

Abstract
Growing evidence suggests that T cells may play a critical role in combating severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Hence, COVID-19 vaccines that can elicit a robust T cell response may be particularly important. The design, development and experimental evaluation of such vaccines is aided by an understanding of the landscape of T cell epitopes of SARS-CoV-2, which is largely unknown. Due to the challenges of identifying epitopes experimentally, many studies have proposed the use of in silico methods. Here, we present a review of the in silico methods that have been used for the prediction of SARS-CoV-2 T cell epitopes. These methods employ a diverse set of technical approaches, often rooted in machine learning. A performance comparison is provided based on the ability to identify a specific set of immunogenic epitopes that have been determined experimentally to be targeted by T cells in convalescent COVID-19 patients, shedding light on the relative performance merits of the different approaches adopted by the in silico studies. The review also puts forward perspectives for future research directions.
Keywords: Coronavirus, COVID-19, Computational prediction, SARS-CoV, Peptide-HLA binding, Immunogenicity, Allergenicity, Toxicity, Reverse vaccinology, Immunoinformatics
Abbreviations: ANN, artificial neural network; CoV, coronavirus; COVID-19, coronavirus disease 2019; CTL, cytotoxic T lymphocytes; E, envelope; GISAID, global initiative on sharing avian influenza database; HIV, human immunodeficiency virus; HLA, human leukocyte antigen; ICS, intracellular cytokine staining; IEDB, immune epitope database and analysis resource; IFN-γ, interferon-gamma; M, membrane; ML, Machine learning; MERS, middle east respiratory syndrome; MHC, major histocompatibility complex; N, nucleocapsid; ORF, open reading frame; PBMCs, peripheral blood mononuclear cells; S, spike; SARS, severe acute respiratory syndrome; TCR, T cell receptor; ViPR, virus pathogen database and analysis resource
Graphical abstract

1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of the ongoing COVID-19 pandemic, has resulted in major loss of human life worldwide. At present, prior to the mass distribution of functioning vaccines, governments have mainly resorted to traditional public health measures to control the spread of COVID-19, such as social distancing, quarantine, mask mandates, and community containment. The quest for an effective COVID-19 functioning vaccine has been pursued at an unprecedented pace, with two vaccines already being authorized for emergency use by the U.S. Food and Drug Administration (FDA) [1] (in less than a year), and more than 300 vaccine candidates currently under trial [2].
Vaccines can stimulate both cellular and humoral responses, mediated by T cells and antibodies respectively. Thus far, the majority of COVID-19 vaccine candidates (including the approved ones) are based on the spike (S) protein [2], which is the major target of neutralizing antibodies [[3], [4], [5]]. However, there is growing evidence to suggest that stimulating broad T cell responses via vaccines may also be important [6,7]. This is supported by SARS-CoV-2 immunological studies reporting robust T cell responses to be correlated with favourable disease outcome [[8], [9], [10]] and being potentially long-lasting [8]. Emerging data also suggests the coordinated response of neutralizing antibodies and T cells to be protective against COVID-19 [2,11]. Systematic longitudinal SARS-CoV-2 immunological studies on larger cohorts will help to further corroborate these findings, but nevertheless, they support the notion that T cells, may play a prominent role in conferring protection against COVID-19.
There is a large diversity of SARS-CoV-2 vaccines currently under trial and these have been developed using both conventional and modern approaches [12]. Vaccines developed using conventional approaches, based on using a live-attenuated or inactivated virus, have been effective in preventing infectious diseases caused by viruses such as polio, measles, rubella, chickenpox, and many others. These approaches, however, have not produced successful vaccines against other lethal viruses such as human immunodeficiency virus (HIV), hepatitis C virus, and dengue virus. Modern vaccine technologies (e.g., peptide, subunit, DNA/RNA) provide promising alternatives. In fact, for the case of SARS-CoV-2, the majority of vaccines in the advanced phase II or III of clinical trials are based on modern vaccine approaches [2]. These approaches introduce specific parts of the virus or their genes into the body to produce a targeted immune response. As a natural infection pathway is not followed, they can suffer from weak immunogenicity, though immunogenicity may be boosted by augmenting with appropriately developed adjuvants [13,14].
Like for other viruses, T cells get activated by recognizing SARS-CoV-2 peptides (short linear amino acid sequences) presented on the surface of infected cells via human leukocyte antigen (HLA) molecules (Fig. 1 ). The goal of a COVID-19 vaccine is to mimic this procedure by stimulating immune cells that specifically recognize SARS-CoV-2, thereby preparing the immune system to combat the virus upon natural infection. A common challenge faced by modern vaccine approaches is to identify specific SARS-CoV-2 peptides that are capable of inducing a robust and protective T cell immune response. This is an important task in the overall vaccine development pipeline as it provides the basic recommendations for all downstream efforts to be subsequently pursued in vaccine synthesis, laboratory testing, and clinical trials. Experimentally identifying SARS-CoV-2 peptides that elicit T cell responses is difficult, due in part to the enormous number of possible choices to test, and to the high genetic variability of major histocompatibility complex (MHC) genes that code for HLA molecules.
Fig. 1.

Schematic illustration of T cell responses against SARS-CoV-2 and T cell epitope prediction using in silico approaches. (A) Viral peptides, derived from SARS-CoV-2 proteins after multiple intra-cellular processing steps, are presented on the surface of infected cells and antigen presenting cells via HLA class I and class II molecules, respectively. Naïve T cells, specialized in distinguishing foreign-peptides from self-peptides via training in the thymus, scan these peptide-HLA complexes to determine if the peptides belong to a foreign microbe. Recognition of a foreign-peptide leads to activation, proliferation, and differentiation of naïve T cells into effector cells. There are two main types of effector T cells: CD8+ T cells (or cytotoxic T lymphocytes; CTLs) that get activated by viral peptides bound to HLA class I molecules and help in killing the SARS-CoV-2 infected cells (right panel), while CD4+ T cells (or helper T lymphocytes) get activated by peptides bound to HLA class II molecules and help in further enhancing SARS-CoV-2-specific CD8+ T cell and antibody responses (left panel). These adaptive immune cells, activated by peptide-HLA complexes, can collectively mount a potent immune response against SARS-CoV-2. (B) In silico approaches analyze SARS-CoV-2 protein sequences to predict a number of potential HLA-I and HLA-II epitopes that can be used to guide experiments to characterize T cell responses in COVID-19 patients and to inform SARS-CoV-2 vaccine design.
While each person has 12 unique types of HLA alleles, currently more than 27,000 known HLA alleles are listed in the immune polymorphism database [15], and these vary in their peptide binding specificities. With the availability of a large amount of data related to peptide-HLA binding, numerous attempts to solve the problem of T cell epitope identification (i.e., predicting peptides capable of eliciting T cell response) have been proposed that leverage this data through in silico methods [[16], [17], [18], [19]]. For SARS-CoV-2, very soon after the first genetic sequences were made available in January 2020, in silico methods began to be employed to predict and recommend T cell epitopes as potential targets for a SARS-CoV-2 vaccine (Fig. 1). In addition to guiding vaccine development, many of these predictions have been helpful in informing experimental studies directed towards understanding immune responses naturally elicited in convalescent COVID-19 patients (Fig. 1).
This review discusses the rationale and features of the in silico methods and tools that have been employed so far for SARS-CoV-2 T cell epitope prediction. As we describe, a diverse set of computational techniques have been employed, often exploiting machine learning approaches, and in some cases exploiting the expected cross-reactivity of epitopes between genetically similar viruses. These in silico methods and tools have often been developed independently and in many cases have been trained using datasets related to other viruses or other microbes, thereby making it difficult to understand the relative performance of the epitope predictions for SARS-CoV-2. To help shed light on these questions, this review presents a comparison of the predictions of 61 SARS-CoV-2 in silico studies, revealing commonalities and differences among the specific SARS-CoV-2 epitopes predicted by different methods. We also assess and compare the predictions when applied to emerging data from nine experimental studies that have identified SARS-CoV-2 T cell epitopes targeted in convalescent COVID-19 patients. Insights into the current state of SARS-CoV-2 T cell epitope prediction are also put forward, together with perspectives on future research directions and opportunities.
2. In silico methods used for SARS-CoV-2 T cell epitope prediction
We queried PUBMED on 8 September 2020 using the search terms “T cell, covid-19, epitopes, computational, and in silico”, which produced a list of 40 publications. After excluding those that did not report SARS-CoV-2 epitopes, this list was reduced to 31 publications (entries 1 to 31 in Table 1 ). Using the same search terms in Google Scholar on 8 September 2020, we gathered an additional 34 publications, giving a total of 65 SARS-CoV-2 in silico epitope prediction studies (Table 1). These studies can be broadly grouped into two classes based on their rationale for epitope prediction: those that predict SARS-CoV-2 epitopes using SARS-CoV immunological data by exploiting the genetic similarity between the two viruses (Ahmed et al. [20], Lee et al. [21], Grifoni et al. [22], and Ranga et al. [23]), and those that apply peptide-HLA binding prediction methods (the remaining 61 studies). We review and discuss each of these approaches in the following.
Table 1.
List of reviewed in silico SARS-CoV-2 T cell epitope prediction studies.
| No. | Study labela | HLA-I epitope predictionb | HLA-II epitope predictionc | Immunogenicityd | IFN-γ productione | Conservation | Allergenicityf | Toxicityg | Autoimmunity | Vaccine construct |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ahmed2020 [20] | Using SARS-CoV immunological data | Using SARS-CoV immunological data | - | - | Y | - | - | - | - |
| 2 | Grifoni2020h [22] | Using SARS-CoV immunological data, NetMHCpan-4.0 | Using SARS-CoV immunological data, Tepitool | - | - | - | - | - | - | - |
| 3 | Ranga2020 [23] | Using SARS-CoV immunological data, NetCTL-1.2 | - | - | - | - | - | - | - | - |
| 4 | Lee2020h [21] | Using SARS-CoV immunological data, NetMHCpan-4.0 | Using SARS-CoV immunological data | iPred | - | - | - | - | - | - |
| 5 | Baruah2020 [112] | NetCTL-1.2, NetChop, CTLPred | - | - | IFNepitope | - | - | - | - | - |
| 6 | Crooke2020 [96] | NetCTL-1.2, NetMHCpan-4.0 | NetMHCIIpan-3.2 | Vaxijen-2.0 | - | - | AllerCatPro | ToxinPred | - | - |
| 7 | Ojha2020 [113] | NetCTL-1.2 | IEDB (method NSi) | - | - | - | - | - | - | Y |
| 8 | Wang2020 [114] | NetMHCpan-4.0 | IEDB (recommended) | Vaxijen-2.0 | - | - | - | - | Y | - |
| 9 | Poran2020 [115] | HLAthena | NeonMHC2 | Response against few predicted epitopes tested in recovered patients | - | - | - | - | Y | - |
| 10 | UlQamar2020 [116] | IEDB (consensus) | IEDB (consensus) | Vaxijen-2.0 | - | - | AllerTOP-2.0 | NS | - | Y |
| 11 | Gupta2020vr [117] | NetCTLpan-1.1 | IEDB (recommended) | Vaxijen-2.0 | IFNepitope | - | AllerTOP-2.0 | ToxinPred | - | - |
| 12 | Enayatkhani2020 [118] | RANKPEP | RANKPEP | - | - | AllerTOP-2.0 | - | - | Y | |
| 13 | Ong2020 [119] | Vaxign, IEDB (consensus) | Vaxign, IEDB (consensus) | - | - | - | - | - | Y | - |
| 14 | Abdelmageed 2020 [120] | IEDB (consensus) | IEDB (recommended) | - | - | - | - | - | - | - |
| 15 | Mukherjee2020 [121] | Tepitool, NetMHCpan-4.0, nHLAPred, CTLPred | Tepitool | Vaxijen-2.0 | - | Y | AllerTOP-2.0, AlgPred | ToxinPred | Y | - |
| 16 | Vashi2020 [122] | IEDB (method NS) | IEDB (method NS) | - | - | Y | - | - | - | - |
| 17 | Ahmad2020 [123] | MHCPred | MHCPred | Vaxijen-2.0 | - | - | AllerTOP-2.0 | - | Y | Y |
| 18 | Naz2020 [124] | Tepitool | IEDB (recommended) | Vaxijen-2.0, Calis et al. |
- | - | AllerTOP-2.0 | - | - | Y |
| 19 | Chen2020 [125] | NetMHCpan-4.0 | IEDB (recommended) | Vaxijen-2.0, Calis et al. |
- | - | AllerTOP-2.0 | ToxinPred | - | Y |
| 20 | Martin2020 [126] | NetCTL-1.2 | IEDB (recommended) | Vaxijen-2.0 | IFNepitope | - | AllerTOP-2.0 | ToxinPred | - | Y |
| 21 | Dong2020 [127] | NetCTL-1.2 | IEDB (consensus) | - | IFNepitope | - | - | - | - | Y |
| 22 | Ghafouri2020 [128] | IEDB (method NS) | IEDB (method NS) | Vaxijen-2.0 | - | - | AllerTOP-2.0 | ToxinPred | - | Y |
| 23 | Banerjee2020 [129] | NetCTL-1.2 | NetMHCII-2.3 | - | - | - | - | - | - | Y |
| 24 | Samad2020 [130] | NetCTL-1.2 | IEDB (consensus) | Vaxijen-2.0, Calis et al. |
IFNepitope | - | AllerTOP-2.0 | ToxinPred | - | Y |
| 25 | Bhatnager2020 [131] | NetMHCpan-4.0, CTLPred | IEDB (recommended 2.2) | IFNepitope | - | AlgPred, AllergenFP | NS | - | Y | |
| 26 | Devi2020 [132] | NetCTL-1.2 | IEDB (consensus) | Vaxijen-2.0, | IFNepitope | - | AllerTOP-2.0 | ToxinPred | - | Y |
| 27 | AbrahamPeele2020 [133] | NetCTL-1.2 | IEDB (method NS) | Vaxijen-2.0, Calis et al. |
IFNepitope | - | AllerTOP-2.0 | ToxinPred | - | Y |
| 28 | Ismail2020 [134] | NetMHC-4.0, , MHCPred | IEDB (consensus), MHCPred | Vaxijen-2.0 | IFNepitope | - | AllerTOP-2.0 | ToxinPred | Y | Y |
| 29 | Jakhar2020 [135] | NetCTL-1.2, IEDB (method NS) | NetMHCIIpan-3.0 | Vaxijen-2.0, Calis et al. |
IFNepitope | Y | - | ToxinPred | - | Y |
| 30 | Panda2020 [136] | NetCTL-1.2 | - | Vaxijen-2.0 | - | - | - | - | - | - |
| 31 | Campbell2020 [137] | pVACtools | pVACtools | - | - | - | - | - | - | - |
| 32 | Tilocca2020 [138] | IEDB (method NS) | IEDB (method NS) | - | - | - | - | - | - | - |
| 33 | Santoni2020 [139] | NetMHC-4.0, NetCTL-1.2 | - | - | - | - | - | - | Y | - |
| 34 | Dijkstra2020 [140] | NetMHC | - | - | - | - | - | - | - | - |
| 35 | Prachar2020 [83] | NetMHC-4.0 | NetMHCII-2.3 | - | - | - | - | - | - | - |
| 36 | Ramaiah2020 [141] | - | IEDB (consensus) | - | - | - | - | - | - | - |
| 37 | Gupta2020 [142] | NetMHCpan-4.0 | Sturniolo method | Vaxijen-2.0, Calis et al. |
AllerTOP-2.0 | ToxinPred | ||||
| 38 | Srivastava2020 [143] | IEDB (consensus) | SMM-align, Sturniolo method | - | IFNepitope | - | ToxinPred | - | Y | |
| 39 | Mitra2020 [144] | NetMHC-4.0, NetCTL-1.2, IEDB (consensus) | MHCPred, NetMHCIIpan-3.2, IEDB (consensus) | Vaxijen-2.0 | IFNepitope | - | AllerTOP, AlgPred | ToxinPred | Y | Y |
| 40 | Singh2020 [145] | NetCTL-1.2, IEDB (consensus) | NetMHCIIpan-3.2 | Vaxijen-2.0 | IFNepitope | - | AllerTOP-2.0 | Y | Y | |
| 41 | Saha2020 [146] | ProPred1 | ProPred | Vaxijen-2.0 | - | - | - | - | - | Y |
| 42 | Nerli2020 [147] | NetMHCpan-4.0 | - | Electrostatic surface potential | - | - | - | - | - | - |
| 43 | Liu2020 [108] | NetMHCpan-4.0, MHCflurry | NetMHCIIpan-4.0 | - | - | - | - | - | Y | - |
| 44 | Khan2020 [148] | NetCTL-1.2 | PREDIVAC | Calis et al. | - | - | AlgPred | ToxinPred | Y | - |
| 45 | Banerjee2020a [149] | - | IEDB (method NS) | Vaxijen-2.0 | - | - | - | - | - | - |
| 46 | Bojin2020 [150] | IEDB (method NS) | IEDB (method NS) | - | - | - | - | - | - | - |
| 47 | NazneenAkhand 2020 [151] | IEDB (method NS) | IEDB (method NS) | Vaxijen-2.0 | IFNepitope | - | AllergenFP, AllerTOP | ToxinPred | - | Y |
| 48 | Feng2020 [152] | NetMHCpan, iNeo-Pred | - | NS | - | - | - | NS | Y | - |
| 49 | Bhattacharya2020 [153] | ProPred1 | ProPred | Vaxijen-2.0 | - | - | - | - | - | Y |
| 50 | Chauhan2020 [154] | NetCTL-1.2, | IEDB (consensus), NetMHCIIpan-3.2 | Vaxijen-2.0 | IFNepitope | - | AlgPred, AllerTOP-2.0 | - | - | Y |
| 51 | Fast2020 [155] | NetMHCpan-4.0 | MARIA | - | - | - | - | - | - | - |
| 52 | Joshi2020 [156] | NetMHC-4.0, MHCPred | NetMHCIIpan-3.2, MHCPred | Vaxijen-2.0 | - | - | - | ToxinPred | - | - |
| 53 | Kar2020 [157] | NetCTL-1.2, IEDB (consensus) | NetMHCIIpan-3.2 | Vaxijen-2.0, Calis et al. |
IFNepitope | - | AllerTOP-2.0, AllergenFP | - | - | Y |
| 54 | Qamar2020 [158] | IEDB (consensus) | IEDB (consensus) | Vaxijen-2.0 | - | Y | AllergenFP | ToxinPred | - | - |
| 55 | Ahammad2020 [159] | NetCTL-1.2 | IEDB (consensus) | Vaxijen-2.0, Calis et al. |
IFNepitope | - | AllerTOP-2.0 | ToxinPred | Y | Y |
| 56 | Kiyotani2020 [160] | NetMHCpan-4.0, NetMHC-4.0 | NetMHCIIpan-3.1 | - | - | - | - | - | - | - |
| 57 | Sarkar2020 [161] | NetMHCpan-4.0 | IEDB (Sturniolo) | Vaxijen-2.0 | - | - | AllerTOP-2.0, AllergenFP | ToxinPred | Y | Y |
| 58 | Romero-Lopez2020 [162] | Tepitool | IEDB (recommended 2.2) | Calis et al. | - | - | - | - | - | - |
| 59 | Sanami2020 [163] | ProPred1 | ProPred | Vaxijen-2.0 | IFNepitope | - | AllerTOP-2.0 | ToxinPred | Y | Y |
| 60 | Kalita2020 [164] | NetCTL-1.2, IEDB (method NS) | NetMHCIIpan-3.0 | - | IFNepitope | - | - | ToxinPred | - | Y |
| 61 | Rahman2020 [165] | SMM | SMM-align | - | IFNepitope | - | - | - | Y | |
| 62 | Lin2020 [166] | IEDB (consensus) | IEDB (method NS) | Vaxijen-2.0 | - | - | AllergenFP | ToxinPred | - | - |
| 63 | Yarmarkovich2020 [167] | NetMHC-4.0 | NetMHCII-2.3 | - | - | - | - | - | Y | - |
| 64 | Yazdani2020 [168] | NetMHC-4.0, CTLPred | IEDB (consensus), RANKPEP | Vaxijen-2.0 | IFNepitope | - | AllergenFP | - | - | Y |
| 65 | Lucchese2020 [169] | Identified pentamers from the proteome | - | - | - | - | - | - | Y | - |
Studies that used SARS-CoV immunological data for prediction [[1], [2], [3], [4]] are shown in bold font, while all other studies that used peptide-HLA binding prediction methods are in regular font. In silico studies [[1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30]] were obtained from PubMed, while the remaining studies [[31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [62], [63], [64], [65]] were obtained by searching Google Scholar.
HLA-I epitope prediction: ProPred1 [54], SMM [58,59], CTLPred [51], Tepitool [66], nHLAPred [67], NetMHC-4.0 [40], NetChop [48], NetCTL-1.2 [49], NetMHCpan [37], HLAthena [17], MHCflurry [41], NetMHCpan-4.0 [16], iNeo-Pred [152], Vaxign [57], RANKPEP [52], pVACtools [65], NetMHC [36], NetCTLpan-1.1 [39], MHCPred [62], IEDB (consensus) [63].
HLA-II epitope prediction: MHCPred [62], NeonMHC2 [19], NetMHCII-2.3 [42], NetMHCIIpan-3.0 [43], NetMHCIIpan-3.1 [44], NetMHCIIpan-3.2 [42], NetMHCIIpan-4.0 [18], SMM-align [60], RANKPEP [52], Sturniolo method [61], ProPred [55], pVACtools [65], PREDIVAC [56], IEDB (recommended) [43], MARIA [45], IEDB (consensus) [64], Vaxign [57], Tepitool [66], IEDB (recommended 2.2) [43].
Immunogenicity: Vaxijen-2.0 [79], Calis et al. [80], iPred [81]; Electrostatic surface potential [147].
IFN-γ production: IFNepitope [85].
Toxicity: ToxinPred [98].
Lee2020 [21] and Grifoni2020 [22] reported two sets of epitopes: one set based on using SARS-CoV immunological data and the other using NetMHCpan-4.0. For the purpose of this analysis, we have only considered the set of epitopes predicted using SARS-CoV immunological data.
NS: Not specified.
2.1. Methods that exploit immunological data of SARS-CoV
Compared to other human coronaviruses, early studies using phylogenetic analysis [24,25] suggested SARS-CoV-2 to be most similar to SARS-CoV, the virus that caused the 2003 SARS outbreak. In fact, the genetic similarity of SARS-CoV-2 and SARS-CoV was found to be quite high in the structural proteins (~76% in S and >90% in N, M, and E proteins) [20], which were known to induce robust and long-lasting T cell immunity against SARS-CoV [26]. Motivated by the high genetic similarity of SARS-CoV with SARS-CoV-2, multiple in silico studies [[20], [21], [22], [23]] used the information of T cell epitopes available from previous SARS-CoV immunological studies to predict likely targets of SARS-CoV-2 T cell responses (Table 1). This approach is well motivated, with cross-reactive T cell epitopes being reported previously for genetically similar viruses [27], including zika virus, dengue virus, and other flaviviruses [[28], [29], [30], [31]]. Interestingly, this was also the basic idea behind the first successful vaccine against an infectious disease, developed by Jenner in 1796, which induced protective immunity against the smallpox virus through inoculation with a related cowpox virus.
For identifying the SARS-CoV T cell epitopes likely to generate cross-reactive immune responses against SARS-CoV-2, Ahmed et al. [20] scanned all available SARS-CoV T cell epitopes in the ViPR database [32] that had been determined previously by using experimental positive HLA binding or T cell assays, and identified epitopes that had an exact match in the SARS-CoV-2 sequences available at that time. Subsequent studies by Lee et al. [21], Grifoni et al. [22], and Ranga et al. [23] used the SARS-CoV epitopes as well as peptide-HLA binding prediction methods (discussed in the next subsection) and proposed the common ones as potential SARS-CoV-2 epitopes.
The sequence data of SARS-CoV-2 continues to be deposited into public databases at an unprecedented rate, with the number of complete genome sequences available for SARS-CoV-2 in the GISAID database exceeding 65,000 (as of September 2020), much greater than that available for many other viruses (e.g., ~12,900 whole genome sequences are publicly available for HIV [33], and ~4400 for the hepatitis C virus [34]). Taking advantage of this data, the authors of [20] later proposed a web-based platform, COVIDep [35], for reporting SARS-CoV T cell epitopes as potential vaccine targets for SARS-CoV-2 based on the latest sequence data available. Compared to [20], which reported SARS-CoV epitopes that were fully conserved within the SARS-CoV-2 sequences available in February 2020, COVIDep provides a parameter that enables identification of SARS-CoV epitopes that are identical among a desired fraction of the latest available SARS-CoV-2 sequence data.
The epitopes predicted using SARS-CoV immunological information have been used by multiple experimental studies for SARS-CoV-2 to probe the immune responses in convalescent COVID-19 patients. As will be discussed in Section 4, many epitopes predicted using this approach have been found to elicit a cross-reactive T cell response in patients.
2.2. Methods based on prediction of peptide-HLA binding
The large majority of in silico SARS-CoV-2 vaccine design studies so far have predicted T cell epitopes using existing peptide-HLA binding prediction methods (see Table 1). A number of these studies also performed a subsequent refinement step where the set of epitopes was narrowed down by running computational tests to identify those capable of eliciting a robust and safe T cell response. In some cases, the refined set of predicted epitopes were used to design a vaccine construct by including appropriate signal sequences, proteasomal cleavage sites, and linkers, and these were further tested in silico for features such as immunogenicity, safety, and structural stability.
2.2.1. Initial epitope prediction methods
The in silico search for SARS-CoV-2 T cell epitopes has benefitted from years of research in developing peptide-HLA binding algorithms. These methods have matured over time thanks to increased availability of experimental data and methodological advancements. Most methods are specialized for predicting either HLA class I-restricted epitopes (i.e., CD8+ T cell epitopes) or HLA class II-restricted epitopes (i.e., CD4+ T cell epitopes), while some methods have been developed for predicting epitopes for both HLA classes. Based on the methodology employed, these T cell epitope prediction methods can be broadly divided into two groups: Machine learning (ML) based methods and (non-ML) bioinformatics based methods.
The current state-of-the-art ML epitope prediction methods utilize artificial neural networks (ANN). Such methods that have been used for SARS-CoV-2 HLA-I epitope prediction include NetMHC [36], NetMHCpan [37,38], NetCTLpan-1.1 [39], NetMHC-4.0 [40], HLAthena [17], MHCflurry [41] and NetMHCpan-4.0 [16]. Corresponding methods that have been used for prediction of SARS-CoV-2 HLA-II epitopes include NetHMCII-2.3 [42], NetMHCIIpan-3.0 [43], NetMHCIIpan-3.1 [44], NetMHCIIpan-3.2 [42], NetMHCIIpan-4.0 [18], NeonMHC2 [19] and MARIA [45] (for a historical perspective on the evolution of these methods, see [46]). The suffix -pan in the names of these methods indicates pan-specificity; i.e., the ability to predict peptide-HLA binding for a large set of alleles within the HLA class, including the ones that are absent in the training set. This feature has been made possible by integrating information about the amino acids characterizing the HLA binding groove in training the ANN [47].
The earlier ANN-based methods such as NetMHC, NetMHCpan, NetHMCII-2.3, NetMHCIIpan-3.0, NetMHCIIpan-3.1, etc., were mostly trained using data obtained from HLA binding assays, which characterize the binding affinity of synthetic peptides to HLA molecules. The most recent ANN-based methods, such as MHCflurry, NetMHCpan-4.0, and NetMHCIIpan-4.0, additionally employ data from HLA ligand elution assays, which use advances in mass spectrometry techniques to isolate a large number of peptides that are naturally processed and presented by human cells expressing a single HLA. A few SARS-CoV-2 epitope prediction studies such as HLAthena and NeonMHC2 have also used methods that were trained solely on HLA ligand elution assay data. However, it has been demonstrated for both HLA classes that the ANN models trained using both types of data provide superior performance to models trained on one type of data only [16].
Some methods used by SARS-CoV-2 epitope prediction studies include the HLA-I specific methods NetChop [48] and NetCTL-1.2 [49], which incorporated additional intracellular factors involved in HLA antigen presentation in an attempt to improve peptide-HLA binding prediction. These factors include proteasomal cleavage sites and transport efficiency of TAP (the transporter associated with antigen processing) in antigen presenting cells [50]. However, inclusion of these factors has been found to show only a marginal improvement in peptide-HLA binding prediction over the ANN-based methods trained solely on HLA binding assays [48]. While almost all recent epitope prediction methods are ANN-based, a few early ones (such as CTLPred [51] and RANKPEP [52]) were based on an alternative ML approach, support vector machines, and have also been used by SARS-CoV-2 studies (Table 1).
Distinct from the ML methods just described, several SARS-CoV-2 studies have used (non-ML) bioinformatics methods to predict SARS-CoV-2 epitopes. These methods use position-specific scoring functions that assume each position in a peptide to be independently interacting with HLA. They generally assign a score to the studied peptide according to position-specific amino acid features such as amino acid frequencies or amino acid physicochemical profiles (e.g., obtained from BLOSUM matrices [53]) at specific peptide positions. Such methods that have been used to predict SARS-CoV-2 epitopes include ProPred1 [54] for predicting HLA-I epitopes, ProPred [55] and Predivac [56] for predicting HLA-II epitopes, and Vaxign [57] for predicting epitopes for both HLA classes. Some bioinformatics methods also use scoring functions involving the interactions of pairs of peptide positions with the HLA. Methods in this category that have been used to predict SARS-CoV-2 epitopes include SMM [58,59] for predicting HLA-I epitopes, SMM-Align [60] and the method by Sturniolo [61] for predicting HLA-II epitopes, and MHCPred [62] for predicting epitopes restricted by both HLA classes. In general, these bioinformatics methods work for a limited set of HLA alleles, with the exception of Predivac [56] and the method by Sturniolo [61], both of which are pan-specific.
Several SARS-CoV-2 studies have used the analysis resource provided by the immune epitope database (IEDB) [43] for epitope prediction. The IEDB provides a collection of several of the above-mentioned prediction methods, and also recommends a best performing method for each HLA class. Some SARS-CoV-2 studies have used the IEDB’s HLA class-specific consensus methods, which predict peptide-HLA binding for their respective HLA class based on the consensus of several prediction methods [63,64]. A few studies have also used pipeline tools like pVACtools [65] and TepiTool [66], that allow users to predict epitopes for both HLA classes from a set of pre-defined ML and bioinformatics methods. Another method used for predicting SARS-CoV-2 epitopes is nHLAPred [67], which uses a combination of ML and bioinformatics methods to predict HLA-I-restricted epitopes.
The variety of peptide-HLA binding prediction methods used by in silico studies to predict SARS-CoV-2 epitopes raises questions as to whether the predictions of these studies are overlapping or distinct, and whether there are specific methods that appear most appropriate for predicting SARS-CoV-2 epitopes. We explore these questions subsequently in Sections 3 and 4, where we show that multiple common SARS-CoV-2 epitopes have indeed been predicted by independent in silico studies, while also identifying methods whose predicted epitopes are found to have induced T cell responses in convalescent COVID-19 patients.
2.2.2. Refinement of predicted epitopes
Beyond epitope prediction based on peptide-HLA binding, a number of SARS-CoV-2 in silico studies used computational tools to screen the predicted epitopes for their immunogenicity, and for their ability to elicit a robust and safe T cell response. A brief description of these tools and the rationale for using them is discussed below.
2.2.2.1. Screening predicted epitopes for their immunogenicity and robustness
Presentation of a peptide by an HLA molecule, while necessary for inducing a T cell response, does not guarantee T cell recognition and activation. That is, presentation does not imply that the peptide will be immunogenic. Thus, it is important to assess the immunogenicity of the predicted epitopes obtained from peptide-HLA binding prediction methods. The specific factors that differentiate an immunogenic HLA-presented peptide from a non-immunogenic one are still not well known, though a number of factors have been suggested to be the cause of this difference. For example, immunogenicity of a peptide may increase due to abundance of peptide-HLA complexes displayed on cells [68,69], early expression of the protein to which the peptide belongs [63,70,71], competition with other peptide-HLA complexes for stimulating T cells [72,73], and low genetic similarity of the peptide to a self-peptide (i.e., a host derived peptide) [[74], [75], [76], [77], [78]].
Several existing computational tools have been used to assess the immunogenicity of the SARS-CoV-2 epitopes obtained from peptide-HLA binding prediction methods. Of these, Vaxijen-2.0 [79] is the most commonly used and can predict immunogenicity of both HLA-I and HLA-II epitopes. This method was originally developed to predict protein immunogenicity by accounting for higher order interactions between protein sequence positions and exploiting the physicochemical properties (hydrophobicity, molecular size, polarity) of amino acids. It was trained on a set of known immunogenic and non-immunogenic proteins of viral, bacterial and tumor origins. Another common method that has been used for determining immunogenicity of HLA-I-restricted peptides in SARS-CoV-2 studies is the one available on the IEDB (Calis et al. [80]). This method was developed by comparing a set of immunogenic and non-immunogenic presented peptides compiled from multiple experimental sources. Specifically, immunogenicity of presented peptides was found to be largely dependent on the amino acids present at positions 4-6 of the peptide, and specific physicochemical properties of the amino acids, having large and aromatic side chains, in the peptide. A model exploiting these features was then developed to predict immunogenicity of a given HLA-I-restricted peptide.
In addition to Vaxijen-2.0 and Calis et al., one SARS-CoV-2 study [21] used iPred [81] for immunogenicity prediction, which is also based on physicochemical properties of the amino acids in the peptide. A novel method to predict the immunogenicity of SARS-CoV-2 HLA-I restricted peptides was also proposed in Gao et al. [82] which utilizes a physics-based model and takes into account factors such as peptide-HLA binding affinity and similarity of a peptide with pathogen-derived and human-derived peptides. For model training and testing, a well-characterized dataset of immunogenic HIV T cell epitopes was used. This model was reported to outperform Calis et al. [80] in predicting immunogenic epitopes for the HIV dataset. Gao et al. used this method to filter the SARS-CoV-2 epitopes proposed in Ahmed et al. [20] and Prachar et al. [83] based on their predicted immunogenicity.
For the specific case of peptides presented by HLA class II, the activated CD4+ T cells differentiate into T helper (Th) cells of various types having distinct effector functions [72]. Of particular importance are the Th1 and Th2 cells. Th1 cells secrete the interferon-gamma (IFN-γ) cytokine and promote cell-mediated immunity, while Th2 cells secrete the interleukin-4 cytokine and promote humoral immunity [84]. For inducing robust cell-mediated immunity, multiple in silico SARS-CoV-2 vaccine design studies have further screened the predicted immunogenic HLA-II peptides to identify those that are likely to induce IFN-γ (Table 1). This was done primarily using the computational method IFNepitope [85], an ML approach that uses a dataset comprising peptides experimentally determined to induce IFN-γ, along with peptides that induce cytokines other than IFN-γ. This method was trained on data of IFN-γ inducing epitopes obtained from IEDB.
In addition to screening for immunogenicity, to ensure that stimulated T cell responses are robust to genetic variations arising during viral evolution, it is important to identify epitopes that are highly conserved. For SARS-CoV-2, while the mutation rate appears low due to the presence of a genetic proof-reading exoribonuclease nsp14 protein [86], it is still important to consider conservation of epitopes to avoid mutations that accumulate in the population [87]. Thus, a few in silico SARS-CoV-2 vaccine design studies have considered the conservation of predicted immunogenic epitopes among the available SARS-CoV-2 sequences (or a subset of them) for providing vaccine target recommendations (Table 1). The majority of the in silico SARS-CoV-2 studies have computed the conservation of predicted epitopes using in-house code, while a few have used the epitope conservancy tool available at the IEDB [43].
2.2.2.2. Screening predicted epitopes for their allergenicity, toxicity and autoimmunity
Vaccines are generally administered to otherwise healthy individuals as a preventive measure against disease. A crucial factor when selecting epitopes for a vaccine, apart from the ability of the epitopes to elicit a protective immune response, is whether or not they have any associated safety concern. Any adverse reaction caused by a vaccine is likely to contribute to anti-vaccine sentiment and potentially lead to loss of public trust in immunization programs [88,89]. Even though any potential vaccine would ultimately need to undergo rigorous in vivo and in vitro safety trials, computational tools can help, as a first step, to screen for potential safety concerns. Here, we give a brief review of computational tools that have been used for assessing allergenicity, toxicity, and autoimmunity of SARS-CoV-2 epitopes. Almost all studies proposing an in silico vaccine perform these tests on the vaccine construct, while some also perform these at the initial epitope selection step (see Table 1).
Allergenicity of a substance is its potential to cause hypersensitivity in an individual by evoking an immune response [90]. The reason why certain proteins or peptides cause an allergic reaction in humans is not precisely known. Several SARS-CoV-2 studies have used more than one computational tool to test for allergenicity of predicted epitopes (see Table 1). These include AlgPred [91], AllerTOP [92], AllerTOP-2.0 [93], AllergenFP [94], and AllerCatPro [95]. AlgPred is a suite of multiple allergenicity prediction approaches based on motif alignment, support vector machines, and hybrid approaches; AllerTOP, AllerTOP-2.0, and AllergenFP are three related methods based on the physicochemical similarity of the considered protein with known allergens; while AllerCatPro is a recently developed method that uses 3D structure information along with sequence similarity with known allergens to predict allergenicity. In a comparative test, AllerCatPro was shown to have superior performance to the other methods in identifying allergens [95]. However, of the 65 in silico studies considered in this review (Table 1), only one study [96] used AllerCatPro for allergenic screening of predicted SARS-CoV-2 epitopes.
Toxicity is the capacity of a substance to damage a living organism by interacting with biomolecules and disrupting normal cellular functions [97]. The effects of this disruption may range from slight symptoms like nausea in mild cases, through to death in severe cases. SARS-CoV-2 studies that tested for toxicity of predicted epitopes have used ToxinPred [98], a support vector machine based method that uses a position-specific scoring function to predict peptide toxicity from sequence information. ToxinPred has been trained on a set of known toxic (of bacterial and animal origin) and non-toxic peptides obtained from the Universal Protein Resource [99]. While a number of SARS-CoV-2 in silico studies have used ToxinPred (Table 1), the accuracy of these toxicity predictions for viral epitopes has yet to be confirmed.
Autoimmunity refers to the process of the body launching an immune response against its own healthy self [100]. In the context of T cells, this would constitute a T cell mounting a response against a self-peptide. The exact causal mechanism that triggers such a response in not known, though there is evidence that it may be triggered in some individuals in the aftermath of a viral or bacterial infection [101,102]. While evidence for a vaccine-induced autoimmune response has not been found in controlled studies, occasional case reports of such an occurrence are present in the literature [103,104]. This gives rise to some concern that if a vaccine contains viral epitopes similar to self-peptides, it may evoke an autoimmune response [105,106]. To account for this, some of the SARS-CoV-2 studies have checked for sequence similarity between the human proteome and viral epitopes to identify epitopes that may potentially induce an autoimmune reaction [107]. The computational hurdle here is the large size of the human proteome which makes testing multiple epitopes for sequence similarity challenging. However, of all possible SARS-CoV-2 peptides of lengths 8-10 (HLA-I restricted) and 13-20 (HLA-II restricted), only ~0.03% were found to match exactly with the human proteome [108], suggesting that autoimmunity may not be a common issue for SARS-CoV-2.
Although the above-mentioned screening tests are well motivated, the ability of the specific tools used to effectively screen SARS-CoV-2 epitopes for immunogenicity, induction of IFN-γ, allergenicity, and toxicity still remains unclear. In an attempt to shed some light on the predictive nature of these tools, in Section 4 we analyze and compare their predictions when applied to epitopes of SARS-CoV-2 that have been confirmed experimentally in patients.
2.2.3. Design of a vaccine construct
While some of the SARS-CoV-2 in silico studies only identified a list of epitopes, several went a step further to propose a multi-epitope vaccine construct (see Table 1). Briefly, this entailed selecting appropriate linkers, which play a key role in the structural stability of the vaccine, and adjuvants, which help to boost the immune response. These in silico designs spanned a host of modern vaccine technologies, e.g., subunit vaccines, peptide vaccines, RNA and DNA vaccines [109,110]. Almost all SARS-CoV-2 studies that presented an in silico design of a multi-epitope vaccine construct also performed in silico tests for immunogenicity, conservation, allergenicity, and toxicity using the tools reviewed above, regardless of whether or not these tests were performed at the epitope level. As mentioned above, the majority of these in silico tools were originally developed to analyze proteins, and thus they may be more appropriate for analyzing multi-epitope vaccine constructs than individual epitopes. In addition, physicochemical composition, secondary and tertiary structure predictions, as well as molecule docking studies with human immune receptors were also investigated for the proposed constructs. None of these studies, however, tested their predictions experimentally. As the focus of the current review is on epitope identification, we refer the interested reader to [111] for more details on vaccine constructs.
3. Comparison of studies predicting SARS-CoV-2 epitopes
To compare the predictions of the in silico studies (listed in Table 1), the predicted epitopes and their specific HLA allele associations [170] were compiled. In cases where the specific HLA allele information was unavailable, we recorded it as “NA”. For a meaningful comparison, we focused only on the predicted T cell epitopes comprising 8 to 20 amino acids, representing the typical combined range of CD4+ and CD8+ T cell epitopes [171,172]. This excluded Enayatkhani et al. [118], Yarmarkovich et al. [167], Yazdani et al. [168], and Lucchese et al. [169] from the analysis, giving a remaining set of 61 studies (Fig. 2 ).
Fig. 2.

Summary and comparison of 61 in silico studies that have predicted SARS-CoV-2 T cell epitopes. (Top left panel) Heatmap shows the fraction of common epitopes predicted across each pair of studies. The fraction is computed relative to the number of epitopes predicted by the study indicated in each row (the total number of epitopes predicted in each study are shown within parentheses on the right). Four in silico studies that used SARS-CoV immunological data are indicated in bold font. Of the epitopes predicted by these studies, only the ones predicted based on homology with SARS-CoV epitopes were included. Study labels indicated in the figure correspond to those in Table 1. (Top right panel) Bar plots show the fraction of predicted epitopes for each HLA class in each study, with the total number shown within parentheses. (Bottom left panel) Heatmap shows the number of predicted epitopes derived from each SARS-CoV-2 protein for each in silico study. Each column in this heatmap corresponds to the study mentioned at the top of each column in the top left panel heatmap. Missing tiles indicate no predicted epitopes. (Bottom right panel) Bar plots show the fraction of predicted epitopes, across studies, derived from each SARS-CoV-2 protein, with the total number shown within parentheses. Predicted epitopes were assigned HLA class based on the HLA allele (bearing 4-digit resolution or higher) reported against them; or as “NA” otherwise.
The number of T cell epitopes predicted by these SARS-CoV-2 studies varied widely (minimum = 1 epitope; maximum = 3407 epitopes), even for studies that used the same peptide-HLA prediction method (Fig. 2). This can be attributed to differences in subsequent prediction refinement steps and the study objectives. Studies reporting a very limited number of T cell epitopes (<10), e.g., Joshi et al. [156], Rahman et al. [165], Khan et al. [148], Jakhar et al. [135], Samad et al. [130], were primarily focused on designing a short vaccine construct for eliciting a targeted immune response. Most of these studies refined the set of initial epitopes obtained by peptide-HLA binding prediction methods based on the tests described in Section 2.2.2. In contrast, the studies that predicted a large set of epitopes (>400) had objectives such as identifying all possible epitopes having high coverage in a specific ethnic population (Feng et al. [152]), identifying epitopes recognized by a large number of HLA alleles including rare ones (Campbell et al. [137]), or finding all possible epitopes binding to a specific allele even with low predicted affinity (Nerli et al. [147]). These did not perform any refinement of the set of epitopes obtained using the peptide-HLA binding methods.
A number of the SARS-CoV-2 studies had a sizeable overlap among their predicted set of epitopes (Fig. 2). This is not surprising since many had used similar computational pipelines (Table 1). For example, the 4 studies based on exploiting SARS-CoV immunological data all relied on sequence similarity of SARS-CoV-2 epitopes with known SARS-CoV epitopes, resulting in considerable overlap among their predicted set of epitopes (Fig. 2). Similarly, there was overlap among predictions of several studies based on peptide-HLA binding prediction (Fig. 2), which mostly used a similar set of methods for epitope prediction (Fig. 3 ). The ones that stand out among the latter group of studies for having a large overlap with multiple others include Feng et al. [152], Campbell et al. [137], and Nerli et al. [147] (Fig. 2), which may be attributed to the large number of epitopes that they predicted.
Fig. 3.
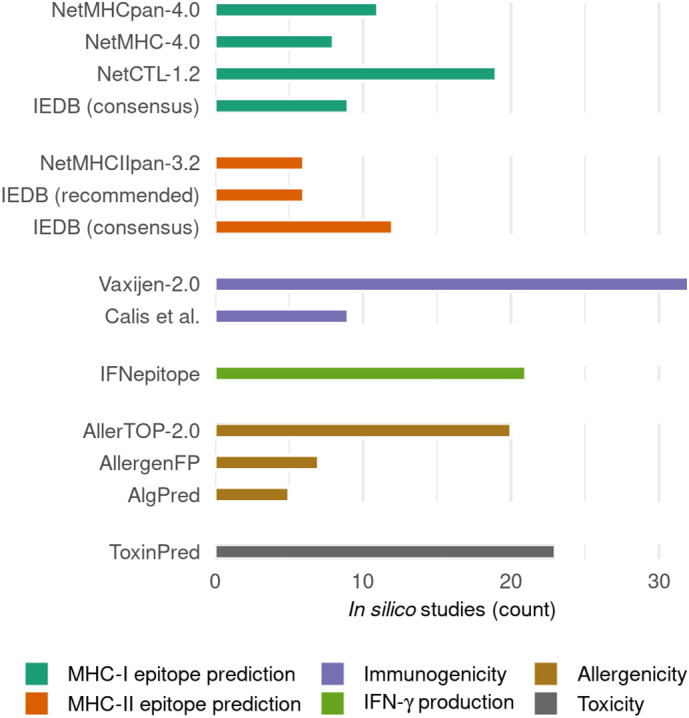
Common in silico prediction methods that have been used by the reviewed SARS-CoV-2 studies. Only methods that were explicitly mentioned by at least 5 in silico SARS-CoV-2 studies (Table 1) are shown here. The methods are grouped according to the category shown in the legend.
The in silico predicted epitopes lie within each of the 12 SARS-CoV-2 proteins. The structural S protein, which is also one of the most immunogenic proteins of SARS-CoV-2, was the most commonly analyzed protein (Fig. 2). Surprisingly, the other structural proteins (M, N and E), also reported to be immunogenic in SARS-CoV-2 [6] as well as SARS-CoV [173,174], were analyzed by only a few studies. The N protein, in particular, is highly expressed [115], and the epitopes derived from this protein may be especially worthy of further experimental investigation. Compared to other proteins, the number of epitopes predicted for the longer proteins (ORF1a, ORF1b, and S) was much larger. As expected, the number of unique T cell epitopes predicted per protein across all studies was found to be strongly correlated with the length of protein (r = 0.99).
Similar numbers of unique HLA-I and HLA-II restricted epitopes were predicted by the SARS-CoV-2 studies listed in Table 1 (2,239 HLA-1 and 2,580 HLA-II), while a good number of epitopes (464) had missing (“NA”) HLA class restriction. For the studies predicting epitopes exclusively based on peptide-HLA binding methods, it is surprising to observe epitopes having unknown HLA class restriction. This is because almost all prediction methods (discussed in Section 2.2.1) require specifying the HLA allele for predicting associated epitopes. However, a close inspection of the related studies revealed that this was due to either non-reporting of HLA allele restriction of the predicted epitopes (e.g., Sarkar et al. [161]), or cases where only the number of unique HLA alleles associated with the predicted epitopes were reported without specifying the individual correspondences (e.g., Sanami et al. [163]). As for the studies leveraging the immunological data of SARS-CoV from public databases, the SARS-CoV epitopes having no HLA class information were assigned the “NA” HLA class.
4. Correspondence between predicted and experimentally-determined T cell epitopes
With a large number of studies predicting SARS-CoV-2 T cell epitopes using different computational pipelines (involving specific epitope prediction and refinement methods), it is difficult to assess their accuracy solely based on their performance in predicting epitopes of other organisms [175]. For this purpose, “ground truth” information of experimentally-determined SARS-CoV-2 T cell epitopes is required. This data has started to emerge from immunological assays that analyze immune responses in COVID-19 patients. As of 8 September 2020, we found eight experimental studies [8,9,115,[176], [177], [178], [179], [180]], reviewed in [181], as well as an additional study [182], that reported positive T cell immune responses from blood samples of convalescent COVID-19 patients against epitopes derived from SARS-CoV-2 proteins. Compiling data from these nine studies yielded a total of 324 (unique) epitopes. These 324 epitopes present a sampling (albeit not comprehensive) of the landscape of epitopes targeted by COVID-19 patients, and hence they provide an initial basis for which to conduct a comparative analysis of different in silico epitope prediction methods.
The nine experimental studies measured T cell responses after stimulation of the peripheral blood mononuclear cells (PBMCs) from convalescent COVID-19 patients. Of these, some studies obtained a set of peptides to synthesize using one or more of the in silico epitope prediction methods, which were then used to stimulate the PBMCs. Three such studies [8,9,180] used NetMHCpan-4.0, two studies [177,182] used NetMHC4.0, while one study [115] used HLAThena and NeonMHC2 to select the set of peptides to synthesize. Two of these studies [8,182] also investigated a few epitopes that were predicted in [20,22] using SARS-CoV immunological data. This set of immunological studies demonstrates the application of multiple in silico T cell epitope prediction methods in guiding experimental investigations. Specifically, immune responses are measured against a reduced set of defined peptides predicted in silico which helps in identifying precise epitopes. In alternative experimental studies, stimulation was done using pools of overlapping k-mer peptides. One such study [176] synthesized pools of 15-mer overlapping peptides to measure T cell responses, while another study [179] synthesized pools of 15-18-mer overlapping peptides in addition to epitopes predicted using SARS-CoV immunological data [20]. While using pools of overlapping peptides does not reveal the precise “epitope” stimulating the T cell response, it still provides the information of immunogenic peptides encompassing the epitopes. Lastly, one study [178] employed a new experimental framework called T-Scan [183] to identify SARS-CoV-2 epitopes.
We compared the 324 experimentally-determined epitopes (including both precise epitopes and immunogenic peptides) against all unique 8-20 residue-long in silico predicted T cell epitopes (5273) (Fig. 2). We found that 309 of the experimentally-determined epitopes encompassed at least a single predicted epitope, while 163 of these epitopes matched identically to predicted ones. Looking closely at those in silico studies that predicted at least one of the 163 identically-matched epitopes, we observed that studies which used SARS-CoV immunological data had collectively a higher hit rate (proportion of identically-matched epitopes in the set of predicted epitopes) (25%) as compared to the studies that used peptide-HLA binding prediction-based methods (3%). This difference in hit rate points to the usefulness of using SARS-CoV data to predict immune targets for SARS-CoV-2.
Most in silico prediction studies as well as experimental studies were biased towards certain HLA alleles. Together, the nine experimental studies reported T cell epitopes associated with 18 different HLA alleles, with the largest proportion of the reported epitopes (35/116) associated with the HLA-A*02:01 allele. This is not surprising since HLA-A*02:01 is the most prevalent HLA-I allele globally [184]. Similarly, roughly a third of the epitopes predicted by in silico methods were also associated with HLA-A*02:01. Thus, the experimentally-determined and predicted epitopes associated with HLA-A*02:01 represent a reasonable dataset for assessing the predictions of SARS-CoV-2 in silico studies. Altogether, 33 of the 35 experimentally-determined HLA-A*02:01-associated epitopes matched identically with predicted epitopes, and these were derived from five proteins (S, ORF1a, N, ORF3a and M) (Fig. 4 ). Most of these epitopes (27/33) were reported to elicit a T cell response in multiple convalescent COVID-19 patients, with 10 of them eliciting responses in more than 5 patients. This suggests the potential immunodominance of these epitopes [185,186] across the segment of population bearing the HLA-A*02:01 allele (Fig. 4). Of these 33 epitopes, in silico studies based on peptide-HLA binding prediction and those leveraging SARS-CoV immunological data predicted 32 and 17, respectively. After grouping the in silico studies based on the peptide-HLA prediction tool used, those involving NetMHCpan-4.0 (despite the differences in refinement steps) appeared to predict most (30/33) of the epitopes (Table 2 ). However, the method leveraging SARS-CoV immunological data had the highest hit rate in predicting experimentally-determined HLA-A*02:01-associated epitopes (14.8%) (Table 2).
Fig. 4.

Summary of the experimentally-determined HLA-A*02:01-associated SARS-CoV-2 epitopes that were also predicted by in silico studies. (Left panel) List of 33 experimentally-determined HLA-A*02:01-associated epitopes that matched identically with epitopes predicted by in silico studies. (Middle panel) Number of convalescent COVID-19 patients bearing the HLA-A*02:01 allele whose blood sample responded (filled bar) and did not respond (empty bar) upon stimulation with the epitope. (Right panel) Number of in silico studies that predicted the epitope in the context of HLA-A*02:01. Orange represents the number of studies that used SARS-CoV immunological data, while purple represents the number of studies based on peptide-HLA binding prediction. The labels of the in silico studies (Table 1) predicting each epitope are listed on the right. Epitopes are colored according to the SARS-CoV-2 protein from which they are derived (counts shown in legend) and ordered in descending order of the number of patients whose samples responded. The two experimentally-determined HLA-A*02:01 epitopes which did not match identically with any epitope predicted by in silico studies were 906YLFDESGEFKL916 in ORF1a and 20FLAFVVFL27 in E. These epitopes were reported to induce a T cell response in 9/36 and 2/3 COVID-19 convalescent patients, respectively.
Table 2.
Approaches adopted by in silico studies that predicted at least half of the experimentally-determined HLA-A*02:01-associated epitopes.
| No. | In silico studies | Approach | Total number of predicted epitopes | Number of predicted epitopes matching experimentally-determined epitopes | Hit ratea |
|---|---|---|---|---|---|
| 1 | Nerli2020 [147], Wang2020 [114], Bhatnager2020 [131] | Based on peptide-HLA binding prediction (involving NetMHCpan-4.0) | 722 | 30 | 4.2% |
| 2 | Ahmed2020 [20], Grifoni2020 [22], Ranga2020 [23], Lee2020 [21] | Using SARS-CoV immunological data | 115 | 17 | 14.8% |
Hit rate represents the positive predicted value (i.e., ratio of the number of predicted epitopes matching experimentally-determined epitopes to the total number of in silico predicted epitopes).
A large fraction of these experimentally-determined epitopes (27/33) were predicted to promiscuously bind to multiple HLA alleles by in silico methods (Table 3 ). For example, the S-derived epitope 269YLQPRTFLL277 was collectively predicted by 12 in silico methods to bind to 43 HLA alleles in addition to HLA-A*02:01. This HLA promiscuity of the experimentally-determined epitopes predicted by in silico methods (Table 3) can help guide future experimental studies to identify the potential immunodominance of these epitopes across a large segment of the global population carrying different HLA alleles.
Table 3.
Distinct HLA alleles predicted, across in silico studies, to be associated with the 33 experimentally-determined HLA-A*02:01-restricted SARS-CoV-2 epitopes.
| No. | Epitopea | Protein |
In silico prediction methods (count) |
In silico predicted HLAs |
|---|---|---|---|---|
| 1 | 139LLYDANYFL147 | ORF3a | 1 | A*02:01 |
| 2 | 3886KLWAQCVQL3894 | ORF1a | 3 | A*02:01, A*02:02, A*02:03, A*02:06, A*02:121, A*02:131, A*02:141, A*02:158, A*02:173, A*02:181, A*02:196, A*02:205, A*02:214, A*02:228, A*02:238, A*02:25, A*02:257, A*02:266, A*02:70, A*02:71, A*02:73, A*02:85, A*02:95, A*03:01, A*31:01 |
| 3 | 269YLQPRTFLL277 | S | 12 | A*01:01, A*02:01, A*02:02, A*02:03, A*02:06, A*02:07, A*02:12, A*02:13, A*02:131, A*02:142, A*02:150, A*02:170, A*02:180, A*02:187, A*02:196, A*02:205, A*02:214, A*02:233, A*02:238, A*02:247, A*02:257, A*02:44, A*02:50, A*02:54, A*02:69, A*02:71, A*02:73, A*02:85, A*23:01, A*24:02, A*32:01, B*07:02, B*08:01, B*08:22, B*08:38, B*08:41, B*08:56, C*03:03, C*03:04, C*04:01, C*06:02, C*07:02, C*12:03, C*14:02 |
| 4 | 4094ALWEIQQVV4102 | ORF1a | 3 | A*02:01, A*02:02, A*02:03, A*02:06, A*02:11, A*02:121, A*02:139, A*02:141, A*02:150, A*02:16, A*02:173, A*02:181, A*02:19, A*02:196, A*02:205, A*02:214, A*02:228, A*02:238, A*02:25, A*02:257, A*02:266, A*02:50, A*02:70, A*02:71, A*02:73, A*02:85, A*02:95, A*68:02, A*69:01, B*15:01 |
| 5 | 1000RLQSLQTYV1008 | S | 6 | A*02:01, A*02:02, A*02:03, A*02:06, A*02:07, A*68:02 |
| 6 | 222LLLDRLNQL230 | N | 6 | A*02:01, A*02:02, A*02:03, A*02:04, A*02:07, A*02:11, A*02:13, A*02:132, A*02:141, A*02:150, A*02:16, A*02:173, A*02:181, A*02:19, A*02:196, A*02:205, A*02:214, A*02:228, A*02:238, A*02:25, A*02:262, A*02:54, A*02:70, A*02:71, A*02:73, A*02:85, A*02:95, A*03:01, A*23:01, A*32:01, B*08:01, B*08:22, B*08:38, B*08:41, B*08:56, B*14:02, B*46:01, B*51:01, C*01:02, C*02:02, C*03:02, C*03:03, C*03:04, C*03:71, C*04:01, C*05:01, C*06:02, C*07:02, C*08:02, C*12:03, C*14:02, C*14:03, C*17:01 |
| 7 | 2332ILFTRFFYV2340 | ORF1a | 4 | A*02:01, A*02:03, A*02:06, A*02:09, A*02:107, A*02:118, A*02:121, A*02:131, A*02:141, A*02:158, A*02:160, A*02:173, A*02:181, A*02:187, A*02:196, A*02:205, A*02:214, A*02:228, A*02:238, A*02:25, A*02:257, A*02:266, A*02:28, A*02:29, A*02:30, A*02:31, A*02:33, A*02:34, A*02:40, A*02:42, A*02:51, A*02:58, A*02:59, A*02:60, A*02:61, A*02:66, A*02:67, A*02:68, A*02:70, A*02:71, A*02:73, A*02:74, A*02:85, A*02:95, B*08:01 |
| 8 | 107YLYALVYFL115 | ORF3a | 4 | A*02:01, A*02:02, A*02:09, A*02:11, A*02:12, A*02:121, A*02:131, A*02:141, A*02:158, A*02:16, A*02:173, A*02:181, A*02:187, A*02:196, A*02:205, A*02:214, A*02:228, A*02:238, A*02:25, A*02:263, A*02:266, A*02:54, A*02:58, A*02:69, A*02:71, A*02:73, A*02:85, A*02:95, A*33:08, A*74:04, B*40:13 |
| 9 | 72ALSKGVHFV80 | ORF3a | 3 | A*02:01, A*02:50 |
| 10 | 1220FIAGLIAIV1228 | S | 8 | A*02:01, A*02:02, A*02:03, A*02:06, A*02:07, A*02:131, A*02:150, A*02:170, A*02:179, A*02:187, A*02:196, A*02:205, A*02:214, A*02:228, A*02:238, A*02:248, A*02:257, A*02:50, A*02:69, A*02:71, A*02:85, A*02:95, A*68:02, A2, B*46:01, C*03:04, C*12:03, C*15:02 |
| 11 | 221LLLLDRLNQL230 | N | 2 | A*02:01 |
| 12 | 3403FLNGSCGSV3411 | ORF1a | 4 | A*02:01, A*02:02, A*02:03, A*02:06, A*02:16, A*02:171, A*02:50, A*68:02 |
| 13 | 417KIADYNYKL425 | S | 8 |
A*02:01, A*02:05, A*02:06, A*02:07, A*02:102, A*02:104, A*02:128, A*02:131, A*02:142, A*02:155, A*02:161, A*02:173, A*02:186, A*02:187, A*02:196, A*02:209, A*02:22, A*02:229, A*02:243, A*02:25, A*02:262, A*02:50, A*02:69, A*02:90, A*02:95, A*11:01, A*24:02, A*31:01, A*32:01, B*07:02, B*08:01, B*27:05, B*35:01, B*38:01, B*39:02, C*04:01, C*07:02, C*15:02, Cw*04:01 |
| 14 | 821LLFNKVTLA829 | S | 4 | A*02:01, A*02:03, A*02:06, A*02:07, B*08:01 |
| 15 | 424KLPDDFTGCV433 | S | 3 | A*02:01 |
| 16 | 825FGDDTVIEV833 | ORF1a | 2 | A*02:01, C*05:21, C*05:30 |
| 17 | 3467VLAWLYAAV3475 | ORF1a | 3 | A*02:01, A*02:02, A*02:03, A*02:06, A*02:11, A*02:148, A*02:22, A*02:230, A*02:253, A*02:258, A*68:02 |
| 18 | 3639FLLPSLATV3647 | ORF1a | 5 | A*02:01, A*02:02, A*02:03, A*02:06, A*02:11, A*02:12, A*02:13, A*02:132, A*02:141, A*02:158, A*02:16, A*02:173, A*02:181, A*02:19, A*02:196, A*02:205, A*02:214, A*02:228, A*02:238, A*02:25, A*02:263, A*02:27, A*02:28, A*02:50, A*02:51, A*02:54, A*02:61, A*02:70, A*02:71, A*02:73, A*02:85, A*02:99, A*68:02 |
| 19 | 1062FLHVTYVPA1070 | S | 5 | A*02:01, A*02:03, A*02:06, A*02:07, B*54:01 |
| 20 | 983RLDKVEAEV991 | S | 6 | A*02:01, A*02:02, A*02:03, A*02:06, A*02:07, A*02:16, A*68:02, C*04:01 |
| 21 | 995RLITGRLQSL1004 | S | 2 | A*02:01 |
| 22 | 26FLFLTWICL34 | M | 4 | A*02:01, A*02:07 |
| 23 | 338KLDDKDPNF346 | N | 2 | A*02:01, C*05:33 |
| 24 | 386KLNDLCFTNV395 | S | 3 | A*02:01, A*02:03 |
| 25 | 112YLGTGPEAGL121 | N | 1 | A*02:01 |
| 26 | 316GMSRIGMEV324 | N | 5 | A*02:01, A*02:03, A*02:50 |
| 27 | 219LALLLLDRL227 | N | 1 | A*02:01 |
| 28 | 202KIYSKHTPI210 | S | 5 | A*02:01, A*02:07, A*30:01, A*32:01, C*03:04, C*15:02 |
| 29 | 857GLTVLPPLL865 | S | 4 | A*02:01, A*02:07 |
| 30 | 958ALNTLVKQL966 | S | 5 | A*02:01, A*02:03 |
| 31 | 996LITGRLQSL1004 | S | 4 | A*02:01, B*07:02, B*08:01, C*03:03, C*03:04, C*12:03 |
| 32 | 1185RLNEVAKNL1193 | S | 6 | A*02:01, A*02:03, A*02:11, A*02:128, A*02:171, A*02:196, A*02:230, A*02:238, A*02:253, A*02:258, A*02:99, A*32:01, B*27:20 |
| 33 | 976VLNDILSRL984 | S | 7 | A*01:01, A*02:01, A*02:03, A*02:06, A*02:07, A*02:11, A*02:13, A*02:132, A*02:148, A*02:151, A*02:171, A*02:186, A*02:19, A*02:196, A*02:209, A*02:22, A*02:230, A*02:238, A*02:253, A*02:258, A*02:52, A*02:54, A*02:70, A*02:71, A*02:73, A*02:85, A*02:99, A*03:01, A*11:01, A*23:01, A*24:02, A*26:01, A*30:01, A*30:02, A*31:01, A*32:01, A*33:01, A*68:01, A*68:02, B*07:02, B*08:01, B*15:01, B*35:01, B*40:01, B*44:02, B*44:03, B*46:01, B*51:01, B*53:01, B*57:01, B*58:01, C*04:01, C*05:04, C*05:23, C*05:33 |
Epitopes are listed in the same order as in Fig. 4.
We used the compiled experimental SARS-CoV-2 data to assess the predictions of various refinement tools employed in SARS-CoV-2 in silico studies (Table 1). We selected the refinement tools for which a web-server was available and used the recommended parameters on the web-server of each tool to analyze the experimentally-determined SARS-CoV-2 epitopes. This compiled data of experimentally-determined epitopes, obtained from positive T cell responses in convalescent COVID-19 patients, serves as a reasonable ground truth for the tools predicting immunogenicity.
For Vaxijen-2.0 [79], the most commonly used tool by SARS-CoV-2 in silico studies for screening epitopes for immunogenicity (Fig. 3), we obtained predictions for the experimentally-determined epitopes using the available web-server by selecting the organism as “viruses”, as recommended by the authors. Our analysis revealed that Vaxijen-2.0 classified only ~56% (182/324) of the experimentally-determined epitopes as immunogenic (Fig. 5 ). Unlike Vaxijen-2.0, Calis et al. [80] was developed for predicting immunogenicity of only HLA class I epitopes and does not perform binary classification. Instead, it provides a score to each epitope, with a high score representing high confidence in the epitope being immunogenic. We considered all epitopes predicted to have positive scores as immunogenic and vice versa, in accordance with the majority of SARS-CoV-2 in silico studies that used Calis et al. to predict epitope immunogenicity (Table 1). Assessing the immunogenicity of 98 HLA class I experimentally-determined epitopes, Calis et al. predicted only ~63% (62/98) of them to be immunogenic (Fig. 5). To investigate the performance of these methods in more detail, we tested their accuracy in predicting the top 10 HLA-A*02:01-associated immunodominant epitopes (Fig. 4). In this case, Vaxijen-2.0 and Calis et al. predicted 30% (3/10) and 60% (6/10) of these to be immunogenic. Hence, the most commonly-used methods for immunogenicity prediction incorrectly classified over a third of the 328 experimentally-determined epitopes as non-immunogenic. The accuracy of these methods does not appear to improve even for predicting the highly immunogenic epitopes, highlighting their suboptimal performance.
Fig. 5.
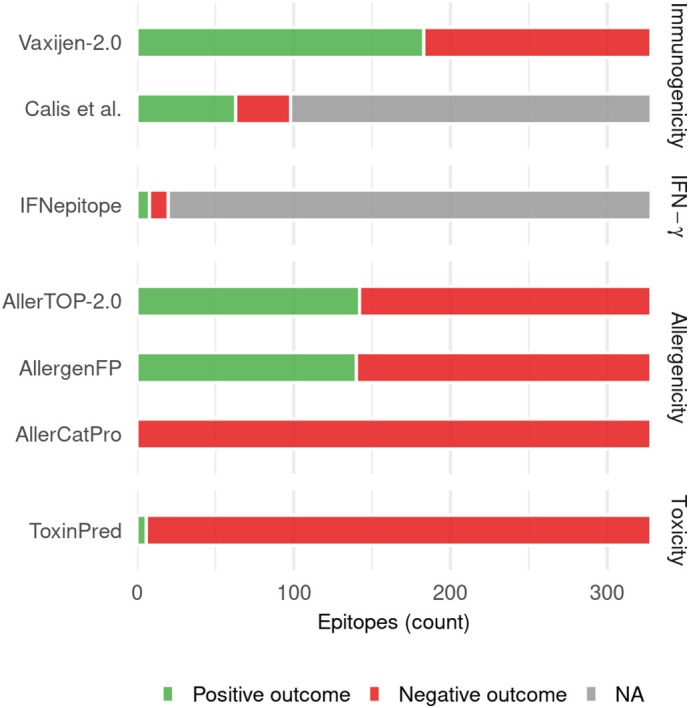
Results obtained for the 324 experimentally-determined SARS-CoV-2 T cell epitopes [8,9,115,[176], [177], [178], [179], [180],182] when they were provided as input to the computational tools most commonly used in the reviewed SARS-CoV-2 in silico studies for refinement of epitopes obtained by peptide-HLA binding prediction methods. Positive outcomes indicate the number of epitopes the computational tool predicts to have the characteristic (immunogenicity, IFN-γ production, allergenicity, toxicity) being tested, and vice versa for negative outcomes. “NA” indicates the number of epitopes that could not be analyzed by the specific tool. In case of Calis et al. [80], the method is applicable to HLA-I epitopes only, while in the case of IFNepitope, this was because only a subset of the experimentally-determined epitopes had IFN-γ production information available.
For a selected subset (20) of HLA-II restricted experimentally-determined epitopes, the recognizing CD4+ T cells were confirmed to be producing IFN-γ using flow cytometry [9]. We used this subset of epitopes to assess performance of the IFNepitope tool [85] commonly used in SARS-CoV-2 in silico studies (Fig. 3). The predictions of IFNepitope for the selected experimentally-determined epitopes were obtained from the associated webserver using the recommended approach (motif and SVM hybrid) and model (IFN-γ vs non-IFN-γ). This analysis showed that IFNepitope correctly predicted only 40% (8/20) of the experimentally-determined IFN-γ producing SARS-CoV-2 epitopes (Fig. 5).
In contrast to immunogenicity and IFN-γ production, no information was available regarding allergenicity and toxicity of the experimentally-determined epitopes from the immunological studies. Thus, no ground truth information is available to assess the performance of tools predicting these epitope characteristics. Nevertheless, we can still analyze the experimentally-determined epitopes using these tools and, at least for the case of allergenicity prediction tools, compare their relative predictions. We used the default parameter settings in the webservers for all allergenicity and toxicity prediction tools. Our analysis showed that AllerTOP-2.0 [93] and AllergenFP [94], the two most commonly used tools for determining allergenicity (Fig. 3), predicted a high fraction ~43% (142/328 and 140/328, respectively) of the experimentally-determined epitopes to be allergenic, with less than half of these (66) being commonly predicted by both methods. Hence, the variation in predictions of these methods was high. This disparity was even more evident for the AllerCatPro method [95], which predicted all experimentally-determined epitopes to be non-allergenic (Fig. 5). Hence, due to the wide variation in predictions and a lack of experimental data to validate them, the practical applicability of the allergenicity tools for SARS-CoV-2 remains unclear and further investigation is required. Lastly, in terms of toxicity prediction, ToxinPred [98] was the only tool used to predict toxicity of epitopes by the SARS-CoV-2 in silico studies (Table 1). Our analysis revealed that it predicted 98% (322/328) of the experimentally-determined epitopes to be non-toxic. However, similar to the case of allergenicity predicting tools, evaluating the accuracy of these toxicity predictions is not possible at present due to a lack of relevant experimental information.
5. Summary and perspectives
In silico epitope identification is an important component in the vaccine development pipeline as it provides recommendations for immune targets that may be exploited by vaccine designs. It is also very helpful for guiding immunological assays designed to understand T cell responses elicited by vaccines or those mounted naturally against COVID-19 infections. This review has made vivid the large amount of work that has been done already in predicting and analyzing epitopes of SARS-CoV-2, with a focus on T cells. The 65 studies that we have reviewed employed different computational approaches, along with an impressive array of computational tools.
The aim of this review was not only to summarize the methods that have been employed so far, but also to provide a comparative analysis of their epitope predictions, as well as to offer insights into the performance of the different approaches. The ability to test prediction accuracy hinges on the availability of experimental ground truth data, which is rapidly evolving but still remains limited. Data limitations precluded, for example, the performance evaluation of tools that predict epitope safety features. However, data from nine independent immunological studies of convalescent COVID-19 patients provided a set of T cell epitopes that offered a means to test the basic ability of the in silico T cell prediction methods for identifying SARS-CoV-2 epitopes reported to be immunogenic.
The fact that the large majority (>95%) of the experimentally-determined epitopes for HLA-A*02:01 were identical to an epitope predicted by at least one in silico method offers strong evidence for the practical significance of these methods in identifying immunogenic T cell epitopes in the context of SARS-CoV-2. While the comparison carried out here cannot ascertain which prediction method performed better (given that many studies differed in their prediction refinement step and objectives), we could still compare the hit rates of in silico studies grouped according to their common underlying approach for predicting SARS-CoV-2 epitopes (Table 2). This analysis showed that for HLA-A*02:01 (the HLA allele with the largest number of epitopes available), the two approaches with the highest hit rates in predicting the set of experimentally-determined SARS-CoV-2 epitopes were peptide-HLA binding prediction using NetMHCpan-4.0 and the approach that leveraged SARS-CoV immunological data. Hence, these distinct approaches both appear to be well supported for their further use in guiding additional epitope identification for SARS-CoV-2, and for their application to identify epitopes for other viruses.
Our analysis has provided insights into the computational tools that have been used by a number of SARS-CoV-2 in silico studies to further refine the set of predicted epitopes based on specific features. Most notably, the observation that the most commonly used tools for predicting epitope immunogenicity identified almost one-third of the experimentally-determined immunogenic epitopes to be non-immunogenic points to their suboptimality in relation to SARS-CoV-2. Similarly, the performance of the tool that has been used for screening HLA class II epitopes for inducing IFN-γ production was also found to be suboptimal. It should be recognized however, that these in silico screening tools are general-purpose tools that were developed more than seven years ago. For SARS-CoV-2, there appears to be significant room for improvement, and more specialized tools (e.g., Gao et al. [82]) may be more effective. While several of the surveyed studies used in silico tools to assess the safety (allergenicity, toxicity) of SARS-CoV-2 epitopes, the accuracy of these predictions could not be validated due to the lack of experimental data. Discordance in the prediction of some of these safety assessment tools (Section 4) also highlights the need for further research and systematic experimental validation. This is an important research direction since the utility of such in silico tools in pre-clinical trials is gaining recognition by both regulatory bodies and funding agencies [187,188], as it is in line with the principles of 3Rs (replacement, reduction, refinement) for humane animal research.
Several experimentally-determined epitopes associated with HLA-A*02:01 appear to be immunodominant across multiple convalescent COVID-19 patients. Interestingly, the majority of these epitopes were predicted to have promiscuous HLA association by multiple methods. This suggests that vaccines designed to target such epitopes have the potential to provide high population coverage. However, the promiscuity of these epitopes remains to be verified experimentally, and this would appear to be an important direction for future studies.
In this review we have focused on T cells, which form one arm of the adaptive immune system. The other arm, comprising antibodies produced by B cells, is also important for preventing viral infection. In fact, recent experimental studies have suggested that protection against SARS-CoV-2 may be mediated collectively by both T cells and antibodies [11]. There have been extensive efforts in characterizing the neutralizing antibodies against SARS-CoV-2 [[3], [4], [5]], as well as in identifying the B cell epitopes which may be targeted by neutralizing antibodies. Of the in silico SARS-CoV-2 studies that have been reviewed (Table 1), many had also predicted B cell epitopes for potentially eliciting a neutralizing antibody response. Some of these epitope predictions, particularly those made by methods leveraging SARS-CoV data [20,22], have also been observed experimentally [35,[189], [190], [191], [192], [193]]. The development of in silico methods to identify B cell epitopes for SARS-CoV-2 is currently an active area of research. Like for the case of T cells, ML methods may also be considered for predicting B cell epitopes (e.g., [194]), however developing predictive models for B cells is more complicated since the predicted epitopes must fold into conformations that are similar to the native protein for eliciting an antibody response.
For SARS-CoV-2, as well as other coronaviruses, one feature that simplifies the identification of potentially robust epitopes is the fact that the genetic variation is quite low. For example, almost all (~99%) of the epitopes that were predicted by a study in early 2020 [24] are still highly conserved (>99%) within SARS-CoV-2 sequences [35], despite a three orders of magnitude increase in the amount of sequence data available. Hence, based on our current understanding, T cell escape by genetic variation may not be a significant factor for SARS-CoV-2. This is in contrast to other viruses that are highly mutable, such as HIV and hepatitis C virus, for which more elaborate computational methods have been developed to facilitate robust T cell epitope identification and to aid vaccine design [[195], [196], [197], [198], [199], [200], [201], [202], [203], [204], [205], [206], [207]].
Generally speaking, the knowledge being gained through the broad application of in silico T cell epitope prediction methods and tools to SARS-CoV-2 can help guide further studies aimed at epitope determination and vaccine design for various other viruses. For example, the observed cross-reactivity of T cell epitopes between SARS-CoV and SARS-CoV-2 motivate studies that seek to identify epitopes that are genetically similar across a spectrum of coronaviruses (e.g., SARS-CoV, MERS-CoV, SARS-CoV-2, common cold human coronaviruses, as well as animal coronaviruses). The identification of such epitopes could help guide “pan-coronavirus” vaccine designs that are aimed at safeguarding against both current human coronaviruses and novel coronaviruses that may leap from other species to infect humans in the future [208,209]. An increased understanding of the landscape of SARS-CoV-2 immunogenic T cell epitopes targeted by COVID-19 patients would open up the space of possibilities to explore, and this could play an important role in the search for a pan-coronavirus vaccine.
Acknowledgments
The authors were supported by the General Research Fund of the Hong Kong Research Grants Council (RGC) [Grant No. 16204519 and 16201620]. S.F.A. was additionally supported by the Hong Kong Ph.D. Fellowship Scheme (HKPFS).
References
- 1.FDA COVID-19 vaccines. 2020. https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/covid-19-vaccines (accessed December 30, 2020)
- 2.Le T.T., Cramer J.P., Chen R., Mayhew S. Evolution of the COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020;19:667–668. doi: 10.1038/d41573-020-00151-8. [DOI] [PubMed] [Google Scholar]
- 3.Yong S.E.F., Anderson D.E., Wei W.E., Pang J., Chia W.N., Tan C.W., Teoh Y.L., Rajendram P., Toh M.P.H.S., Poh C., Koh V.T.J., Lum J., Suhaimi N.-A.M., Chia P.Y., Chen M.I.-C., Vasoo S., Ong B., Leo Y.S., Wang L., Lee V.J.M. Connecting clusters of COVID-19: an epidemiological and serological investigation. Lancet Infect. Dis. 2020;20:809–815. doi: 10.1016/s1473-3099(20)30273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zohar T., Alter G. Dissecting antibody-mediated protection against SARS-CoV-2. Nat. Rev. Immunol. 2020;20:392–394. doi: 10.1038/s41577-020-0359-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu F., Wang A., Liu M., Wang Q., Chen J., Xia S., Ling Y., Zhang Y., Xun J., Lu L., Jiang S., Lu H., Wen Y., Huang J. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. MedRxiv. 2020 doi: 10.1101/2020.03.30.20047365. [DOI] [Google Scholar]
- 6.Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S., Marrama D., de Silva A.M., Frazier A., Carlin A.F., Greenbaum J.A., Peters B., Krammer F., Smith D.M., Crotty S., Sette A. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiskopf D., Schmitz K.S., Raadsen M.P., Grifoni A., Okba N.M.A., Endeman H., van den Akker J.P.C., Molenkamp R., Koopmans M.P.G., van Gorp E.C.M., Haagmans B.L., de Swart R.L., Sette A., de Vries R.D. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.abd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snyder T.M., Gittelman R.M., Klinger M., May D.H., Osborne E.J., Taniguchi R., Zahid H.J., Kaplan I.M., Dines J.N., Noakes M.N., Pandya R., Chen X., Elasady S., Svejnoha E., Ebert P., Pesesky M.W., De Almeida P., O’Donnell H., DeGottardi Q., Keitany G., Lu J., Vong A., Elyanow R., Fields P., Greissl J., Baldo L., Semprini S., Cerchione C., Mazza M., Delmonte O.M., Dobbs K., Carreño-Tarragona G., Barrio S., Imberti L., Sottini A., Quiros-Roldan E., Rossi C., Biondi A., Bettini L.R., D’Angio M., Bonfanti P., Tompkins M.F., Alba C., Dalgard C., Sambri V., Martinelli G., Goldman J.D., Heath J.R., Su H.C., Notarangelo L.D., Martinez-Lopez J., Carlson J.M., Robins H.S. Magnitude and dynamics of the T-cell response to SARS-CoV-2 infection at both individual and population levels. MedRxiv. 2020 doi: 10.1101/2020.07.31.20165647. [DOI] [Google Scholar]
- 9.Nelde A., Bilich T., Heitmann J.S., Maringer Y., Salih H.R., Roerden M., Lübke M., Bauer J., Rieth J., Wacker M., Peter A., Hörber S., Traenkle B., Kaiser P.D., Rothbauer U., Becker M., Junker D., Krause G., Strengert M., Schneiderhan-Marra N., Templin M.F., Joos T.O., Kowalewski D.J., Stos-Zweifel V., Fehr M., Rabsteyn A., Mirakaj V., Karbach J., Jäger E., Graf M., Gruber L.-C., Rachfalski D., Preuß B., Hagelstein I., Märklin M., Bakchoul T., Gouttefangeas C., Kohlbacher O., Klein R., Stevanović S., Rammensee H.-G., Walz J.S. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat. Immunol. 2021;22:74–85. doi: 10.1038/s41590-020-00808-x. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X., Tan Y., Ling Y., Lu G., Liu F., Yi Z., Jia X., Wu M., Shi B., Xu S., Chen J., Wang W., Chen B., Jiang L., Yu S., Lu J., Wang J., Xu M., Yuan Z., Zhang Q., Zhang X., Zhao G., Wang S., Chen S., Lu H. Viral and host factors related to the clinical outcome of COVID-19. Nature. 2020;583:437–440. doi: 10.1038/s41586-020-2355-0. [DOI] [PubMed] [Google Scholar]
- 11.Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J., Kato Y., Crotty E.G., Kim C., Rawlings S.A., Mateus J., Tse L.P.V., Frazier A., Baric R., Peters B., Greenbaum J., Ollmann Saphire E., Smith D.M., Sette A., Crotty S. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell. 2020:1–17. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586:516–527. doi: 10.1038/s41586-020-2798-3. [DOI] [PubMed] [Google Scholar]
- 13.Reed S.G., Orr M.T., Fox C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013;19:1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 14.Vartak A., Sucheck S. Recent advances in subunit vaccine carriers. Vaccines. 2016;4:12. doi: 10.3390/vaccines4020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson J., Barker D.J., Georgiou X., Cooper M.A., Flicek P., Marsh S.G.E. IPD-IMGT/HLA database. Nucleic Acids Res. 2020;48:D948–D955. doi: 10.1093/nar/gkz950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abelin J.G., Keskin D.B., Sarkizova S., Hartigan C.R., Zhang W., Sidney J., Stevens J., Lane W., Zhang G.L., Eisenhaure T.M., Clauser K.R., Hacohen N., Rooney M.S., Carr S.A., Wu C.J. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–326. doi: 10.1016/j.immuni.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reynisson B., Alvarez B., Paul S., Peters B., Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–W454. doi: 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abelin J.G., Harjanto D., Malloy M., Suri P., Colson T., Goulding S.P., Creech A.L., Serrano L.R., Nasir G., Nasrullah Y., McGann C.D., Velez D., Ting Y.S., Poran A., Rothenberg D.A., Chhangawala S., Rubinsteyn A., Hammerbacher J., Gaynor R.B., Fritsch E.F., Greshock J., Oslund R.C., Barthelme D., Addona T.A., Arieta C.M., Rooney M.S. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity. 2019;51:766–779.e17. doi: 10.1016/j.immuni.2019.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Ahmed S.F., Quadeer A.A., McKay M.R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. 2020;12:254. doi: 10.3390/v12030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyun-Jung Lee C., Koohy H. In silico identification of vaccine targets for 2019-nCoV. F1000Research. 2020;9:145. doi: 10.12688/f1000research.22507.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grifoni A., Sidney J., Zhang Y., Scheuermann R.H., Peters B., Sette A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe. 2020;27:1–10. doi: 10.1016/j.chom.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranga V., Niemelä E., Tamirat M.Z., Eriksson J.E., Airenne T.T., Johnson M.S. Immunogenic SARS-CoV-2 epitopes: in silico study towards better understanding of COVID-19 disease—paving the way for vaccine development. Vaccines. 2020;8:408. doi: 10.3390/vaccines8030408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., Chen J., Meng Y., Wang J., Lin Y., Yuan J., Xie Z., Ma J., Liu W.J., Wang D., Xu W., Holmes E.C., Gao G.F., Wu G., Chen W., Shi W., Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., Chen H.-D., Chen J., Luo Y., Guo H., Jiang R.-D., Liu M.-Q., Chen Y., Shen X.-R., Wang X., Zheng X.-S., Zhao K., Chen Q.-J., Deng F., Liu L.-L., Yan B., Zhan F.-X., Wang Y.-Y., Xiao G.-F., Shi Z.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng O.-W., Chia A., Tan A.T., Jadi R.S., Leong H.N., Bertoletti A., Tan Y.-J. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine. 2016;34:2008–2014. doi: 10.1016/j.vaccine.2016.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welsh R.M., Che J.W., Brehm M.A., Selin L.K. Heterologous immunity between viruses. Immunol. Rev. 2010;235:244–266. doi: 10.1111/j.0105-2896.2010.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen J., Tang W.W., Sheets N., Ellison J., Sette A., Kim K., Shresta S. Identification of Zika virus epitopes reveals immunodominant and protective roles for dengue virus cross-reactive CD8+ T cells. Nat. Microbiol. 2017;2 doi: 10.1038/nmicrobiol.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slon Campos J.L., Mongkolsapaya J., Screaton G.R. The immune response against flaviviruses. Nat. Immunol. 2018;19:1189–1198. doi: 10.1038/s41590-018-0210-3. [DOI] [PubMed] [Google Scholar]
- 30.Ahmed S.F., Quadeer A.A., Barton J.P., McKay M.R. Cross-serotypically conserved epitope recommendations for a universal T cell-based dengue vaccine. PLoS Negl. Trop. Dis. 2020;14 doi: 10.1371/journal.pntd.0008676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adikari T.N., Di Giallonardo F., Leung P., Grifoni A., Sette A., Weiskopf D., Bull R.A., Luciani F. Conserved epitopes with high HLA-I population coverage are targets of CD8+ T cells associated with high IFN-γ responses against all dengue virus serotypes. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-77565-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pickett B.E., Sadat E.L., Zhang Y., Noronha J.M., Squires R.B., Hunt V., Liu M., Kumar S., Zaremba S., Gu Z., Zhou L., Larson C.N., Dietrich J., Klem E.B., Scheuermann R.H. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012;40:D593–D598. doi: 10.1093/nar/gkr859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuiken C., Leitner T., Hahn B., Mullins J., Wolinsky S., Foley B., Apetrei C., Mizrachi I., Rambaut A., Korber B. 2010. HIV Sequence Compendium. [Google Scholar]
- 34.Singer J.B., Thomson E.C., McLauchlan J., Hughes J., Gifford R.J. GLUE: a flexible software system for virus sequence data. BMC Bioinformatics. 2018;19:532. doi: 10.1186/s12859-018-2459-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmed S.F., Quadeer A.A., McKay M.R. COVIDep: a web-based platform for real-time reporting of vaccine target recommendations for SARS-CoV-2. Nat. Protoc. 2020;15:2141–2142. doi: 10.1038/s41596-020-0358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nielsen M., Lundegaard C., Worning P., Lauemøller S.L., Lamberth K., Buus S., Brunak S., Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoof I., Peters B., Sidney J., Pedersen L.E., Sette A., Lund O., Buus S., Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nielsen M., Lundegaard C., Blicher T., Lamberth K., Harndahl M., Justesen S., Røder G., Peters B., Sette A., Lund O., Buus S. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One. 2007;2 doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stranzl T., Larsen M.V., Lundegaard C., Nielsen M. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics. 2010;62:357–368. doi: 10.1007/s00251-010-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andreatta M., Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32:511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Donnell T.J., Rubinsteyn A., Bonsack M., Riemer A.B., Laserson U., Hammerbacher J. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 2018;7:129–132.e4. doi: 10.1016/j.cels.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 42.Jensen K.K., Andreatta M., Marcatili P., Buus S., Greenbaum J.A., Yan Z., Sette A., Peters B., Nielsen M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154:394–406. doi: 10.1111/imm.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karosiene E., Rasmussen M., Blicher T., Lund O., Buus S., Nielsen M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics. 2013;65:711–724. doi: 10.1007/s00251-013-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andreatta M., Karosiene E., Rasmussen M., Stryhn A., Buus S., Nielsen M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics. 2015;67:641–650. doi: 10.1007/s00251-015-0873-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen B., Khodadoust M.S., Olsson N., Wagar L.E., Fast E., Liu C.L., Muftuoglu Y., Sworder B.J., Diehn M., Levy R., Davis M.M., Elias J.E., Altman R.B., Alizadeh A.A. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019;37:1332–1343. doi: 10.1038/s41587-019-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peters B., Nielsen M., Sette A. T cell epitope predictions. Annu. Rev. Immunol. 2020;38:123–145. doi: 10.1146/annurev-immunol-082119-124838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lundegaard C., Lamberth K., Harndahl M., Buus S., Lund O., Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008;36:W509–W512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nielsen M., Lundegaard C., Lund O., Keşmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- 49.Larsen M.V., Lundegaard C., Lamberth K., Buus S., Lund O., Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peters B., Bulik S., Tampe R., van Endert P.M., Holzhütter H.-G. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol. 2003;171:1741–1749. doi: 10.4049/jimmunol.171.4.1741. [DOI] [PubMed] [Google Scholar]
- 51.Bhasin M., Raghava G.P.S. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine. 2004;22:3195–3204. doi: 10.1016/j.vaccine.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 52.Dönnes P., Elofsson A. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinformatics. 2002;3 doi: 10.1186/1471-2105-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh H., Raghava G.P.S. ProPred1: prediction of promiscuous MHC Class-I binding sites. Bioinformatics. 2003;19:1009–1014. doi: 10.1093/bioinformatics/btg108. [DOI] [PubMed] [Google Scholar]
- 55.Singh H., Raghava G.P.S. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236–1237. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- 56.Oyarzún P., Ellis J.J., Bodén M., Kobe B. PREDIVAC: CD4+ T-cell epitope prediction for vaccine design that covers 95% of HLA class II DR protein diversity. BMC Bioinformatics. 2013;14:52. doi: 10.1186/1471-2105-14-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He Y., Xiang Z., Mobley H.L.T. Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J. Biomed. Biotechnol. 2010;2010:1–15. doi: 10.1155/2010/297505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peters B., Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. 2005;6 doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peters B., Tong W., Sidney J., Sette A., Weng Z. Examining the independent binding assumption for binding of peptide epitopes to MHC-I molecules. Bioinformatics. 2003;19:1765–1772. doi: 10.1093/bioinformatics/btg247. [DOI] [PubMed] [Google Scholar]
- 60.Nielsen M., Lundegaard C., Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8:238. doi: 10.1186/1471-2105-8-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sturniolo T., Bono E., Ding J., Raddrizzani L., Tuereci O., Sahin U., Braxenthaler M., Gallazzi F., Protti M.P., Sinigaglia F., Hammer J. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat. Biotechnol. 1999;17:555–561. doi: 10.1038/9858. [DOI] [PubMed] [Google Scholar]
- 62.Guan P., Doytchinova I.A., Zygouri C., Flower D.R. MHCPred: a server for quantitative prediction of peptide-MHC binding. Nucleic Acids Res. 2003;31:3621–3624. doi: 10.1093/nar/gkg510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moutaftsi M., Peters B., Pasquetto V., Tscharke D.C., Sidney J., Bui H.-H., Grey H., Sette A. A consensus epitope prediction approach identifies the breadth of murine CD8+ T-cell responses to vaccinia virus. Nat. Biotechnol. 2006;24:817–819. doi: 10.1038/nbt1215. [DOI] [PubMed] [Google Scholar]
- 64.Wang P., Sidney J., Dow C., Mothé B., Sette A., Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 2008;4 doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hundal J., Kiwala S., McMichael J., Miller C.A., Xia H., Wollam A.T., Liu C.J., Zhao S., Feng Y.Y., Graubert A.P., Wollam A.Z., Neichin J., Neveau M., Walker J., Gillanders W.E., Mardis E.R., Griffith O.L., Griffith M. PVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res. 2020;8:409–420. doi: 10.1158/2326-6066.CIR-19-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paul S., Sidney J., Sette A., Peters B. TepiTool: a pipeline for computational prediction of T cell epitope candidates. Curr. Protoc. Immunol. 2016;114:8.19.1–18.19.24. doi: 10.1002/cpim.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lata S., Bhasin M., Raghava G.P.S. In: Immunoinformatics. Flower D.R., editor. Humana Press; Totowa, NJ: 2007. Application of machine learning techniques in predicting MHC binders; pp. 201–215. [DOI] [PubMed] [Google Scholar]
- 68.Pang K.C., Sanders M.T., Monaco J.J., Doherty P.C., Turner S.J., Chen W. Immunoproteasome subunit deficiencies impact differentially on two immunodominant influenza virus-specific CD8+ T cell responses. J. Immunol. 2006;177:7680–7688. doi: 10.4049/jimmunol.177.11.7680. [DOI] [PubMed] [Google Scholar]
- 69.Gallimore A. Protective immunity does not correlate with the hierarchy of virus-specific cytotoxic T cell responses to naturally processed peptides. J. Exp. Med. 1998;187:1647–1657. doi: 10.1084/jem.187.10.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oseroff C., Kos F., Bui H.-H., Peters B., Pasquetto V., Glenn J., Palmore T., Sidney J., Tscharke D.C., Bennink J.R., Southwood S., Grey H.M., Yewdell J.W., Sette A. HLA class I-restricted responses to vaccinia recognize a broad array of proteins mainly involved in virulence and viral gene regulation. Proc. Natl. Acad. Sci. 2005;102:13980–13985. doi: 10.1073/pnas.0506768102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng M., Jin N., Zhang H., Jin M., Lu H., Ma M., Li C., Yin G., Wang R., Liu Q. Construction and immunogenicity of a recombinant fowlpox virus containing the capsid and 3C protease coding regions of foot-and-mouth disease virus. J. Virol. Methods. 2006;136:230–237. doi: 10.1016/j.jviromet.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 72.Owen J.A., Punt J., Stranford S.A. 7th ed. W. H. Freeman; New York: 2013. Kuby Immunology. [Google Scholar]
- 73.Zhang N., Bevan M.J. CD8+ T cells: foot soldiers of the immune system. Immunity. 2011;35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Košmrlj A., Chakraborty A., Kardar M., Shakhnovich E. Thymic selection of T-cell receptors as an extreme value problem. Phys. Rev. Lett. 2009;103 doi: 10.1103/PhysRevLett.103.068103. [DOI] [PubMed] [Google Scholar]
- 75.Kosmrlj A., Read E.L., Qi Y., Allen T.M., Altfeld M., Deeks S.G., Pereyra F., Carrington M., Walker B.D., Chakraborty A.K. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature. 2010;465:350–354. doi: 10.1038/nature08997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rolland M., Nickle D.C., Deng W., Frahm N., Brander C., Learn G.H., Heckerman D., Jojic N., Jojic V., Walker B.D., Mullins J.I. Recognition of HIV-1 peptides by host CTL is related to HIV-1 similarity to human proteins. PLoS One. 2007;2 doi: 10.1371/journal.pone.0000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gebe J.A., Yue B.B., Unrath K.A., Falk B.A., Nepom G.T. Restricted autoantigen recognition associated with deletional and adaptive regulatory mechanisms. J. Immunol. 2009;183:59–65. doi: 10.4049/jimmunol.0804046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Calis J.J.A., de Boer R.J., Keşmir C. Degenerate T-cell recognition of peptides on MHC molecules creates large holes in the T-cell repertoire. PLoS Comput. Biol. 2012;8 doi: 10.1371/journal.pcbi.1002412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Doytchinova I.A., Flower D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Calis J.J.A., Maybeno M., Greenbaum J.A., Weiskopf D., De Silva A.D., Sette A., Keşmir C., Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013;9 doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pogorelyy M.V., Fedorova A.D., McLaren J.E., Ladell K., Bagaev D.V., Eliseev A.V., Mikelov A.I., Koneva A.E., Zvyagin I.V., Price D.A., Chudakov D.M., Shugay M. Exploring the pre-immune landscape of antigen-specific T cells. Genome Med. 2018;10:68. doi: 10.1186/s13073-018-0577-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao A., Chen Z., Segal F.P., Carrington M., Streeck H., Chakraborty A.K., Julg B. Predicting the immunogenicity of T cell epitopes: from HIV to SARS-CoV-2. BioRxiv. 2020 doi: 10.1101/2020.05.14.095885. [DOI] [Google Scholar]
- 83.Prachar M., Justesen S., Steen-Jensen D.B., Thorgrimsen S., Jurgons E., Winther O., Bagger F.O. Identification and validation of 174 COVID-19 vaccine candidate epitopes reveals low performance of common epitope prediction tools. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-77466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schroder K., Hertzog P.J., Ravasi T., Hume D.A. Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 85.Dhanda S.K., Vir P., Raghava G.P.S. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct. 2013;8:30. doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ma Y., Wu L., Shaw N., Gao Y., Wang J., Sun Y., Lou Z., Yan L., Zhang R., Rao Z. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc. Natl. Acad. Sci. 2015;112:9436–9441. doi: 10.1073/pnas.1508686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., Hastie K.M., Parker D.G. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020:1–16. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Larson H.J., Hartigan-Go K., de Figueiredo A. Vaccine confidence plummets in the Philippines following dengue vaccine scare: why it matters to pandemic preparedness. Hum. Vaccines Immunother. 2019;15:625–627. doi: 10.1080/21645515.2018.1522468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Larson H.J., Cooper L.Z., Eskola J., Katz S.L., Ratzan S. Addressing the vaccine confidence gap. Lancet. 2011;378:526–535. doi: 10.1016/S0140-6736(11)60678-8. [DOI] [PubMed] [Google Scholar]
- 90.Sicherer S.H., Sampson H.A. Food allergy. J. Allergy Clin. Immunol. 2010;125:S116–S125. doi: 10.1016/j.jaci.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 91.Saha S., Raghava G.P.S. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34:W202–W209. doi: 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dimitrov I., Flower D.R., Doytchinova I. AllerTOP - a server for in silico prediction of allergens. BMC Bioinformatics. 2013;14:S4. doi: 10.1186/1471-2105-14-S6-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dimitrov I., Bangov I., Flower D.R., Doytchinova I. AllerTOP v.2 - a server for in silico prediction of allergens. J. Mol. Model. 2014;20:2278. doi: 10.1007/s00894-014-2278-5. [DOI] [PubMed] [Google Scholar]
- 94.Dimitrov I., Naneva L., Doytchinova I., Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30:846–851. doi: 10.1093/bioinformatics/btt619. [DOI] [PubMed] [Google Scholar]
- 95.Maurer-Stroh S., Krutz N.L., Kern P.S., Gunalan V., Nguyen M.N., Limviphuvadh V., Eisenhaber F., Gerberick G.F. AllerCatPro—prediction of protein allergenicity potential from the protein sequence. Bioinformatics. 2019;35:3020–3027. doi: 10.1093/bioinformatics/btz029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Crooke S.N., Ovsyannikova I.G., Kennedy R.B., Poland G.A. Immunoinformatic identification of B cell and T cell epitopes in the SARS-CoV-2 proteome. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-70864-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baker D.J. Toxic Trauma. Springer International Publishing; Cham: 2016. Chapter 3: The classification and properties of toxic hazards; pp. 25–47. [DOI] [Google Scholar]
- 98.Gupta S., Kapoor P., Chaudhary K., Gautam A., Kumar R., Raghava G.P.S. In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8 doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Consortium T.U. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nagy Z.A. A History of Modern Immunology. Elsevier; 2014. Chapter 10: autoimmunity. [DOI] [Google Scholar]
- 101.Janeway C., Travers P., Walport M., Shlomchik M. Immunobiology: The Immune System in Health and Disease. Garland Science; 2001. Chapter 13: autoimmunity and transplantation. [Google Scholar]
- 102.Oldstone M.B.A. Molecular mimicry: its evolution from concept to mechanism as a cause of autoimmune diseases. Monoclon. Antib. Immunodiagn. Immunother. 2014;33:158–165. doi: 10.1089/mab.2013.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schattner A. Consequence or coincidence? The occurrence, pathogenesis and significance of autoimmune manifestations after viral vaccines. Vaccine. 2005;23:3876–3886. doi: 10.1016/j.vaccine.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 104.Nakayama T. Causal relationship between immunological responses and adverse reactions following vaccination. Vaccine. 2019;37:366–371. doi: 10.1016/j.vaccine.2018.11.045. [DOI] [PubMed] [Google Scholar]
- 105.Seib K.L., Dougan G., Rappuoli R. The key role of genomics in modern vaccine and drug design for emerging infectious diseases. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Amela I., Cedano J., Querol E. Pathogen proteins eliciting antibodies do not share epitopes with host proteins: a bioinformatics approach. PLoS One. 2007;2 doi: 10.1371/journal.pone.0000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kanduc D. Peptide cross-reactivity: the original sin of vaccines. Front. Biosci. 2012;S4:341. doi: 10.2741/s341. [DOI] [PubMed] [Google Scholar]
- 108.Liu G., Carter B., Bricken T., Jain S., Viard M., Carrington M., Gifford D.K. Computationally optimized SARS-CoV-2 MHC class I and II vaccine formulations predicted to target human haplotype distributions. Cell Syst. 2020;11:131–144.e6. doi: 10.1016/j.cels.2020.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rauch S., Jasny E., Schmidt K.E., Petsch B. New vaccine technologies to combat outbreak situations. Front. Immunol. 2018;9 doi: 10.3389/fimmu.2018.01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brisse M., Vrba S.M., Kirk N., Liang Y., Ly H. Emerging concepts and technologies in vaccine development. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.583077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Loomis R.J., Johnson P.R. Emerging vaccine technologies. Vaccines. 2015;3:429–447. doi: 10.3390/vaccines3020429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baruah V., Bose S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J. Med. Virol. 2020;92:495–500. doi: 10.1002/jmv.25698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ojha R., Gupta N., Naik B., Singh S., Verma V.K., Prusty D., Prajapati V.K. High throughput and comprehensive approach to develop multiepitope vaccine against minacious COVID-19. Eur. J. Pharm. Sci. 2020;151 doi: 10.1016/j.ejps.2020.105375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang D., Mai J., Zhou W., Yu W., Zhan Y., Wang N., Epstein N.D., Yang Y. Immunoinformatic analysis of T- and B-cell epitopes for SARS-CoV-2 vaccine design. Vaccines. 2020;8:355. doi: 10.3390/vaccines8030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Poran A., Harjanto D., Malloy M., Arieta C.M., Rothenberg D.A., Lenkala D., van Buuren M.M., Addona T.A., Rooney M.S., Srinivasan L., Gaynor R.B. Sequence-based prediction of SARS-CoV-2 vaccine targets using a mass spectrometry-based bioinformatics predictor identifies immunogenic T cell epitopes. Genome Med. 2020;12:70. doi: 10.1186/s13073-020-00767-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Qamar M.T. ul, Rehman A., Ashfaq U.A., Awan M.Q., Fatima I., Shahid F., Chen L.-L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-CoV-2: immunoinformatics and in silico approaches. BioRxiv. 2020 doi: 10.1101/2020.02.28.970343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gupta A.K., Khan M.S., Choudhury S., Mukhopadhyay A., Sakshi A. Rastogi, Thakur A., Kumari P., Kaur M., Shalu C. Saini, Sapehia V., Barkha P.K. Patel, Bhamare K.T., Kumar M. CoronaVR: a computational resource and analysis of epitopes and therapeutics for severe acute respiratory syndrome coronavirus-2. Front. Microbiol. 2020;11:1–21. doi: 10.3389/fmicb.2020.01858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Enayatkhani M., Hasaniazad M., Faezi S., Gouklani H., Davoodian P., Ahmadi N., Einakian M.A., Karmostaji A., Ahmadi K. Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: an in silico study. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2020.1756411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ong E., Wong M.U., Huffman A., He Y. COVID-19 coronavirus vaccine design using reverse vaccinology and machine learning. Front. Immunol. 2020;11:1581. doi: 10.3389/fimmu.2020.01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abdelmageed M.I., Abdelmoneim A.H., Mustafa M.I., Elfadol N.M., Murshed N.S., Shantier S.W., Makhawi A.M. Design of a multiepitope-based peptide vaccine against the E protein of human COVID-19: an immunoinformatics approach. Biomed. Res. Int. 2020;2020:1–12. doi: 10.1155/2020/2683286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mukherjee S., Tworowski D., Detroja R., Mukherjee S.B., Frenkel-Morgenstern M. Immunoinformatics and structural analysis for identification of immunodominant epitopes in SARS-CoV-2 as potential vaccine targets. Vaccines. 2020;8:290. doi: 10.3390/vaccines8020290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vashi Y., Jagrit V., Kumar S. Understanding the B and T cell epitopes of spike protein of severe acute respiratory syndrome coronavirus-2: a computational way to predict the immunogens. Infect. Genet. Evol. 2020;84 doi: 10.1016/j.meegid.2020.104382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ahmad S., Navid A., Farid R., Abbas G., Ahmad F., Zaman N., Parvaiz N., Azam S.S. Design of a novel multi epitope-based vaccine for pandemic coronavirus disease (COVID-19) by vaccinomics and probable prevention strategy against avenging zoonotics. Eur. J. Pharm. Sci. 2020;151 doi: 10.1016/j.ejps.2020.105387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Naz A., Shahid F., Butt T.T., Awan F.M., Ali A., Malik A. Designing multi-epitope vaccines to combat emerging coronavirus disease 2019 (COVID-19) by employing immuno-informatics approach. Front. Immunol. 2020;11:1663. doi: 10.3389/fimmu.2020.01663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen H.-Z., Tang L.-L., Yu X.-L., Zhou J., Chang Y.-F., Wu X. Bioinformatics analysis of epitope-based vaccine design against the novel SARS-CoV-2. Infect. Dis. Poverty. 2020;9:88. doi: 10.1186/s40249-020-00713-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martin W.R., Cheng F. A rational design of a multi-epitope vaccine against SARS-CoV-2 which accounts for the glycan shield of the spike glycoprotein. ChemRxiv. 2020 doi: 10.26434/chemrxiv.12770225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dong R., Chu Z., Yu F., Zha Y. Contriving multi-epitope subunit of vaccine for COVID-19: immunoinformatics approaches. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.01784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ghafouri F., Cohan R.A., Noorbakhsh F., Samimi H., Haghpanah V. An in-silico approach to develop of a multi-epitope vaccine candidate against SARS-CoV-2 envelope (E) protein. Res. Sq. 2020 doi: 10.21203/rs.3.rs-30374/v1. [DOI] [Google Scholar]
- 129.Banerjee S., Majumder K., Gutierrez G.J., Gupta D., Mittal B. Immuno-informatics approach for multi-epitope vaccine designing against SARS-CoV-2. BioRvix. 2020 doi: 10.1101/2020.07.23.218529. [DOI] [Google Scholar]
- 130.Samad A., Ahammad F., Nain Z., Alam R., Imon R.R., Hasan M., Rahman M.S. Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J. Biomol. Struct. Dyn. 2020;162:1–17. doi: 10.1080/07391102.2020.1792347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bhatnager R., Bhasin M., Arora J., Dang A.S. Epitope based peptide vaccine against SARS-COV2: an immune-informatics approach. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2020.1787227. [DOI] [PubMed] [Google Scholar]
- 132.Devi A., Chaitanya N.S.N. In silico designing of multi-epitope vaccine construct against human coronavirus infections. J. Biomol. Struct. Dyn. 2020:1–15. doi: 10.1080/07391102.2020.1804460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Abraham Peele K., Srihansa T., Krupanidhi S., Vijaya Sai A., Venkateswarulu T.C. Design of multi-epitope vaccine candidate against SARS-CoV-2: a in-silico study. J. Biomol. Struct. Dyn. 2020:1–9. doi: 10.1080/07391102.2020.1770127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ismail S., Ahmad S., Azam S.S. Immunoinformatics characterization of SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. J. Mol. Liq. 2020;314 doi: 10.1016/j.molliq.2020.113612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jakhar R., Kaushik S., Gakhar S.K. 3CL hydrolase-based multiepitope peptide vaccine against SARS-CoV-2 using immunoinformatics. J. Med. Virol. 2020:2114–2123. doi: 10.1002/jmv.25993. [DOI] [PubMed] [Google Scholar]
- 136.Panda P.K., Arul M.N., Patel P., Verma S.K., Luo W., Rubahn H.G., Mishra Y.K., Suar M., Ahuja R. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 2020;6:1–15. doi: 10.1126/sciadv.abb8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Campbell K.M., Steiner G., Wells D.K., Ribas A., Kalbasi A. Prioritization of SARS-CoV-2 epitopes using a pan-HLA and global population inference approach. BioRxiv. 2020 doi: 10.1101/2020.03.30.016931. [DOI] [Google Scholar]
- 138.Tilocca B., Soggiu A., Sanguinetti M., Babini G., De Maio F., Britti D., Zecconi A., Bonizzi L., Urbani A., Roncada P. Immunoinformatic analysis of the SARS-CoV-2 envelope protein as a strategy to assess cross-protection against COVID-19. Microbes Infect. 2020;22:182–187. doi: 10.1016/j.micinf.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Santoni D., Vergni D. In the search of potential epitopes for Wuhan seafood market pneumonia virus using high order nullomers. J. Immunol. Methods. 2020;481–482 doi: 10.1016/j.jim.2020.112787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dijkstra J.M., Hashimoto K. Expected immune recognition of COVID-19 virus by memory from earlier infections with common coronaviruses in a large part of the world population. F1000Research. 2020;9 doi: 10.12688/f1000research.23458.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ramaiah A., Arumugaswami V. Insights into cross-species evolution of novel human coronavirus 2019-nCoV and defining immune determinants for vaccine development. BioRxiv. 2020 doi: 10.1101/2020.01.29.925867. [DOI] [Google Scholar]
- 142.Gupta E., Mishra R.K., Niraj R.R.K. Identification of potential vaccine candidates against SARS-CoV-2, A step forward to fight novel coronavirus 2019-nCoV: a Reverse Vaccinology Approach. BioRxiv. 2020 doi: 10.1101/2020.04.13.039198. [DOI] [Google Scholar]
- 143.Srivastava S., Verma S., Kamthania M., Kaur R., Badyal R.K., Saxena A.K., Shin H.-J., Kolbe M., Pandey K.C. Structural basis for designing multiepitope vaccines against COVID-19 infection: in silico vaccine design and validation. JMIR Bioinforma. Biotechnol. 2020;1 doi: 10.2196/19371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mitra D., Pandey J., Swaroop S. Multi-epitope-based peptide vaccine design against SARS-CoV-2 using its spike protein. BioRxiv. 2020 doi: 10.1101/2020.04.23.055467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Singh A., Thakur M., Sharma L.K., Chandra K. Designing a multi-epitope peptide-based vaccine against SARS-CoV-2. BioRxiv. 2020 doi: 10.1101/2020.04.15.040618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Saha R., Prasad B.V.L.S. In silico approach for designing of a multi-epitope based vaccine against novel Coronavirus (SARS-CoV-2) BioRxiv. 2020 doi: 10.1101/2020.03.31.017459. [DOI] [Google Scholar]
- 147.Nerli S., Sgourakis N.G. Structure-based modeling of SARS-CoV-2 peptide/HLA-A02 antigens. BioRxiv. 2020 doi: 10.1101/2020.03.23.004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Khan A., Alam A., Imam N., Siddiqui M.F., Ishrat R. Design of an epitope-based peptide vaccine against the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): a vaccine informatics approach. BioRxiv. 2020 doi: 10.1101/2020.05.03.074930. 2020.05.03.074930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Banerjee A., Santra D., Maiti S. Energetics and IC50 based epitope screening in SARS CoV-2 (COVID 19) spike protein by immunoinformatic analysis implicating for a suitable vaccine development. J. Transl. Med. 2020;18:281. doi: 10.1186/s12967-020-02435-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bojin F., Gavriliuc O., Margineanu M., Paunescu V. Design of an epitope-based synthetic long peptide vaccine to counteract the novel China coronavirus (2019-nCoV) 2020. https://www.preprints.org/manuscript/202002.0102/v1 Preprints.
- 151.Nazneen Akhand M.R., Azim K.F., Hoque S.F., Moli M.A., Das Joy B., Akter H., Afif I.K., Ahmed N., Hasan M. Genome based evolutionary study of SARS-CoV-2 towards the prediction of epitope based chimeric vaccine. BioRxiv. 2020 doi: 10.1101/2020.04.15.036285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Feng Y., Qiu M., Zou S., Li Y., Luo K., Chen R., Sun Y., Wang K., Zhuang X., Zhang S., Chen S., Mo F. Multi-epitope vaccine design using an immunoinformatics approach for 2019 novel coronavirus in China (SARS-CoV-2) BioRxiv. 2020 doi: 10.1101/2020.03.03.962332. [DOI] [Google Scholar]
- 153.Bhattacharya M., Sharma A.R., Patra P., Ghosh P., Sharma G., Patra B.C., Lee S., Chakraborty C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-CoV-2): immunoinformatics approach. J. Med. Virol. 2020;92:618–631. doi: 10.1002/jmv.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chauhan V., Rungta T., Rawat M., Goyal K., Gupta Y., Singh M.P. Excavating SARS-coronavirus 2 genome for epitope-based subunit vaccine synthesis using immunoinformatics approach. J. Cell. Physiol. 2020 doi: 10.1002/jcp.29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fast E., Altman R.B., Chen B. Potential T-cell and B-cell epitopes of 2019-nCoV. BioRxiv. 2020;XXX:1–9. doi: 10.1101/2020.02.19.955484. [DOI] [Google Scholar]
- 156.Joshi A., Joshi B.C., Mannan M.A., Kaushik V. Epitope based vaccine prediction for SARS-CoV-2 by deploying immuno-informatics approach. Informatics Med. Unlocked. 2020;19 doi: 10.1016/j.imu.2020.100338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kar T., Narsaria U., Basak S., Deb D., Castiglione F., Mueller D.M., Srivastava A.P. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-67749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Qamar M.T. ul, Shahid F., Ashfaq U.A., Aslam S., Fatima I., Fareed M.M., Zohaib A., Chen L.-L. 2020. Structural modeling and conserved epitopes prediction against SARS-COV-2 structural proteins for vaccine development; pp. 1–33. [DOI] [Google Scholar]
- 159.Ahammad I., Lira S.S. Designing a novel mRNA vaccine against SARS-CoV-2: an immunoinformatics approach. Int. J. Biol. Macromol. 2020;162:820–837. doi: 10.1016/j.ijbiomac.2020.06.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kiyotani K., Toyoshima Y., Nemoto K., Nakamura Y. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. J. Hum. Genet. 2020;65:569–575. doi: 10.1038/s10038-020-0771-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sarkar B., Ullah M.A., Johora F.T., Taniya M.A., Araf Y. Immunoinformatics-guided designing of epitope-based subunit vaccines against the SARS Coronavirus-2 (SARS-CoV-2) Immunobiology. 2020;225 doi: 10.1016/j.imbio.2020.151955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.J.-Z.L. Romero-López, Carnalla-Cortés M, Pacheco-Olvera DL, Ocampo M, Oliva-Ramírez J, Moreno-Manjón J, Bernal-Alferes B, García-Latorre E, Domínguez-López ML, Arturo Reyes-SandovalPrediction of SARS-CoV2 spike protein epitopes reveals HLA-associated susceptibility, Res. Sq. (n.d.) 1–20. doi.10.21203/rs.3.rs-25844/v1.
- 163.Sanami S., Zandi M., Pourhossein B., Mobini G.-R., Safaei M., Abed A., Arvejeh P.M., Chermahini F.A., Alizadeh M. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int. J. Biol. Macromol. 2020 doi: 10.1016/j.ijbiomac.2020.07.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kalita P., Padhi A.K., Zhang K.Y.J., Tripathi T. Design of a peptide-based subunit vaccine against novel coronavirus SARS-CoV-2. Microb. Pathog. 2020;145 doi: 10.1016/j.micpath.2020.104236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rahman N., Ali F., Basharat Z., Shehroz M., Khan M.K., Jeandet P., Nepovimova E., Kuca K., Khan H. Vaccine design from the ensemble of surface glycoprotein epitopes of SARS-CoV-2: an immunoinformatics approach. Vaccines. 2020;8:1–17. doi: 10.3390/vaccines8030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lin L., Ting S., Yufei H., Wendong L., Yubo F., Jing Z. Epitope-based peptide vaccines predicted against novel coronavirus disease caused by SARS-CoV-2. Virus Res. 2020;288 doi: 10.1016/j.virusres.2020.198082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yarmarkovich M., Warrington J.M., Farrel A., Maris J.M. Identification of SARS-CoV-2 vaccine epitopes predicted to induce long-term population-scale immunity. Cell Reports Med. 2020 doi: 10.1016/j.xcrm.2020.100036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yazdani Z., Rafiei A., Yazdani M., Valadan R. Design an efficient multi-epitope peptide vaccine candidate against SARS-CoV-2: an in silico analysis. Infect. Drug Resist. 2020;13:3007–3022. doi: 10.2147/IDR.S264573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lucchese G. Epitopes for a 2019-nCoV vaccine. Cell. Mol. Immunol. 2020;17:539–540. doi: 10.1038/s41423-020-0377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nunes E., Heslop H., Fernandez-Vina M., Taves C., Wagenknecht D.R., Eisenbrey A.B., Fischer G., Poulton K., Wacker K., Hurley C.K., Noreen H., Sacchi N. Definitions of histocompatibility typing terms. Blood. 2011;118:e180–e183. doi: 10.1182/blood-2011-05-353490. [DOI] [PubMed] [Google Scholar]
- 171.O’Brien C., Flower D.R., Feighery C. Peptide length significantly influences in vitro affinity for MHC class II molecules. Immunome Res. 2008;4:6. doi: 10.1186/1745-7580-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Trolle T., McMurtrey C.P., Sidney J., Bardet W., Osborn S.C., Kaever T., Sette A., Hildebrand W.H., Nielsen M., Peters B. The length distribution of class I–restricted T cell epitopes is determined by both peptide supply and MHC allele–specific binding preference. J. Immunol. 2016;196:1480–1487. doi: 10.4049/jimmunol.1501721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li C.K.-F., Wu H., Yan H., Ma S., Wang L., Zhang M., Tang X., Temperton N.J., Weiss R.A., Brenchley J.M., Douek D.C., Mongkolsapaya J., Tran B.-H., Lin C.-L.S., Screaton G.R., Hou J.-L., Mcmichael A.J., Xu X.-N. T cell responses to whole SARS coronavirus in humans. J. Immunol. 2008;181:5490–5500. doi: 10.4049/jimmunol.181.8.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Channappanavar R., Fett C., Zhao J., Meyerholz D.K., Perlman S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J. Virol. 2014;88:11034–11044. doi: 10.1128/jvi.01505-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Roider J., Meissner T., Kraut F., Vollbrecht T., Stirner R., Bogner J.R., Draenert R. Comparison of experimental fine-mapping to in silico prediction results of HIV-1 epitopes reveals ongoing need for mapping experiments. Immunology. 2014;143:193–201. doi: 10.1111/imm.12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Le Bert N., Tan A.T., Kunasegaran K., Tham C.Y.L., Hafezi M., Chia A., Chng M.H.Y., Lin M., Tan N., Linster M., Chia W.N., Chen M.I., Wang L., Ooi E.E., Kalimuddin S., Tambyah P.A., Low J.G., Tan Y., Bertoletti A. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584:457–462. doi: 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- 177.Chour W., Xu A.M., Ng A.H.C., Choi J., Xie J., Yuan D., Lee J.K., Delucia D.C., Edmark R., Jones L., Schmitt T.M., Chaffee M.E., Duvvuri V., Greenberg P.D., Murray K., Wallick J., Algren H.A., Berrington W.R., O’Mahoney D.S., Goldman J.D., Heath J.R. Shared antigen-specific CD8+ T cell responses against the SARS-CoV-2 spike protein in HLA A*02:01 COVID-19 participants. MedRxiv. 2020 doi: 10.1101/2020.05.04.20085779. 2020.05.04.20085779. [DOI] [Google Scholar]
- 178.Ferretti A.P., Kula T., Wang Y., Nguyen D.M.V., Weinheimer A., Dunlap G.S., Xu Q., Nabilsi N., Perullo C.R., Cristofaro A.W., Whitton H.J., Virbasius A., Olivier K.J., Buckner L.R., Alistar A.T., Whitman E.D., Bertino S.A., Chattopadhyay S., MacBeath G. Unbiased screens show CD8+ T cells of COVID-19 patients recognize shared epitopes in SARS-CoV-2 that largely reside outside the spike Protein. Immunity. 2020;53:1095–1107. doi: 10.1016/j.immuni.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., López-Camacho C., Slon-Campos J., Zhao Y., Stuart D.I., Paesen G.C., Grimes J.M., Antson A.A., Bayfield O.W., Hawkins D.E.D.P., Ker D.-S., Wang B., Turtle L., Subramaniam K., Thomson P., Zhang P., Dold C., Ratcliff J., Simmonds P., de Silva T., Sopp P., Wellington D., Rajapaksa U., Chen Y.-L., Salio M., Napolitani G., Paes W., Borrow P., Kessler B.M., Fry J.W., Schwabe N.F., Semple M.G., Baillie J.K., Moore S.C., Openshaw P.J.M., Ansari M.A., Dunachie S., Barnes E., Frater J., Kerr G., Goulder P., Lockett T., Levin R., Zhang Y., Jing R., Ho L.-P., Cornall R.J., Conlon C.P., Klenerman P., Screaton G.R., Mongkolsapaya J., McMichael A., Knight J.C., Ogg G., Dong T. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Shomuradova A.S., Vagida M.S., Sheetikov S.A., Zornikova K.V., Kiryukhin D., Titov A., Peshkova I.O., Khmelevskaya A., Dianov D.V., Malasheva M., Shmelev A., Serdyuk Y., Bagaev D.V., Pivnyuk A., Shcherbinin D.S., Maleeva A.V., Shakirova N.T., Pilunov A., Malko D.B., Khamaganova E.G., Biderman B., Ivanov A., Shugay M., Efimov G.A. SARS-CoV-2 epitopes are recognized by a public and diverse repertoire of human T cell receptors. Immunity. 2020;53:1245–1257.e5. doi: 10.1016/j.immuni.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Quadeer A.A., Ahmed S.F., McKay M.R. Epitopes targeted by T cells in convalescent COVID-19 patients. BioRxiv. 2020 https://www.biorxiv.org/content/10.1101/2020.08.26.267724v1 2020.08.26.267724. [Google Scholar]
- 182.Schulien I., Kemming J., Oberhardt V., Wild K., Seidel L.M., Killmer S., Sagar F. Daul, Lago M. Salvat, Decker A., Luxenburger H., Binder B., Bettinger D., Sogukpinar O., Rieg S., Panning M., Huzly D., Schwemmle M., Kochs G., Waller C.F., Nieters A., Duerschmied D., Emmerich F., Mei H.E., Schulz A.R., Llewellyn-Lacey S., Price D.A., Boettler T., Bengsch B., Thimme R., Hofmann M., Neumann-Haefelin C. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat. Med. 2020 doi: 10.1038/s41591-020-01143-2. [DOI] [PubMed] [Google Scholar]
- 183.Kula T., Dezfulian M.H., Wang C.I., Abdelfattah N.S., Hartman Z.C., Wucherpfennig K.W., Lyerly H.K., Elledge S.J. T-Scan: a genome-wide method for the systematic discovery of T cell epitopes. Cell. 2019;178 doi: 10.1016/j.cell.2019.07.009. 1016-1028.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gonzalez-Galarza F.F., Christmas S., Middleton D., Jones A.R. Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res. 2011;39:D913–D919. doi: 10.1093/nar/gkq1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tian Y., Grifoni A., Sette A., Weiskopf D. Human T cell response to dengue virus infection. Front. Immunol. 2019;10:1–9. doi: 10.3389/fimmu.2019.02125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Fleri W., Paul S., Dhanda S.K., Mahajan S., Xu X., Peters B., Sette A. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 2017;8 doi: 10.3389/fimmu.2017.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Busquet F., Hartung T., Pallocca G., Rovida C., Leist M. Harnessing the power of novel animal-free test methods for the development of COVID-19 drugs and vaccines. Arch. Toxicol. 2020;94:2263–2272. doi: 10.1007/s00204-020-02787-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.FDA Development and Licensure of Vaccines to Prevent COVID-19. 2020. https://www.fda.gov/media/139638/download (accessed September 8, 2020)
- 189.Ng L.F., Naqiah Amrun S., Lee C.Y.-P., Lee B., Fong S.-W., Young B.E., Sin-Ling Chee R., Kim-Wah Yeo N., Torres-Ruesta A., Carissimo G., Meng Poh C., Wei Chang Z., Zirui Tay M., Chan Y.-H., Chen M.I.-C., Tambyah P.A., Kalimuddin S., Pada S., Tan S.Y., Jin Sun L., Leo Y.-S., Lye D.C., Renia L. Linear B-cell epitopes in the spike and nucleocapsid proteins as markers of SARS- CoV-2 exposure and disease severity. EBioMedicine. 2020;58:102911. doi: 10.1016/j.ebiom.2020.102911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li Y., Ma M., Lei Q., Wang F., Lai D., Hou H., Xu Z., Zhang B., Chen H., Yu C., Xue J., Zheng Y., Wang X., Jiang H., Zhang H., Qi H., Guo S., Zhang Y., Lin X., Yao Z., Wu J., Sheng H., Sun Z., Fan X., Tao S. Linear epitope landscape of SARS-CoV-2 spike protein constructed from 1,051 COVID-19 patients. SSRN Electron. J. 2020 doi: 10.2139/ssrn.3671941. 2020.07.13.20152587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Pinto D., Park Y., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., Peter A., Guarino B., Spreafico R., Cameroni E., Case J.B., Chen R.E., Havenar-Daughton C., Snell G., Telenti A., Virgin H.W., Lanzavecchia A., Diamond M.S., Fink K., Veesler D., Corti D. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020 doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- 192.Poh C.M., Carissimo G., Wang B., Amrun S.N., Lee C.Y.-P., Chee R.S.-L., Fong S.-W., Yeo N.K.-W., Lee W.-H., Torres-Ruesta A., Leo Y.-S., Chen M.I.-C., Tan S.-Y., Chai L.Y.A., Kalimuddin S., Kheng S.S.G., Thien S.-Y., Young B.E., Lye D.C., Hanson B.J., Wang C.-I., Renia L., Ng L.F.P. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 2020;11:2806. doi: 10.1038/s41467-020-16638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Quadeer A.A. Immunodominant epitopes based serological assay for detecting SARS-CoV-2 exposure: promises and challenges. EBioMedicine. 2020;59 doi: 10.1016/j.ebiom.2020.102947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jespersen M.C., Peters B., Nielsen M., Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45:W24–W29. doi: 10.1093/nar/gkx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chakraborty A.K., Barton J.P. Rational design of vaccine targets and strategies for HIV: a crossroad of statistical physics, biology, and medicine. Reports Prog. Phys. 2017;80 doi: 10.1088/1361-6633/aa574a. [DOI] [PubMed] [Google Scholar]
- 196.Gaiha G.D., Rossin E.J., Urbach J., Landeros C., Collins D.R., Nwonu C., Muzhingi I., Anahtar M.N., Waring O.M., Piechocka-Trocha A., Waring M., Worrall D.P., Ghebremichael M.S., Newman R.M., Power K.A., Allen T.M., Chodosh J., Walker B.D. Structural topology defines protective CD8+ T cell epitopes in the HIV proteome. Science. 2019;364:480–484. doi: 10.1126/science.aav5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sohail M.S., Louie R.H.Y., McKay M.R., Barton J.P. MPL resolves genetic linkage in fitness inference from complex evolutionary histories. Nat. Biotechnol. 2020 doi: 10.1038/s41587-020-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Quadeer A.A., Louie R.H.Y., McKay M.R. Identifying immunologically-vulnerable regions of the HCV E2 glycoprotein and broadly neutralizing antibodies that target them. Nat. Commun. 2019;10:2073. doi: 10.1038/s41467-019-09819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Quadeer A.A., Barton J.P., Chakraborty A.K., McKay M.R. Deconvolving mutational patterns of poliovirus outbreaks reveals its intrinsic fitness landscape. Nat. Commun. 2020;11:377. doi: 10.1038/s41467-019-14174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ahmed S.F., Quadeer A.A., Morales-Jimenez D., McKay M.R. Sub-dominant principal components inform new vaccine targets for HIV Gag. Bioinformatics. 2019;35:3884–3889. doi: 10.1093/bioinformatics/btz524. [DOI] [PubMed] [Google Scholar]
- 201.Sohail M.S., Quadeer A.A., McKay M.R. How genetic sequence data can guide vaccine design. IEEE Potentials. 2020;39:31–37. doi: 10.1109/MPOT.2020.2967896. [DOI] [Google Scholar]
- 202.Ferguson A.L., Mann J.K., Omarjee S., Ndung’u T., Walker B.D., Chakraborty A.K. Translating HIV sequences into quantitative fitness landscapes predicts viral vulnerabilities for rational immunogen design. Immunity. 2013;38:606–617. doi: 10.1016/j.immuni.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Quadeer A.A., Morales-Jimenez D., McKay M.R. Co-evolution networks of HIV/HCV are modular with direct association to structure and function. PLOS Comput. Biol. 2018;14 doi: 10.1371/journal.pcbi.1006409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Quadeer A.A., Louie R.H.Y., Shekhar K., Chakraborty A.K., Hsing I.-M., McKay M.R. Statistical linkage analysis of substitutions in patient-derived sequences of genotype 1a hepatitis C virus nonstructural protein 3 exposes targets for immunogen design. J. Virol. 2014;88:7628–7644. doi: 10.1128/JVI.03812-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dahirel V., Shekhar K., Pereyra F., Miura T., Artyomov M., Talsania S., Allen T.M., Altfeld M., Carrington M., Irvine D.J., Walker B.D., Chakraborty A.K. Coordinate linkage of HIV evolution reveals regions of immunological vulnerability. Proc. Natl. Acad. Sci. 2011;108:11530–11535. doi: 10.1073/pnas.1105315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hart G.R., Ferguson A.L. Computational design of hepatitis C virus immunogens from host-pathogen dynamics over empirical viral fitness landscapes. Phys. Biol. 2018;16 doi: 10.1088/1478-3975/aaeec0. [DOI] [PubMed] [Google Scholar]
- 207.Louie R.H.Y., Kaczorowski K.J., Barton J.P., Chakraborty A.K., McKay M.R. Fitness landscape of the human immunodeficiency virus envelope protein that is targeted by antibodies. Proc. Natl. Acad. Sci. 2018;115:E564–E573. doi: 10.1073/pnas.1717765115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Latinne A., Hu B., Olival K.J., Zhu G., Zhang L., Li H., Chmura A.A., Field H.E., Zambrana-Torrelio C., Epstein J.H., Li B., Zhang W., Wang L.-F., Shi Z.-L., Daszak P. Origin and cross-species transmission of bat coronaviruses in China. Nat. Commun. 2020;11:4235. doi: 10.1038/s41467-020-17687-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 209.Menachery V.D., Yount B.L., Debbink K., Agnihothram S., Gralinski L.E., Plante J.A., Graham R.L., Scobey T., Ge X.-Y., Donaldson E.F., Randell S.H., Lanzavecchia A., Marasco W.A., Shi Z.-L., Baric R.S. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015;21:1508–1513. doi: 10.1038/nm.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]