

Abstract
Intermolecular C–C bond-forming reactions are underdeveloped transformations in the field of biocatalysis. Here we report a photoenzymatic intermolecular hydroalkylation of olefins catalyzed by flavin-dependent ‘ene’reductases. Radical initiation occurs via photoexcitation of a rare high-order enzyme-templated charge-transfer complex that forms between an alkene, α-chloroamide, and flavin hydroquinone. This unique mechanism ensures that radical formation only occurs when both substrates are present within the protein active site. This active site can control the radical terminating hydrogen atom transfer, enabling the synthesis of enantioenriched γ-stereogenic amides. This work highlights the potential for photoenzymatic catalysis to enable new biocatalytic transformations via previously unknown electron transfer mechanisms.
Graphical Abstract
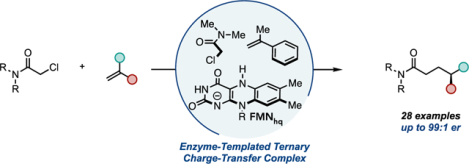
Intermolecular C–C bond-forming reactions are essential tools for the construction of societally valuable organic molecules.1 Enzymes are attractive catalysts for these transformations because of their ability to control reactive intermediates with unique precision. Unfortunately, the scope of natural C–C bond-forming enzymatic reactions is limited to relatively few retrosynthetic disconnections and often display limited substrate promiscuity.1d This substrate specificity results from the requirement for two substrates to bind and form reactive intermediates simultaneously within a protein active site. While some enzymes ensure co-localization through intricate gating mechanisms,2 the most substrate promiscuous enzymes form long-lived reactive intermediates or assume reactive conformations with substrates displaying a high affinity for the protein active site. This approach enables coupling with a reasonably broad collection of substrates that possess only modest active site affinities. This catalytic strategy is central to many of the most commonly used C–C bond-forming enzymes, such as aldolases (Figure 1A),1d, 3 carboligases,4 and artificial metalloenzymes.5 While long-lived intermediates are amenable to this approach, it is incompatible with more transient species.
Figure 1.

Mechanisms of Enzymatic Intermolecular C–C Bond Formation
Organic radicals are versatile intermediates capable of undergoing a variety of synthetically valuable chemical reactions.6 However, these species can be challenging to utilize for radical biocatalytic reactions as a function of their short lifetimes. Our group has recently developed modular mechanisms for forming radical intermediates within enzyme active sites to address long-standing selectivity challenges in the radical literature.7 During this time, we found that organohalides will form charge-transfer (CT) complexes with flavin hydroquinone (FMNhq) within the active sites of ‘ene’-reductases (EREDs).8 Upon photoexcitation, both nucleophilic and electrophilic radical intermediates are generated that engage in various intramolecular C–C bond-forming reactions, often with high levels of enantio- and diastereoselectivity.
Given the variety of radical cyclizations available to these enzymes, we questioned whether they could also catalyze intermolecular reductive coupling with alkenes.9 A challenge to achieving an intermolecular reaction is undesired hydrodehalogenation if radical formation occurs in the absence of the coupling partner (Figure 1B) As EREDs are only known to possess a single substrate-binding site, it was unclear at the onset how gating of radical formation would occur.10,11
We initiated our studies by exploring the coupling of N,N-dimethylchloroamide 1 with three and a half equivalents of α-methylstyrene 2 under cyan light irradiation (λmax = 497 nm) with a small collection of EREDs (Table S1). In a concurrent study by Huang et. al, this substrate was reported to be unreactive.9 We found that many EREDs can catalyze the coupling reaction with only trace formation of the undesired hydrodehalogenated product 4. The EREDs from Gluconobacter oxydans, with a previously identified beneficial mutation (GluER-T36A), afforded amide 3 in 80% yield with high levels of enantioselectivity for the R-enantiomer (Table 1, Entry 1). The S-enantiomer of product 3 can be accessed using the homolog from N. punctiforme (NostocER) (Table 1, Entry 2). Reaction optimization revealed that both enzymes could catalyze the reaction in near quantitative yield by adding 10% (v/v) DMSO (Table 1, Entries 3–4). Under these conditions, reactions could be carried out with as little as 0.5 mol % lyophilized enzyme with little change in the yield (Table S3). The reaction can be run with either purified protein or lyophilized cell-free lysate, providing identical levels of enantioselectivity, with only a modest decrease in yield when run on a preparative scale, highlighting the transformation’s robust nature (Table 1, entries 5–6).12 Overall, this reaction demonstrates the ability of EREDs to precisely control hydrogen atom transfer to prochiral radicals, a distal site that has proven challenging to control with conventional catalytic methodologies.13
Table 1.
Enzyme Screen
| Enantiomeric Ratio (e.r.) | |||
|---|---|---|---|
| 1a | GluER-T36A | 80 | 97:3 |
| 2a | NostocER | 50 | 10:90 |
| 3b | GluER-T36A | 99 | 99:1 |
| 4b | NostocER | 96 | 10:90 |
| 5c | GluER-T36A | 77 (61)d | 99:1 |
| 6e | GluER-T36A | 87 | 99:1 |

1 (10.0 μmol, 2.0 mg), ‘ene’-reductase (0.050 μmol), NADP+ (0.10 μmol), GDH-105 (0.5 mg/rxn), glucose (60 μmol), Tris (100 mM, pH=9.0), 36 hours, 25 ˚C.
1 (20.0 μmol, 4.0 mg), ‘ene’-reductase (0.10 μmol), NADP+ (0.10 μmol), GDH-105 (1.0 mg/rxn), glucose (60 μmol), Tris (100 mM, pH=9.0), DMSO (10%(v/v)), 24 hours, 25 ˚C
optimized conditions with cell-free lysate.
Preparative gram scale reaction using cell-free lysate.
Preparative scale reaction run on 0.2 mmol scale, 40.0 mg using purified enzyme.
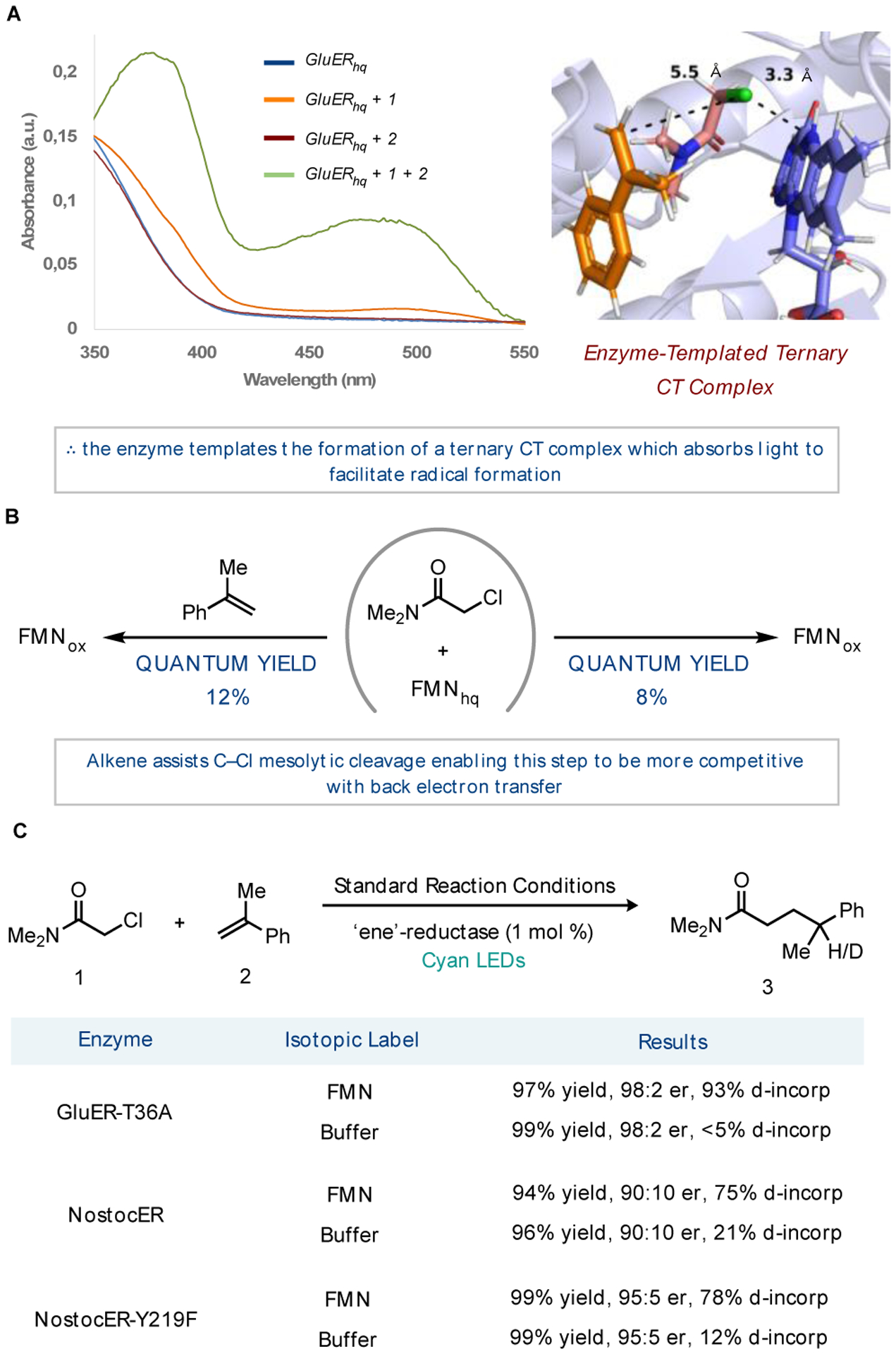
The most striking feature of this reaction was the lack of hydrodehalogenated product 4, the expected product if alkene is not present within the protein active site during radical formation.14 A series of mechanistic experiments were conducted to understand the origin of this selectivity. We began by probing whether a CT complex was responsible for radical formation. When GluER-T36A was reduced with sodium dithionite, negligible absorption was observed above 400 nm, consistent with the absorption features of FMNhq (Figure 2A). Upon the addition of chloroamide 1, a new absorption feature is observed at 500 nm. This feature is less pronounced when compared to amides containing tethered alkenes.8 Addition of α-methylstyrene 2 to the ternary CT-complex of GluER-T36A, FMNhq, and chloroamide 1 pronounced a new absorption feature at 380 and 500 nm. When this mixture was passed through molecular weight cutoff filters to remove all small molecules, the absorption feature is lost, and FMNhq was reformed (Supplemental Figure 4). Nearly identical spectral features were observed when the same experiments were run with NostocER, and no CT complex was observed in the absence of protein (Supplemental Figure 6 and 8).
Figure 2.
Mechanistic information
These experiments provide strong evidence that a quaternary CT complex between the protein, FMNhq, chloroamide 1, and α-methylstyrene 2 is responsible for the observed reactivity. This unexpected complex provides an explanation for the lack of hydrodehalogenated product, as radical formation is limited to the situation where both substrates are present within the enzyme active site. Moreover, the similarity between the absorption features of flavin semiquinone (FMNsq) and the quaternary CT complex implies a large degree of charge transfer in the ground state. Docking both substrates into the enzyme active site provided a potential binding pose, with TD-DFT calculations supporting the increased absorptivity of the quaternary complex by comparison to the ternary one lacking alkene (Figure 2a and Supplemental Figures 41 and 43). This represents a rare example of a quaternary CT complex.15,16
Transient absorption spectroscopy was conducted to better understand charge transfer dynamics in the presence and absence of styrene (Figure 2B). When GluER is reduced with dithionite and chloroamide 1 is added to the mixture, we observe a charge-transfer state with a lifetime of 9 ps that decays to the flavin quinone with a quantum yield of 8%.17 In contrast, when the same experiment is run in the presence of α-methylstyrene, the lifetime of the charge transfer state remains 5 ps, but decays instead to the flavin semiquinone, which exhibits a lifetime of 45 ps before ultimately forming flavin quinone on a 300 ps timescale. The quantum yield of this process significantly higher at 12%. These results suggest that α-methylstyrene facilitates C–Cl mesolytic cleavage for productive chemistry.18
With an understanding of both the mechanism of radical formation and the lifetime of radical intermediates, we next focused on the mechanism of radical termination.8 A series of isotope incorporation experiments were conducted with both GluER-T36A and NostocER. When reactions were run with glucose-1-d1, leading to deuteration of the flavin N5-position, with GluER-T36A, 93% deuterium incorporation was observed at the γ-position of the product (Figure 2C). In contrast, when reactions are run with glucose-1-H1 in deuterated buffer, to in situ label tyrosine OH protons, <5% deuterium incorporation is observed. These results suggest that with GluER-T36A, radical termination occurs almost exclusively through hydrogen atom transfer from FMNsq.
NostocER provided slightly different results when the same experiments were conducted. When reactions were run with isotopically labeled flavin, only 75% deuterium incorporation was observed at the γ-position of the product while the experiments with deuterated buffer afforded 21% deuterium incorporation (Figure 2C). These experiments suggest radical termination via hydrogen atom transfer from flavin and tyrosine are competitive.19 These competing mechanisms of radical termination potentially account for the lower levels of enantioselectivity observed with NostocER. We hypothesized that mutation of these tyrosines to phenylalanine would shut down the tyrosine termination pathway to favor hydrogen atom transfer from flavin. We selected two tyrosines within the active site and found that mutation of Y219 to phenylalanine (Y219F) afforded a variant that provided improved yields and enantioselectivities (Table S5). Re-evaluating the degree of deuterium incorporation for this mutant revealed the undesired tyrosine termination pathway was significantly decreased.20 (Figure 2c).
With an improved mechanistic understanding in hand, we explored the transformation’s scope and limitations (Figure 3). A variety of tertiary amides are tolerated by the reaction, with pyrrolidine, Weinreb, and benzyl amides affording product in high yields and enantioselectivities (Figure 3, 16–18). In general, GluER-T36A afforded products with higher levels of enantioselectivity than NostocER. Pleasingly, α,-difluorochloroamides are tolerated and afford product in high yield and enantioselectivity with NostocER with no observed defluorination (Figure 3, 19). Secondary amides are effective (Figure 3, 20), although primary amides were less reactive (Supplemental Figure 2). Finally, α-chloroacetophenone was an effective coupling partner, providing product in high yield and selectivity with GluER-T36A (Figure 3, 21). UV-vis experiments to probe the spectral features of a possible CT complex were complicated by ground state oxidation of flavin hydroquinone (Supplemental Figure 6). As visible light irradiation affords higher yields than reactions run in the dark,21 it is possible that a CT complex is formed but is obscured by competing ground state reduction of the substrate.
Figure 3.

Substrate Scope
Next, we explored the scope of the alkene coupling partner with the α-chloroamide. Despite their differing impact on the electronics of the styrenyl alkene, methoxy substituents at the para-, meta-, and ortho- positions are well tolerated and afford high yields and selectivity when using NostocER (Figure 3, 22a-e). Electron-withdrawing substituents, such as bromide and trifluoromethyl are also accepted with products formed in excellent yields and selectivity.22 Regarding alkene substituents, we found that larger aliphatic groups at the α-position are accommodated, providing products with excellent enantioselectivity levels (Figure 3, 23a-b). Trifluoromethyl groups are also compatible, although yields are more modest (Figure 3, 23c).23 Ester substitutents are also tolerated, affording product in high yields with good levels of enantioselectivity (Figure 3, 23d). Finally, unsubstituted styrene is reactive (Figure 3, 24).
Beyond simple styrenyl alkenes, we found that this chemistry accommodates an even broader range of alkenes (Figure 4). For instance, these enzymes are tolerant of electron-deficient heterocycles such as pyridines and pyrazine (Figure 4, 25–27). When using α-methylvinylpyridines, the product is afforded in high yields with excellent levels of enantioselectivity. Interestingly, NostocER can differentiate sterically similar phenyl and pyridine rings to furnish products with promising levels of enantioselectivity. Beyond aromatic substituents, aliphatic alkenes are also tolerated. Protected allylic amines and unprotected allylic alcohols are also reactive, with NostocER providing the best yields (Figure 4, 28, 29). Finally, enamides, vinyl ethers, and vinyl acetates are competent coupling partners (Figure 4, 30–33).
Figure 4.

Non-styrenyl alkenes
To further demonstrate the synthetic utility of this reaction, we explored intermediates or products that could be diverted to provide other useful products. We found that using α-bromostyrene with N,N-dimethylchloroamide, a lactone is formed in high yields with excellent levels of enantioselectivity (Figure 5, 34). This likely forms via initial formation of the coupled γ-bromo product followed by intramolecular SN2 cyclization. We also found that electron-rich enamides can function as coupling partners to generate in situ formation of ketones, providing an effective method for preparing 1,4-dicarbonyls (Figure 5, 35). This product is likely formed from oxidation of the α-amido radical via the intermediate semiquinone followed by hydrolysis.
Figure 5.
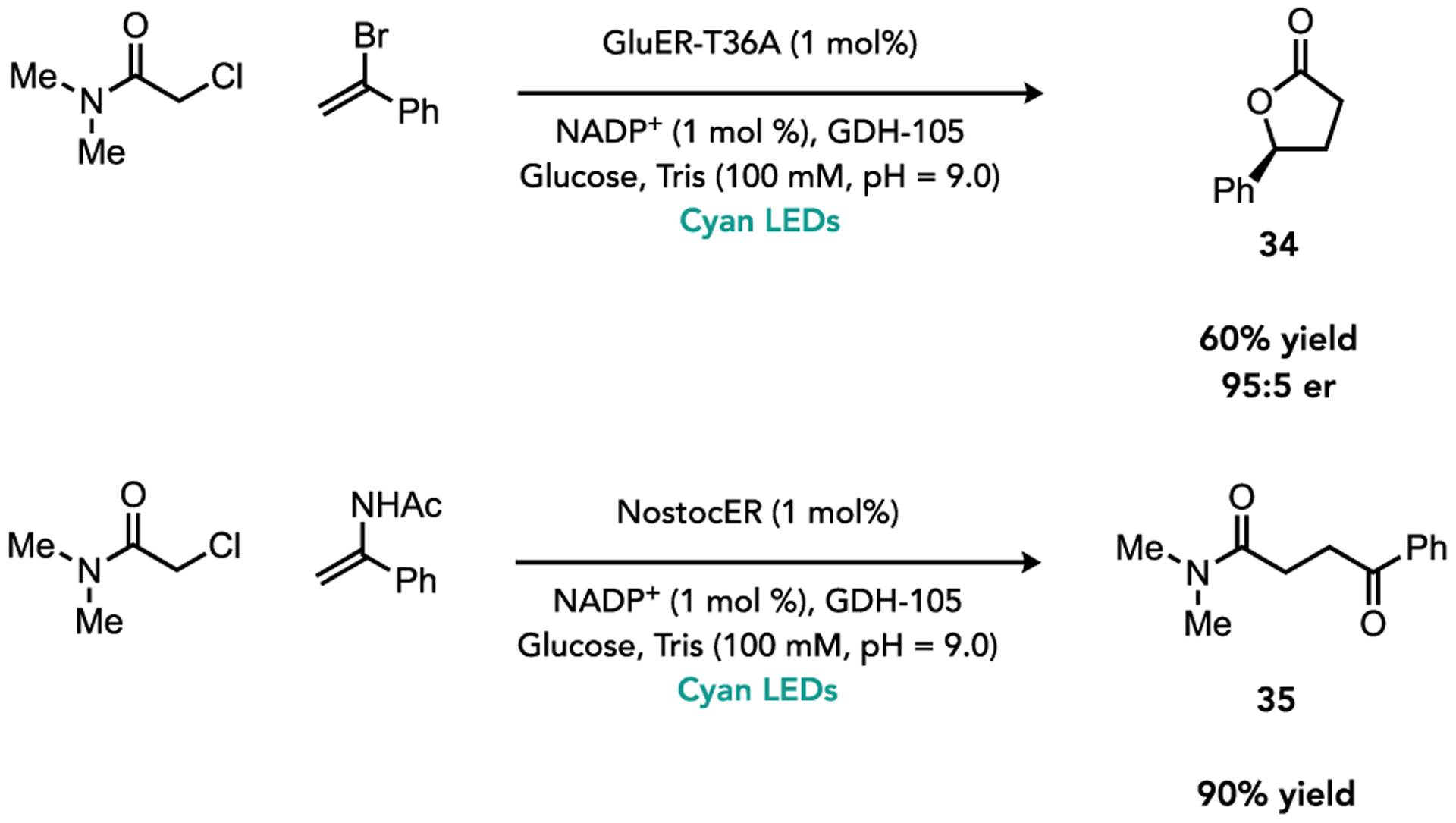
Lactone and Ketone Synthesis
In conclusion, we have discovered an effective method for preparing γ-stereogenic amides using photoenzymatic catalysis. This represents a non-natural mechanism of intermolecular C–C bond formation that we anticipate being useful in preparing various synthetically valuable motifs. The unique quaternary CT-complex provides an unprecedented mechanism for gating radical formation.
Supplementary Material
ACKNOWLEDGMENT
Reaction optimization and substrate scope explorations were supported by the National Institutes of Health (NIH) National Institute of General Medical Sciences (NIGMS) (R01 GM127703) and Bristol-Myers-Squibb as part of the Princeton Catalysis Initiative (T.K. H.). Mechanistic experiments, including and transient absorption and UV-Vis spectroscopy was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy (DOE) through grant DE-SC0019370. We acknowledge the MacMillan Group for the use of their instrumentation. D.G.O. acknowledges support from the Postgraduate Scholarships Doctoral Program of the Natural Sciences and Engineering Research Council of Canada. C.G.P. acknowledges the NSF-GRFP for support. J.S.D. acknowledges support from the Bristol-Myers-Squibb Graduate Fellowship.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.(a) Luh T-Y; Leung M.-k.; Wong K-T, Transition Metal-Catalyzed Activation of Aliphatic C−X Bonds in Carbon−Carbon Bond Formation. Chem. Rev 2000, 100, 3187–3204. [DOI] [PubMed] [Google Scholar]; (b) Varun BV; Dhineshkumar J; Bettadapur KR; Siddaraju Y; Alagiri K; Prabhu KR, Recent advancements in dehydrogenative cross coupling reactions for CC bond formation. Tet. Lett 2017, 58, 803–824. [Google Scholar]; (c) Reckenthäler M; Griesbeck AG, Photoredox Catalysis for Organic Syntheses. Adv. Synth. Catal 2013, 355, 2727–2744. [Google Scholar]; (d) Schmidt NG; Eger E; Kroutil W, Building Bridges: Biocatalytic C–C-Bond Formation toward Multifunctional Products. ACS Catal 2016, 6, 4286–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Turner NJ; O’Reilly E, Biocatalytic retrosynthesis. Nat. Chem. Biol 2013, 9, 285–288. [DOI] [PubMed] [Google Scholar]; (b) Zetzsche LE; Narayan ARH, Broadening the scope of biocatalytic C–C bond formation. Nat. Rev. Chem 2020, 4, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rétey J, Enzymic Reaction Selectivity by Negative Catalysis or How Do Enzymes Deal with Highly Reactive Intermediates? Angew. Chem. Int. Ed 1990, 29, 355–361. [Google Scholar]
- 3.Windle CL; Müller M; Nelson A; Berry A, Engineering aldolases as biocatalysts. Curr. Opin. Chem. Biol 2014, 19, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Müller M; Sprenger GA; Pohl M, CC bond formation using ThDP-dependent lyases. Curr. Opin. Chem. Biol 2013, 17, 261–270. [DOI] [PubMed] [Google Scholar]
- 5.(a) Chen K; Arnold FH, Engineering new catalytic activities in enzymes. Nat. Catal 2020, 3, 203–213. [Google Scholar]; (b) Thomas CM; Ward TR, Artificial metalloenzymes: proteins as hosts for enantioselective catalysis. Chem. Soc. Rev 2005, 34, 337–346. [DOI] [PubMed] [Google Scholar]; (c) Davis HJ; Ward TR, Artificial Metalloenzymes: Challenges and Opportunities. ACS Cent. Sci 2019, 5, 1120–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lewis RD; Garcia-Borrás M; Chalkley MJ; Buller AR; Houk KN; Kan SBJ; Arnold FH, Catalytic iron-carbene intermediate revealed in a cytochrome c carbene transferase. Proc. Natl. Acad. Sci 2018, 115, 7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hayashi T; Tinzl M; Mori T; Krengel U; Proppe J; Soetbeer J; Klose D; Jeschke G; Reiher M; Hilvert D, Capture and characterization of a reactive haem-carbenoid complex in an artificial metalloenzyme. Nat. Catal 2018, 1, 578–584. [Google Scholar]
- 6.(a) Studer A; Curran DP, Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]; (b) Beckwith ALJ, Centenary Lecture. The pursuit of selectivity in radical reactions. Chem. Soc. Rev 1993, 22, 143–151. [Google Scholar]; (c) Romero NA; Nicewicz DA, Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]; (d) Skubi KL; Blum TR; Yoon TP, Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Emmanuel MA; Greenberg NR; Oblinsky DG; Hyster TK, Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 2016, 540, 414–417 [DOI] [PubMed] [Google Scholar]; (b) Biegasiewicz KF; Cooper SJ; Emmanuel MA; Miller DC; Hyster TK, Catalytic promiscuity enabled by photoredox catalysis in nicotinamide-dependent oxidoreductases. Nat. Chem 2018, 10, 770–775. [DOI] [PubMed] [Google Scholar]; (c) Sandoval BS; Kurtoic SI; Chung MM; Biegasiewicz KF; Hyster TK, Photoenzymatic Catalysis Enables Radical-Mediated Ketone Reduction in Ene-Reductases. Angew. Chem. Int. Ed 2019, 58, 8714. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Black MJ; Biegasiewicz KF; Meichan AJ; Oblinsky DG; Kudisch B; Scholes GD; Hyster TK, Asymmetric redox-neutral radical cyclization catalyzed by flavin-dependent ‘ene’-reductases. Nat. Chem 2020, 12, 71–75 [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nakano Y; Black MJ; Meichan AJ; Sandoval BA; Chung MM; Biegasiewicz KF; Zhu T; Hyster TK, Photoenyzmatic Hydrogenation of Heteroaromatic Olefins using ‘Ene’-Reductases with Photoredox Catalysts. Angew. Chem. Int. Ed 2020, 59, 10484–10488. [DOI] [PubMed] [Google Scholar]
- 8.(a) Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK, Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Clayman PD; Hyster TK, Photoenzymatic Generation of Unstabilized Alkyl Radicals: An Asymmetric Reductive Cyclization J. Am. Chem. Soc 2020, 142, 15673–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.During preparation of the manuscript, a similar intermolecular hydroalkylation was reported by the group of Huimin Zhao; see:; Huang X; Wang B; Wang Y; Jiang G; Feng J; Zhao H, Photoenzymatic enantioselective intermolecular radical hydroalkylation. Nature 2020, 584, 69–74. [DOI] [PubMed] [Google Scholar]
- 10.Adalbjörnsson BV; Toogood HS; Fryszkowska A; Pudney CR; Jowitt TA; Leys D; Scrutton NS, Biocatalysis with Thermostable Enzymes: Structure and Properties of a Thermophilic ‘ene’-Reductase related to Old Yellow Enzyme. ChemBioChem 2010, 11, 197–207. [DOI] [PubMed] [Google Scholar]
- 11.(a) Alkema WBL; Dijkhuis A-J; de Vries E; Janssen DB, The role of hydrophobic active-site residues in substrate specificity and acyl transfer activity of penicillin acylase. European Journal of Biochemistry 2002, 269 (8), 2093–2100. [DOI] [PubMed] [Google Scholar]; (b) Nagy EZA; Tork SD; Lang PA; Filip A; Irimie FD; Poppe L; Toşa MI; Schofield CJ; Brem J; Paizs C; Bencze LC, Mapping the Hydrophobic Substrate Binding Site of Phenylalanine Ammonia-Lyase from Petroselinum crispum. ACS Catal 2019, 9, 8825–8834. [Google Scholar]; (c) Zhang W; Ma M; Huijbers MME; Filonenko GA; Pidko EA; van Schie M; de Boer S; Burek BO; Bloh JZ; van Berkel WJH; Smith WA; Hollmann F, Hydrocarbon Synthesis via Photoenzymatic Decarboxylation of Carboxylic Acids. J. Am. Chem. Soc 2019, 141, 3116–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.When the reaction is run open to air, the yield is decreased to 15%
- 13.Zou Y-Q; Hörmann FM; Bach T, Iminium and enamine catalysis in enantioselective photochemical reactions. Chem. Soc. Rev 2018, 47, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Only trace quantities of the hydrodehalogenated product are observed with N-methyl-N-benzyl chloroamide 7 is used as a substrate in the absence of alkene, suggesting this complex plays only a minor role in radical formation (Supplemental Figure 1).
- 15.For a proposed ternary CT complex in solution; see:; Kumar R; Flodén NJ; Whitehurst WG; Gaunt MJ, A general carbonyl alkylative amination for tertiary amine synthesis. Nature 2020, 581, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]; For examples of quaternary CT complexes in solid state; see:; Wiscons RA; Coropceanu V; Matzger AJ, Quaternary Charge-Transfer Solid Solutions: Electronic Tunability through Stoichiometry. Chem. Mater 2019, 31, 6598–6604. [Google Scholar]; Fu M-C; Shang R; Zhao B; Wang B; Fu Y, Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide. Science 2019, 363 (6434), 1429. [DOI] [PubMed] [Google Scholar]; For templated CT complexes; see:; Biedermann F; Scherman OA, Cucurbit[8]-uril Mediated Donor-Acceptor Ternary Complexes: A Model System for Studying Charge-Transfer Interactions. J. Phys. Chem. B 2012, 116, 2842–2849. [DOI] [PubMed] [Google Scholar]
- 16.A competition experiment between p-Me-α-methyl styrene and p-CF3-α-methyl styrene afforded almost exclusive formation of the p-Me with both NostocER and GluER-T36A, suggesting more electron rich alkenes more readily form quaternary CT complexes or that corresponding CT complex is more reactive. See Supplemental Figure 34.
- 17.Experiments of irradiating the ERED with just chloroamide show flavin alkylation (Supplemental Figure 38 and 39). When alkene is added the formation of alkylated flavin is limited. This suggests this is a catalyst degradation pathway
- 18.Similar results were observed with NostocER (Supplemental Figure 5).
- 19.(a) de Crécy-Lagard V; Nizam S; Gazara RK; Verma S; Singh K; Verma PK, Comparative Structural Modeling of Six Old Yellow Enzymes (OYEs) from the Necrotrophic Fungus Ascochyta rabiei : Insight into Novel OYE Classes with Differ-ences in Cofactor Binding, Organization of Active Site Resi-dues and Stereopreferences. PLoS ONE 2014, 9, e95989. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chaparro-Riggers JF; Rogers TA; Vazquez-Figueroa E; Polizzi KM; Bommarius AS, Comparison of Three Enoate Reductases and their Potential Use for Biotransformations. Adv. Synth. Catal 2007, 349, 1521–1531 [Google Scholar]
- 20.The lack of complete suppression of deuterium incorporation when the reaction is run in D2O suggests additional active site tyrosines also act as competitive hydrogen atom sources, likely with the same facial selectivity as the flavin.
- 21.Reaction in the dark with GluER T36A affords product in 3% yield with additional hydrodehalogenated product is formed (Supplemental Figure 3)
- 22.Mutating tyrosine 219, the residue we hypothesize to function as a competitive hydrogen atom source, to phenylalanine (NostocER-Y219F) can provide improved enantioselectivity.
- 23.A small amount of defluorinated product is observed, potentially formed via a reduction/elimination mechanism.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.