

Abstract
Cardiovascular calcification is an insidious form of ectopic tissue mineralization that presents as a frequent comorbidity of atherosclerosis, aortic valve stenosis, diabetes, renal failure, and chronic inflammation. Calcification of the vasculature and heart valves contributes to mortality in these diseases. An inability to clinically image or detect early microcalcification coupled with an utter lack of pharmaceutical therapies capable of inhibiting or regressing entrenched and detectable macrocalcification has led to a prominent and deadly gap in care for a growing portion of our rapidly-aging population. Recognition of this mounting concern has arisen over the past decade, and led to a series of revolutionary works that has begun to pull back the curtain on the pathogenesis, mechanistic basis, and causative drivers of cardiovascular calcification. Central to this progress is the discovery that calcifying extracellular vesicles act as active precursors of cardiovascular microcalcification in diverse vascular beds. More recently, the ‘omics revolution has resulted in the collection and quantification of vast amounts of molecular-level data. As the field has become poised to leverage these resources for drug discovery, new means of deriving relevant biological insights from these rich and complex datasets have come into focus through the careful application of systems biology and network medicine approaches. As we look onward towards the next decade, we envision a growing need to standardize approaches to study this complex and multi-faceted clinical problem, and expect that a push to translate mechanistic findings into therapeutics will begin to finally provide relief for those impacted by this disease.
Keywords: Calcification, Extracellular Vesicles, Atherosclerosis, Aortic Stenosis, Multi-Omics, Standardization
Subject Terms: Atherosclerosis, Valvular Heart Disease
Graphical Abstract
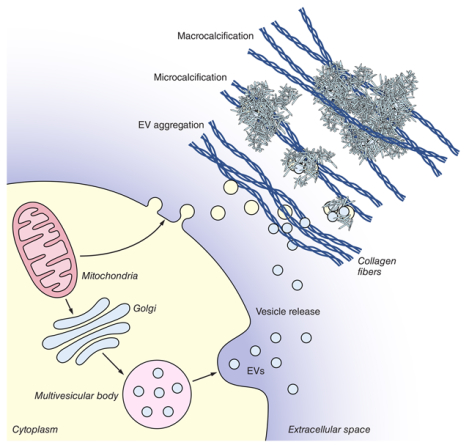
Cardiovascular calcification: a driver of morbidity and mortality
Cardiovascular calcification correlates strongly with cardiovascular events and predicts morbidity and mortality.1 While aortic valve macrocalcification impairs the movement of valve leaflets resulting in valve stenosis and heart failure, arterial microcalcification may cause plaque rupture at interface areas between high- and low-density tissue, resulting in myocardial infarction. There are currently no FDA approved drugs to prevent or treat calcification, and there is a lack of clinical imaging modalities to detect early subclinical calcification.
Molecular imaging identifies microcalcification and osteogenic activity
Beginning just over a decade ago, a series of papers from our group demonstrated the power of a high resolution optical near-infrared fluorescence (NIRF) molecular imaging to detect the earliest stages of calcification in atherosclerotic plaques2 and aortic valves3 undetectable by routine histological methods (e.g. von Kossa, Alizarin Red) and conventional imaging modalities (e.g. computed tomography (CT)). Advanced nanotechnology enabled the creation of an imaging agent targeting hydroxyapatite by chemically conjugating bisphosphonate with a fluorochrome4 and offered a cellular-resolution tool to identify preclinical microcalcifications (1–5 μm) and detect osteogenic activity associated with activity of alkaline phosphatase, a key contributor to early calcification. Importantly, this translational imaging technology not only provided a sensitive diagnostic tool for detection of microcalcification in vivo, ex vivo and in vitro but also offered functional information through the capability to simultaneously visualize and quantify one or more biological processes in small animals using tracers labeled with dyes operating in distinct near-infrared spectra (e.g., calcification and inflammation or inflammation and proteolytic activity). Using this dual imaging technique, our in vivo longitudinal studies in a mouse model of atherosclerosis extended a paradigm of inflammation-dependent calcification, as opposed to the long-held belief that calcification was an inevitable degenerative consequence of aging. We proposed that in the initiation stage, inflammation precedes calcification, and activated macrophages induce osteogenic transformation of vascular wall cells (e.g., smooth muscle cells (SMC)) through the release of osteogenic factors. In the second stage – propagation, inflammation and calcification develop in parallel within close proximity. This stage is characterized by formation of calcifying cell-derived extracellular vesicles. A striking overlap of macrophage and osteogenic NIRF signals at this stage provided a basis for our hypothesis that macrophage themselves undergo calcification via the release of pro-osteogenic extracellular vesicles (EVs).5,6 During these earlier stages of calcification which are closely associated with inflammation, macrophage-targeting therapies may retard osteogenic activity and could prevent clinical complications, if sensitive diagnostic tools can identify such subclinical lesions early enough. In contrast, advanced stage calcification often associates with few if any macrophages – while such lesions are readily detected by conventional imaging modalities, they are, as it were, set in stone and may be more difficult to treat. More recently, investigators have used positron emission tomography (PET)/CT imaging to confirm the phenomenon of inflammation-dependent calcification in human patients in the vasculature7 and aortic valves.8 In addition, we have shown that (18)F-fluoride PET signal in PET-positive, CT-negative regions of human atherosclerotic plaques correlates with osteogenic activity detected by NIRF imaging and is the result of developing microcalcifications.9
Extracellular vesicles as precursor of microcalcification
Microcalcifications were first described by Anderson and Kim in vasculature and aortic valves, respectively, but the knowledge was limited to transmission electron microscopy of calcified tissue.10,11 Visualizing the formation of subcellular microcalcification (<1μm) in vivo in either mouse or human tissues remains challenging, as this process lays below the resolution of current imaging modalities. To provide a time course of microcalcification biogenesis through observations in 3D, we developed a collagen hydrogel model that recapitulates the fibrous cap of atherosclerotic plaque or fibrotic valve tissue. By seeding with EVs isolated from calcifying cells (e.g., SMCs, valvular interstitial cells (VICs)), one can observe a dynamic process of microcalcification formation within a short 3–5 day time course. Using this platform in concert with high-resolution structure illumination microscopy and NIRF calcium tracer, we demonstrated for the first time how microcalcifications form at the level of a single EV, and proposed a sequential course of microcalcification biogenesis.12 In addition to molecular imaging utilizing calcium tracers, new technologies such as 2-photon microscopy and density-dependent scanning electron microscopy have been used to detect subcellular size microcalcifications and calcifying vesicle aggregates within the tissue sections.12,13 It has been recently reported that EVs released from SMCs and macrophages could serve as building blocks for vascular calcification.5,12,14 Depending on size and type, EVs are classified as exosomes (10–100 nm); microparticles (100–500 nm) and apoptotic bodies (up to 1000 nm). They possess a metabolically active outer membrane that protects their cargo, consisting of proteins, microRNA and other components of the parental cell. Vesicles are released by diverse cells in the body, then can be found in various tissues and fluids, and they participate in both physiological and pathological processes. Cell-derived tissue-entrapped EVs, particularly those in the size range of exosomes, may penetrate through the endothelial barrier and join the pool of EVs in the circulation, thereby serving as putative biomarkers of disease. On the other hand, larger vesicles (100–300 nm) are more likely to be trapped within collagen fibers and other extracellular matrix components, then calcify through aggregation and subsequent nucleation of hydroxyapatite. Notably, calcifying EVs have been identified in calcifying atherosclerotic lesions, aortic valves and medial layer of arteries of patients with chronic kidney disease.5,12,14–16
Contribution of macrophages
The mechanisms of EV-associated calcification are still under investigation, and little is known about the contributions of macrophages to this process. Macrophages are a heterogeneous population, which brings complexity to the pathogenesis of cardiovascular calcification.17 In addition to established roles of macrophages in atherosclerotic plaque progression and promotion of mineral resorption through osteoclastogenesis, some macrophage subpopulations may initiate cardiovascular calcification through the release of calcifying EVs. Our early molecular imaging studies corroborated by histological analyses established the hypothesis that macrophages directly contribute to formation of microcalcification, as intravital microscopy of mouse atheroma showed that NIRF osteogenic signal correlates highly with macrophages.2 Histology further confirmed that early calcification of mouse atherosclerotic plaques associates with macrophage accumulation, and unstable human atherosclerotic lesions contain macrophages and microcalcifications while lacking SMCs. In vitro validation studies using the mouse RAW 264.7 cell line revealed that pro-inflammatory, classically activated macrophages release EVs with high calcification potential, express exosomal markers CD9 and TSG101, and contain pro-inflammatory and pro-thrombotic calcium binding S100A9, also known as migration inhibitory factor-related protein 14 (MRP14) or calgranulin B.18 EVs containing S100A9 formed a complex with Annexin 5 and phosphatidylserine, thereby contributing to accelerated microcalcification formation.5 We recently confirmed this hypothesis in human tissue, human primary macrophages and in an in vivo model of accelerated calcification in diabetic apoE-deficient mice.6 Proteomics of human carotid plaques obtained from diabetic patients showed that S100A9 associates with osteogenic activity. Moreover, in human macrophages treated with high glucose, recombinant S100A9 induced the expression of pro-inflammatory and osteogenic factors, and the release of EVs with high alkaline phosphatase activity. Addition of a receptor for advanced glycation end products (RAGE) antagonist or silencing macrophages with S100A9 siRNA abolished these responses, suggesting that stimulation of the S100A9-RAGE axis by hyperglycemia favors a pro-calcific environment. In vivo molecular imaging further demonstrated that streptozotocin-induced diabetic apoE−/−S100a9−/− mice and mice treated with S100a9 siRNA encapsulated in macrophage-targeted lipid nanoparticles (LNPs) have decreased microcalcification formation in atherosclerotic lesions. We thus concluded that in the diabetic milieu, macrophages release calcific EVs via mechanisms implicating the S100A9-RAGE pathway. Severe pro-inflammatory conditions such as diabetes, where healing is delayed due to exaggerated inflammation, may thus induce further microcalcification formation and fibrous cap thinning thereby contributing to plaque destabilization. Overall, these studies expand the role of macrophages, that were traditionally known as precursors of osteoclast-like cells responsible for mineral resorption, towards a definition as a cell type that can also contribute to both mineral clearance and calcium deposition.
Impact of vascular smooth muscle cells and valvular interstitial cells
Vascular smooth muscle cells (SMCs) are well known contributors to cardiovascular calcification.19–21 However, the transition of SMCs or valvular interstitial cells (VICs) to osteoblast-like cells through osteogenic gene expression resulting in mature bone formation occurs in only 10–13% of vasculature (SMCs) and valves (VICs)22,23 while the remainder undergo either dystrophic calcification (e.g. calcification of apoptotic or dead cells) or through calcification of cell-derived EVs.12,14 Our in vitro experiments utilizing human primary SMCs cultured in osteogenic conditions demonstrated that these cells release abundant amount of calcifying EVs as detected by NIRF calcium tracer, and simultaneously secret fibrillar collagen type 1 as gauged by CNA35-OG488 fluorescent probe.12,24 The key results of these in vitro experiments also led to our new understanding of the formation of macro- and microcalcification. We noted that when calcifying NIRF particles aggregate below dense collagen fibers, they generate macrocalcifications; while when collagen fibers are sparse, EVs could freely move in between collagen fibers, their aggregation is abrogated, and they remain as EVs or form microcalcifications.
We next sought to understand how aggregation of calcifying EVs contributes to formation of microcalcification and whether there is a target molecule responsible for the EV aggregation. To do this, we first characterized EVs isolated from human SMCs and VICs cultured in osteogenic conditions using western blot, nanoparticle tracking analysis and electron microscopy, and then performed label-free quantitative proteomics.25 Annexin A1 (ANXA1), a protein associated with calcium binding and intracellular endosomal transport was identified as a potential tethering protein responsible for EV aggregation and microcalcification formation. To identify which EV subpopulation (e.g. exosome vs microparticle) contributes to ANXA1-associated aggregation, we performed multi-color single-EV microarray or single-EV analysis. Using this advanced method, we showed that ANXA1 siRNA treatment resulted in a strong reduction of ANXA1 positive signal on SMC- and VIC-EVs, and confirmed a calcium dependency of ANXA1 by visualizing reduction of ANXA1 positive signal by chelating calcium either prior or post-binding to EVs on microarray chips. We further showed that ANXA1 colocalizes with microcalcifications and localizes to EVs in atherosclerotic human plaques and stenotic aortic valves using immunofluorescence and transmission electron microscopy. To confirm that ANXA1 can induce EV aggregation, we generated artificial phosphatidylserine vesicles and incubated them with immunopurified green fluorescence protein (GFP)-tagged human ANXA1. Our experiments showed that GFP-tagged ANXA1 is enriched at tethered membrane contact sites of individual vesicles. Moreover, addition of N-terminal ANXA1 neutralizing antibodies suppressed vesicle aggregation. Silencing ANXA1 in SMCs and VICs cultured under osteogenic conditions reduced tissue nonspecific alkaline phosphatase (TNAP) activity, and addition of recombinant human ANXA1 to the culture media reversed the calcification response. Most importantly, ANXA1 knockdown suppressed human EV-mediated microcalcification formation in cell-free 3D collagen hydrogels containing EVs isolated from calcifying vascular SMCs and VICs. These in vitro observations were confirmed independently by corroborative high-resolution microscopy methods such as density-dependent scanning electron microscopy in human atherosclerotic plaques and stenotic aortic valves, and transmission electron microscopy in mouse lesions. Together these data demonstrate that ANXA1 promotes EV aggregation and microcalcification formation.
A new biology of Sortilin - a novel player in vascular calcification
We now have a clearer understanding of how microcalcifications form as calcifying EVs are released from the parental cell into the ECM, followed by aggregation, merging, nucleation of hydroxyapatite, and eventual growth of microcalcification. However, the means by which these EVs gain their mineralization properties was still unknown until recently. Kapustin et al. reported that SMCs stimulated with high levels of calcium and phosphate, mimicking a condition found in patients with chronic kidney disease, produce calcification-prone EVs.14 These EVs are rich in phosphatidylserines, and their loading with inhibitors of calcification (e.g. fetuin-A) is attenuated.16 Calcification and coagulation appear to be linked by these calcifying EVs, as deposition of EV-derived prothrombin plays a role in modulation of vascular calcification and thrombus formation.26 In atherosclerosis, a condition associated with inflammation-induced osteogenic changes in SMCs, the sorting receptor sortilin contributes to the loading of active TNAP into EVs through intracellular trafficking mechanisms.15 TNAP is a key osteogenic protein and contributes to initiation of calcification by hydrolyzing the calcification inhibitor pyrophosphate, which produces the free phosphate required for mineralization. We showed that sortilin is phosphorylated by serine kinases CK2 and FAM20c in the Golgi apparatus, where it binds to active TNAP. The phosphorylated at C-terminus sortilin-TNAP complex is taken up by recycling endosomes via Rab11-dependent trafficking and then loaded into EVs. Importantly, dimerization of sortilin regulates its trafficking to EVs through the formation of homodimers with intermolecular disulfate bonds at 10CC domain and Cys783.27 We validated these in vitro studies in SMCs with two animal models of atherosclerosis: sortilin−/−/ldlr−/− mice and sortilin−/− mice injected with PCSK9 adeno-associated virus (AAV).15,28 NIRF molecular imaging corroborated with histology showed reduction of calcification in sortilin-deficient mice in both models. Moreover, we further confirmed the role of sortilin in vascular calcification in human patients by showing that independently of C-reactive protein and low-density lipoprotein cholesterol, serum sortilin correlates with aortic calcification and cardiovascular risk in a cohort of 830 men.29 These studies demonstrate that sortilin is a mechanistically informative and diagnostically useful biomarker of vascular calcification.
Based on the totality of these studies, we now propose our model of cell-derived EV microcalcification formation (Figure 1). Under diabetic conditions, the S100A9-RAGE axis drives release of TNAP-containing EVs from macrophages;6 macrophages also release S100A9-containing EVs with high calcification potential under calcifying conditions.5 In SMCs, osteogenic conditions alter mitochondrial calcium signaling, which increases TNAP activation.30 Dimerization of sortilin regulates its trafficking into EVs, where it drives TNAP loading and increases EV calcification potential.15,27 After release from the cell, ANXA1 promotes aggregation of EVs trapped in collagen-rich extracellular matrix.25 Finally, aggregated EVs nucleate hydroxyapatite and form microcalcifications, which then grow and advance to macrocalcification.12
Figure 1: Extracellular vesicles are drivers of cardiovascular calcification.
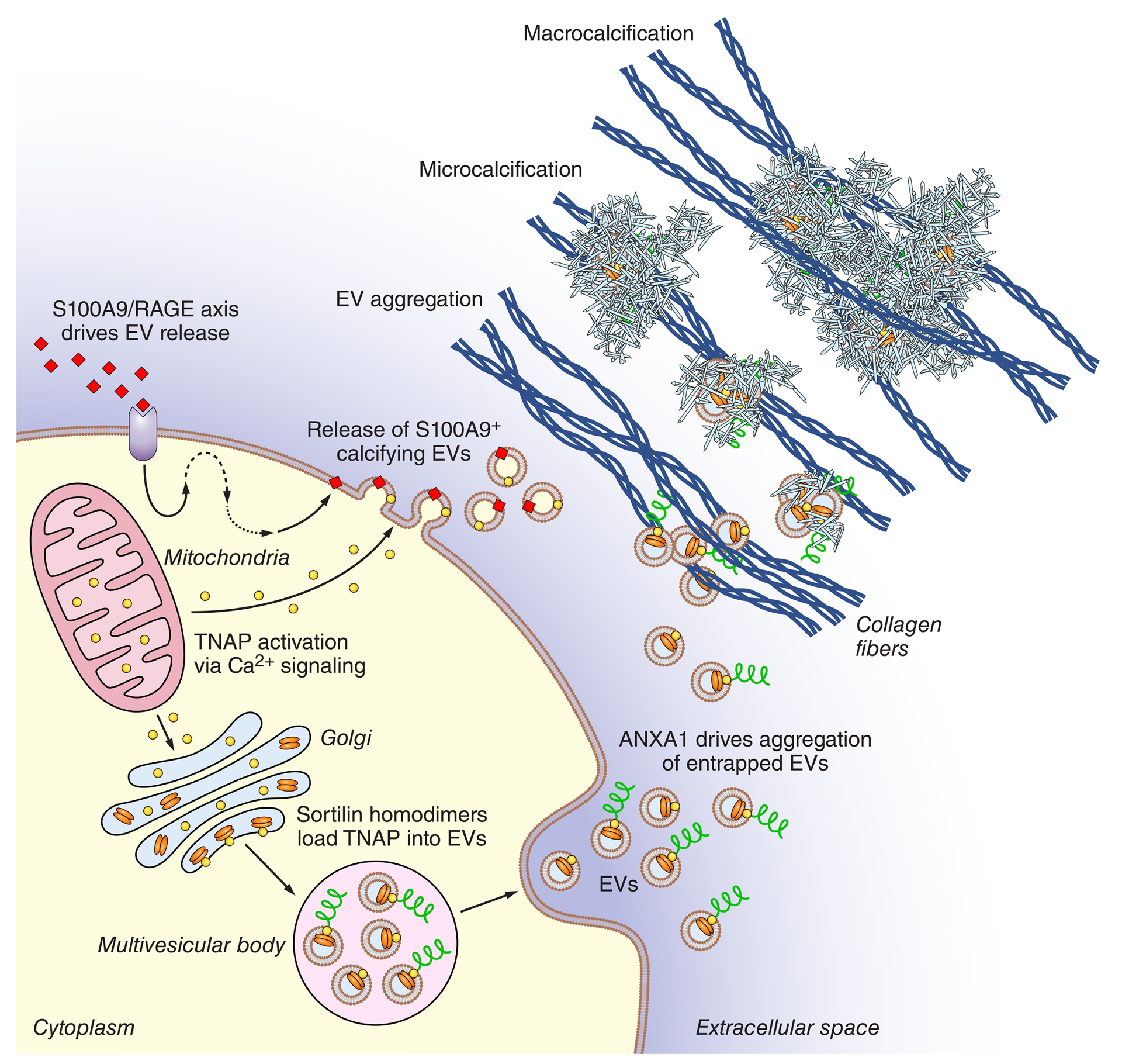
Under diabetic conditions, the S100A9-RAGE axis drives release of tissue non-specific alkaline phosphatase (TNAP)-containing extracellular vesicles (EVs) from macrophages;6 macrophages also release S100A9-containing EVs with high aggregation and calcific potential under calcifying conditions.5 In smooth muscle cells, osteogenic conditions alter mitochondrial calcium signaling which increases TNAP activation.30 Dimerization of sortilin regulates its trafficking into EVs, where it drives TNAP loading and increases EV calcification potential.15,27 After release from the cell, ANXA1 promotes aggregation of EVs trapped in collagen-rich extracellular matrix.25 Aggregated EVs nucleate hydroxyapatite and form microcalcifications, which then grow and progress to macrocalcification.12 Extraction and enrichment of calcifying EVs followed by proteomic study of their cargoes is a promising approach for drug target discovery.61,67 Adapted.25
Other players recently implicated in cardiovascular calcification
As described above, collagen accumulation is intimately associated with the progression of vascular microcalcification and atherosclerotic plaque stability. Recent studies of the collagen receptor discoidin domain receptor family, member 1 (DDR1) show that it regulates amorphous collagen deposition, TNAP activity, release of calcifying EVs from vascular SMCs, and aortic calcification in hyperlipidemic mice through the TGF-β pathway.31 Mechanistically, DDR1 also modulates vascular calcification via the PI3K/Akt pathway upstream of RUNX2 under diabetic conditions, and acts as a matrix stiffness mechanosensor via the RhoA axis to drive osteogenesis in vascular SMCs.32,33 Biomineralization of the collagenous ECM in bone and the vasculature is also regulated by poly(ADP-ribose) (PAR). In response to oxidative stress or DNA damage, vascular SMC poly(ADP-ribose) polymerases (PARPs) deposit extracellular PAR, which binds the ECM and forms spherical sub-micron-sized calcified particles with a high affinity for collagen fibril hole zones. The resultant accumulation of these bodies generates bone-like mineralization of collagen fibers, while PARP inhibitors demonstrate the therapeutic relevance of this mineralization mode.34
The vascular and valvular endothelium is an important shear-sensitive regulator of calcification. In human aortic valve endothelial cells, protective hemodynamic forces activate NOTCH signaling, which inhibits osteoblastic gene networks including matrix gla protein and connexin 40.35 Indeed, investigation of Notch and Notch ligands is likely to be key to therapeutic strategies, as loss of Notch signaling upstream of nitric oxide signaling through second messenger cGMP modulates valve endothelial and interstitial cell phenotypes, induces impaired valve morphogenesis, and VIC calcification.36 Further confirmation of putative roles for cGMP signaling in valvular homeostasis emerged from studies of c-type natriuretic peptide, which induces cGMP signaling via the particulate guanylate cyclase NPR2 receptor, inhibits myofibrogenesis and VIC calcification in vitro, and whose deficiency in mice induces bicuspid aortic valves, CAVD, and ascending aortic dilatations.37
Retinoic acid, a metabolite of vitamin A, regulates mineralization through control of osteogenic transcriptional programs. Therapeutic applications of cyclic retinoids in humans have been problematic due to their adverse impact on triglyceride levels and impairment of liver cholesterol metabolism. In contrast, the synthetic acyclic retinoid peretinoin does not appear to harbor these off-target effects in vitro, suppresses human SMC and human VIC calcification by attenuating TNAP activity RUNX2 expression, and does not inhibit bone osteoblast mineralization – indicating the potential lack of non-specific effects on skeletal mineralization in response to treatment of vascular calcification by peretinoin.38 Others have focused therapeutic development efforts on direct inhibition of mineral formation: phytate is a potent calcification inhibitor, and acts by directly binding to growth sites on hydroxyapatite crystals. In rats, the phytate-based compound SNF472 prevents vitamin D3- or adenine-induced cardiovascular calcification.39 Strikingly, a phase 2 trial of year-long administration of SNF472 in 274 patients has demonstrated significant attenuation of coronary artery and aortic valve calcification vs. placebo control in patients with end-stage kidney disease, though any impact on cardiovascular events remains to be assessed.40
Standardization of study design, nomenclature, and translation
As calcium-based mineralization is a critical component of vertebrate physiology and disease, it is not surprising that this process is active, dynamic, multi-factorial, and tightly governed by external stimuli, multiple cell types, and numerous regulatory signaling cascades. As a result, pathological calcification progresses via different mechanisms depending on the site, species, and disease comorbidities. In 2017 we defined a set of study design requirements that aimed to standardize the investigation of cardiovascular calcification in order to better define the context, models, and methodologies that are employed.41 These ranged from careful selection of cell culture models in order to maintain relevance to the in vivo disease process of interest (e.g. hyperphosphatemic vs. inflammatory, passage-dependency, etc.), disclosure of all components used to induce calcification in cell culture models (including phosphate sources), and ensuring careful and thoughtful translation of findings between vascular and valvular calcification. In such a manner, we aimed to improve replicability and translational potential of findings in order to accelerate therapeutic development. Many of these recommendations have gained traction in recent years.42–45 Standardization is especially important in the context of EV-associated calcification. The term “extracellular vesicles” refers collectively to a group of secreted, membrane-bound particles that contains cell-specific and often overlapping subclasses described as exosomes, ectosomes, microvesicles, microparticles, multivesicular bodies, matrix vesicles, and apoptotic bodies. With multiple routes of biogenesis, a wide variation in size (30–1000 nm in diameter), unique protein markers, and alterations in cargoes between vesicle classes, careful characterization of vesicle populations is necessary.46 Since 2014, the Minimal Information for Studies of Extracellular Vesicles (MISEV) guidelines have defined the nomenclature, collection, enrichment, characterization, and functional analysis of extracellular vesicle studies as a broad field – these extensive sets of practical best practices have rightfully become de facto requirements for publication.47 Importantly, In the context of cardiovascular calcification, both macrophages, SMCs, and VICs release EVs on the order of 30–300 nm in diameter. Notably, despite overlapping size ranges, calcifying EVs released from macrophages appear to have microvesicular (budding/fission) origins,5 while those from vascular SMCs originate as exosomes that are packaged within multivesicular bodies.14 Careful adherence to the MISEV guidelines during the study of calcifying EV populations and reporting of said results will be important to addressing the relative contributions of macrophage-, SMC-, and VIC-derived calcifying EVs to the genesis and growth of cardiovascular microcalcifications, to dissect the roles of circulating and tissue-resident EVs, and to address the functional impact of these diverse particle populations on recipient cells throughout the cardiovascular system.48
Omics, systems biology, and network medicine in the pursuit of drug discovery
As is clear from the above discussion, the onset and progression of cardiovascular calcification is a multifactorial, context-dependent process that occurs in complex tissues. The lack of pharmacotherapeutics to address this mineralization in the clinic is due in large part to poor mechanistic understanding. An effective drug discovery strategy (Figure 2) must combine efficient diagnostic tools with a focused search for target compounds and a sound validation strategy; a key step in this cycle is the application of omics strategies to the holistic study of complex biological systems. Multi-omics, as a whole, encompasses the large-scale study of the (epi)genome, transcriptome, proteome, and metabolome, respectively. Genome-wide association studies (GWAS) have identified genetic variants, or SNPs, in close proximity to the cell cycle regulators CDKN2A and CDKN2B and the phosphatase regulator PHACTR1 to be significantly associated with coronary artery calcification,49 while GWAS on patients with valvular calcification found the LPA gene to be a replicable risk-associated allele in multiple ethnic groups and SNPs in IL1F9 are associated with mitral annular calcification.50 Epigenetic regulation of cardiovascular calcification has received relatively little study to date, though genome-wide studies have found numerous differentially-methylated sites between normal and stenotic aortic valves.51 Moving down the line, transcriptomics have connected dysregulated inhibitors of calcification such as osteopontin and matrix γ-carboxyglutamate protein to uremic vascular calcification52 and incriminated several noncoding RNAs in regulation of RUNX2-driven arterial calcification.53 Mass spectrometric investigations of the proteome have found NDRG-2 and collagen VI to be in close proximity with calcific nodules in human valvular calcification,54 while targeted plasma proteomics have identified a set of 35 plasma biomarkers which predict high-risk coronary plaques.55 Metabolomics remains in its infancy in our field, though it has recently moved beyond biomarker discovery in biofluids to tissue pathogenesis; metabolites from the cholesterol, purine, pyrimidine, and ceramide pathways were significantly enriched in mature atherosclerotic plaques.56
Figure 2: Cardiovascular calcification drug discovery pipeline.
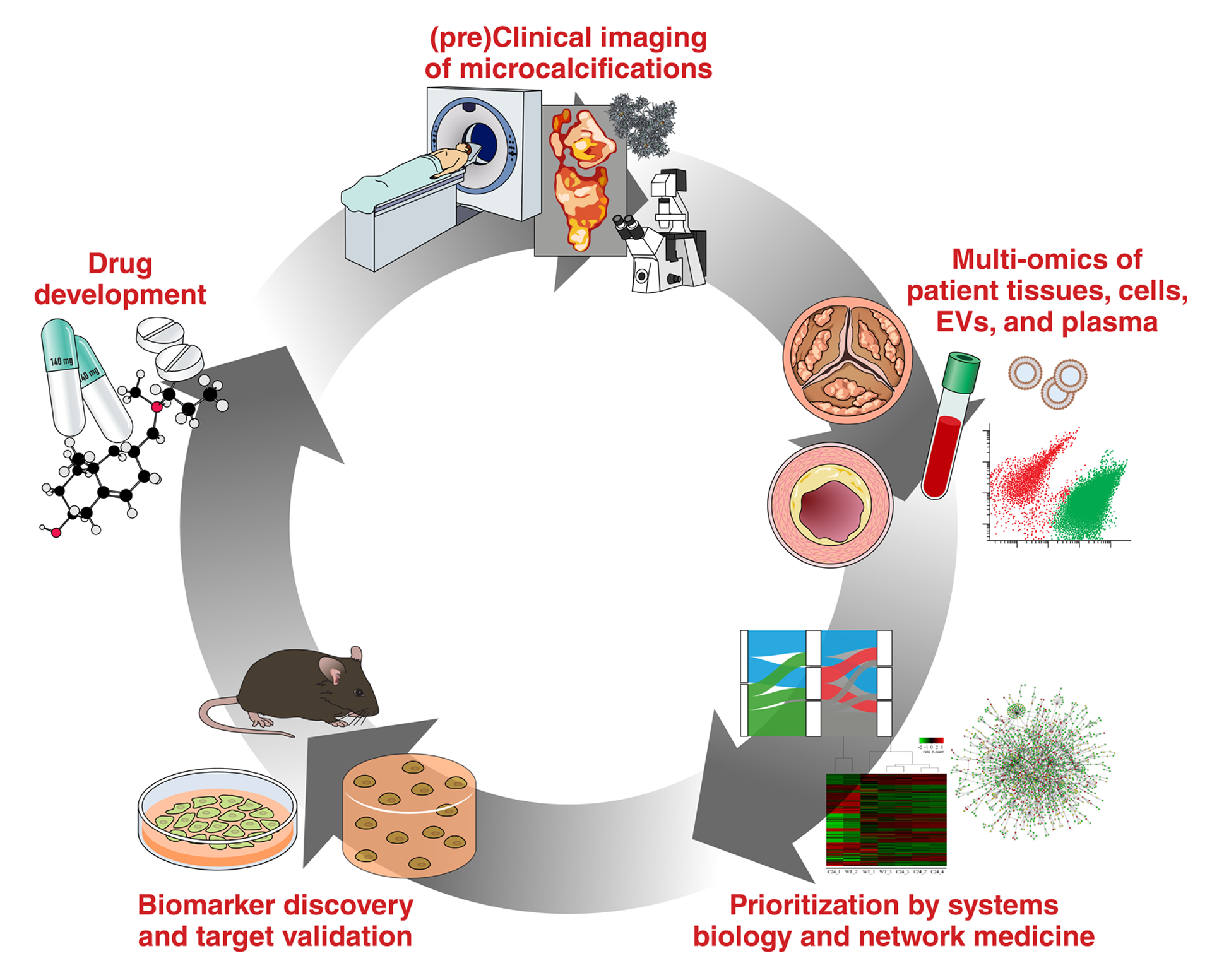
Microcalcifications are detected and quantified by pre-clinical or clinical imaging modalities (e.g. molecular imaging or PET/CT).9 Genomics, transcriptomics, proteomics, lipidomics, and metabolomics (as a whole: multi-omics) of normal and diseased human tissues, plasma, and extracellular vesicles (EVs) can be employed to identify potential drug and biomarker targets in the context of cardiovascular calcification.60,61 Systems biology and network medicine approaches are then used to prioritize these molecules for further investigation, and provide novel insights into disease etiology, pathogenesis, and progression.60,61 High-priority therapeutic targets and biomarker candidates are validated in two- or three-dimensional in vitro models or in vivo/ex vivo disease models.45,68 Drugs are then designed to target validated candidates, either through high-throughput screening or computational drug discovery. Lastly, drug efficacy is assessed by imaging for evidence of inhibition or regression of microcalcification. Adapted44.
The potential of these omic approaches is massive, but the interpretation of these data into coherent and validatable biological understanding suitable for mechanistic insight and drug target prioritization remains an area of major importance and keen study. How do we effectively pair these holistic data-driven investigations with reductionist hypothesis-driven approaches? Much of the work to date has focused on pathway or gene ontology enrichment, where biological molecules are annotated to familiar cellular components, molecular functions, or biological processes. Networks are also employed to present complex interactions in a quantitative manner that enables inference or prediction of key disease-related molecules, biomarkers, and therapeutic candidates. Gene set enrichment and functional network analyses of carotid endarterectomy transcriptomics has implicated hypoxia, chemokines, calcification, cytoskeletal force production, and ECM homeostasis in symptomatic vs. asymptomatic plaques.57 Others have discovered a shift in aortic transcriptional programs from vascular homeostasis towards an osteochondrocytic profile via the application of differential expression analysis, gene ontology cluster analysis and pathway enrichment analysis to a rat model of chronic uremia and renal dysfunction-induced vascular calcification.52 As the depth of individual omics datasets grows, and as more omics modalities become concurrently available to any one laboratory, “integrative omics” or “multi-omics” have become an important aspect of these studies. This approach pairs two or more omics “layers” (e.g. genomics and transcriptomics, transcriptomics and proteomics, etc.) in order to more fully capture the biological complexity of human disease. Early work to incorporate multi-omics integration for cardiovascular calcification was performed by Guauque-Olarte et al., where a pair of GWAS studies conducted on patients with aortic stenosis were combined and the resultant susceptibility genes were analyzed by RNA-seq. Alongside this, gene expression profiling on a second set of stenotic and control aortic valves was performed for expression quantitative trait loci (eQTL) profiling. By filtering differentially-expressed genes from their transcriptomics data for those that were both in close proximity to GWAS SNPs that were trending towards significance and that were significant eQTLs, the pro-osteogenic transcription factor RUNX2 was identified as a potential driver of calcific aortic valve disease (CAVD), while the voltage-dependent calcium channel CACNA1C was found to be a novel susceptibility gene.58 Integrative omics have also been employed on human plasma. The combination of plasma proteomics and metabolomics was utilized to differentiate the molecular drivers of aortic valve regurgitation vs. those of CAVD.59 In our studies, we have focused on the usage of multi-omics and systems biology techniques to both derive novel insights into disease pathobiology, and to identify potential targets for therapeutic intervention. We have recently embraced a network medicine approach to multi-omics integration. After conducting both transcriptomics and proteomics on spatially- and temporally-discretized samples of valvular tissue from patients with CAVD, we identified mutually-exclusive sets of genes and proteins that characterized each anatomical region of the valve leaflet and stage of disease. Enriched pathway networks revealed large clusters of pathways that predominated the biological response to disease within each region of the valve, and mapping of stage/layer-specific proteins onto the global human protein-protein interaction network revealed fibronectin-1 as a key molecular driver of CAVD progression. Such insight would not have been found by analysis of the transcriptomics or proteomics layers on their own.60 In a similar vein, we developed a means to extract tissue-entrapped EVs from fibrocalcific human atherosclerotic plaques and calcified aortic valves via a combination of density-gradient separation and mass spectrometry-guided EV enrichment. Subsequent network-based integration on proteomics and miRNA-sequencing of the tissue-entrapped EV vesiculome revealed that plaque- and valve-derived EV cargoes shared an overall ability to regulate Rho GTPase and MAPK intracellular signaling cascades, while EV proteins and miRs differentially modulated cellular contraction and p53-mediated transcriptional regulation in diseased vascular vs. valvular tissue.61
It has been known for some time that cardiovascular calcification occurs in complex tissues, which harbor multiple cell types of both tissue-resident and circulating origins. Which subsets of cells are mere bystanders, which are working to protect tissues from pathological changes, and which are actively driving disease? These questions are of fundamental importance to the development and targeting of successful therapeutic strategies, but until recently omics approaches were largely limited to the study of bulk cell and tissue samples, where information on cellular heterogeneity was lost to averaging. Technological advances have popularized and made widely accessible the progression of omics investigations from that of global gene/protein levels down to the level of the single cell. Single-cell RNA sequencing (scRNA-seq) typically involves separation of cells into individual droplets, followed by barcoding, library formation, and sequencing; barcoding data is then used to deconvolve the resultant dataset on a per-cell basis. While powerful and promising, care must be taken to avoid artifactual alteration of the transcriptome when applying scRNA-seq to study tissue-derived cell populations. Notably, the study of cardiovascular calcification at the single-cell level is hampered by technical challenges centered around rapid, robust and non-biased extraction of cells from fibrotic and/or calcified tissues and cell cultures. In terms of protein expression, the majority of studies to date have employed mass cytometry (CyTOF). Simplistically, CyTOF is flow cytometry using mass spectroscopy instead of fluorescence detection where cells are labeled with heavy-metal conjugated antibodies against proteins of interest. Due to dramatically-improved spectral overlap vs. that of flow cytometry, CyTOF is currently capable of quantitative measurements across over 130 channels.62 Order-of-magnitude improvements in the depth of the single-cell proteome are on the horizon: promising new approaches combine automated single-cell picking and highly-sensitive tandem mass tagging.63 Much of the work to date has targeted vascular atherosclerotic disease, with a particular focus on immune cell phenotypes in murine aortic plaques.64 Under high-fat diet challenge, scRNA-seq of macrophages from Ldlr−/− mouse aortas and chow-fed controls revealed a population of Trem2-enriched osteoclast-like cells found almost exclusively in atherosclerotic aortas, and which is purported to play a role in lesion calcification.65 In a seminal work combining single-cell transcriptomic and proteomic approaches in human atherosclerotic plaques and peripheral blood mononuclear cells, Fernandez et al. exposed roles for distinct T cell and macrophage subpopulations in the progression of symptomatic atherosclerosis.66 As economies of scale make these techniques widely available in the future, they promise to fundamentally change the study of cardiovascular pathobiology and the way in which pharmacotherapies are targeted to diseased tissues.
Conclusions
The studies described herein have led to a new paradigm, which places EVs front-and-center in the initiation and pathogenesis of cardiovascular calcification. EVs are building blocks of microcalcification and actively participate in the process of mineral formation in the cardiovascular milieu. Osteogenic conditions drive activation of TNAP,30 which is then loaded into EVs through sortilin-mediated trafficking.15,27 The resultant EVs are capable of active mineralization upon release.5,15 When SMC and VIC-derived EVs are trapped by collagen fibers in the extracellular space, they aggregate due to tethering interactions of ANXA125 and nucleate hydroxyapatite to produce microcalcifications.12 While EVs are intimately implicated in the initiation and pathogenesis of cardiovascular calcification, they and their cargoes may also hold clues to treating this disease. We have pioneered enrichment and multi-omics methodologies to enable comprehensive analysis of calcifying EV cargoes produced by both in vitro cell culture models25,67 and fibrocalcific tissues61, with the goal of accelerating translational drug discovery for this unmet need.
The cardiovascular calcification field as a whole has progressed rapidly over the past decade thanks to the exciting efforts of many investigators across the world. This community’s widespread embrace of cutting-edge technological advances in imaging, molecular biology, omics, systems biology, computation, disease modeling, screening, and drug development has driven the investigation of cardiovascular calcification from that of straightforward histological characterization towards a deep and mechanistic understanding of the complex molecular bases of this disease. These studies bring us ever-closer to the promise of efficacious therapeutic interventions. In order to achieve this end goal, a number of fascinating questions remain to be answered: Do EVs modulate osteoblast/osteoclast cross-talk and control vascular tissue mineralization or, perhaps most excitingly, reversion of calcification? Can we find new means of analyzing heterogenous calcifying EV populations produced by differing routes of biogenesis from within a mineralizing plaque that contains multiple cell types? Will technologies with single-EV resolution of the miRNAome, proteome, and lipidome enable a clear understanding of the functional impact of the calcifying EV cargoes, or vesiculome, on recipient cells in the cardiovascular milieu? Such questions will need to be accompanied by further revolutions in imaging modalities that enable in vivo tracking of vesicles in order to prove mechanism/causality, identify origin and localization, or confirm therapeutic efficacy. In the end, the safe and tissue-specific inhibition/regression of cardiovascular mineralization or stabilization of vulnerable atherosclerotic plaques may rest on a deep mechanistic understanding of the physicochemical mechanisms of mineral formation, and an ability to adopt those EV-associated pathways for disruption of hydroxyapatite nucleation or delivery of therapeutics.
Highlights.
Ectopic cardiovascular calcification is a common comorbidity of atherosclerosis, aortic valve stenosis, diabetes, renal failure, and chronic inflammation, and contributes substantially towards mortality in these diseases. There are no pharmaceutical treatments able to inhibit or regress calcification.
Over the past decade, the discovery that calcifying extracellular vesicles are released by vascular and valvular cells, and that those vesicles act as both the physical constituents and active mineral-forming units of cardiovascular microcalcifications has revolutionized our understanding of this disease.
The field’s rapid progression and embrace of cutting-edge technological advances in imaging, molecular biology, omics, systems biology, computation, disease modeling, screening, and drug development are fueling mechanistic insights into EV-mediated cardiovascular calcification. These findings promise to underpin future development of successful pharmacotherapies.
Sources of Funding
This study was supported by NIH grants R01 HL114805, R01 HL119798, R01 HL136431, R01 HL141917, and R01 HL147095.
Abbreviations
- AAV
adeno-associated virus
- ANXA1
annexin A1
- CAVD
calcific aortic valve disease
- CT
computed tomography
- CyTOF
cytometry by time-of-flight; mass cytometry
- DDR1
discoidin domain receptor family, member 1 eQTL
- EV
extracellular vesicle
- GFP
green fluorescence protein
- GWAS
genome-wide association study
- LNP
lipid nanoparticle
- MISEV
Minimal Information for Studies of Extracellular Vesicles
- MRP14
migration inhibitory factor-related protein 14
- NIRF
near-infrared fluorescence
- PAR
poly(ADP-ribose)
- PARP
poly(ADP-ribose) polymerases
- PET
positron emission tomography
- RAGE
receptor for advanced glycation end products
- SMC
smooth muscle cell
- VIC
valvular interstitial cells
Footnotes
Disclosures
None.
References
- 1.Chen J, Budoff MJ, Reilly MP, et al. Coronary artery calcification and risk of cardiovascular disease and death among patients with chronic kidney disease. JAMA Cardiol. 2017;2:635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850 [DOI] [PubMed] [Google Scholar]
- 3.Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–386 [DOI] [PubMed] [Google Scholar]
- 4.Zaheer A, Lenkinski RE, Mahmood A, Jones AG, Cantley LC, Frangioni JV. In vivo near-infrared fluorescence imaging of osteoblastic activity. Nat Biotechnol. 2001;19:1148–1154 [DOI] [PubMed] [Google Scholar]
- 5.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage-derived matrix vesicles: An alternative novel mechanism for microcalcification in atherosclerotic plaques. Circulation research. 2013;113:72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawakami R, Katsuki S, Travers R, et al. S100a9-rage axis accelerates formation of macrophage-mediated extracellular vesicle microcalcification in diabetes mellitus. Arteriosclerosis, thrombosis, and vascular biology. 2020;40:1838–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdelbaky A, Corsini E, Figueroa AL, Fontanez S, Subramanian S, Ferencik M, Brady TJ, Hoffmann U, Tawakol A. Focal arterial inflammation precedes subsequent calcification in the same location: A longitudinal fdg-pet/ct study. Circulation. Cardiovascular imaging 2013;6:747–754 [DOI] [PubMed] [Google Scholar]
- 8.Dweck MR, Jones C, Joshi NV, et al. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation. 2012;125:76–86 [DOI] [PubMed] [Google Scholar]
- 9.Creager MD, Hohl T, Hutcheson JD, et al. (18)f-fluoride signal amplification identifies microcalcifications associated with atherosclerotic plaque instability in positron emission tomography/computed tomography images. Circulation. Cardiovascular imaging 2019;12:e007835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanimura A, McGregor DH, Anderson HC. Matrix vesicles in atherosclerotic calcification. Proc Soc Exp Biol Med. 1983;172:173–177 [DOI] [PubMed] [Google Scholar]
- 11.Kim KM. Calcification of matrix vesicles in human aortic valve and aortic media. Fed Proc. 1976;35:156–162 [PubMed] [Google Scholar]
- 12.Hutcheson JD, Goettsch C, Bertazzo S, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nature materials. 2016;15:335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertazzo S, Gentleman E, Cloyd KL, Chester AH, Yacoub MH, Stevens MM. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nature materials. 2013;12:576–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapustin AN, Chatrou ML, Drozdov I, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circulation research. 2015;116:1312–1323 [DOI] [PubMed] [Google Scholar]
- 15.Goettsch C, Hutcheson JD, Aikawa M, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. The Journal of clinical investigation. 2016;126:1323–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circulation research. 2011;109:e1–12 [DOI] [PubMed] [Google Scholar]
- 17.Rogers MA, Aikawa M, Aikawa E. Macrophage heterogeneity complicates reversal of calcification in cardiovascular tissues. Circulation research. 2017;121:5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croce K, Gao H, Wang Y, Mooroka T, Sakuma M, Shi C, Sukhova GK, Packard RR, Hogg N, Libby P, Simon DI. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120:427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM. Phosphate regulation of vascular smooth muscle cell calcification. Circulation research. 2000;87:E10–17 [DOI] [PubMed] [Google Scholar]
- 20.Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine wnt signals. The Journal of clinical investigation. 2005;115:1210–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sage AP, Lu J, Tintut Y, Demer LL. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011;79:414–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohler ER 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528 [DOI] [PubMed] [Google Scholar]
- 23.Torre M, Hwang DH, Padera RF, Mitchell RN, VanderLaan PA. Osseous and chondromatous metaplasia in calcific aortic valve stenosis. Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology. 2016;25:18–24 [DOI] [PubMed] [Google Scholar]
- 24.Krahn KN, Bouten CV, van Tuijl S, van Zandvoort MA, Merkx M. Fluorescently labeled collagen binding proteins allow specific visualization of collagen in tissues and live cell culture. Anal Biochem. 2006;350:177–185 [DOI] [PubMed] [Google Scholar]
- 25.Rogers MA, Buffolo F, Schlotter F, et al. Annexin a1-dependent tethering promotes extracellular vesicle aggregation revealed with single-extracellular vesicle analysis. Sci Adv. 2020;6:eabb1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapustin AN, Schoppet M, Schurgers LJ, Reynolds JL, McNair R, Heiss A, Jahnen-Dechent W, Hackeng TM, Schlieper G, Harrison P, Shanahan CM. Prothrombin loading of vascular smooth muscle cell-derived exosomes regulates coagulation and calcification. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:e22–e32 [DOI] [PubMed] [Google Scholar]
- 27.Itoh S, Mizuno K, Aikawa M, Aikawa E. Dimerization of sortilin regulates its trafficking to extracellular vesicles. The Journal of biological chemistry. 2018;293:4532–4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goettsch C, Hutcheson JD, Hagita S, Rogers MA, Creager MD, Pham T, Choi J, Mlynarchik AK, Pieper B, Kjolby M, Aikawa M, Aikawa E. A single injection of gain-of-function mutant pcsk9 adeno-associated virus vector induces cardiovascular calcification in mice with no genetic modification. Atherosclerosis. 2016;251:109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goettsch C, Iwata H, Hutcheson JD, O’Donnell CJ, Chapurlat R, Cook NR, Aikawa M, Szulc P, Aikawa E. Serum sortilin associates with aortic calcification and cardiovascular risk in men. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:1005–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers MA, Maldonado N, Hutcheson JD, Goettsch C, Goto S, Yamada I, Faits T, Sesaki H, Aikawa M, Aikawa E. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circulation research. 2017;121:220–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krohn JB, Hutcheson JD, Martinez-Martinez E, Irvin WS, Bouten CV, Bertazzo S, Bendeck MP, Aikawa E. Discoidin domain receptor-1 regulates calcific extracellular vesicle release in vascular smooth muscle cell fibrocalcific response via transforming growth factor-beta signaling. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lino M, Wan MH, Rocca AS, Ngai D, Shobeiri N, Hou G, Ge C, Franceschi RT, Bendeck MP. Diabetic vascular calcification mediated by the collagen receptor discoidin domain receptor 1 via the phosphoinositide 3-kinase/akt/runt-related transcription factor 2 signaling axis. Arteriosclerosis, thrombosis, and vascular biology. 2018;38:1878–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ngai D, Lino M, Rothenberg KE, Simmons CA, Fernandez-Gonzalez R, Bendeck MP. Ddr1 (discoidin domain receptor-1)-rhoa (ras homolog family member a) axis senses matrix stiffness to promote vascular calcification. Arteriosclerosis, thrombosis, and vascular biology. 2020;40:1763–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller KH, Hayward R, Rajan R, et al. Poly(adp-ribose) links the DNA damage response and biomineralization. Cell Rep. 2019;27:3124–3138 e3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White MP, Theodoris CV, Liu L, Collins WJ, Blue KW, Lee JH, Meng X, Robbins RC, Ivey KN, Srivastava D. Notch1 regulates matrix gla protein and calcification gene networks in human valve endothelium. Journal of molecular and cellular cardiology. 2015;84:13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, LaHaye S, Tao G, Lucchesi PA, Lincoln J, Lilly B, Garg V. Endothelial nitric oxide signaling regulates notch1 in aortic valve disease. Journal of molecular and cellular cardiology. 2013;60:27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blaser MC, Wei K, Adams RLE, Zhou YQ, Caruso LL, Mirzaei Z, Lam AY, Tam RKK, Zhang H, Heximer SP, Henkelman RM, Simmons CA. Deficiency of natriuretic peptide receptor 2 promotes bicuspid aortic valves, aortic valve disease, left ventricular dysfunction, and ascending aortic dilatations in mice. Circulation research. 2018;122:405–416 [DOI] [PubMed] [Google Scholar]
- 38.Rogers MA, Chen J, Nallamshetty S, Pham T, Goto S, Muehlschlegel JD, Libby P, Aikawa M, Aikawa E, Plutzky J. Retinoids repress human cardiovascular cell calcification with evidence for distinct selective retinoid modulator effects. Arteriosclerosis, thrombosis, and vascular biology. 2020;40:656–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrer MD, Ketteler M, Tur F, Tur E, Isern B, Salcedo C, Joubert PH, Behets GJ, Neven E, D’Haese PC, Perello J. Characterization of snf472 pharmacokinetics and efficacy in uremic and non-uremic rats models of cardiovascular calcification. PLoS One. 2018;13:e0197061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raggi P, Bellasi A, Bushinsky D, Bover J, Rodriguez M, Ketteler M, Sinha S, Salcedo C, Gillotti K, Padgett C, Garg R, Gold A, Perello J, Chertow GM. Slowing progression of cardiovascular calcification with snf472 in patients on hemodialysis: Results of a randomized phase 2b study. Circulation. 2020;141:728–739 [DOI] [PubMed] [Google Scholar]
- 41.Hutcheson JD, Blaser MC, Aikawa E. Giving calcification its due: Recognition of a diverse disease: A first attempt to standardize the field. Circulation research. 2017;120:270–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holmar J, Noels H, Bohm M, Bhargava S, Jankowski J, Orth-Alampour S. Development, establishment and validation of in vitro and ex vivo assays of vascular calcification. Biochem Biophys Res Commun. 2020;530:462–470 [DOI] [PubMed] [Google Scholar]
- 43.Gayrard N, Muyor K, Notarnicola C, Duranton F, Jover B, Argiles A. Optimisation of cell and ex vivo culture conditions to study vascular calcification. PLoS One. 2020;15:e0230201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogers MA, Aikawa E. Cardiovascular calcification: Artificial intelligence and big data accelerate mechanistic discovery. Nature reviews. Cardiology. 2019;16:261–274 [DOI] [PubMed] [Google Scholar]
- 45.Goto S, Rogers MA, Blaser MC, Higashi H, Lee LH, Schlotter F, Body SC, Aikawa M, Singh SA, Aikawa E. Standardization of human calcific aortic valve disease in vitro modeling reveals passage-dependent calcification. Frontiers in cardiovascular medicine. 2019;6:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blaser MC, Aikawa E. Roles and regulation of extracellular vesicles in cardiovascular mineral metabolism. Frontiers in cardiovascular medicine. 2018;5:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (misev2018): A position statement of the international society for extracellular vesicles and update of the misev2014 guidelines. Journal of extracellular vesicles. 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blaser MC, Aikawa E. Differential mirna loading underpins dual harmful and protective roles for extracellular vesicles in atherogenesis. Circulation research. 2019;124:467–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Donnell CJ, Kavousi M, Smith AV, et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation. 2011;124:2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. The New England journal of medicine. 2013;368:503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gosev I, Zeljko M, Duric Z, Nikolic I, Gosev M, Ivcevic S, Besic D, Legcevic Z, Paic F. Epigenome alterations in aortic valve stenosis and its related left ventricular hypertrophy. Clin Epigenetics. 2017;9:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rukov JL, Gravesen E, Mace ML, Hofman-Bang J, Vinther J, Andersen CB, Lewin E, Olgaard K. Effect of chronic uremia on the transcriptional profile of the calcified aorta analyzed by rna sequencing. Am J Physiol Renal Physiol. 2016;310:F477–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nanoudis S, Pikilidou M, Yavropoulou M, Zebekakis P. The role of micrornas in arterial stiffness and arterial calcification. An update and review of the literature. Front Genet. 2017;8:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mourino-Alvarez L, Iloro I, de la Cuesta F, et al. Maldi-imaging mass spectrometry: A step forward in the anatomopathological characterization of stenotic aortic valve tissue. Scientific reports. 2016;6:27106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bom MJ, Levin E, Driessen RS, et al. Predictive value of targeted proteomics for coronary plaque morphology in patients with suspected coronary artery disease. EBioMedicine. 2019;39:109–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vorkas PA, Shalhoub J, Isaac G, Want EJ, Nicholson JK, Holmes E, Davies AH. Metabolic phenotyping of atherosclerotic plaques reveals latent associations between free cholesterol and ceramide metabolism in atherogenesis. J Proteome Res. 2015;14:1389–1399 [DOI] [PubMed] [Google Scholar]
- 57.Perisic L, Aldi S, Sun Y, et al. Gene expression signatures, pathways and networks in carotid atherosclerosis. J Intern Med. 2016;279:293–308 [DOI] [PubMed] [Google Scholar]
- 58.Guauque-Olarte S, Messika-Zeitoun D, Droit A, et al. Calcium signaling pathway genes runx2 and cacna1c are associated with calcific aortic valve disease. Circulation. Cardiovascular genetics 2015;8:812–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mourino-Alvarez L, Baldan-Martin M, Gonzalez-Calero L, et al. Patients with calcific aortic stenosis exhibit systemic molecular evidence of ischemia, enhanced coagulation, oxidative stress and impaired cholesterol transport. International journal of cardiology. 2016;225:99–106 [DOI] [PubMed] [Google Scholar]
- 60.Schlotter F, Halu A, Goto S, et al. Spatiotemporal multi-omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation. 2018;138:377–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blaser MC, Buffolo F, Halu A, et al. Conserved and divergent modulation of calcification in atherosclerosis and aortic valve disease by tissue extracellular vesicles. bioRxiv. 202004.02.022525, Preprint [Google Scholar]
- 62.Matos TR, Liu H, Ritz J. Research techniques made simple: Experimental methodology for single-cell mass cytometry. J Invest Dermatol. 2017;137:e31–e38 [DOI] [PubMed] [Google Scholar]
- 63.Budnik B, Levy E, Harmange G, Slavov N. Scope-ms: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018;19:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winkels H, Ehinger E, Vassallo M, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell rna-sequencing and mass cytometry. Circulation research. 2018;122:1675–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A. Single-cell rna-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circulation research. 2018;122:1661–1674 [DOI] [PubMed] [Google Scholar]
- 66.Fernandez DM, Rahman AH, Fernandez NF, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hutcheson JD, Goettsch C, Pham T, Iwashita M, Aikawa M, Singh SA, Aikawa E. Enrichment of calcifying extracellular vesicles using density-based ultracentrifugation protocol. Journal of extracellular vesicles. 2014;3:25129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van der Valk D, van der Ven C, Blaser M, et al. Engineering a 3d-bioprinted model of human heart valve disease using nanoindentation-based biomechanics. Nanomaterials. 2018;8:296. [DOI] [PMC free article] [PubMed] [Google Scholar]