

Abstract
Background
The aim of the current study was to investigate and track the SARS-CoV-2 in Iranian Coronavirus Disease 2019 (COVID-19) patients using molecular and phylogenetic methods.
Methods
We enrolled seven confirmed cases of COVID-19 patients for the phylogenetic assessment of the SARS-CoV-2 in Iran. The nsp-2, nsp-12, and S genes were amplified using one-step RT-PCR and sequenced using Sanger sequencing method. Popular bioinformatics software were used for sequences alignment and analysis as well as phylogenetic construction.
Results
The mean age of the patients in the present study was 60.42 ± 9.94 years and 57.1% (4/7) were male. The results indicated high similarity between Iranian and Chinese strains. We could not find any particular polymorphisms in the assessed regions of the three genes. Phylogenetic trees by neighbor-joining and maximum likelihood method of nsp-2, nsp-12, and S genes showed that there are not any differences between Iranian isolates and those of other countries.
Conclusion
As a preliminary phylogenetic study in Iranian SARS-CoV-2 isolates, we found that these isolates are closely related to the Chinese and reference sequences. Also, no sensible differences were observed between Iranian isolates and those of other countries. Further investigations are recommended using more comprehensive methods and larger sample sizes.
Keywords: Phylogeny, COVID-19, Viral infection, Pandemic
Highlights
-
•
SARS-2 genome showed one genetic pattern.
-
•
Iran has the same sequence of SARS-2 like other countries isolates.
-
•
SNPs in nsp-2 did not show any polymorphisms between this study isolates and other countries.
1. Introduction
Coronaviruses are responsible for respiratory tract infections (RTIs). In the December of 2019, cases of pneumonia with unknown origin were reported in Wuhan, China (Bradburne et al., 1967; Hui et al., 2020; van der Hoek, 2007; van der Hoek et al., 2004; Woo et al., 2005; Zaki et al., 2012). The disease started spreading in the China first and then around world. Until March 20, 2020, this virus resulted in 234′073 confirmed cases and 9′840 deaths worldwide; the statistics are 18′407 confirmed cases and 1′284 deaths in Iran [7]77. This is while the number of studies on this novel coronavirus is growing all around the world (Biscayart et al., 2020; Bogoch et al., 2020; Lu et al., 2020).
Coronaviruses can infect the respiratory and intestinal epithelial cells and generates a wide range of the clinical presentations in humans (Wevers and van der Hoek, 2009). Coronaviruses are classified in Nidovirales order and Cornidoviridae suborder. Orthocoronavirinae subfamily in Coronaviridae family is divided into four genera. Severe acute respiratory syndrome-related coronavirus type 2 (SARS-CoV-2), and other SARS-like coronaviruses are classified in Sarbecovirs subgenus in Betacoronaviruses genera (Luk et al., 2019; van Boheemen et al., 2012). Endemic coronaviruses, such as HCoV-NL63, HCoV-229E, HCoV-OC43, and HKU1 can induce upper respiratory tract infections (UTRI) with mild respiratory manifestation in immune competent adults while infection with the epidemic coronaviruses, like SARS-CoV and Middle East respiratory syndrome-related coronavirus (MERS-CoV), could lead to the lower respiratory tract infections (LRTI) as well as serious respiratory manifestations (Forni et al., 2017; Su et al., 2016). In endemic areas for coronaviruses, it is suggested that the HCoV-229E and HCoV-OC43 are responsible for 15–29% of the common cold cases, while during the epidemic coronaviruses, there is another scenario at work (Monto, 1974). The SARS-CoV epidemic started from Guangdong, China, in 2002–2003 and resulted in 8′089 infected cases and a 9.6% mortality rate (Zhong et al., 2003). Also, the MERS-CoV in 2012 firstly started in Middle East and infected 2′465 cases demonstrating a high mortality (Azhar et al., 2019; Reusken et al., 2013). All the mentioned pandemics as well as the current pandemic condition with SARS-CoV-2 reflect the importance of following coronaviruses in human population. The aim of the current study was to investigate and track the SARS-CoV-2 in Iranian Coronavirus Disease 2019 (COVID-19) patients using molecular and phylogenetic methods.
2. Materials and methods
2.1. Cases
We enrolled seven cases between February–March 2020, with the clinical presentations including fever, cough, and radiologic features of the COVID-19 who referred to Firoozgar Hospital, Iran University of Medical Science, Tehran, Iran, after obtaining the written consent from all of the enrolled cases. The study was approved by the Ethical committee of the Iran University of Medical Science, Tehran, Iran (ethical code no. IR.IUMS.REC.1398.1344). The laboratory records including biochemical and hematological parameters, blood gases analyses, and cardiac markers were recorded. Also, all virological findings were obtained and used for further investigations.
2.2. SARS-CoV-2 pathogen detection
Respiratory samples, including the nasopharyngeal and throat swabs, were obtained from the enrolled patients. The SARS-CoV-2 primary detections were performed using the standard real-time PCR method explained below using Rotor-Gene-Q 6000 thermocycler (Corbett, Australia) according to the recommended protocol. After the SARS-CoV-2 was confirmed in cases, the samples were stored in −70 °C for further analysis.
Briefly, we used LightMix SarbecoV E-gene plus EAV control (Roche, Germany) by using 5.4 μl deionized DNase RNase free water, 0.5 μl primer probe mix, 4 μl Roche MasterMix, 0.1 μl RT emzyme and 10 μl extracted RNA or control. Heating protocol was 1 cycle at 55 °C 3 s, and 95 °C 30s; 45 cycles at 95 °C 3 s, and 60 °C 12 s; and a cycle at 40 °C 10s.
2.3. RT-PCR and sequencing
The RNA was extracted from the clinical samples using the Extraction of viral DNA/RNA kit (FAVORGEN Biotech Corporation, Taiwan) based on the manufactures' instructions. All the extracted RNAs were evaluated for the quality of the extraction via spectrophotometry method using NanoDrop ND-1000® (Thermo Fisher Scientific Inc., Waltham, MA, USA). PrimeScript One Step RT-PCR Kit (Takara Bio, Japan) was used for the synthesis of complementary DNA (cDNA) and PCR according to the manufactures' instructions.
We used the specific primers for amplifying the RdRp (RNA dependent RNA polymerase), the S protein, and nsp-2 (non-structural protein 2)(Chan et al., 2020b; Nao et al., 2020). Conventional PCR method was used for amplification of 344 bp region of RNA-dependent RNA polymerase (RdRp) gene; for the 158 bp region of Spike (S) protein; and a nested-PCR for amplification of 346 and 322 bp region of nsp-2 gene (Table 1 ).
Table 1.
Primers were used for amplification of RdRp, nsp-2 and S genes of SARS-CoV-2.
| Gene | Primers | Sequence 5′ → 3′ | Product size | |
|---|---|---|---|---|
| RdRp | – | Forward | CAAGTGGGGTAAGGCTAGACTTT | 344 bp |
| Reverse | ACTTAGGATAATCCCAACCCAT | |||
| Nsp-2 | Outer | Forward | CTCGAACTGCACCTCATGG | 346 bp |
| Reverse | CAGAAGTTGTTATCGACATAGC | |||
| Inner | Forward | ACCTCATGGTCATGTTATGG | 322 bp | |
| Reverse | GACATAGCGAGTGTATGCC | |||
| Spike | – | Forward | CCTACTAAATTAAATGATCTCTGCTTTACT | 158 bp |
| Reverse | CAAGCTATAACGCAGCCTGTA |
A total volume of 50 ul Master Mix was performed using 25ul 2× EnzymeMix (Takara Bio, Japan) for one-step RT PCR of RdRp, S and the first round of nsp-2 PCR, and 2× Super PCR Mastermix (Yekta Tajhiz Azma Co., Iran) for the second round of PCR for nsp-2 gene amplification, 1ul (10 pMol concentration) of each primers, 4 μl (0.2–0.5 μM concentration) of extracted specimens, and distilled RNase/DNase free water added to reach the rest of total volume.
A Bio-Rad (T100™ Thermal Cycler) was performed for heating program. Heating protocol, for RdRp, S, and the first round of nsp-2 PCR, was 1 step at 50 °C 40 min, 94 °C 5 min, 40 cycles at 94 °C 30s, 58 °C 30s, 72 °C 30s, and 1 step at 72 °C 5 min. The second round of PCR for nsp-2 gene included 94 °C 3 min, 94 °C 20s, 58 °C 20s, 72 °C 20s, and 72 °C 3 min. Visualization of PCR products was performed using 1.5% concentration of agarose gel into an electrophoresis system and stained using safe stain (YTA, Yekta Tajhiz Azma Co., Iran) behind UV translaminator.
2.4. Phylogenetic assessment
PCR products underwent purification using High Pure PCR Product Purification Kit (Roche Diagnostic GmbH, Mannheim, Germany) corresponding to the manufacturer's instructions. Also, an ABI 3730 XL sequencer was used for bidirectional sequencing. Raw data were trimmed and analyzed using popular bioinformatics software CLC workbench 5, and Basic Local Alignment Search Tool (BLAST). All sequences were submitted into GeneBank database (https://www.ncbi.nlm.nih.gov/genbank/) and released accession numbers were used to draw phylogenetic tree. SARS-CoV-2 reference sequence (NC_045512.2) and other coronavirus sequences were obtained from NCBI (https://blast.ncbi.nlm.nih.gov) database. Moreover, MEGA X software was used for multiple sequence alignment (MSA) and the phylogenetic trees were drown using 1000 replicate bootstrap method. In addition, the neighbor joining and maximum likelihood methods were carried out for similarity assessment and evolution analysis.
3. Results
The mean age ± SD of the patients in the present study was 60.42 ± 9.94 years and 57.1% of patients were male. The mean of the white blood cell count (WBC) and platelets count were 6.97 ± 4.01 × 1000 cells/mm3 and 181.4 ± 28.87 × 1000 Platelets/mm3, respectively. Furthermore, the differential WBC indicated that the polymorph nuclear leukocytes (PMN) and lymphocytes were 75 ± 11.8% and 18 ± 8.9%, respectively. The investigations of the inflammatory markers were assessed by qualitative C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). The CRP was positive in all patients and the ESR was 33.2 ± 20.4 mm per hour (mm/h). Also, the sequence analyses were performed on 3 out of 7 cases due to insufficient quality of the rest of the samples (Fig. 1 ).
Fig. 1.

Left: Primary Real-time PCR results of seven positive samples included for analysis. The yellow curve is positive control and the pink one is negative (NTC) control. Right: PCR products of specimen's candidate for sequencing. 346 and 322 bp bands are the first and the second round for nsp-2 amplifications; 462 bp band is internal control of one-step RT PCR kit (Takara Bio, Japan). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Obtained sequence was used for multiple sequence alignment (MSA) as illustrated in Fig. 2 . The MSA analysis could not show any particular polymorphism in the assessed regions of all three genes (only nsp-2 is illustrated in Fig. 2). Furthermore, the neighbor joining phylogenetic analysis showed the highest similarity between Iranian sequences for nsp-2 and the obtained sequence from Wuhan of Hubei province, China, as illustrated in Fig. 3 . Also, the neighbor joining phylogenetic analysis showed the most similarity for RdRp between Iranian sequences and those from China and USA (Fig. 4 ). Also, the same results were observed in the assessment of the S gene (Fig. 5 ). The phylogenetic assessment indicated that the bat coronaviruses and other SARS-related bat coronaviruses are highly similar to each other regarding their ancestors (Fig. 6 ).
Fig. 2.

MSA investigation for SNPs in nsp-2 did not show any polymorphisms, the red nucleotides show conservation between this study isolates and prototype or other countries isolated sequences. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.

Phylogenetic tree for 322 nucleotides of the nsp-2 gene of the SARS-CoV-2 using neighbor joining method and 1000 bootstrap, the red triangle shows the reference sequence for the SARS-CoV-2 and the blue triangle indicates Iranian isolates. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.

Phylogenetic tree for the 338 nucleotides of RdRp gene of the SARS-CoV-2 using neighbor joining method and 1000 bootstrap, the red triangle shows the reference sequence for the SARS-CoV-2 and the blue triangle indicates Iranian isolates. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 5.

Phylogenetic tree for 157 nucleotides of the S gene of the SARS-CoV-2 using neighbor joining method and 1000 bootstrap, the red triangle shows the reference sequence for the SARS-CoV-2 and the blue triangle indicates Iranian isolates. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 6.
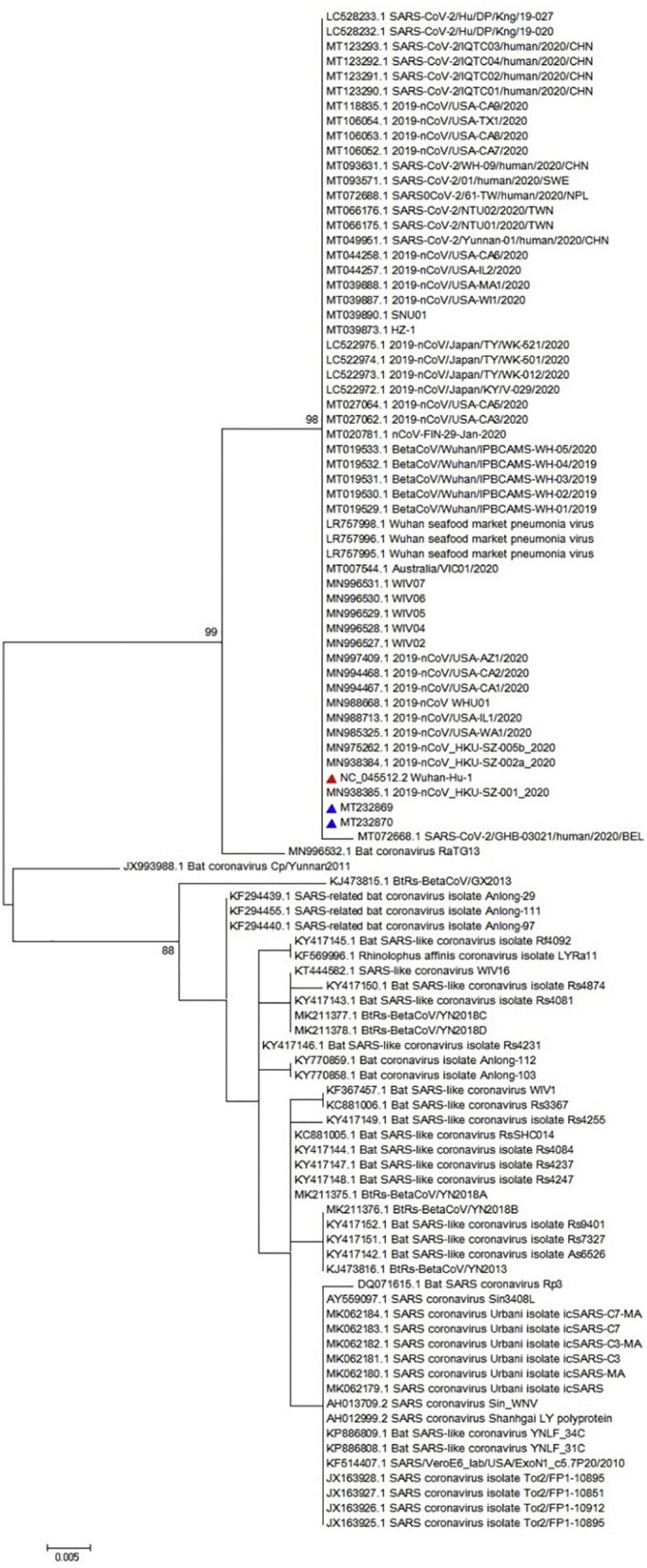
Phylogenetic tree for 338 nucleotide of the RdRp gene of the SARS-CoV-2 using maximum likelihood method and 1000 bootstrap, the red triangle shows the reference sequence for the SARS-CoV-2 and the blue triangle indicates Iranian isolates. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
The COVID-19 statistics show a high distribution of this disease in Iran and all around the word [7]77. The SARS-CoV-2 shows a great potential for the transmission due to its highly contagious nature (Zhu et al., 2020). Considering the importance of the public knowledge about the SARS-CoV-2 epidemiologic and its phylogenetic analysis, in the present preliminary study in Iran, we aimed to investigate and track the SARS-CoV-2 in Iranian COVID-19 patients using molecular and phylogenetic methods.
The neighbor joining phylogenetic analysis demonstrated the highest similarity between Iranian sequences for nsp-2 and the obtained sequence from Wuhan of Hubei province, China (NC_045512) as well as a familial cluster of SARS-CoV-2 in Hong Kong- Shenzhen Hospital, Shenzhen, Guangdong, China, whose direct relationship with the seafood market of Wuhan of Hubei province, China (MN975262) (Chan et al., 2020b) is confirmed, as illustrated in Fig. 3. Also, the neighbor joining phylogenetic analysis showed the most similarity for RdRp with obtained sequence from MN938384 (Chan et al., 2020b) and MN985325, which were the first obtained sequences from the USA (Fig. 4). Analysis of the S sequence showed a high similarity with NC_045512 and MN938384 sequences (Fig. 5). The maximum likelihood phylogenetic indicates that the Bat coronavirus RaTG13 (MN996532), Bat coronavirus Cp/Yunnan2011 (JX993988), and other SARS-related bat coronaviruses have the highest similarity revealing a close relationship among their ancestors considering the RdRp (Fig. 6).
As a major consideration, coronaviruses encode six main and variable numbers of accessory open reading frames (ORFs), and in case of the SARS-CoV-2, from 5′ to 3’ ORFs including ORF1a/b, spike (S) protein or surface glycoprotein, membrane (M), envelope (E) and nucleocapsid (N) main ORFs, and 3a, 6, 7a, 7b, 8, and 10 accessories (Chen et al., 2020; Hussain et al., 2005; Wu et al., 2020). The S glycoprotein is responsible for the attachment of the virus into the host cell. The S glycoprotein is cleaved to S1 and S2 in SARS-CoV and SARS-CoV-2 by host TMPRSS2 serine protease (Glowacka et al., 2011; Hoffmann et al., 2020). The S1 domain is vital for the virus attachment as receptor in host cell and the S2 domain is useful for the membrane fusion (Glowacka et al., 2011; Hoffmann et al., 2020). The SARS-CoV and SASR-CoV-2 use the Angiotensin Converting Enzyme 2 (ACE2) for the attachment (Liu et al., 2020). The conducted studies reported that the SARS-CoV-2 S protein shows 80% and 76% similarity with bat-SL-CoVZXC21 and SARS-CoV, respectively (Chan et al., 2020a). Regardless of S protein, the ORF 1a/b of the virus encodes 16 nonstructural proteins, which is known as nsp (nonstructural protein). The nsp-12 of the SARS-CoV-2 plays a role as RNA dependent RNA polymerase, while the nsp-2 function is not clear yet (Chan et al., 2020a). Our current phylogenetic study showed that the Iranian isolates have the most similarity with the Chinese isolates. The study conducted by Eden et al. (Eden et al., 2020) showed that the isolated sequences from the Australian and New Zealand patients who travelled from Iran showed a clade different from those of other circulating strains. They also suggested that three major substitutions in the coding and non-coding regions of the nsp-2 include G1397A, T28688C, and G29742T in this particular clade. The results of the current study did not show any differences or clades in the assessed sequences between Iranian strains and those obtained from other countries in the phylogenetic study of the RdRp, nsp-2, and S proteins. However, it should be noted that the Eden et al. (Eden et al., 2020) phylogenetic study was conducted using the whole genome of the virus in contrast to our study. The MSA assessment of the nsp-2 for the three suggested substitutions by Eden et al. could not be assessed due to differences between our sequencing regions. Furthermore, Bal et al. (Bal et al., 2020) could find three nucleotide variations in some of the assessed samples in France. They suggested that these variations might be associated with intra host evolution of the virus. In another study, conducted by Sardar et al (Sardar et al., 2020), it is suggested that some of the mutations in isolated viruses could be unique. For instance, a substation in S protein C24351T is unique in Indian isolates.
A major limitation of the current study was the limited region targeted for the nucleotide sequencing. Using high throughput methods and next generation sequencing (NGS) could result in a great advancement in the assessment of these variations (Xiao et al., 2020). These comprehensive methods can also further our knowledge on Iranian SARS-CoV-2 isolates.
5. Conclusion
In conclusion, the results of the present study indicated that the SARS-CoV-2 sequences from Iran have great similarity with Chinese and reference sequences although limited regions were sequenced and no variations were observed among them. To sum up, we found no identified differences between Iranian isolates and those from other regions. The current investigation was conducted as a preliminary study in Iran; further studies are to be performed to increase our knowledge regarding SARS-CoV-2 isolates.
Declaration of Competing Interest
There is no conflict of interests between authors in the present study.
Acknowledgments
Acknowledgement
This study was supported by a grant (code no. 98-5-30-17541) from Iran University of Medical Sciences, Tehran, Iran.
References
- Azhar E.I., Hui D.S.C., Memish Z.A., Drosten C., Zumla A. The Middle East respiratory syndrome (MERS) Infect. Dis. Clin. N. Am. 2019;33:891–905. doi: 10.1016/j.idc.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal A., Destras G., Gaymard A., Bouscambert-Duchamp M., Valette M., Escuret V., Frobert E., Billaud G., Trouillet-Assant S., Cheynet V., Brengel-Pesce K., Morfin F., Lina B., Josset L. Molecular characterization of SARS-CoV-2 in the first COVID-19 cluster in France reveals an amino-acid deletion in nsp2 (Asp268Del) Clin. Microbiol. Infect. 2020 doi: 10.1016/j.cmi.2020.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscayart C., Angeleri P., Lloveras S., Chaves T.D.S.S., Schlagenhauf P., Rodríguez-Morales A.J. The next big threat to global health? 2019 novel coronavirus (2019-nCoV): what advice can we give to travellers? – interim recommendations January 2020, from the Latin-American society for travel medicine (SLAMVI) Travel Med. Infect. Dis. 2020;33 doi: 10.1016/j.tmaid.2020.101567. 101567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoch I.I., Watts A., Thomas-Bachli A., Huber C., Kraemer M.U., Khan K. Pneumonia of unknown etiology in wuhan, china: potential for international spread via commercial air travel. Journal of Travel Medicine. 2020 doi: 10.1093/jtm/taaa008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boheemen S., de Graaf M., Lauber C., Bestebroer T.M., Raj V.S., Zaki A.M., Osterhaus A.D., Haagmans B.L., Gorbalenya A.E., Snijder E.J. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio. 2012;3 doi: 10.1128/mBio.00473-12. e00473–00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradburne A., Bynoe M., Tyrrell D. Effects of a“ new” human respiratory virus in volunteers. Br. Med. J. 1967;3:767. doi: 10.1136/bmj.3.5568.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F.-W., Kok K.-H., Zhu Z., Chu H., To, K.K.-W, Yuan S., Yuen K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerging Microbes & Infections. 2020;9:221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F.-W., Yuan S., Kok K.-H., To, K.K.-W, Chu H., Yang J., Xing F., Liu J., Yip C.C.-Y., Poon R.W.-S. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Liu Q., Guo D. Coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92(4):418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden J.-S., Rockett R., Carter I., Rahman H., de Ligt J., Hadfield J., Storey M., Ren X., Tulloch R., Basile K. An emergent clade of SARS-CoV-2 linked to returned travellers from Iran. bioRxiv. 2020;6(1) doi: 10.1093/ve/veaa027. veaa027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forni D., Cagliani R., Clerici M., Sironi M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017;25:35–48. doi: 10.1016/j.tim.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowacka I., Bertram S., Müller M.A., Allen P., Soilleux E., Pfefferle S., Steffen I., Tsegaye T.S., He Y., Gnirss K. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011;85:4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L. Human coronaviruses: what do they cause? Antivir. Ther. 2007;12:651. [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J., Wolthers K.C., Wertheim-van Dillen P.M., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nat. Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020 doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui D., Madani T., Ntoumi F., Kock R., Dar O., Ippolito G., Mchugh T., Memish Z., Drosten C., Zumla A. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health-the latest 2019 novel coronavirus outbreak in Wuhan, China. International journal of infectious diseases: IJID: official publication of the International Society for Infectious Diseases. 2020;91:264. doi: 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S., Chen Y., Yang Y., Xu J., Peng Y., Wu Y., Li Z., Zhu Y., Tien P., Guo D. Identification of novel subgenomic RNAs and noncanonical transcription initiation signals of severe acute respiratory syndrome coronavirus. J. Virol. 2005;79:5288–5295. doi: 10.1128/JVI.79.9.5288-5295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Xiao X., Wei X., Li J., Yang J., Tan H., Zhu J., Zhang Q., Wu J., Liu L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020;92(6):595–601. doi: 10.1002/jmv.25726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Stratton C.W., Tang Y.W. Outbreak of pneumonia of unknown Etiology in Wuhan China: the Mystery and the Miracle. J. Med. Virol. 2020 doi: 10.1002/jmv.25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk H.K., Li X., Fung J., Lau S.K., Woo P.C. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infection, Genetics and Evolution. 2019 doi: 10.1016/j.meegid.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monto A.S. Medical reviews. Coronaviruses. The Yale journal of biology and medicine. 1974;47:234. [PMC free article] [PubMed] [Google Scholar]
- Nao N., Shirato K., Katano H., Matsuyama S., Takeda M. 2020. Detection of Second Case of 2019-nCoV Infection in Japan (Corrected Version) [Google Scholar]
- Reusken C.B., Haagmans B.L., Müller M.A., Gutierrez C., Godeke G.-J., Meyer B., Muth D., Raj V.S., Smits-De Vries L., Corman V.M. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect. Dis. 2013;13:859–866. doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar R., Satish D., Birla S., Gupta D. Comparative analyses of SAR-CoV2 genomes from different geographical locations and other coronavirus family genomes reveals unique features potentially consequential to host-virus interaction and pathogenesis. bioRxiv. 2020 doi: 10.1016/j.heliyon.2020.e04658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S., Wong G., Shi W., Liu J., Lai A.C., Zhou J., Liu W., Bi Y., Gao G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wevers B.A., van der Hoek L. Recently discovered human coronaviruses. Clin. Lab. Med. 2009;29:715–724. doi: 10.1016/j.cll.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Chu C.-M., Chan K.-H., Tsoi H.-W., Huang Y., Wong B.H., Poon R.W., Cai J.J., Luk W.-K. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A., Peng Y., Huang B., Ding X., Wang X., Niu P., Meng J., Zhu Z., Zhang Z., Wang J. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe. 2020 doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, M., Liu, X., Ji, J., Li, M., Li, J., Yang, L., Sun, W., Ren, P., Yang, G., Zhao, J., 2020. Multiple approaches for massively parallel sequencing of HCoV-19 genomes directly from clinical samples. bioRxiv. [DOI] [PMC free article] [PubMed]
- Zaki A.M., Van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zhong N., Zheng B., Li Y., Poon L., Xie Z., Chan K., Li P., Tan S., Chang Q., Xie J. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet. 2003;362:1353–1358. doi: 10.1016/S0140-6736(03)14630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]