highly enriched in primary mouse NPCs and performed combinatory testing of these surface markers toward purifying hNPC fractions. We found that EpCAM can selectively exclude a proportion of SIX2– populations (Figure S5A) while NGFR can select for SIX2+ cells (Figure S5B). A combination of EpCAM–/NGFR+ was able to enrich the SIX2+ population from primary human fetal kidney cells (Figure S5C). Enriched human NPCs could be stably expanded as 3D aggregates in NPSR medium for up to 2 months without losing the expression of SIX2. However, cultured hNPCs grow slowly and showed minimal response to spinal cord induction (data not shown), suggesting species differences in niche signals for human and mouse NPC self-renewal. To improve the culture of hNPCs, we tested additional factors. We found that supplementation of inhibitors for the Smad1/5/8 and Smad2/3 pathways to NPSR medium could support robust derivation and propagation of hNPCs. In this culture condition (designated as human NPSR or hNPSR medium), hNPCs grew fast and were routinely passaged every 4–5 days at a 1:10 ratio (Figures 6C and 6D). Importantly, hNPCs could be maintained long-term in the hNPSR medium (>7 months and >50 passages at the time of writing) with homogeneous expression of NPC marker genes SIX2, SALL1, CITED1, and PAX2 (Figures 6E and 6F). Of note, the hNPSR/3D culture condition enabled the derivation of human NPC lines with similar growth rates and gene expression from different human gestational ages ranging from 9 to 17 weeks (summarized in Figure 6G), demonstrating the robustness of hNPSR culture (Figure 6A).
Figure 6. Derivation and Stable Long-Term Expansion of Human NPCs.

(A) Schematic of purification and derivation of human NPC lines from human fetal kidney.
(B) Immunofluorescence analyses of an 11-week human fetal kidney section. Scale bars, 100 μm.
(C) Bright-field images showing cultured human NPC aggregates within one passage cycle. Scale bars, 200 μm.
(D) Growth curve of cultured human NPCs within one passage cycle.
(E) Immunofluorescence analyses of cultured human NPCs (P50). Scale bars, 100 μm.
(F) Quantification of data in (E). Data presented as mean ± SD.
(G) Summary of human NPC line derivation efficiency.
See also Figure S5.
To test the nephrogenic potential of long-term cultured hNPC lines, we first employed the spinal cord induction assay. Within as few as 7 days after induction, hNPCs readily gave rise to numerous tubular structures (Figure 7A). Immunostaining confirmed the proper differentiation of hNPCs into all major segments of the human nephron, including PODXL+ glomeruli, LTL+ proximal tubules, and CDH1+ Henle’s loop/distal tubules (Figure 7B). Similar to the mouse, we found that treatment of hNPCs with CH/F2 for 1–2 days followed by additional 5–7 days autonomous differentiation could efficiently generate human nephron organoids (Figures 7C, 7D, S6A, and S6B). Importantly, all hNPC lines derived from different gestational stages efficiently differentiate to nephron organoids even after >50 passages in culture (summarized in Figure 7E). When we transplanted hNPSR-cultured hNPCs to the mouse omentum together with the spinal cord, they were able to differentiate in vivo to major segments of the nephron (Figures 7F, 7G, S6C, and S6D). Similarly, we observed lucifer-yellow dye accumulation in the cysts formed from the transplants (Figure S6E), with the cyst fluid enriched in creatinine (Figure S6F), indicating that urine filtration capability might be acquired in these transplants. These results demonstrate that the hNPSR/3D culture enables the derivation and long-term expansion of hNPC lines with in vitro and in vivo nephrogenic potential (Table S2).
Figure 7. Cultured Human NPC Lines Maintain Nephrogenic Potential In Vitro and In Vivo.
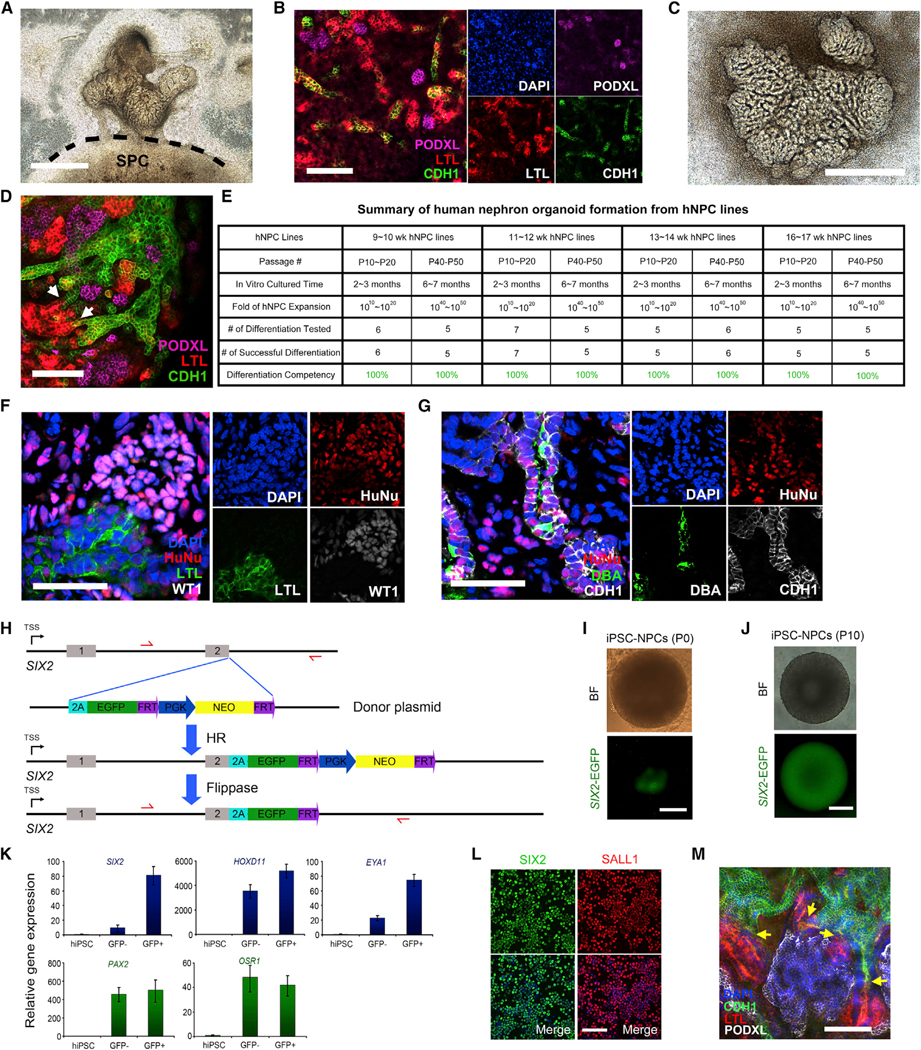
(A) Bright-field image of the spinal cord induction assay. hNPC line (P43) was co-cultured with E12.5 spinal cord for 7 days. Dashed line indicates the boundary between spinal cord and differentiated hNPCs. SPC, spinal cord. Scale bar, 1 mm.
(B) Whole-mount immunofluorescence analyses of nephron organoid in (A). Scale bar, 100 μm.
(C) Bright-field image of the nephron organoids derived in chemically defined condition (Figure S6A) for 8 days. Scale bar, 1 mm.
(D) Whole-mount immunofluorescence analysis of nephron organoid in (C). Scale bar, 100 μm.
(E) Summary of human nephron organoid formation efficiency from cultured human NPC lines.
(F and G) Immunofluorescence analyses of renal structures derived from human NPCs (P40–P50) (P43) after 3 weeks of omentum transplantation. Scale bars, 50 μm.
(H) Schematic of the SIX2-EGFP reporter iPSC line generation. 2A-EGFP-PGK-Neo cassette was inserted downstream of the last exon of endogenous SIX2 gene by transcription activator-like effector nuclease (TALEN)-mediated homologous recombination (HR). Stable single-cell colonies were obtained following neomycin selection. Flippase was then expressed in these single-cell clones via transient transfection to excise the FRT-flanked PGK-Neo cassette.
(I) Bright-field (BF) and fluorescence images show that after differentiation (P0), clusters of SIX2-EGFP+ cells were formed from SIX2-EGFP knockin reporter hiPSC line. Scale bar, 200 μm.
(J) Bright-field (BF) and fluorescence images show that after ten passages of cultured hiPSC-NPCs in the hNPSR/3D condition (~2 months), the SIX2-EGFP signal was maintained. Scale bar, 200 μm.
(K) qRT-PCR analyses in undifferentiated hiPSCs (control), SIX2-EGFP–, and SIX2-EGFP+ cells after differentiation (P0). Data are presented as mean ± SD.
(L) Cytospin immunostaining in cultured hiPSC-NPCs (P10). Scale bar, 100 μm.
(M) Whole-mount immunofluorescence analysis of nephron organoids derived from cultured hiPSC-NPCs (P10). The yellow arrows indicate the connecting sites of CDH1+ Henle’s loop/distal tubule and LTL+ proximal tubule. Scale bar, 100 μm.
See also Figure S6.
To broaden the utility of hNPSR/3D culture, we attempted the derivation of human induced pluipotent stem cell (hiPSC)-derived NPC lines. Due to the low efficiency of NPC differentiation in our hands using existing protocols and/or contaminating cells from other lineages, we failed to obtain hiPSC-derived NPC lines as unfractionated mixture or after EpCAM–/NGFR+ enrichment (data not shown). To purify NPCs from the mixture of differentiated cells, we resorted to the generation of a SIX2-EGFP reporter hiPSC line by knocking in an EGFP cassette under the control of endogenous SIX2 promoter (Figures 7H and S6G–S6I). Following differentiation (Taguchi et al., 2014), we purified SIX2-EGFP– and SIX2-EGFP+ cells by FACS. We found that while OSR1 and PAX2 were expressed at similar levels, SIX2, EYA1, and HOXD11 were expressed at much higher levels in SIX2-EGFP+ cells than SIX2-EGFP– cells (Figures 7I and 7K). By using the hNPSR/3D condition, we successfully established an hNPC line from sorted SIX2-EGFP+ cells. At the time of writing, the hiPSC-derived NPC line stably maintained the GFP signal as well as SIX2 and SALL1 expression after 2 months of culture (Figures 7J and 7L). More importantly, the hiPSC-derived NPC line could differentiate into major nephron structures upon spinal cord induction (Figure 7M). These results demonstrate that the hNPSR/3D culture conditions can help expansion of human PSC (hPSC)-derived NPCs.
DISCUSSION
3D Aggregate Culture Facilitates Self-Renewal of Mammalian NPCs In Vitro
Here, we report on a NPSR/3D culture system that facilitates long-term self-renewal of mouse and human nephron progenitors in vitro. Consistent with our observations, 2-day aggregate formation and clump passaging employed by Tanigawa et al. also significantly increased the lifespan of monolayer cultured NPCs from 7 days to ~19 days (Tanigawa et al., 2016), highlighting that cell-cell contact likely plays a critical role. It will be interesting to further explore the mechanophysical properties of 3D versus 2D cultured NPCs and determine whether they are involved in dictating the distinct phenotypes observed here, in particular the in vivo differentiation potential. Toward these and other goals, future investigations are warranted to gain a more complete mechanistic understanding of NPC self-renewal.
A Robust and Chemically Defined NPSR Culture Condition
Although necessary, 3D culture alone is not sufficient for long-term self-renewal of isolated NPCs. Through screening for a selected list of growth factors and small molecules guided by previous developmental studies, we were able to narrow down a factor cocktail that, when combined with 3D format, was sufficient for propagating functional NPCs. BMP, fibroblast growth factor (FGF), and Wnt shown to play key roles in NPCs self-renewal, survival, and differentiation, are included in the NPSR medium. Our NPSR culture sheds light on the role LIF plays in maintaining NPC identity in addition to its well-known function in regulating the mesenchymal-epithelial transition (MET) (Barasch et al., 1999), in agreement with recent reports (Tanigawa et al., 2015, 2016). It should be noted that withdrawal of any component in the NPSR medium compromised the self-renewal, potency, or survival of cultured NPCs. Thus, our chemically defined NPSR culture appears to provide a minimal synthetic niche environment for the robust propagation of functional NPCs. An unexpected observation was that single NPCs could not survive in the NPSR/3D condition, providing additional support for survival cues conferred by cell-cell contact and/or paracrine factors. Additional explorations along this line may help further improve NPC culture.
In contrast to mouse NPCs, human NPCs are poorly studied due to the limited access to primary human fetal kidney tissue. The slower growth kinetics, lower propensity toward differentiation, and inability for long-term propagation of human NPCs in mouse NPSR culture suggest that species differences exist. Interestingly, although dispensable for mouse NPCs, additional dual inhibition of Smad2/3 and Smad1/5/8 pathways stabilized human NPCs in a more proliferative state. More importantly, functional human NPCs could be maintained in the hNPSR culture long-term. In contrast to the mouse, the inhibition of Smad2/3 signaling pathway appears to be conducive to long-term culture of human organoids from various tissues, including small intestine, colon, pancreas, and liver (Sato and Clevers, 2015). How this signaling pathway contributes to species-specific differences is an interesting question that warrants future investigation.
NPC-Based Disease Modeling and Drug Screening Platforms
The methodologies introduced here represent a proof of concept for the derivation of NPC lines from both wild-type and transgenic mouse strains. When combined with state-of-art nuclease-based gene editing technologies such as CRISPR-Cas9 (Mali et al., 2013), our findings constitute the basis of a technological platform that could be further expanded and enhanced by generation of kidney specific knockout and knockin disease models as well as gene-corrected lines. Wide implementation of this platform may help enhance the current knowledge of mammalian, and especially human kidney morphogenesis, modeling of kidney-related human diseases, diagnostics, and eventually cell-replacement therapies.
EXPERIMENTAL PROCEDURES
Mouse and Human NPC Lines: Derivation and Culture
For detailed protocol information, please refer to Supplemental Experimental Procedures. Six2tm3(EGFP/cre/ERT2)Amc (Six2GCE) mice were purchased from The Jackson Laboratory (stock 009600) (Kobayashi et al., 2008). Mouse fetal/neonatal kidneys were dissociated to single cells. Six2-GFP+ cells were purified by FACS. 3,000 sorted GFP+ cells were seeded into one well of U-bottom low-attachment 96-well plate in mouse NPSR medium containing BMP7, FGF2, heparin, Y27632, mouse LIF, and CHIR99021. Medium was changed every 2 days. For passaging, NPC aggregates were dissociated into single cells and passaged at ratios of 1: 20 to 1:40 every 4–6 days. For ICR and NEP25 mice (Matsusaka et al., 2005), the CDP method was used to derive NPC lines. Human NPCs were enriched by FACS using Fluor-conjugated cell-surface antibodies against human EpCAM and NGFR. 10,000 cells were seeded into one well of 96-well U-bottom low-attachment plate in human NPSR medium containing BMP7, FGF2, heparin, Y27632, human LIF, CHIR99021, LDN193189, and A83–01. Medium was changed every 2 days, and the aggregates were dissociated and split at 1:10 every 4–5 days. All of the animal experiments were performed under the ethical guidelines of the Salk Institute, and animal protocols were reviewed and approved by the Salk Institute Institutional Animal Care and Use Committee (IACUC). Experiments with human samples were approved by the Institutional Review Board (IRB) Committee.
Nephron Organoid Generation
For detailed protocol information, please refer to Supplemental Experimental Procedures. NPC aggregates were placed onto transwell inserts at the air-liquid interface with culture medium at the bottom containing CHIR99021 and FGF2 for the first 2 days to generate FS-NPCs. The culture medium was then switched to basal medium containing 5% knockout serum replacement (KSR) for another 5–10 days to generate nephron organoids.
Spinal Cord Induction Assay
For detailed protocol information, please refer to Supplemental Experimental Procedures. E11.5–E13.5 mouse spinal cords were manually dissected and cut into small pieces. NPC aggregates were placed in direct contact with the dorsal side of the spinal cords on air-liquid interface of transwell inserts. Culture medium containing 10% FBS was added to the bottom chamber.
Supplementary Material
Highlights.
Derivation and long-term culture of mouse and human NPC lines
Rapid and efficient nephron organoid formation from mouse and human NPC lines
Long-term cultured NPC lines show nephrogenic potential in vivo
NPC lines enable gene editing and disease modeling
ACKNOWLEDGMENTS
We would like to thank M. Ku and M. Chang of the H.A. and Mary K. Chapman Charitable Foundations Genomic Sequencing Core for performing RNA-seq, J.O. of Human Embryonic Stem Cell Core Facility of Sanford Consortium for Regenerative Medicine for FACS, and M. Schwarz and P. Schwarz for administrative help. T.A. was supported by The Kyoto University Foundation, The Nagai Foundation Tokyo, The Kidney Foundation, Japan (grant JKFB15-4), and the UCAM. M.L and Y.X. were supported by a California Institute for Regenerative Medicine (CIRM) Training Grant fellowship. H.K. Liao was supported by Universidad Cató lica San Antonio de Murcia (UCAM). E.B. was supported by a Catharina Foundation fellowship. Work in the laboratory of J.C.I.B. was supported by UCAM (mouse work), Fundacion Dr. Pedro Guillen, The G. Harold and Leila Y. Mathers Charitable Foundation, The Leona M. and Harry B. Helmsley Charitable Trust (2012-PG-MED002), and The Moxie Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, six figures, and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2016.07.016.
ACCESSION NUMBERS
The accession number for the RNA-seq data reported in this paper is GEO: GSE78772.
REFERENCES
- Araoka T, Mae S, Kurose Y, Uesugi M, Ohta A, Yamanaka S, and Osafune K. (2014). Efficient and rapid induction of human iPSCs/ESCs into nephrogenic intermediate mesoderm using small molecule-based differentiation methods. PLoS ONE 9, e84881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak H, Huh SH, Chen S, Jeanpierre C, Martinovic J, Parisot M, Bole-Feysot C, Nitschke P, Salomon R, Antignac C, et al. (2012). FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev. Cell 22, 1191–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barasch J, Yang J, Ware CB, Taga T, Yoshida K, Erdjument-Bromage H, Tempst P, Parravicini E, Malach S, Aranoff T, and Oliver JA (1999). Mesenchymal to epithelial conversion in rat metanephros is induced by LIF. Cell 99, 377–386. [DOI] [PubMed] [Google Scholar]
- Brown AC, Muthukrishnan SD, and Oxburgh L. (2015). A synthetic niche for nephron progenitor cells. Dev. Cell 34, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TJ, Park JS, Hayashi S, Majumdar A, and McMahon AP (2005). Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 9, 283–292. [DOI] [PubMed] [Google Scholar]
- Cheng HT, Miner JH, Lin M, Tansey MG, Roth K, and Kopan R. (2003). Gamma-secretase activity is dispensable for mesenchyme-to-epithelium transition but required for podocyte and proximal tubule formation in developing mouse kidney. Development 130, 5031–5042. [DOI] [PubMed] [Google Scholar]
- Costantini F, and Kopan R. (2010). Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev. Cell 18, 698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep CQ, Ma D, Deo RC, Holm TM, Naylor RW, Arora N, Wingert RA, Bollig F, Djordjevic G, Lichman B, et al. (2011). Identification of adult nephron progenitors capable of kidney regeneration in zebrafish. Nature 470, 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressler GR (2006). The cellular basis of kidney development. Annu. Rev. Cell Dev. Biol 22, 509–529. [DOI] [PubMed] [Google Scholar]
- Dudley AT, Godin RE, and Robertson EJ (1999). Interaction between FGF and BMP signaling pathways regulates development of metanephric mesenchyme. Genes Dev. 13, 1601–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imberti B, Tomasoni S, Ciampi O, Pezzotta A, Derosas M, Xinaris C, Rizzo P, Papadimou E, Novelli R, Benigni A, et al. (2015). Renal progenitors derived from human iPSCs engraft and restore function in a mouse model of acute kidney injury. Sci. Rep 5, 8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kispert A, Vainio S, and McMahon AP (1998). Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development 125, 4225–4234. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, and McMahon AP (2008). Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuure S, Popsueva A, Jakobson M, Sainio K, and Sariola H. (2007). Glycogen synthase kinase-3 inactivation and stabilization of beta-catenin induce nephron differentiation in isolated mouse and rat kidney mesenchymes. J. Am. Soc. Nephrol 18, 1130–1139. [DOI] [PubMed] [Google Scholar]
- Lam AQ, Freedman BS, Morizane R, Lerou PH, Valerius MT, and Bonventre JV (2014). Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J. Am. Soc. Nephrol 25, 1211–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little MH (2016). Growing kidney tissue from stem cells: how far from “party trick” to medical application? Cell Stem Cell 18, 695–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little MH, and McMahon AP (2012). Mammalian kidney development: principles, progress, and projections. Cold Spring Harb. Perspect. Biol 4, a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mae S, Shono A, Shiota F, Yasuno T, Kajiwara M, Gotoda-Nishimura N, Arai S, Sato-Otubo A, Toyoda T, Takahashi K, et al. (2013). Monitoring and robust induction of nephrogenic intermediate mesoderm from human pluripotent stem cells. Nat. Commun 4, 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusaka T, Xin J, Niwa S, Kobayashi K, Akatsuka A, Hashizume H, Wang QC, Pastan I, Fogo AB, and Ichikawa I. (2005). Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J. Am. Soc. Nephrol 16, 1013–1023. [DOI] [PubMed] [Google Scholar]
- Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, and Bonventre JV (2015). Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol 33, 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishinakamura R. (2016). Stem cells and renal development in 2015: advances in generating and maintaining nephron progenitors. Nat. Rev. Nephrol 12, 67–68. [DOI] [PubMed] [Google Scholar]
- Park JS, Valerius MT, and McMahon AP (2007). Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 134, 2533–2539. [DOI] [PubMed] [Google Scholar]
- Reginensi A, Clarkson M, Neirijnck Y, Lu B, Ohyama T, Groves AK, Sock E, Wegner M, Costantini F, Chaboissier MC, and Schedl A. (2011). SOX9 controls epithelial branching by activating RET effector genes during kidney development. Hum. Mol. Genet 20, 1143–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, and Clevers H. (2015). SnapShot: growing organoids from stem cells. Cell 161, 1700–1700.e1. [DOI] [PubMed] [Google Scholar]
- Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, and Oliver G. (2006). Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 25, 5214–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, and Nishinakamura R. (2014). Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 14, 53–67. [DOI] [PubMed] [Google Scholar]
- Takasato M, Er PX, Becroft M, Vanslambrouck JM, Stanley EG, Elefanty AG, and Little MH (2014). Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol 16, 118–126. [DOI] [PubMed] [Google Scholar]
- Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, Parton RG, Wolvetang EJ, Roost MS, Chuva de Sousa Lopes SM, and Little MH (2015). Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526, 564–568. [DOI] [PubMed] [Google Scholar]
- Tanigawa S, and Perantoni AO (2016). Modeling renal progenitors: defining the niche. Differentiation 91, 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa S, Sharma N, Hall MD, Nishinakamura R, and Perantoni AO (2015). Preferential propagation of competent SIX2+ nephronic progenitors by LIF/ROCKi treatment of the metanephric mesenchyme. Stem Cell Rep. 5, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa S, Taguchi A, Sharma N, Perantoni AO, and Nishinakamura R. (2016). Selective in vitro propagation of nephron progenitors derived from embryos and pluripotent stem cells. Cell Rep., Published online April 13, 2016. 10.1016/j.celrep.2016.1003.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyohara T, Mae S, Sueta S, Inoue T, Yamagishi Y, Kawamoto T, Kasahara T, Hoshina A, Toyoda T, Tanaka H, et al. (2015). Cell therapy using human induced pluripotent stem cell-derived renal progenitors ameliorates acute kidney injury in mice. Stem Cells Transl. Med 4, 980–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PH, and Brown PA (1996). The relationship between enzymuria and kidney enzyme activities in experimental gentamicin nephrotoxicity. Ren. Fail 18, 899–909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.