

Abstract
Recent studies have profiled the innate immune signatures in patients infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and suggest that cellular responses to viral challenge may affect disease severity. Yet the molecular events that underlie cellular recognition and response to SARS-CoV-2 infection remain to be elucidated. Here, we find that SARS-CoV-2 replication induces a delayed interferon (IFN) response in lung epithelial cells. By screening 16 putative sensors involved in sensing of RNA virus infection, we found that MDA5 and LGP2 primarily regulate IFN induction in response to SARS-CoV-2 infection. Further analyses revealed that viral intermediates specifically activate the IFN response through MDA5-mediated sensing. Additionally, we find that IRF3, IRF5, and NF-κB/p65 are the key transcription factors regulating the IFN response during SARS-CoV-2 infection. In summary, these findings provide critical insights into the molecular basis of the innate immune recognition and signaling response to SARS-CoV-2.
Keywords: SARS-CoV-2, lung epithelial cells, interferon, MDA5, IRF3, IRF5, NF-κB/p65
Graphical Abstract
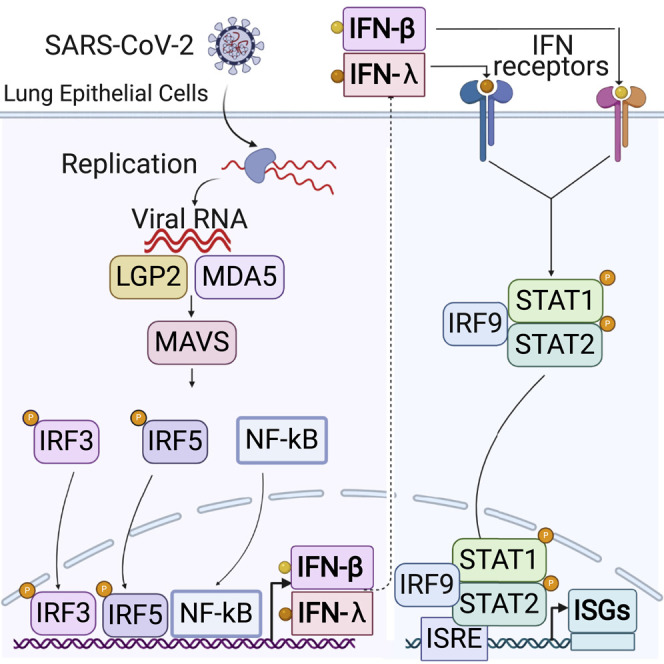
The molecular events that underlie innate immune recognition and response to SARS-CoV-2 infection remain unclear. Yin et al. report that SARS-CoV-2 replication induces a delayed interferon (IFN) response that is triggered by sensing of viral RNA through the MDA5 pattern recognition receptor.
Introduction
A novel coronavirus termed severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was identified as the causative agent of the severe CoV disease 2019 (COVID-19) pandemic, which is still an ongoing global health emergency (Felgenhauer et al., 2020; Lei et al., 2020; Wu et al., 2020a; Zhou et al., 2020a). Since late December 2019, this virus has affected 216 countries and nearly 65 million people worldwide, according to the COVID-19 Situation Dashboard of the World Health Organization. There have been no US Food and Drug Administration (FDA)-approved drugs or vaccines for the treatment or prevention of SARS-CoV-2 or any of the other CoVs, however, emergency approvals of several vaccines and drugs, including remdesivir, have provided important prophylactic and therapeutic options to combat this pandemic disease (Aschenbrenner, 2020; Grein et al., 2020) (Rubin and Longo, 2020).
The innate immune system plays a central role in clearance of viral infections (García-Sastre, 2017; Takeuchi and Akira, 2009). Upon the sensing of viral infections, through the recognition of pathogen-associated molecular patterns (PAMPs), host cells quickly turn on multiple signaling cascades, leading to the transcriptional induction of type I and type III interferons (IFNs) (Lazear et al., 2019). Both types of IFNs bind to their respective receptors to trigger activation of the JAK-STAT pathway, which in turn drives the expression of hundreds of IFN-stimulated genes (ISGs) that directly and indirectly exert antiviral activities through many different mechanisms (Lazear et al., 2019; Stanifer et al., 2019). Transcriptome analysis revealed that COVID-19 patients developed elevated cytokine and IFN signatures in the lungs (Zhou et al., 2020b). In particular, the initial IFN response appears to be higher in patients with mild to moderate disease, while it was reduced in those in critical condition (Hadjadj et al., 2020). Immune profiling of 113 COVID-19 patients with moderate and severe disease further revealed IFNs were elevated in COVID-19 patients throughout the course of disease (Lucas et al., 2020). Viral load appears to be highly correlated with the levels of IFNs and tumor necrosis factor alpha (TNF-α), suggesting that viral load may drive cytokine production (Lucas et al., 2020). More recently, two independent studies demonstrated that IFN signaling is important in defense against SARS-CoV-2 and that limiting this response through either inherited deleterious variants or autoantibodies leads to severe COVID-19 (Bastard et al., 2020; Zhang et al., 2020b). Although the specific molecular mechanisms involved in SARS-CoV-2-mediated IFN induction are yet unknown, these studies underscore the significance of IFN signaling in controlling disease severity and immunopathology.
SARS-CoV-2 is an enveloped, positive-sense, single-stranded RNA betacoronavirus that replicates primarily in ciliated cells of the nasal and bronchiolar epithelium and type 2 pneumocyte cells in the alveolar regions (Gordon et al., 2020). After binding the host receptor ACE2 to facilitate entry (Hoffmann et al., 2020), the incoming viral particle is uncoated, and the viral RNA is translated to generate the replication complexes, resulting in genome replication and progeny virion production (Aschenbrenner, 2020). Analogous to other CoVs such as SARS-CoV-1 and Middle East respiratory syndrome CoV (MERS-CoV), double-stranded RNA (dsRNA) intermediates are produced during replication. These dsRNA intermediates are thought to be recognized by the primary RNA patten recognition receptors (PRRs) such as the RIG-I-like receptors (RLRs), RIG-I, MDA5, and LGP2, which signal through MAVS, or the Toll-like receptors (TLRs), specifically TLR3, which signals through TIR domain-containing adaptor-inducing IFN-β (TRIF) (Frieman et al., 2008). Previous studies indicated that the specific PRR that senses viral RNA is both virus and cell type dependent (Frieman et al., 2008; Hoffmann et al., 2015; Li et al., 2010; Roth-Cross et al., 2008). For example, MDA5 has been shown to act as the primary sensor for the recognition of mouse hepatitis virus (MHV) in macrophages (Roth-Cross et al., 2008). In addition, SARS-CoV-infected mice likely require MAVS signaling pathways to induce the IFN response, but MERS-CoV, on the other hand, signals through TLR7 (Channappanavar et al., 2019).
Here, we investigated the molecular mechanism of IFN induction in an epithelial cell line (Calu-3) and induced pluripotent stem cell (iPSC)-derived airway epithelial cells during SARS-CoV-2 infection. We find that compared with Sendai virus (SeV) infection or polyI:C transfection, SARS-CoV-2 infection induces a delayed increase of IFNs, ISGs, and inflammatory cytokines, resulting in limited infection and spread in lung epithelial cells. To determine the pathway by which SARS-CoV-2 is recognized in lung epithelial cells, a panel of PRRs previously reported to be involved in viral RNA sensing were investigated in the context of SARS-CoV-2 infection (Jensen and Thomsen, 2012; Lee et al., 2019). Interestingly, MDA5, LGP2, and NOD1, but not RIG-I, are required for recognition of SARS-CoV-2 in lung epithelial cells. Moreover, IRF3, IRF5, and RelA (p65) act as the key transcription factors that trigger the induction of IFNs. These findings provide insight into how the host senses SARS-CoV-2 to trigger the innate immune response in infected lung epithelial cells.
Results
SARS-CoV-2 Infection Triggers a Delayed Innate Immune Response in Calu-3 Cells
To investigate the cellular response to SARS-CoV-2 infection, we infected Calu-3 cells, an airway epithelial cell line, with SARS-CoV-2 (Figure 1 A). Viral replication was then assessed by quantifying the SARS-CoV-2 NP-positive cells through immunostaining and intracellular viral RNA using qRT-PCR. The SARS-CoV-2-positive cells were readily detected at 24 h post-infection (hpi), peaked at 48 hpi, and then decreased at 72 hpi (Figure 1B). An analogous transient increase is observed from the quantification of the intracellular viral mRNA. Viral mRNA levels peaked at around 24 hpi and then declined over the course of infection (Figure 1C). Of note, the number of viable cells at 96 hpi was significantly smaller compared with the early time points, indicating that SARS-CoV-2 infection exhibits some cytotoxic effects in Calu-3 cells (Figure 1B). We reasoned that the decrease in SARS-CoV-2 replication at later time points might be due to the activation of IFN pathways. To this end, we measured the mRNA levels of type I and type III IFNs, as well as ISGs, and inflammatory cytokines over the course of SARS-CoV-2 infection using qRT-PCR. The mRNA expression of IFN-β and two major type III IFNs, including IFN-λ1 and IFN-λ2/3, were all significantly elevated in infected cells, while the expression of IFN-α1 and IFN-λ4 did not increase up to 24 hpi (Figure 1D). Western blot analysis further revealed that the protein levels of ISG56 were increased in infected cells, compared to the mock-infected cells (Figure 1E). Intriguingly, stimulation with RLRs agonists such as polyI:C/3p-hpRNA transfection and SeV infection rapidly activated the signaling transduction in Calu-3 cells (Figure S1A). IFN-β mRNA peaked at 12 hpi, then declined in SeV infected cells but was only weakly induced at 12 hpi and further increased at 24 hpi in SARS-CoV-2-infected cells (Figure S1B). SARS-CoV-2-dependent IFN induction was accompanied by a weak induction of IFIT1, ISG15, IL6, and cxcl10 mRNAs, likely due to the blockade of STAT1/2 signaling mediated by virus infection (Figures S1C–S1E) (Miorin et al., 2020, Xia et al., 2020). To further confirm that the induction of IFNs was indeed due to replication and sensing of viral RNA, we infected Calu-3 cells with SARS-CoV-2 at different MOIs in the presence of remdesivir, which inhibits SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) function. As expected, we found that treatment with remdesivir almost completely abolished viral RNA replication (Figure 1F). The expression levels of IFNs and ISGs were concomitantly reduced to basal levels in the cells treated with remdesivir (Figure 1G; Figure S1F), suggesting that newly synthesized viral genomes are required to elicit a robust cellular immune response. Collectively, these results demonstrate that SARS-CoV-2 replication induces a delayed innate immune response in Calu-3 cells and that it is delayed relative to canonical stimulatory treatments such as polyI:C/3p-hpRNA and SeV infection.
Figure 1.

SARS-CoV-2 Infection Triggers the Innate Immune Response in Calu-3 Cells
(A) Calu-3 cells (1 × 105) were infected with SARS-CoV-2 with MOI = 0.125. At the indicated time points post-infection, cells were fixed, immunostained with rabbit-anti-SARS-CoV-2 NP antibody (green) and DAPI (blue), and imaged using an IC200 high-content imager. Representative immunofluorescence images are shown. Scale bar, 100 μm.
(B) The percentage of infection was calculated as the ratio between the number of infected cells stained for SARS-CoV-2 NP and the total amount of cells stained with DAPI. Data are from four independent experiments with three technical replicates and show mean ± SEM. The red line represents the relative cell numbers over time.
(C) Intracellular viral RNA was measured using qRT-PCR using primers targeting the N2 regions used by the Centers for Disease Control and Prevention (CDC) assay. The results show the mean ± SEM of four independent experiments.
(D) The kinetics of SARS-CoV-2 infection-induced expression of mRNAs encoding different IFNs in Calu-3 cells is displayed. Data are expressed as fold change relative to mock-infected cells and show the mean ± SD of four independent experiments.
(E) Immunoblots of ISG56, SARS-CoV-2 spike protein, and β-actin in Calu-3 cells infected with SARS-CoV-2 are shown. Calu-3 cells (1 × 106) were infected with SARS-CoV-2 with MOI = 1. At the indicated time points post-infection, cells were collected for western blot analysis with indicated antibodies.
(F) Calu-3 cells (1 × 105) were infected with SARS-CoV-2 at the indicated MOI in the presence of remdesivir for 48 h. Intracellular viral RNA was measured in infected Calu-3 cells with or without remdesivir treatment using qRT-PCR using primers targeting the N2 regions.
(G) IFN induction in SARS-CoV-2-infected Calu-3 cells was quantified using qRT-PCR. Data are expressed as fold change relative to mock-infected cells.
SARS-CoV-2 Induces IFN Responses in iPSC-Derived Airway Epithelial Cells
To further confirm the IFN response results obtained from Calu-3 cells, we used iPSC-derived airway epithelial cells that exhibit phenotypic similarities to mature lung epithelium (Konishi et al., 2016; Wong et al., 2012). The differentiated airway cultures were characterized by expression of acetylated α-tubulin (Ac-TUB; a marker for ciliated cells), MUC5B (a marker for goblet cells), and cystic fibrosis transmembrane conductance regulator (CFTR), which expressed predominantly on ciliated cells (Figure S2A). Quantification of the expression of markers for each cell type revealed that the largest population of the differentiated airway cultures was ciliated cells: FOXJ1 for ciliated cells (32.3% ± 2.1%), MUC5B for goblet cells (2.59% ± 0.36%), CCSP for secretory (club) cells (rare but existing), p63 for basal cells (1.28% ± 0.62%), and CHGA for neuroendocrine cells (3.86% ± 0.95%) (Figure S2B). These cells highly expressed ACE2 and TMPRSS2 proteins, which are indispensable for SARS-CoV-2 infection (Figure S2C). As expected, SARS-CoV-2 could productively infect differentiated airway cultures, as evidenced by the fact that the different MOIs yielded a dose-dependent difference in infection as monitored by NP staining and qPCR for viral RNA. Treatment with remdesivir dramatically reduced infection, as expected (Figures 2 A–2C; Figure S3A). We next assessed the innate immune response in cells infected with SARS-CoV-2 by measuring the expression levels of IFNs, ISGs, and inflammatory cytokines. As observed in Calu-3 cells, IFN mRNAs, including IFN-β, IFN-λ1, and IFN-λ2/3, were strongly induced (Figure 2D), while levels of IFN-α1 and IFN-λ4 were unaffected (Figure S3B). The mRNA expression of a number of ISGs (e.g., IFIT1, IFITM1, and ISG15) and proinflammatory cytokines (e.g., IL6, IL8, and IL1β) were also elevated, although the magnitude of induced proinflammatory cytokine expression was much lower compared with the expression of IFNs or ISGs (Figures S3C–S3D). Suppression of viral replication by remdesivir drastically reduced the induction of IFNs and ISGs, as well as proinflammatory cytokines, suggesting that the replication of SARS-CoV-2 RNA was required for the induction of IFNs and ISGs in iPSC-derived airway epithelial cells (Figures S3B–S3D). To further investigate these results, we evaluated the kinetics of IFN mRNA and protein expression in airway epithelium during infection using qRT-PCR and ELISA, respectively. Increases in IFN-β, IFN-λ1, and IFN-λ2/3 at both mRNA and protein levels were observed in infected cells starting at 24 hpi. No detectable upregulation was found at the early time points (2 and 10 hpi), despite increases in viral mRNA levels at these time points (Figures 2E–2G). Together, these results corroborate the data collected from Calu-3 cells and published data (Emanuel et al., 2020; Lei et al., 2020; Ravindra et al., 2020) showing that SARS-CoV-2 replication in airway epithelial cells induces a delayed innate immune response.
Figure 2.

SARS-CoV-2 Infection Induces IFN Signaling in iPSC-Derived Airway Epithelium
(A) iPSC-derived airway epithelium was infected with SARS-CoV-2 at the indicated MOI for 48 h before fixation. The cells were immunostained with rabbit-anti-SARS-CoV-2 NP antibody and imaged using an IC200 high-content imager. Representative immunofluorescence images are shown. Scale bar, 100 μm.
(B and C) Intracellular and extracellular viral RNA was measured using qRT-PCR using primers targeting the N2 regions used by the CDC assay. Data are from three technical replicates.
(D) IFN mRNA levels in SARS-CoV-2-infected epithelial cells were quantified using qRT-PCR. Data are expressed as fold change relative to mock-infected cells.
(E) The kinetics of intracellular viral mRNA in the infected cells. The results show the mean ± SD of two independent experiments.
(F) The kinetics of IFN mRNAs in the cells upon SARS-CoV-2 infection. Data are expressed as fold change relative to mock-infected cells.
(G) Kinetics of IFN secretion responses in infected cells. Concentrations of IFN-β and IFN-λ in the culture supernatants were measured using specific ELISA, respectively. The results show the mean ± SEM of the average of the duplicates in each of two independent experiments.
IFN Production Inhibits SARS-CoV-2 Replication
It was recently demonstrated that exogenously supplied recombinant IFNs exhibit greater inhibitory activity against SARS-CoV-2 in vitro compared with SARS-CoV-1 (Felgenhauer et al., 2020), suggesting that SARS-CoV-2 is more sensitive to IFN. We confirmed these observations in Calu-3 cells infected with SARS-CoV-2 in the presence of IFN treatment (Figure S4A). To better understand the role of IFN signaling activation in the SARS-CoV-2 life cycle, we transiently knocked down the key components involved in IFN signaling, including IFNAR1 (a type I IFN receptor), IL10RB (a type III IFN receptor), STAT1, STAT2, and IRF9, by small interfering RNA (siRNA) delivery into Calu-3 cells. The efficacy of knockdown for these target genes was evaluated using western blot (Figure 3 A). siRNA-mediated silencing of these genes greatly suppressed the cellular responsiveness to IFN-β and IFN-λ treatment respectively, which allowed us to assess the role of the signaling and the ISG response in SARS-CoV-2 infection (Figure 3B). Forty-eight hours after siRNA transfection, Calu-3 cells were challenged with SARS-CoV-2 for 48 h prior to imaging and RNA extraction. IFIT1 mRNA induction was quantified using qRT-PCR, and viral replication was also evaluated accordingly (Figures 3C and 3D). Compared with the cells transfected with scrambled siRNA, IFIT1 mRNA induction was greatly reduced in the cells depleted of IFNAR1, STAT1, STAT2, or IRF9 and to a lesser extent in cells depleted of IL10RB (Figure 3C). Concordantly, depletion of these genes resulted in an increased viral titer in the collected supernatants (Figure 3D; Figure S4B). Accordingly, the percentage of SARS-CoV-2 NP-positive cells were ~2- to 3-fold greater upon depletion of these genes involved in IFN signaling (Figures 3E and 3F). These results suggest that both type I and type III IFNs induced by infection limit virus replication and spread in lung epithelial cells.
Figure 3.

IFN Production Inhibits SARS-CoV-2 Replication
(A) Immunoblots of IFNAR1, IL10RB, STAT1, STAT2, IRF9, and actin in Calu-3 cells transfected with indicated pooled siRNA for 48 h are shown.
(B) Calu-3 cells were transfected with siRNAs. Forty-eight hours post-transfection, the cells were treated with IFN-β (1,000 U/mL) or IFN-λ (1,700 U/mL) for 8 h prior to RNA extraction. IFIT1 mRNA was quantified using qRT-PCR. Data are expressed as fold change relative to non-treated cells.
(C) Calu-3 cells were transfected with siRNAs. At 48 h post-transfection, the cells were infected with SARS-CoV-2 at MOI = 0.125 for a further 48 h. IFIT1 mRNA was quantified using qRT-PCR. Data are expressed as fold change relative to non-treated cells.
(D) Viral titer in the collected supernatants were determined by plaque assay in Vero E6 cells. Data show mean ± SD from one representative experiment in triplicate (n = 3) of two independent experiments.
(E) The siRNA-transfected Calu-3 cells were infected with SARS-CoV-2 with MOI = 0.125. At 48 h post-infection, cells were fixed, immunostained with rabbit-anti-SARS-CoV-2 NP antibody (green) and DAPI (blue), and imaged using an IC200 high-content imager. Representative immunofluorescence images are shown. Scale bar, 100 μm.
(F) The percentage of infection was calculated as the ratio between the number of infected cells stained for SARS-CoV-2 NP and the total amount of cells stained with DAPI. Data are from three independent experiments with three technical replicates.
A Survey of Viral RNA Sensors Responsible for the IFN Response to SARS-CoV-2 Infection
SARS-CoV-2 genome replication generates viral RNA transcripts that may be recognized by viral RNA sensors such as RIG-I, MDA5, and TLR3 (Birra et al., 2020). To determine which PRRs are specifically involved in recognition of SARS-CoV-2 RNA in lung epithelial cells, we selected 16 viral RNA sensors for knockdown alongside MAVS, the key adaptor responsible for RLR signaling activation. Calu-3 cells were transiently transfected with pooled siRNAs targeting the selected genes. 48 h post-transfection, SARS-CoV-2 (MOI = 0.125) was added to the plates and incubated for 48 h. Detectable silencing of target genes was confirmed at the mRNA level using qRT-PCR (Figure 4 A). In the context of SARS-CoV-2 infection, depletion of MDA5, LGP2, and NOD1 drastically reduced IFN-β mRNA expression (Figure 4B), whereas depletion of other genes, including RIG-I and TLR3, had no effect on the IFN response, suggesting that MDA5, LGP2, and NOD1 are the predominant receptors involved in innate immune sensing of SARS-CoV-2 infection. As expected, the number of infected cells and viral titers in the supernatants were increased in cells depleted of MDA5, LGP2, and NOD1 (Figure 4C; Figures S5A and S5B). Surprisingly, depletion of LRRFIP1, a dsRNA-binding protein had no effect on IFN-β production, but the SARS-CoV-2 infection was significantly increased compared with parental cells, indicating that LRRFIP1 modulates SARS-CoV-2 infection in an IFN-independent manner. Collectively, these results suggest that innate immune sensing of SARS-CoV-2 infection is regulated by multiple factors, including MDA5, LGP2, and NOD1.
Figure 4.

MDA5/LGP2 Are the Dominant RNA Sensors Responsible for Innate Immune Induction in Calu-3 Cells Infected with SARS-CoV-2
(A) siRNA-mediated knockdown efficiency in Calu-3 cells was evaluated using qRT-PCR with specific primers.
(B) The siRNA-transfected Calu-3 cells were infected with SARS-CoV-2 with MOI = 0.125. At 48 h post-infection, the total RNA was extracted using the NucleoSpin 96 RNA extraction kit. The IFN-β mRNA was quantified by quantitative RT-PCR. Data are expressed as fold change relative to non-treated cells.
(C) The percentage of infection was calculated as the ratio between the number of infected cells stained for SARS-CoV-2 NP and the total amount of cells stained with DAPI. Data are from three independent experiments with three technical replicates.
(D) Immunoblots of parental, MDA5, RIG-I, MAVS, and β-actin in the CRISPR-knockout (KO) Calu-3 cells.
(E) The CRISPR-knockout cells were infected with SARS-CoV-2 with MOI = 0.125. At 48 h post-infection, cells were fixed, immunostained with rabbit-anti-SARS-CoV-2 NP antibody, and imaged using Celigo. The percentage of infection was calculated as the ratio between the number of infected cells stained for SARS-CoV-2 NP and the total amount of cells stained with DAPI. Data are from two independent experiments with three technical replicates.
(F) IFN-β mRNA induction in the CIRPSR-KO cells infected with SARS-CoV-2.
(G) Representative immunofluorescence images are shown.
(H) The CRISPR-KO cells were transfected with either PolyI:C (5 μg/mL) or total RNA extracted from infected cells (10 μg/mL). 8 h post-transfection, the cells were lysed to measure IFN-β mRNA production using qRT-PCR.
To further confirm that MDA5 is essential for sensing of SARS-CoV-2 infection, we knocked out MDA5, RIG-I, and MAVS in Calu-3 cells using CRISPR-Cas9. The expression of targeted genes in knockout cells were evaluated using western blotting (Figure 4D). To further verify that our cells were functionally knocked out for these targets, the cells were either stimulated with polyI:C or infected with SeV, and the production of the IFN-β mRNA was evaluated. As expected, the RIG-I-knockout cells did not respond to SeV infection, whereas MDA5-knockout cells did not respond to polyI:C stimulation. The cells depleted of MAVS also did not respond to either treatment (Figures S5C and S5D). After this confirmation, wild-type (WT) or knockout Calu-3 cells were infected with SARS-CoV-2. Forty-eight hours after infection, the infection of SARS-CoV-2 was determined by assessing NP immunostaining. The number of infected cells was significantly increased in MDA5- or MAVS-knockout cells, correlating with a significant decrease in IFN production. Conversely, depletion of RIG-I has no effect on virus replication or IFN production, which is consistent with the results observed in siRNA-transfected cells (Figures 4E–4G; Figure S5E). To more precisely address the importance of MDA5 in the SARS-CoV-2 infection-induced IFN response, we extracted intracellular RNA from SARS-CoV-2-infected Calu-3 cells and transfected these purified RNAs into naive cells to determine whether the viral RNA from infected cells could directly trigger the IFN response. Total RNA extracted from remdesivir-treated infected cells were used as a negative control. Transfection of the RNA extracted from infected cells into WT cells significantly induced IFN-β, while IFN-β mRNA levels were only slightly increased in the cells transfected with the intracellular RNA collected from infected remdesivir-treated cells (Figure 4H). More important, cells depleted of MDA5 or MAVS are less responsive to the stimulation by the transfected RNA, while RIG-I depletion has no effect. Collectively, these results further indicate that the replication intermediates are the major inducer of IFN through MDA5 signaling.
IRF3, IRF5, and p65 Are Required for IFN Signaling in Response to SARS-CoV-2 Infection
To identify the downstream components of the RLR signaling cascade that control IFN expression, we focused on transcription factors IRF1, IRF3, IRF5, IRF7, and p65. TBK1, a master regulator of innate immune signaling, is also included as a positive control. Consistent with its role in regulating antiviral immunity, siRNA-mediated knockdown of TBK1 almost completely prevented the expression of IFN-β mRNA in response to SARS-CoV-2 infection. Depletion of the transcription factors IRF3, IRF5, and p65 significantly reduced IFN induction, while IRF7 depletion had a limited effect (Figures 5A and 5B). Accordingly, virus replication and viral titers in the supernatants were greatly enhanced in cells depleted of TBK1, IRF3, IRF5, or p65 but not in cells depleted of IRF7 (Figure 5C; Figures S6A and S6B). These results indicate that IRF3, IRF5, and p65 are required for transcriptional activation of IFN expression. Consistent with these results, treatment with MRT67307, a potent kinase inhibitor specifically blocking TBK1 and I-kappa-B kinase epsilon (IKKε), efficiently enhanced SARS-CoV-2 infection in Calu-3 cells in a dose-dependent manner (Figure 5D). To complement these studies on siRNA-mediated knockdown, we monitored the phosphorylation state of these factors in the cells during SARS-CoV-2 infection. Calu-3 cells were infected with SARS-CoV-2 at an MOI of 1.0 and then collected at the indicated time for protein detection using western blotting. The SARS-CoV-2 NP protein was readily detected at 24 hpi and increased at 48 hpi, indicating SARS-CoV-2 productively replicated in these cells. The phosphorylated forms of TBK1 and IRF3 were detected at 24 hpi and then decreased to near basal levels at 48 hpi. Higher levels of phosphorylated p65 were still detected even at 48 hpi, suggesting that the activation of NF-κB signaling is extended in the infected cells, compared with IRF3 activation (Figure 5E). We were unable to monitor the dynamic change of phosphorylated IRF1, IRF5, and IRF7 because of the lack of suitable reagents. We then evaluated the nuclear translocation of these transcription factors in infected Calu-3 cells using confocal imaging. Cells were infected with SARS-CoV-2 at an MOI of 1.0 and then fixed at the indicated time points for detection of dsRNA as a surrogate of SARS-CoV-2 replication (Figure S6C) as well as the transcription factors with specific antibodies. We observed no nuclear accumulation of the detected transcription factors at the early time points post-infection. Significant portions of infected cells showed nuclear translocation of IRF3 (41%), IRF5 (23%), and p65 (48%) at 24 hpi, whereas the percentages of infected cells harboring IRF3 and IRF5 in the nucleus at 48 hpi dropped to 14% and 5%, respectively. Nevertheless, the number of infected cells with p65 in the nucleus slightly increased to 54% at 48 hpi (Figures 5F and 5G). Of note, we observed no nuclear accumulation of IRF7 in SARS-CoV-2-infected cells. Collectively, these results suggest that IRF3, IRF5, and NF-κB are the key transcription factors involved in the IFN response to SARS-CoV-2 infection in Calu-3 cells.
Figure 5.

IRF3, IRF5, and p65 Are Required for IFN Signaling Transduction in Response to SARS-CoV-2 Infection
(A) siRNA-mediated knockdown efficiency in Calu-3 cells was evaluated using qRT-PCR with specific primers.
(B) The IFN-β mRNA in the infected cells transfected with indicated siRNA was quantified using qRT-PCR. Data are expressed as fold change relative to non-treated cells.
(C) The percentage of infection was calculated as the ratio between the number of infected cells stained for SARS-CoV-2 NP and the total amount of cells stained with DAPI. Data are from three independent experiments with three technical replicates.
(D) The infection of SARS-CoV-2 in Calu-3 cells treated with MRT67307 at different concentrations. Calu-3 cells were infected with SARS-CoV-2 with MOI = 0.125 in the presence of MRT67307. At 48 h post-infection, cells were fixed, immunostained with rabbit-anti-SARS-CoV-2 NP antibody, and imaged using Celigo. Data are from two independent experiments with three technical replicates.
(E) Calu-3 cells infected with SARS-CoV-2 at an MOI of 1.0 were collected at indicated time points for detection of protein expression with specific antibodies.
(F) The confocal images showing nuclear translocation of IRF3, IRF5, IRF7, and RELA in the cells infected with SARS-CoV-2 at an MOI of 1.0. Cells were stained with NF-κB p65 (Cell Signaling, 8242), IRF3, or IRF7 along with anti-dsRNA [rJ2] antibody, and DAPI was used to stain the nuclei. Scale bar, 20 μm.
(G) Quantification of nuclear translocation of IRF3, IRF5, IRF7, or p65 from six fields of view collected from two independent experiments conducted as in (F).
Discussion
Compared with the recent epidemic outbreaks of SARS-CoV-1 and MERS-CoV, which had significantly higher case fatality rates, SARS-CoV-2 causes mild or asymptomatic disease in most cases (Petrosillo et al., 2020; Wang et al., 2020; Yu et al., 2020; Yuen et al., 2020). Eighty percent of cases of severe disease have occurred in patients older than 65 years and those with compromised immune systems (Chowdhury et al., 2020; Pormohammad et al., 2020; Zhang et al., 2020a). Importantly, a study of 50 COVID-19 patients with a range of disease severity found that IFN responses were higher in patients with mild to moderate disease but were lower in severely ill patients (Hadjadj et al., 2020). Additional studies found strong expression of numerous ISGs in the lung fluid of COVID-19 patients and infected mice, supporting the notion that the control of SARS-CoV-2 infection in most cases may be due to activation of the innate immune response (Israelow et al., 2020; Zhou et al., 2020b). Consistent with these observations, this study, as have others, demonstrated that lung epithelial cells induce an antiviral state in response to SARS-CoV-2, thereby limiting viral replication (Banerjee et al., 2020; Emanuel et al., 2020; Galani et al., 2021; Ravindra et al., 2020). Similar observations have been made in human intestinal epithelial cells infected with SARS-CoV-2 (Lamers et al., 2020; Stanifer et al., 2020). In addition to IFNs and ISGs, multiple proinflammatory cytokines, including IL6 and IL1β, were induced in the infected cells, but not to high levels, suggesting that epithelial cells might not be major contributors to the inflammatory response upon SARS-CoV-2 infection.
In this study, we observed that the kinetics of IFN production in response to viral challenge were delayed relative to the kinetics of viral replication by several hours. On the contrary, transfection with polyI:C or infection with SeV rapidly induced the IFN response in Calu-3 cells. These results suggest that SARS-CoV-2 has likely developed specific, and likely multiple, evasion strategies to dampen innate immune surveillance, and induction of the response occurs only after these avoidance mechanisms are overcome (Lei et al., 2020). For example, CoV endoribonuclease (EndoU) is essential for suppression of dsRNA-activated early host responses through cleavage of viral RNAs (Ancar et al., 2020; Hackbart et al., 2020; Kindler et al., 2017). Additionally, SARS-CoV-2 inhibits IFN signaling by blocking STAT1/STAT2 phosphorylation and nuclear import (Miorin et al., 2020; Xia et al., 2020), as observed in this study. Further studies are necessary to fully understand the underlying basis SARS-CoV-2 harbors comparable mechanisms to evade pattern recognition responses and IFN signaling transduction.
To date, numerous viral RNA sensors have been reported to initiate the innate immune response by specifically recognizing viral RNAs. Among them, TLRs, including TLR3 and TLR7, and RLRs, including MDA5 and RIG-I, have been documented to play a role in the CoV-induced IFN response in a cell type-dependent manner (Channappanavar et al., 2019; Li et al., 2010; Roth-Cross et al., 2008). We surveyed the impact of depletion of 16 viral RNA sensors potentially involved in sensing of SARS-CoV-2 infection and found that in addition to MDA5, LGP2 and NOD1 are essential, whereas RIG-I has no effect on the induction of IFNs in response to SARS-CoV-2. It remains unclear whether other sensors such as RIG-I have no viral targets or are possibly blocked by viral proteins. RIG-I and MDA5 share structural similarities and stimulate a signaling cascade through MAVS activation. Nonetheless, RIG-I preferentially recognizes RNA with a 5′ triphosphate group, while MDA5 preferentially binds to long dsRNA (Reikine et al., 2014). LGP2 is a helicase related to RIG-I and MDA5 but lacks the N-terminal CARD that is required for IFN induction in infected cells. LGP2 may function to amplify the MDA5-mediated IFN response by increasing the capacity of MDA5 to form stable filaments on short dsRNA (Sanchez David et al., 2019). Surprisingly, NOD1, a well-established intracellular sensor of bacterial peptidoglycans, was also identified to be critical for the IFN response to SARS-CoV-2 infection. It has been reported that NOD1 activation by Tri-DAP (a NOD1 agonist) suppresses human cytomegalovirus (HCMV) infection through IFN induction (Fan et al., 2016). SARS-CoV-2 infection may trigger NOD1 activation, leading to IFN production through the NF-κB pathway activation. Alternatively, NOD1 may regulate the MDA5-mediated signaling activated by SARS-CoV-2, as NOD1 has been shown to promote the IFN production by directly binding viral RNA and modulating the MDA5-MAVS complex formation (Wu et al., 2020b). LRRFIP1, an RNA-binding protein, was also identified as a factor that suppresses SARS-CoV-2 infection, in an IFN-independent manner. Previous reports have suggested that LRRFIP1 mediates the production of IFN via a β-catenin-dependent pathway in macrophages (Yang et al., 2010). Given the RNA-binding capacity of LRRFIP1, we hypothesize that LRRFIP1 inhibits SARS-CoV-2 infection by directly suppressing viral RNA transcription or translation (Takimoto, 2019). Further investigation is needed to understand the functional role of LRRFIP1 in SARS-CoV-2 infection.
In this study, we also examined the role of transcription factors, including IRF1, IRF3, IRF5, IRF7, and p65, in IFN signaling transduction in response to SARS-CoV-2 infection and found that IRF3, IRF5, and p65 play pivotal roles in regulating IFN mRNA production in SARS-CoV-2-infected cells. Intriguingly, IRF7, the master regulator of IFN responses (Honda et al., 2005), was not activated in cells infected with SARS-CoV-2, although its expression was upregulated by IFNs induced by viral infection. As IRF7 contributes mainly to the late-phase induction of IFNs in response to stimulation (Daffis et al., 2008; Ning et al., 2011), it is plausible that the accumulation of viral IFN antagonists at the late time point post-infection could actively suppress IFN signaling, leading to the inactivation of the IRF7-mediated IFN response, as reported recently (Lei et al., 2020). IRF3 and IRF5 are therefore likely to be the primary drivers of IFN induction in SARS-CoV-2-infected lung epithelium. In addition, NF-κB/p65 displays prolonged phosphorylation and nuclear localization upon SARS-CoV-2 infection compared with the shorter activation times of IRF3 and IRF5. Beyond its roles in the IFN and inflammatory responses (Liu et al., 2017), NF-κB activation is also essential for cell survival and proliferation. The extended NF-κB signaling activation could be beneficial for the efficient replication of SARS-CoV-2 by maintaining the viability of the infected cells.
Taken together, these findings indicate that human lung epithelial cells mount a robust cellular antiviral response that is mediated primarily through the MDA5 RNA sensor. Induction of these pathways results in a delayed induction of type I and type III IFN signaling, which ultimately results in restriction of viral replication. Further investigation of the host-pathogen interactions that govern these innate immune signaling circuits will likely provide additional insights into the molecular basis of severe disease outcomes and enable new therapeutic strategies for the treatment of patients with COVID-19.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-SARS-CoV-2 NP antibody | Kwok-Yung Yuen | N/A |
| Mouse anti-SARS-CoV-2 spike antibody [1A9] | GeneTex | Cat# GTX632604; RRID: AB_2864418 |
| Rabbit anti-RIG-I antibody | Enzo Life Sciences | Cat# ALX-210-932-C100; RRID: AB_2052506 |
| Rabbit anti-MDA5 antibody | Enzo Life Sciences | Cat# ALX-210-935-C100; RRID: AB_2051837 |
| Rabbit anti-IRF-9 antibody | Cell Signaling | Cat# 76684; RRID: AB_2799885 |
| Rabbit anti-STAT1 antibody | Cell Signaling | Cat# 14994; RRID: AB_2737027 |
| Rabbit anti-STAT2 antibody | Cell Signaling | Cat# 72604; RRID: AB_2799824 |
| Rabbit anti-ISG56 antibody | Thermo | Cat# PA3-848; RRID: AB_1958733 |
| Mouse anti-IFNAR1 antibody | Santa Cruz | Cat# sc-7391; RRID: AB_2122749 |
| Rabbit anti-IL10RB antibody | Thermo | Cat# 19387-1-AP; RRID: AB_10643371 |
| Rabbit anti-NF-kB p65 antibody | Cell Signaling | Cat# 8242; RRID: AB_10859369 |
| Rabbit anti-Phospho-NF-kB p65 antibody | Cell Signaling | Cat# 3033; RRID: AB_331284 |
| Rabbit anti-IRF3 antibody | Cell Signaling | Cat# 11904; RRID: AB_2722521 |
| Rabbit anti-Phospho-IRF3 antibody | Abcam | Cat# ab76493; RRID: AB_1523836 |
| Rabbit anti-TBK1 antibody | Cell Signaling | Cat# 3504; RRID: AB_2255663 |
| Rabbit anti-Phospho-TBK1 antibody | Cell Signaling | Cat# 5483; RRID: AB_10693472 |
| Rabbit anti-IRF5 antibody | Abcam | Cat# ab181553; RRID: AB_2801301 |
| Rabbit anti-IRF7 antibody | Sigma | Cat# HPA052757; RRID: AB_2681937 |
| Rabbit anti-IRF7 antibody | Cell Signaling | Cat# 4920; RRID: AB_2127551 |
| Rabbit anti-Phospho-IRF7 antibody | Cell Signaling | Cat# 12390; RRID: AB_2797896 |
| Rabbit anti-MAVS antibody | Enzo Life Sciences | Cat# ALX-210-929-C100; RRID: AB_2050916 |
| Mouse anti-dsRNA [rJ2] antibody | Kerafast | Cat# ES2001 |
| Mouse anti-acetylated tubulin antibody | Sigma | Cat# T7451; RRID: AB_609894 |
| Mouse anti-MUC5B antibody | Santa Cruz | Cat# sc-21768; RRID: AB_627975 |
| Rabbit anti-CFTR antibody | Alomone labs | Cat# ACL-006; RRID: AB_2039804 |
| Goat anti-ACE2 antibody | R&D systems | Cat# AF933; RRID: AB_355722 |
| Rabbit anti-TMPRSS2 antibody | Abcam | Cat# ab214462 |
| Rabbit anti-β-actin antibody | Sigma | Cat# A2228; RRID: AB_476697 |
| Rabbit anti-Phospho STAT1 (Y701) antibody | Cell Signaling | Cat# 9167; RRID: AB_561284 |
| Rabbit anti-Phospho STAT2 (Y690) antibody | Cell Signaling | Cat# 88410; RRID: AB_2800123 |
| Mouse anti-FOXJ1 antibody | Thermo | Cat# 14-9965-82; RRID: AB_1548835 |
| Mouse anti-CCSP antibody | R&D | Cat# MAB4218; RRID: AB_2183286 |
| Rabbit anti-p63 antibody | Cell Signaling | Cat# 13109; RRID: AB_2637091 |
| Mouse anti-CHGA antibody | Santa Cruz | Cat# sc-393941; RRID: AB_2801371 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo | Cat# A-11034; RRID: AB_2576217 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo | Cat# A-11011; RRID: AB_143157 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo | Cat# A-11001; RRID: AB_2534069 |
| IRDye® 800CW Donkey anti-Mouse IgG (H + L) | Li-Cor | Cat# 926-68072; RRID: AB_10953628 |
| IRDye® 680RD Donkey anti-Rabbit IgG (H + L) | Li-Cor | Cat# 926-68073; RRID: AB_10954442 |
| Bacterial and Virus Strains | ||
| SARS-CoV-2 USA-WA1/2020 | BEI Resources | NR-52281 |
| Sendai Virus (Cantell Strain) | Charles River | 10100774 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human IFN-α2a | PBL | 11100-1 |
| Human IL-29/IFN Lambda 1 | PBL | 11725–1 |
| High molecular weight (HMW) poly I:C | InvivoGen | tlrl-piclv |
| 3p-hpRNA | InvivoGen | tlrl-hprnalv |
| TransIT®-mRNA Transfection Kit | Mirus | MIR2250 |
| MRT67307 | Invivogen | Inh-met |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo | 13778150 |
| Critical Commercial Assays | ||
| DIY Human IFN Lambda 3/1/2 (IL-28B/29/28A) ELISA Kit | pbl assay science | 61840-1 |
| VeriKine-HS™ Human Interferon Beta TCM ELISA Kit | pbl assay science | 41435-1 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo | 4368813 |
| TaqMan Fast Virus 1-Step Master Mix | Thermo | 4444436 |
| Power SYBR Green Master Mix | Thermo | 4368708 |
| Experimental Models: Cell Lines | ||
| Vero E6 | ATCC | CRL-1586 |
| Calu-3 | ATCC | HTB-55 |
| Oligonucleotides | ||
| Primers for qRT-PCR | This paper | Table S1 |
| Software and Algorithms | ||
| Prism 8 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | N/A |
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sumit K. Chanda (schanda@sbpdiscovery.org).
Materials availability
This study did not generate new unique reagents. Commercially available reagents are indicated in the Key Resources Table.
Data and code availability
This study did not generate/analyze datasets/code.
Experimental Model and Subject Details
Cells and viruses
Vero E6 (ATCC® CRL-1586) were maintained in Dulbecco Modified Eagle medium (DMEM, GIBCO) containing 10% heat-inactivated fetal bovine serum (FBS, GIBCO), 50 U/mL penicillin, 50 μg/mL streptomycin. Calu-3 (ATCC® HTB-55) were maintained in Eagle’s Minimum Essential Medium (EMEM, ATCC) supplemented with 10% heat-inactivated fetal bovine serum (FBS, GIBCO), 50 U/mL penicillin, 50 μg/mL streptomycin.
SARS-CoV-2 USA-WA1/2020 strain was obtained from BEI Resources (NR-52281). The virus was further propagated in Vero E6 cells and stored at −80°C in aliquots. Plaque forming unit (PFU) were performed to titrate the cultured virus. All experiments involving live SARS-CoV-2 followed the approved standard operating procedures of the Biosafety Level 3 facility at Sanford Burnham Prebys Medical Discovery Institute.
Differentiation of human induced pluripotent stem cells (hiPSCs) into airway epithelial cells
Airway epithelial cells were differentiated from B2-3 SFTPC–GFP reporter hiPSCs via NKX2-1+ lung progenitor cells (Gotoh et al., 2014). Briefly, NKX2-1+ lung progenitor cells were induced and isolated by carboxypeptidase M (CPM)-based cell sorting as previously described (Yamamoto et al., 2017). Isolated CPM+ cells were cultured at a density of 9.4 × 105 cells/cm on a culture plate with PneumaCult-ALI Maintenance medium (STEMCELL Technologies) supplemented with 10 μM Y27632(LC Laboratories), 4 μg/ml heparin (Nacalai Tesque) and 1 μM hydrocortisone (Sigma-Aldrich) (Konishi et al., 2016). After two days of culture, the medium was further supplemented with 10 μM DAPT (Wako) and replenished on every other day for a period of one month.
Method Details
CRISPR-Cas9 knockout in Calu-3 cells
Detailed protocols for RNP production have been previously published (Hultquist et al., 2019). Briefly, lyophilized guide RNA (gRNA) and tracrRNA (Dharmacon) were suspended at a concentration of 160 μM in 10 mM Tris-HCL, 150mM KCl, pH 7.4. 5μL of 160μM gRNA was mixed with 5μL of 160μM tracrRNA and incubated for 30 min at 37°C. The gRNA:tracrRNA complexes were then mixed gently with 10μL of 40μM Cas9 (UC-Berkeley Macrolab) to form CRISPR-Cas9 ribonucleoproteins (crRNPs). Five 3.5μL aliquots were frozen in Lo-Bind 96-well V-bottom plates (E&K Scientific) at −80°C until use. Each gene was targeted by 4 independent, multiplexed gRNA derived from the Dharmacon pre-designed Edit-R library for gene knockout. Non-targeting negative control gRNA (Dharmacon, U-007501) was delivered in parallel.
Each electroporation reaction consisted of 2.5x105 Calu-3 cells, 3.5 μL crRNPs, and 20 μL electroporation buffers. Calu-3 cells were grown in fully supplemented MEM (10% FBS, 1xPen/Strep, 1x non-essential amino acids) to 70% confluency, suspended, and counted. crRNPs were thawed and allowed to come to room-temperature. Immediately prior to electroporation, cells were centrifuged at 400xg for 3 min, supernatant was removed by aspiration, and the pellet was resuspended in 20 μL of room-temperature SE electroporation buffer plus supplement (Lonza) per reaction. 20 μL of cell suspension was then gently mixed with each crRNP and aliquoted into a 96-well electroporation cuvette for nucleofection with the 4-D Nucleofector X-Unit (Lonza) using pulse code EO-120. Immediately after electroporation, 80 μL of pre-warmed media was added to each well and cells were allowed to rest for 30 min in a 37°C cell culture incubator. Cells were subsequently moved to 12-well flat-bottomed culture plates pre-filled with 500 μL pre-warmed media. Cells were cultured at 37°C / 5% CO2 in a dark, humidified cell culture incubator for 4 days to allow for gene knockout and protein clearance prior to downstream applications.
siRNA Transfection and SARS-CoV-2 infection
The siRNA transfection was conducted in 384-well plate format. 6,000 Calu-3 cells were plated and transfected with siRNA SMARTpools (Dharmacon) using RNAiMAX (Invitrogen). 48 h post-transfection, cells were infected with SARS-CoV-2 at MOI of 0.125 per well. 48 h post-infection, infected cells were fixed with 5% paraformaldehyde (PFA) for 4 h and permeabilized with 0.4% Triton X-100 for 15 min. After blocking with blocking buffer containing 3% bovine serum albumin (BSA) and 10% goat serum for 30 min, cells were incubated for overnight at 4°C with rabbit-anti-SARS-CoV-2 NP polyclonal antibodies. After three washes with phosphate-buffered saline (PBS), the cells were incubated with Alexa Fluor 488-conjugated goat-anti-rabbit IgG (Thermo Fisher Scientific, USA) for 1 h at room temperature. After three additional washes, antifade-4 6-diamidino-2-phenylindole (DAPI) mounting solution (Sigma) was added to the cells for 30 min before imaging. The fluorescence intensity of cells was scanned and the SARS-CoV-2 NP-positive cells were automatically calculated by Celigo imaging cytometry system (Nexcelom, Lawrence, MA, USA) or using the IC200 imaging system (Vala Sciences, CA, USA).
Real-time qRT-PCR
Total RNA was extracted from Calu-3 cells or iPSC-derived airway epithelial cells with NucleoSpin® 96 RNA kit (Takara) in accordance with the manufacturer’s instructions. The viral mRNA was quantified with TaqPath 1-step RT-qPCR Master Mix (Thermo) using the primer pairs that specifically target viral genes as previously reported (Peña et al., 2016; Vogels et al., 2020). A quantified full-length SARS-CoV-2 RNA or SeV RNA was used as standard.
The interested target genes expression levels were measured by real-time RT-PCR using qScript One-Step SYBR® Green qRT-PCR Kit, Low ROX(QuantaBio) with specific primers listed in Table S1. The mRNA level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in each sample was used for normalization. ΔΔCT was used to calculate the fold changes relative to the controls.
Confocal imaging analysis
The infected cells were fixed with 5% paraformaldehyde overnight at 4°C, and then permeabilized in 1xPBS (Corning) containing 0.4% (v/v) Triton X-100 for 15 min. After permeabilization, the cells were next incubated with PBS containing 3% bovine serum albumin (BSA) and 10% goat serum (Sigma) for 30 min. Primary antibodies were diluted with blocking solution and incubated with cells overnight at 4°C: NF-kB p65 (1:200), IRF3 (1:200), IRF5 (1:100), or IRF-7 (1:100) along with dsRNA [rJ2] antibody (1:100). Cells were then washed three times with 1xPBS and incubated with secondary antibody for 60 min at room temperature. After incubation, cells were washed three time with 1xPBS, and then incubated for 30 min with DAPI. Cells were imaged using Zeiss LSM 710 confocal microscope (Zeiss) with a 40x objective.
Immunoblotting
Cellular lysates were collected on ice in the lysis buffer (100 mM Tris-HCl (pH 7.5), 50 mM NaCl, 5 mM EDTA, and 1% Triton X-100) in the presence of protease inhibitor cocktail (Roche) as described previously. Samples were separated in Novex Tris-Glycine Gels (Thermo Fisher Scientific), and transferred to a PVDF membrane (Bio-rad). The transferred membranes were incubated overnight at 4°C with primary antibody diluted in the Odyssey® Blocking Buffer (LI-COR Biosciences). After three washes with PBS solution with 0.05% TWEEN-20, the membranes were incubated for 1 h with the appropriate secondary antibodies and then scanned with Odyssey Infrared Imaging System (LI-COR Biosciences).
Antiviral activity of IFNs
Calu-3 cells were plated at 6,000 cells/well in a black 384-well plate (Greiner). At 24 h post-seeding, cells were treated with IFN-β or IFN-λ (PBL) at indicated concentrations. After 24 h of treatment, the cells were infected with SARS-CoV-2 at MOI of 0.125 with additional IFNs at corresponding concentrations. At 48 h post-infection, infected cells were fixed with 5% paraformaldehyde (PFA) for 4 h and then immunostained with anti-SARS-CoV-2 NP antibody as described above.
ELISA for the detection of supernatant IFN-β or IFN-λ concentrations
IFN-β or IFN-λ1/2/3 in supernatants collected from the infected cells were measured by the human IFN-β (41435-1) or IFN-λ1/2/3 (61840-1) ELISA kits (PBL Assay Science, NJ) following manufacturer’s instructions.
Plaque-forming assay
Viral titers in cell culture supernatant were determined by a modified plaque-forming assay with Vero E6 cells. Briefly, 300,000 Vero E6 cells seeded on 12-well plates were inoculated with 10-fold serial dilutions of culture supernatants collected from the infected cells. After inoculation for 1 h at 37°C, 1 mL of 0.6% microcrystalline cellulose (MCC; Sigma 435244, MO, USA) diluted in serum-free DMEM was added. The plates were then incubated 3 days at 37°C/5% CO2. The MCC was then aspirated out, 10% Neutral Buffered Formalin (NBF) was added for 4 h at room temperature and then removed. For staining of fixed-cell monolayers, 0.4% crystal violet in 20% methanol. Plaques were quantified and recorded as PFU/mL.
Quantification and Statistical Analysis
Statistical parameters including the exact value of n, dispersion and precision measures (mean ± SEM/SD), and statistical significance are reported in the figures and figure legends. Data were shown as mean values with error bars indicating the SEM or SD. Statistical significance between groups was determined with unpaired Student’s t test using GraphPad Prism 8.0 (GraphPad, San Diego, CA). Unless otherwise noted, n.s = not significant; ∗ p < 0.05; ∗∗ p < 0.01.
Acknowledgments
The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281. This work was supported by the following grants to the Sanford Burnham Prebys Medical Discovery Institute: DOD W81XWH-20-1-0270, DHIPC U19 AI118610, Fluomics/NOSI U19 AI135972 and R01 AI127302-01A1, as well as generous philanthropic donations from Dinah Ruch and Susan and James Blair. This work was additionally supported by the following grants to Northwestern University Feinberg School of Medicine: a CTSA supplement to NCATS UL1 TR002389, a CTSA supplement to NUCATS with the generous support of the Dixon family UL1 TR001422, and a Cancer Center supplement P30 CA060553. This work was also partly supported by CRIP (Center for Research for Influenza Pathogenesis), a NIAID-supported Center of Excellence for Influenza Research and Surveillance (CEIRS; contract HHSN272201400008C); NIAID grant U19AI142733; NCI grant U54CA260560; supplements to NIAID grant U19AI135972 and DOD grant W81XWH-20-1-0270; the Defense Advanced Research Projects Agency (HR0011-19-2-0020); and by the generous support of the JPB Foundation, the Open Philanthropy Project (research grants 2020-215611 and 2020-218415), and anonymous donors to A.G.-S. Development and implementation of iPSC technology for production of airway epithelial cells was supported by the Incubation Program of the Office of Society-Academia Collaboration for Innovation, Kyoto University. The funding sources had no role in study design, data collection, analysis, interpretation, or writing of the report. We thank Dr. Ryo Shioda (PerkinElmer) for technical assistance in operating Opera Phenix.
Author Contributions
X.Y., L.R., Y.P., L.M.-S., J.K., Y.Y., K.S., S.G., L.M., P.D.D.J., C.-C.Y., and J.F.H. designed and/or performed experiments. X.Y., L.R., Y.P., J.K., Y.Y., K.S., S.G., and J.F.H. analyzed data. L.M. and A.G.-S. provided critical reagents and intellectual input. S.K.C. and X.Y. oversaw the conception and design of the experiments. X.Y., K.M.H., S.Y., and S.K.C. wrote the manuscript. All authors revised the manuscript.
Declaration of Interests
The García-Sastre laboratory has received research support from Pfizer, Senhwa Biosciences, 7Hills Pharma, Pharmamar, Blade Therapeutics, Avimex, Dynavax, Kenall Manufacturing, and ImmunityBio. A.G.-S. has consulting agreements with the following companies involving cash and/or stock: Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Pagoda, Vaxalto, Accurius, and Esperovax. Y.Y. and S.G. are founders, shareholders, and board members of HiLung Inc. Y.Y. and S.G. have patents (PCT/JP2016/059786) related to this work.
Published: January 12, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108628.
Supplemental Information
References
- Ancar R., Li Y., Kindler E., Cooper D.A., Ransom M., Thiel V., Weiss S.R., Hesselberth J.R., Barton D.J. Physiologic RNA targets and refined sequence specificity of coronavirus EndoU. RNA. 2020;26:1976–1999. doi: 10.1261/rna.076604.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschenbrenner D.S. Remdesivir Receives emergency use authorization for severely ill patients with COVID-19. Am. J. Nurs. 2020;120:26. doi: 10.1097/01.NAJ.0000688196.83625.b1. [DOI] [PubMed] [Google Scholar]
- Banerjee A., Budylowski P., Richard D., Maan H., Aguiar J.A., El-Sayes N., D’Agostino M.R., Tremblay B.J.-M., Afkhami S., Karimzadeh M., et al. Early temporal dynamics of cellular responses to SARS-CoV-2. bioRxiv. 2020 doi: 10.1101/2020.06.18.158154. [DOI] [Google Scholar]
- Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.-H., Zhang Y., Dorgham K., Philippot Q., Rosain J., Béziat V., et al. HGID Lab. NIAID-USUHS Immune Response to COVID Group. COVID Clinicians. COVID-STORM Clinicians. Imagine COVID Group. French COVID Cohort Study Group. Milieu Intérieur Consortium. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birra D., Benucci M., Landolfi L., Merchionda A., Loi G., Amato P., Licata G., Quartuccio L., Triggiani M., Moscato P. COVID 19: a clue from innate immunity. Immunol. Res. 2020;68:161–168. doi: 10.1007/s12026-020-09137-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., Fehr A.R., Zheng J., Wohlford-Lenane C., Abrahante J.E., Mack M., Sompallae R., McCray P.B., Jr., Meyerholz D.K., Perlman S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Invest. 2019;129:3625–3639. doi: 10.1172/JCI126363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury M.A., Hossain N., Kashem M.A., Shahid M.A., Alam A. Immune response in COVID-19: a review. J. Infect. Public Health. 2020;13:1619–1629. doi: 10.1016/j.jiph.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S., Samuel M.A., Suthar M.S., Keller B.C., Gale M., Jr., Diamond M.S. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J. Virol. 2008;82:8465–8475. doi: 10.1128/JVI.00918-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel W., Kirstin M., Vedran F., Asija D., Theresa G.L., Roberto A., Filippos K., David K., Salah A., Christopher B., et al. Bulk and single-cell gene expression profiling of SARS-CoV-2 infected human cell lines identifies molecular targets for therapeutic intervention. bioRxiv. 2020 doi: 10.1101/2020.05.05.079194. [DOI] [Google Scholar]
- Fan Y.-H., Roy S., Mukhopadhyay R., Kapoor A., Duggal P., Wojcik G.L., Pass R.F., Arav-Boger R. Role of nucleotide-binding oligomerization domain 1 (NOD1) and its variants in human cytomegalovirus control in vitro and in vivo. Proc. Natl. Acad. Sci. U S A. 2016;113:E7818–E7827. doi: 10.1073/pnas.1611711113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felgenhauer U., Schoen A., Gad H.H., Hartmann R., Schaubmar A.R., Failing K., Drosten C., Weber F. Inhibition of SARS-CoV-2 by type I and type III interferons. J. Biol. Chem. 2020;295:13958–13964. doi: 10.1074/jbc.AC120.013788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Heise M., Baric R. SARS coronavirus and innate immunity. Virus Res. 2008;133:101–112. doi: 10.1016/j.virusres.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galani I.-E., Rovina N., Lampropoulou V., Triantafyllia V., Manioudaki M., Pavlos E., Koukaki E., Fragkou P.C., Panou V., Rapti V., et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021;22:32–40. doi: 10.1038/s41590-020-00840-x. [DOI] [PubMed] [Google Scholar]
- García-Sastre A. Ten strategies of interferon evasion by viruses. Cell Host Microbe. 2017;22:176–184. doi: 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh S., Ito I., Nagasaki T., Yamamoto Y., Konishi S., Korogi Y., Matsumoto H., Muro S., Hirai T., Funato M., et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Reports. 2014;3:394–403. doi: 10.1016/j.stemcr.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grein J., Ohmagari N., Shin D., Diaz G., Asperges E., Castagna A., Feldt T., Green G., Green M.L., Lescure F.-X., et al. Compassionate use of remdesivir for patients with severe COVID-19. N. Engl. J. Med. 2020;382:2327–2336. doi: 10.1056/NEJMoa2007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackbart M., Deng X., Baker S.C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. U S A. 2020;117:8094–8103. doi: 10.1073/pnas.1921485117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann H.H., Schneider W.M., Rice C.M. Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol. 2015;36:124–138. doi: 10.1016/j.it.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N., Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Hultquist J.F., Hiatt J., Schumann K., McGregor M.J., Roth T.L., Haas P., Doudna J.A., Marson A., Krogan N.J. CRISPR-Cas9 genome engineering of primary CD4+ T cells for the interrogation of HIV-host factor interactions. Nat. Protoc. 2019;14:1–27. doi: 10.1038/s41596-018-0069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelow B., Song E., Mao T., Lu P., Meir A., Liu F., Madel Alfajaro M., Wei J., Dong H., Homer R.J., et al. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. bioRxiv. 2020 doi: 10.1101/2020.05.27.118893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen S., Thomsen A.R. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012;86:2900–2910. doi: 10.1128/JVI.05738-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler E., Gil-Cruz C., Spanier J., Li Y., Wilhelm J., Rabouw H.H., Züst R., Hwang M., V’kovski P., Stalder H., et al. Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog. 2017;13:e1006195. doi: 10.1371/journal.ppat.1006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi S., Gotoh S., Tateishi K., Yamamoto Y., Korogi Y., Nagasaki T., Matsumoto H., Muro S., Hirai T., Ito I., et al. Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Reports. 2016;6:18–25. doi: 10.1016/j.stemcr.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers M.M., Beumer J., van der Vaart J., Knoops K., Puschhof J., Breugem T.I., Ravelli R.B.G., Paul van Schayck J., Mykytyn A.Z., Duimel H.Q., et al. SARS-CoV-2 productively infects human gut enterocytes. Science. 2020;369:50–54. doi: 10.1126/science.abc1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear H.M., Schoggins J.W., Diamond M.S. Shared and distinct functions of type I and type III interferons. Immunity. 2019;50:907–923. doi: 10.1016/j.immuni.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.C., Chathuranga K., Lee J.S. Intracellular sensing of viral genomes and viral evasion. Exp. Mol. Med. 2019;51:1–13. doi: 10.1038/s12276-019-0299-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X., Dong X., Ma R., Wang W., Xiao X., Tian Z., Wang C., Wang Y., Li L., Ren L., et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020;11:3810. doi: 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Liu Y., Zhang X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J. Virol. 2010;84:6472–6482. doi: 10.1128/JVI.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Zhang L., Joo D., Sun S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017;2:17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miorin L., Kehrer T., Sanchez-Aparicio M.T., Zhang K., Cohen P., Patel R.S., Cupic A., Makio T., Mei M., Moreno E., et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. U S A. 2020;117:28344–28354. doi: 10.1073/pnas.2016650117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning S., Pagano J.S., Barber G.N. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12:399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña J., Chen-Harris H., Allen J.E., Hwang M., Elsheikh M., Mabery S., Bielefeldt-Ohmann H., Zemla A.T., Bowen R.A., Borucki M.K. Sendai virus intra-host population dynamics and host immunocompetence influence viral virulence during in vivo passage. Virus Evol. 2016;2:vew008. doi: 10.1093/ve/vew008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosillo N., Viceconte G., Ergonul O., Ippolito G., Petersen E. COVID-19, SARS and MERS: are they closely related? Clin. Microbiol. Infect. 2020;26:729–734. doi: 10.1016/j.cmi.2020.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pormohammad A., Ghorbani S., Baradaran B., Khatami A., J Turner R., Mansournia M.A., Kyriacou D.N., Idrovo J.-P., Bahr N.C. Clinical characteristics, laboratory findings, radiographic signs and outcomes of 61,742 patients with confirmed COVID-19 infection: a systematic review and meta-analysis. Microb. Pathog. 2020;147:104390. doi: 10.1016/j.micpath.2020.104390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindra N.G., Alfajaro M.M., Gasque V., Wei J., Filler R.B., Huston N.C., Wan H., Szigeti-Buck K., Wang B., Montgomery R.R., et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human bronchial epithelial cells. bioRxiv. 2020 doi: 10.1101/2020.05.06.081695. [DOI] [Google Scholar]
- Reikine S., Nguyen J.B., Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front. Immunol. 2014;5:342. doi: 10.3389/fimmu.2014.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth-Cross J.K., Bender S.J., Weiss S.R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 2008;82:9829–9838. doi: 10.1128/JVI.01199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin E.J., Longo D.L. SARS-CoV-2 Vaccination — An Ounce (Actually, Much Less) of Prevention. New England Journal of Medicine. 2020;383:2677–2678. doi: 10.1056/NEJMe2034717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez David R.Y., Combredet C., Najburg V., Millot G.A., Beauclair G., Schwikowski B., Léger T., Camadro J.-M., Jacob Y., Bellalou J., et al. LGP2 binds to PACT to regulate RIG-I- and MDA5-mediated antiviral responses. Sci. Signal. 2019;12:eaar3993. doi: 10.1126/scisignal.aar3993. [DOI] [PubMed] [Google Scholar]
- Stanifer M.L., Pervolaraki K., Boulant S. Differential regulation of type I and type III interferon signaling. Int. J. Mol. Sci. 2019;20:1445. doi: 10.3390/ijms20061445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanifer M.L., Kee C., Cortese M., Zumaran C.M., Triana S., Mukenhirn M., Kraeusslich H.-G., Alexandrov T., Bartenschlager R., Boulant S. Critical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells. Cell Rep. 2020;32:107863. doi: 10.1016/j.celrep.2020.107863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O., Akira S. Innate immunity to virus infection. Immunol. Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto M. Multidisciplinary roles of LRRFIP1/GCF2 in human biological systems and diseases. Cells. 2019;8:108. doi: 10.3390/cells8020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogels C.B.F., Brito A.F., Wyllie A.L., Fauver J.R., Ott I.M., Kalinich C.C., Petrone M.E., Casanovas-Massana A., Catherine Muenker M., Moore A.J., et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 2020;5:1299–1305. doi: 10.1038/s41564-020-0761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Wang Z., Zhao J., Zeng X., Wu M., Wang S., Wang T. Epidemiological and clinical course of 483 patients with COVID-19 in Wuhan, China: a single-center, retrospective study from the mobile cabin hospital. Eur. J. Clin. Microbiol. Infect. Dis. 2020;39:2309–2315. doi: 10.1007/s10096-020-03927-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A.P., Bear C.E., Chin S., Pasceri P., Thompson T.O., Huan L.-J., Ratjen F., Ellis J., Rossant J. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat. Biotechnol. 2012;30:876–882. doi: 10.1038/nbt.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.M., Zhang J., Li P.W., Hu Y.W., Cao L., Ouyang S., Bi Y.H., Nie P., Chang M.X. NOD1 promotes antiviral signaling by binding viral RNA and regulating the interaction of MDA5 and MAVS. J. Immunol. 2020;204:2216–2231. doi: 10.4049/jimmunol.1900667. [DOI] [PubMed] [Google Scholar]
- Xia H., Cao Z., Xie X., Zhang X., Chen J.Y.-C., Wang H., Menachery V.D., Rajsbaum R., Shi P.-Y. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 2020;33:108234. doi: 10.1016/j.celrep.2020.108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y., Gotoh S., Korogi Y., Seki M., Konishi S., Ikeo S., Sone N., Nagasaki T., Matsumoto H., Muro S., et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat. Methods. 2017;14:1097–1106. doi: 10.1038/nmeth.4448. [DOI] [PubMed] [Google Scholar]
- Yang P., An H., Liu X., Wen M., Zheng Y., Rui Y., Cao X. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a β-catenin-dependent pathway. Nat. Immunol. 2010;11:487–494. doi: 10.1038/ni.1876. [DOI] [PubMed] [Google Scholar]
- Yu C., Lei Q., Li W., Wang X., Li W., Liu W. Epidemiological and clinical characteristics of 1663 hospitalized patients infected with COVID-19 in Wuhan, China: a single-center experience. J. Infect. Public Health. 2020;13:1202–1209. doi: 10.1016/j.jiph.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen K.-S., Ye Z.-W., Fung S.-Y., Chan C.-P., Jin D.-Y. SARS-CoV-2 and COVID-19: the most important research questions. Cell Biosci. 2020;10:40. doi: 10.1186/s13578-020-00404-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Yao H., Zhang D., Pan J., Peng E., Huang J., Zhang Y., Xu X., Tian G., Xu H., et al. Epidemiologic and clinical characteristics of 42 deaths caused by SARS-CoV-2 infection in Wuhan, China: a retrospective study. Biosaf. Health. 2020;2:164–168. doi: 10.1016/j.bsheal.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J., Ogishi M., Sabli I.K.D., Hodeib S., Korol C., et al. COVID-STORM Clinicians. COVID Clinicians. Imagine COVID Group. French COVID Cohort Study Group. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort. NIAID-USUHS/TAGC COVID Immunity Group Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Ren L., Zhang L., Zhong J., Xiao Y., Jia Z., Guo L., Yang J., Wang C., Jiang S., et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27:883–890.e2. doi: 10.1016/j.chom.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.