

Abstract
The main protease (3CL Mpro) from SARS–CoV-2, the etiological agent of COVID-19, is an essential enzyme for viral replication. 3CL Mpro possesses an unusual catalytic dyad composed of Cys145 and His41 residues. A critical question in the field has been what the protonation states of the ionizable residues in the substrate-binding active-site cavity are; resolving this point would help understand the catalytic details of the enzyme and inform rational drug development against this pernicious virus. Here, we present the room-temperature neutron structure of 3CL Mpro, which allowed direct determination of hydrogen atom positions and, hence, protonation states in the protease. We observe that the catalytic site natively adopts a zwitterionic reactive form in which Cys145 is in the negatively charged thiolate state and His41 is doubly protonated and positively charged, instead of the neutral unreactive state usually envisaged. The neutron structure also identified the protonation states, and thus electrical charges, of all other amino acid residues and revealed intricate hydrogen-bonding networks in the active-site cavity and at the dimer interface. The fine atomic details present in this structure were made possible by the unique scattering properties of the neutron, which is an ideal probe for locating hydrogen positions and experimentally determining protonation states at near-physiological temperature. Our observations provide critical information for structure-assisted and computational drug design, allowing precise tailoring of inhibitors to the enzyme's electrostatic environment.
Keywords: joint neutron, X-ray crystallography, room temperature, SARS–CoV-2, 3CL main protease, 3CL Mpro, protonation state, hydrogen bond, enzyme mechanism, drug design, neutron diffraction, viral protease, SARS–CoV-2 3CL main protease
COVID-19, a deadly disease caused by the novel coronavirus SARS–CoV-2 (severe acute respiratory syndrome–coronavirus 2), is a pandemic of extraordinary proportions, disrupting social life, travel, and the global economy. The development of vaccines and therapeutic intervention measures promises to mitigate the spread of the virus and to alleviate the burdens COVID-19 has caused in many communities in recent months (1–6). SARS–CoV-2 is a single-stranded positive-sense RNA virus with a genome comprised of ∼30,000 nucleotides. The viral replicase gene encodes two overlapping polyproteins, pp1a and pp1ab, consisting of individual viral proteins essential for replication (7, 8). Each polyprotein must be processed into individual functional proteins, a vital step in the virus life cycle. This is accomplished by a chymotrypsin-like protease, 3CL Mpro or main protease, a hydrolase enzyme that cleaves peptide bonds. The proper functioning of 3CL Mpro is indispensable for SARS–CoV-2 replication, whereas its inhibition leads to the inability to produce mature infectious virions. Thus, the enzyme is considered a promising target for the design and development of SARS–CoV-2–specific protease inhibitors and for repurposing existing clinical drugs (9–15).
SARS–CoV-2 3CL Mpro is a cysteine protease (16, 17) and is catalytically active as a homodimer (Fig. 1). Its amino acid sequence is 96% homologous to the earlier SARS–CoV 3CL Mpro, and the catalytic efficiencies of the two enzymes are similar (10, 11, 18–20). The ∼34-kDa enzyme has three distinct domains: catalytic domains I (residues 8–101) and II (residues 102–184) and the α-helical domain III (residues 201–303), which is required for protein dimerization (10, 11). Importantly, the monomeric enzyme shows no catalytic activity, as was demonstrated for SARS–CoV 3CL Mpro (21–24). The catalytic site of 3CL Mpro employs a noncanonical Cys145–His41 dyad thought to be assisted by a water molecule hydrogen-bonded to the catalytic histidine (18, 25). The cysteine thiol group functions as the nucleophile during the first step of the hydrolysis reaction by attacking the carbon atom of the scissile peptide bond. Substrate hydrolysis requires the catalytic dyad to be in the zwitterionic state with deprotonated Cys145 and protonated His41, which either can be generated through a proton transfer from the Cys145 thiol to the His41 imidazole by a general acid-base mechanism (19) or may already be present before substrate binding (20, 26). However, the protonation states of the 3CL Mpro catalytic site have not been experimentally determined. The enzyme recognizes a general amino acid sequence of Leu-Gln↓Ser-Ala-Gly, where ↓ marks the cleavage site, but displays some substrate sequence promiscuity. The active-site cavity is located on the surface of the protease and can bind substrate residues in positions P1´ through P5 in the substrate-binding subsites S1ʹ–S5, respectively (Fig. 2). Subsites S1, S2, and S4 are shaped into well-formed binding pockets, whereas S1ʹ, S3, and S5 are located on the protein surface with no defined shape. The peptide bond cleavage occurs between the substrate residues at the C-terminal position P1ʹ and N-terminal position P1.
Figure 1.
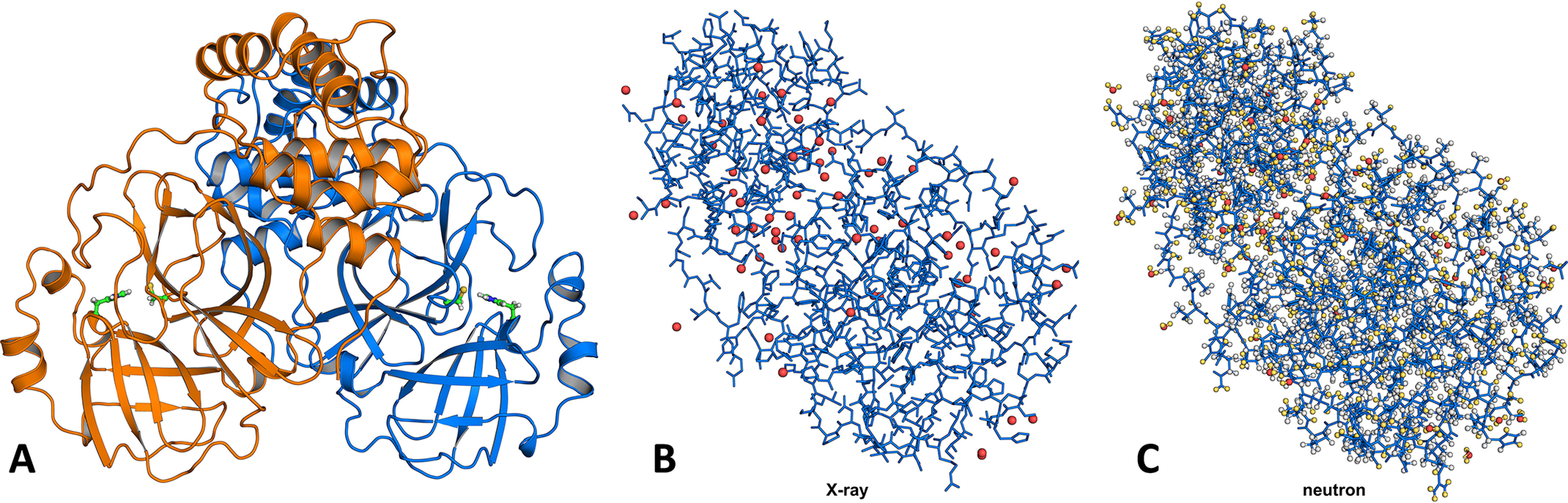
Structure of SARS–CoV-2 3CL Mpro. A, the catalytically active dimer is shown in cartoon representation, with the catalytic dyad residues Cys145 and His41 in ball-and-stick representation. B, the enzyme protomer shown in stick representation demonstrates that X-ray diffraction only provides the positions of heavy atoms, i.e. carbon, nitrogen, oxygen, and sulfur (2380 atoms in one protomer). Water molecules are shown as red spheres. C, the same protomer shown with deuterium atoms colored yellow and hydrogen atoms colored gray illustrates that neutron diffraction provides positions of all atoms in the enzyme (4681 atoms in one protomer). Notice how much busier the protein neutron structure is compared with its X-ray structure because there are twice as many atoms visible when using neutron diffraction data. Only oxygen atoms (red spheres) are visible for water molecules in the X-ray structure, whereas all three atoms are fully visible in the neutron structure.
Figure 2.

SARS–CoV-2 3CL Mpro active site architecture indicating the positions of substrate-binding subsites S1′–S5 and the oxyanion hole.
Current structure-assisted drug design efforts are mainly directed toward reversible and irreversible covalent inhibitors that mimic the protease substrate binding to subsites S1ʹ–S5 in the active-site cavity (9–12, 14), whereas the dimer interface can also be explored for the design of dimerization inhibitors (27, 28). Knowledge of the SARS–CoV-2 3CL Mpro active-site cavity structure at an atomic level of detail, including the actual locations of hydrogen atoms, can provide critical information to improve rational drug design. The presence or absence of hydrogen atoms at specific sites on amino acid residues determines their protonation states and, thus, their electrical charges, defining the electrostatics and hydrogen-bonding interactions. Of note, half of all atoms in protein and small-molecule drugs are hydrogen. X-ray crystallography is typically the standard experimental method for structure-assisted drug design but cannot reliably locate hydrogen atoms in biological macromolecules, leaving significant gaps in our understanding of biological function and drug binding (29). Electron clouds scatter X-rays; thus, scattering power is determined by the number of electrons present in an atom, i.e. by its atomic number. Hydrogen, with just a single electron that often participates in highly polarized chemical bonds, is the weakest possible X-ray scatterer and consequently is invisible in X-ray structures with a few exceptions beyond subatomic resolution (30–32). In contrast, atomic nuclei scatter neutrons, where the scattering power of neutrons is independent of the atomic number. Deuterium, a heavy isotope of hydrogen, scatters neutrons just as well as carbon, nitrogen, and oxygen. Neutron crystallography is capable of accurately determining positions of hydrogen and deuterium atoms and visualizing hydrogen-bonding interactions at moderate resolutions (33–36), where X-rays cannot locate functional hydrogen atoms (30). Moreover, unlike X-rays (37), neutrons cause no direct or indirect radiation damage to protein crystals, permitting diffraction data collection at near-physiological (room) temperature, avoiding possible artifacts induced by the use of cryoprotectant chemicals required for X-ray cryo-crystallographic measurements.
In neutron crystallographic experiments, protein crystals are usually hydrogen/deuterium–exchanged with the heavy water (D2O) to increase the signal-to-noise ratio of the diffraction pattern because hydrogen has a large incoherent scattering cross-section that increases background. Also, the coherent neutron-scattering length of hydrogen is negative (−3.739 fm) and is therefore observed in the neutron-scattering length (or nuclear) density maps as troughs. At moderate resolutions, the negative neutron-scattering length of hydrogen leads to the density cancelation phenomenon observed for CH, CH2, and CH3 groups as hydrogen atoms attached to carbon atoms cannot exchange with deuterium. Conversely, deuterium has a coherent neutron-scattering length of +6.671 fm and, thus, is observed as peaks in nuclear density maps. Because deuterium atoms scatter neutrons just as well as other protein atoms, they can be directly detected in neutron structures at moderate resolutions as low as 2.5–2.6 Å (38, 39). Notably, sulfur has a coherent neutron-scattering length of +2.847 fm, less than half the magnitude of that for carbon, oxygen, nitrogen, and deuterium. Consequently, deprotonated thiol groups (S–) in Cys and side-chain sulfur atoms in Met residues are often not easily visible in nuclear density maps.
We grew neutron-quality crystals of the ligand-free SARS–CoV-2 3CL Mpro at pH 6.6, allowing us to obtain a room-temperature neutron structure of the enzyme refined jointly with a room-temperature X-ray data set collected from the same crystal (Fig. 1) (40). We accurately determined the locations of exchangeable hydrogen atoms that were observed as deuterium attached to electronegative atoms such as oxygen, nitrogen, or sulfur atoms. We discovered that the catalytic dyad comprising residues Cys145 and His41 is in the reactive zwitterionic state having deprotonated negatively charged Cys145 and doubly protonated positively charged His41. Our experimental observations identified the protonation states of all other ionizable amino acid residues, allowing us to accurately map the electric charges and hydrogen-bonding networks in the SARS–CoV-2 3CL Mpro active-site cavity and throughout the enzyme structure. Neutron diffraction data were collected from a hydrogen/deuterium–exchanged SARS–CoV-2 3CL Mpro crystal at pD 7.0 (pD = pH + 0.4) to 2.5 Å resolution (Fig. 3).
Figure 3.
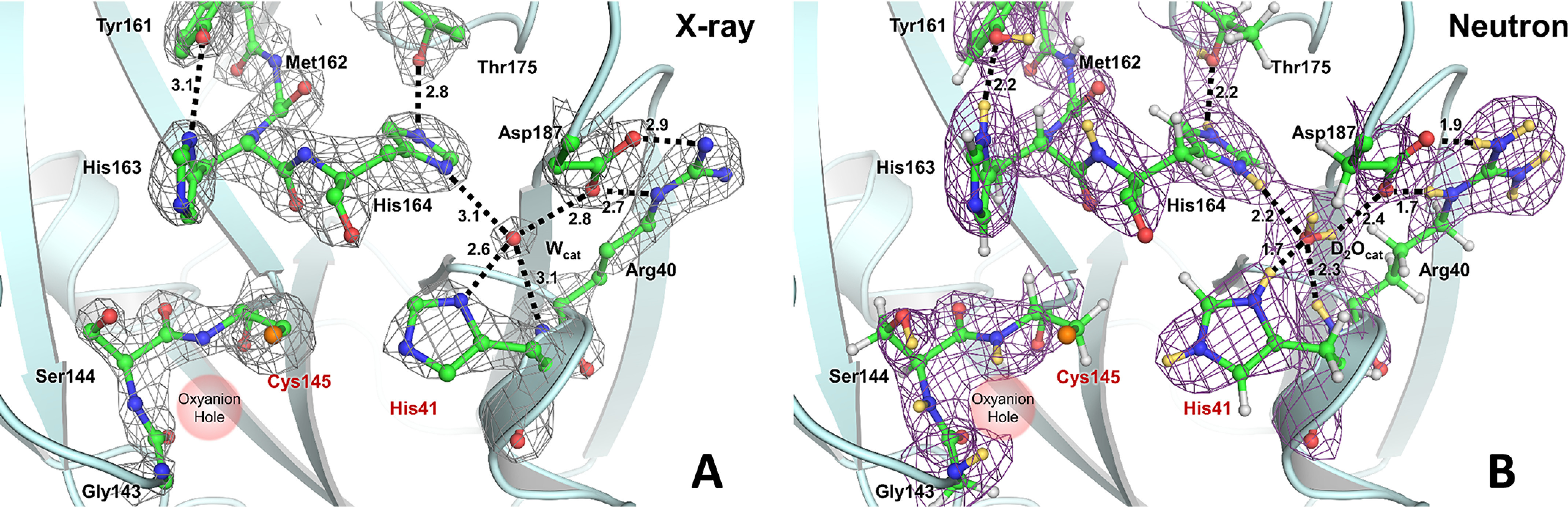
The catalytic site of SARS–CoV-2 3CL Mpro. X-ray diffraction data provide no information on the positions of hydrogen atoms as shown in A, whereas neutron diffraction data provide direct observation of deuterium atoms as shown in B. A, the 2Fo − Fc electron density map is contoured at 2.0 σ level (gray mesh). Distances between the heavy atoms in Ångstroms illustrate possible hydrogen bonds (black dashed lines). B, the 2Fo − Fc nuclear density map is contoured at 2.0 σ level (violet mesh), allowing visualization of the actual protonation states and hydrogen-bonding interactions shown as black dashed lines (deuterium–oxygen distances are shown in Ångstroms). Deuterium atoms are colored yellow, whereas hydrogen atoms are colored light gray.
Results
Protonation states of the catalytic site and nearby residues
The electron density for the catalytic site and the nearby residues of SARS–CoV-2 3CL Mpro is shown in Fig. 3A Although hydrogen-bonding interactions can be inferred from the distances between the heavy atoms, the locations of hydrogen atoms and the protonation states of the amino acid residues can only be assumed. Instead, the nuclear density map shown in Fig. 3B displays the actual positions of exchanged deuterium atoms, accurately visualizing hydrogen-bond donors and acceptors. In the neutron structure, we observed that the catalytic Cys145 thiol is in the deprotonated, negatively charged thiolate state.
In contrast, located 3.9 Å away from Cys145, the catalytic residue His41 is protonated on both Nδ1 and Nε2 nitrogen atoms of the imidazole side chain and is therefore positively charged (Fig. S1). As a result, the catalytic site natively adopts the zwitterionic reactive state required for catalysis (19, 20, 41). His41 is strongly hydrogen-bonded to a water molecule (D2Ocat) that presumably plays the role of the third catalytic residue (18, 25) from a canonical catalytic triad, stabilizing the charge and position of the His41 imidazolium ring. The Nδ1-D…OD2O distance is 1.7 Å. The position of the D2Ocat molecule is stabilized by several more, but possibly weaker, hydrogen bonds with His41 main chain, His164, and Asp187. His164 is doubly protonated and positively charged; it donates a deuterium in a hydrogen bond with the Thr175 hydroxyl within the interior of the protein. As expected, Asp187 is not protonated, is negatively charged, and participates in a strong salt bridge with Arg40. His163 positioned near the catalytic Cys145 is singly protonated and uncharged, making a hydrogen bond with the protonated phenolic side chain of Tyr161, whose OD group is rotated away from the His163 imidazole (Fig. S1). The main-chain deuterium atoms of Gly143, Ser144, and Cys145 comprising the oxyanion hole are also readily visible in the nuclear density (Fig. 3B).
Substrate-binding subsite S1
The P1 group of a substrate, usually Gln, binds in a rather peculiar substrate-binding subsite S1 (Fig. 4A). From one side, it is flanked by residues 140–144, making a turn that creates the oxyanion hole and, on the opposite side, by Met165, Glu166, and His172. The back wall of subsite S1 is created by the side chains of Phe140 and His163. Interestingly, the N terminus of Ser1ʹ of the second protomer within the active 3CL Mpro homodimer reaches in to cap the subsite S1 from the top. In our neutron structure, the N-terminal amine is the protonated, positively charged −ND3+ ammonium cation. It forms three hydrogen bonds, one each with the main chain carbonyl of Phe140, the side chain carboxylate of Glu166, and a D2O molecule. Both histidine residues, His163 and His172, in this subsite are singly protonated and neutral (Fig. S1). The deprotonated Nδ1 of His172 is hydrogen-bonded with the main-chain amide nitrogen of Gly138 with a nitrogen–deuterium distance of 2.2 Å, whereas the protonated Nε2 makes a long, possibly weak, hydrogen-bond interaction with the Glu166 carboxylate with the deuterium–oxygen distance of 2.5 Å.
Figure 4.

The architecture and hydrogen bonds in substrate-binding subsites S1 (A), S2 (B), and S4 (C). The 2Fo − Fc nuclear density map is contoured at 2.0 σ level (violet mesh). Hydrogen bonds are shown as black dashed lines. The deuterium–oxygen distances are in Ångstroms. For clarity, the side chains were omitted for Val186, Arg188, and Gln189. Deuterium atoms are colored yellow, whereas hydrogen atoms are colored light gray.
Substrate-binding subsite S2
Subsite S2 is more hydrophobic than subsite S1, because it binds the hydrophobic residues Leu or Phe in substrate position P2 (Fig. 4B). Subsite S2 is flanked by the π-systems of His41 and the main chains connecting Asp187, Arg188, and Gln189. It is capped by Met165, whereas Met49, situated on the short P2 helix spanning residues Ser46–Leu50, adopts a conformation impeding the entrance to this site. Met49 appears to be conformationally flexible, vacating its position in the ligand-free enzyme to allow various P2 groups to occupy this subsite when inhibitors bind (9–11). Tyr54 serves as the base of subsite S2, and its phenolic hydroxyl group donates its deuterium in a hydrogen bond with the main chain carbonyl of Asp187.
Substrate-binding subsites S3–S5
Among the substrate-binding subsites S3, S4, and S5, only subsite S4 has a well-defined binding pocket architecture. Subsites S3 and S5 are on the protein surface, fully exposed to the bulk solvent, and have ill-defined borders. Subsite S3 is located between residues Glu166 and Gln189 that are >9 Å apart. Subsite S5 is between Pro168 of the β-hairpin flap, spanning residues Met165–His172, and the P5 loop consisting of residues Thr190–Ala194 (Fig. 2). Subsite S4 is formed between a long loop spanning residues Phe185–Ala194 that acts as its base and the β-hairpin flap on the top. The loop turns 180° at Gln189, with its secondary structure being stabilized by hydrogen bonds between the Gln192 side-chain amide and the main chain atoms of Val186. The side chains of Leu167 and Phe185 form the back wall of this site in the protein interior, creating a deep, mainly hydrophobic pocket (Fig. 4C).
Dimer interface
The two protomers in the SARS–CoV-2 3CL Mpro homodimer interact through an extensive dimer interface. Protomer 1 (unprimed residue numbers) forms elaborate networks of hydrogen-bonding interactions with N-terminal residues 1ʹ–16ʹ, a β-strand with residues 118ʹ–125ʹ, and a loop containing residues 137ʹ–142ʹ of protomer 2 (primed residue numbers; Fig. 5). There are also many hydrophobic interactions within the dimer interface. The N termini of the two protomers meet at the start of a short α-helix spanning residues Gly11–Cys16 and Gly11ʹ–Cys16ʹ to form several hydrogen bonds involving the main-chain and side-chain atoms of Ser10, Gly11, Ser10ʹ, Gly11ʹ, and Glu14ʹ (Fig. 5). The N-terminal loop then extends across the face of the opposite protomer to subsite S1. Here, Ala7ʹ, Phe8ʹ, Arg4ʹ, and Ser1ʹ are observed making hydrogen bonds with Val125, Glu290, Lys137, Phe140, and Glu166. Ser123ʹ and Asn119ʹ of protomer 2 β-strand containing residues 118ʹ–125ʹ have direct hydrogen bonds and water-mediated interactions with the C-terminal residues of protomer 1, in addition to the reciprocal hydrogen bonds between Val125' and Ala7 formed because of the 2-fold symmetry of the enzyme dimer (Fig. 5). Similar reciprocity of hydrogen bonding is found for the loop consisting of residues 137ʹ–144ʹ in protomer 2 that interacts with the N-terminal residues Ser1 and Arg4 of protomer 1. Also, the side-chain hydroxyl of Ser139ʹ makes a hydrogen bond with the side-chain amide of Gln299 (Fig. 5).
Figure 5.
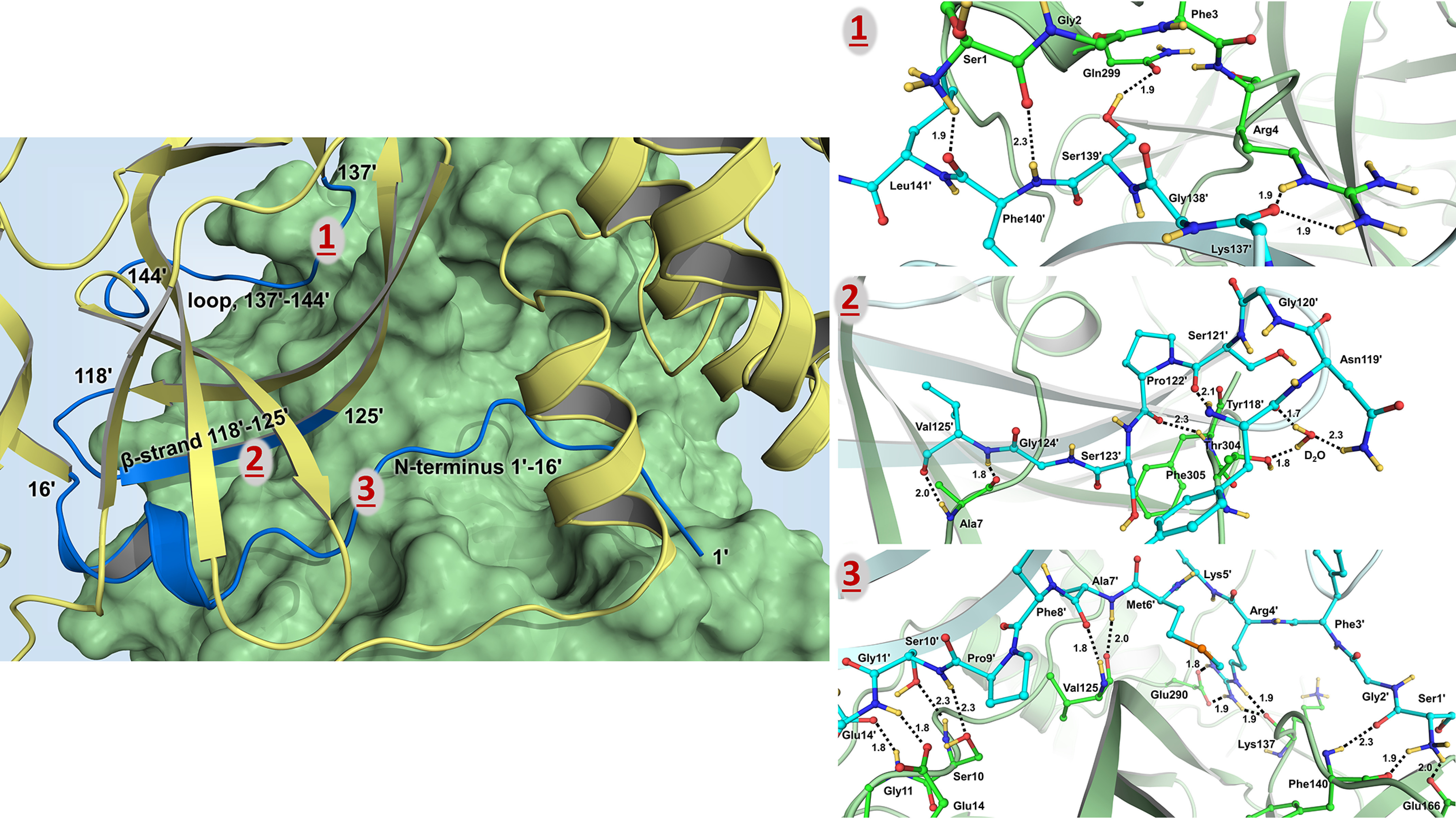
The dimer interface hydrogen bonding and water-mediated interactions visualized in the neutron structure of SARS–CoV-2 3CL Mpro. Left panel, cartoon representation in which protomer 1 (unprimed residues 1–306) is shown as a green surface and protomer 2 (primed residues 1'-306') is colored in yellow. The parts of protomer 2 interacting with protomer 1 are colored blue. Right panels, dimer interface hydrogen bonds made by the N-terminal residues 1′–16′ shown in panel 1; interactions made by a β-strand 118′–125′ shown in panel 2; and interactions made by a loop 137′–141– shown in panel 3. Deuterium atoms are colored yellow, whereas hydrogen atoms are omitted for clarity. Hydrogen bonds are shown as black dashed lines, and the distances are in Ångstroms.
Protonation states of Cys residues
The SARS–CoV-2 3CL Mpro structure contains 12 Cys residues spread throughout the sequence. The reasons for such a large number of cysteines within the enzyme sequence and their functional roles are unknown, except for the catalytic Cys145. By examining the nuclear density, we established the protonation states of all Cys residues in the structure (Figs. S2 and S3). The side chains of residues Cys16, Cys85, Cys117, Cys156, Cys160, Cys265, and Cys300 are protonated thiol groups. Conversely, the remaining Cys residues, Cys22, Cys38, Cys44, Cys128, and Cys145, contain deprotonated thiolates. Sulfur is a soft chemical base, with a large van der Waals radius of 1.84 Å; thus, a deprotonated thiol group is stable in solution at the physiological pH (pKa of free Cys is 8.35). Hence, it is not unexpected to observe deprotonated Cys residues in a protein structure at close-to-physiological pH. In addition, Cys22 and Cys128 are located close to the protein surface, with their negative charges further stabilized by the positive charges of nearby Lys61 and Arg4′ from the second protomer, respectively (Fig. S4). Cys38 and Cys44 are positioned within hydrophobic pockets, with their negative charges shielded from the bulk solvent. Deprotonation of Cys145 is also consistent with it being reactive and oxidation prone, as reported previously (41). Accordingly, because we can unambiguously differentiate between seven protonated and five deprotonated Cys side chains, this provides strong evidence that the catalytic Cys145 is in the reactive negatively charged thiolate form.
Discussion
The COVID-19 pandemic has claimed over a million lives globally to date because of the lack of therapeutic intervention options and vaccines that specifically target the SARS–CoV-2 virus. Rational or structure-assisted and computational drug design efforts could greatly benefit from a detailed structural knowledge of the drug targets, such as the SARS–CoV-2 3CL Mpro enzyme. Considering that half of the protein atoms are hydrogens that play critical roles in various chemistries occurring in enzyme active sites and in inhibitor-binding events, it is of paramount importance to locate hydrogen atoms in a protein to understand enzyme function and to guide drug design. Neutron crystallography is the only technique capable of accurately determining the positions of hydrogen (and deuterium) atoms in biological macromolecules without causing radiation damage to the samples at near-physiological temperature (33). We have succeeded in determining a room-temperature neutron structure of the homodimeric SARS–CoV-2 3CL Mpro enzyme mapping the protonation states of amino acid residues in the active-site cavity and throughout the protein structure. Thus, for the first time, we determined protonation states of all ionizable residues in a cysteine protease and any coronavirus protein. The neutron structure also reveals the elaborate hydrogen-bonding networks formed in the catalytic site, including those in the six substrate-binding subsites, and throughout the dimer interface. Protonation states are determined by the locations of hydrogen atoms, observed in the neutron structure as deuterium after the hydrogen/deuterium exchange was performed. Also, protonation states establish the electrical charges and therefore determine the electrostatic environment in the protein. The implications of knowing the actual protonation states in and around the active site are far-reaching because this information is essential for the structure-assisted and computational drug design. Structure-assisted drug design requires precise tailoring of inhibitor chemical groups to the electrostatic environment of an enzyme's binding site and has to take advantage of hydrogen-bonding opportunities because direct hydrogen bonds between a drug molecule and its protein target are the strongest noncovalent interactions that can dramatically improve the inhibitor-binding affinity. Moreover, if protonation states are not experimentally known and are assumed based on general chemical knowledge, computational methods will produce misleading results.
In the SARS–CoV-2 3CL Mpro neutron structure, we discovered that the catalytic dyad is observed in the reactive zwitterionic state in the crystal at pD 7.0, having a deprotonated negatively charged Cys145 and doubly protonated positively charged His41 (Fig. 3), instead of the catalytically resting (nonreactive) state (26) in which both catalytic residues are neutral (i.e. protonated Cys145 and singly protonated His41). The negative charge on Cys145 thiolate is stabilized by the sulfur atom being a soft chemical base with a large van der Waals radius and by the positive charge on His41 side chain and the oxyanion hole main chain amides positioned in close proximity to the Cys145 side chain. His41 is hydrogen-bonded to the catalytic water molecule held in place by interactions with the doubly protonated His164 and negatively charged Asp87. The substrate-binding subsite S1 contains the protonated positively charged N-terminal amine from protomer 2 and neutral His163. The nonprotonated Nε2 of His163 is exposed and can be utilized as a hydrogen-bond acceptor by an inhibitor P1 substituent (Fig. 4). In the hydrophobic subsite S2, the Tyr54 hydroxyl can act as a hydrogen-bond acceptor for a P2 substituent with a hydrogen-bond donor functionality. Consequently, specific SARS–CoV-2 3CL Mpro protease inhibitor design needs to consider the observed charge distribution in the enzyme active-site cavity, taking a special note that the catalytic site contains a zwitterionic catalytic dyad but appears to be neutral because the opposite charges on Cys145 and His41 located close to each other cancel out.
We also accurately mapped the hydrogen-bonding networks within the dimer interface of SARS–CoV-2 3CL Mpro. Significantly, the N-terminal residues between Ser1 and Cys16 (and Ser1′ and Cys16′) account for the most hydrogen bonds between the two protomers (Fig. 5). There are 13 direct hydrogen bonds formed by each N terminus interacting with the opposite protomer. Ser1 is an essential residue for building the correct architecture of substrate-binding subsite S1. Truncation of the first four N-terminal residues in SARS–CoV 3CL Mpro has previously been shown to disrupt dimerization and dramatically diminish enzyme activity (22). We therefore suggest that the enzyme area that interacts with residues 1–16 from the other protomer may be the most promising part of the dimer interface for designing SARS–CoV-2 3CL Mpro-specific dimerization inhibitors, as was also proposed for the SARS–CoV enzyme (42, 43).
Conclusion
We have successfully determined the neutron structure of the ligand-free SARS–CoV-2 3CL Mpro at near-physiological (room) temperature and pH and mapped the locations of hydrogen atoms (observed as deuterium) in the enzyme. Thus, protonation states and electrical charges of the ionizable residues have been accurately resolved. Establishing the electrostatics in the enzyme active-site cavity and at the dimer interface is critical information to guide the structure-assisted and computational drug design of protease inhibitors, specifically targeting the enzyme from SARS–CoV-2.
Materials and methods
General information
Protein purification columns were purchased from Cytiva (Piscataway, NJ, USA). Crystallization reagents were purchased from Hampton Research (Aliso Viejo, CA, USA). Crystallographic supplies were purchased from MiTeGen (Ithaca, NY, USA) and Vitrocom (Mountain Lakes, NJ, USA).
Cloning, expression, and purification of SARS–CoV-2 3CL Mpro
The 3CL Mpro (Nsp5 Mpro) from SARS–CoV-2 was cloned into pD451-SR plasmid harboring kanamycin resistance (ATUM, Newark, CA, USA), expressed, and purified according to the procedure published elsewhere (25). To make the authentic N terminus of 3CL Mpro, the enzyme sequence is flanked by the maltose-binding protein followed by the protease autoprocessing site SAVLQ↓SGFRK (down arrow indicates the autocleavage site), which corresponds to the cleavage position between NSP4 and NSP5 in the viral polyprotein. To form the authentic C terminus, the enzyme construct codes for the human rhinovirus 3C protease cleavage site (SGVTFQ↓GP) connected to a His6 tag. The N-terminal flanking sequence is autocleaved during the expression in Escherichia coli (BL21 DE3), whereas the C-terminal flanking sequence is removed by the treatment with human rhinovirus 3C protease (Millipore–Sigma). A detailed protocol for the enzyme expression, purification, and crystallization has been published elsewhere (44).
Crystallization
For crystallization, the authentic 3CL Mpro is concentrated to ∼4 mg/ml in 20 mm Tris, 150 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, pH 8.0. The methodology for growing large crystals of 3CL Mpro suitable for neutron diffraction is described as follows. Initial protein crystallization conditions were discovered by screening conducted at the Hauptman–Woodward Medical Research Institute (45). Crystal aggregates were reproduced using the sitting-drop vapor-diffusion method using 25% PEG3350, 0.1 m Bis-Tris, pH 6.5, in 20-μl drops with 1:1 ratio of the protein:well solution. Microseeding using Hampton Research seed beads was performed to grow neutron-quality crystals in Hampton 9-well plates and sandwich box set-ups with 200-μl drops of protein mixed with 18% PEG3350, 0.1 m Bis-Tris, pH 6.0, 3% DMSO at a 1:1 ratio seeded with 1 μl of microseeds at a 1:500 dilution. This condition produced a final pH in the crystallization drop of 6.6 as measured by microelectrode. The crystal tray used to harvest a crystal for neutron diffraction was incubated initially at 18 °C and then gradually lowered to 10 °C over several weeks. The crystal grew to final dimensions of ∼2 × 0.8 × 0.2 mm (∼0.3 mm3) in a triangular plate-like morphology. The crystal was mounted in a quartz capillary accompanied with 18% PEG3350 prepared with 100% D2O and allowed to hydrogen/deuterium exchange for several days before starting the neutron data collection.
Neutron diffraction data collection
The large crystal was screened for diffraction quality using a broad-bandpass Laue configuration using neutrons from 2.8 to 10 Å at the IMAGINE instrument at the high flux isotope reactor at Oak Ridge National Laboratory (46–48). Neutron diffraction data were collected using the macromolecular neutron diffractometer instrument MaNDi at the Spallation Neutron Source (47, 49–51). The crystal was held still at room temperature, and diffraction data were collected for 24 h using all neutrons between 2 and 4.16 Å. Following this, the crystal was rotated by Δϕ = 10°, and a subsequent data frame was collected again for 24 h. A total of 23 data frames were collected in the final neutron data set. Diffraction data were reduced using the Mantid package, with integration carried out using three-dimensional TOF profile fitting (52). Wavelength normalization of the Laue data was performed using the Lauenorm program from the Lauegen suite (53, 54). The neutron data collection statics are shown in Table S1.
X-ray diffraction data collection
The room-temperature diffraction data set was collected from the same crystal used for the neutron data collection using a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator and Osmic VariMax optics. The diffraction images were obtained using an Eiger R 4M hybrid photon counting detector. Diffraction data were integrated using the CrysAlis Pro software suite (Rigaku Inc., The Woodlands, TX). Diffraction data were then reduced and scaled using the Aimless (54) program from the CCP4 suite (55); molecular replacement using PDB code 6WQF (25) was then performed with Molrep (54) from the CCP4 program suite. The protein structure was first refined against the X-ray data using Phenix.refine from the Phenix (56) suite of programs and the Coot (57) molecular graphics program to obtain an accurate model for the subsequent X-ray/neutron joint refinement. The geometry of the final model was then carefully checked with MolProbity (58). The X-ray data collection statics are shown in Table S1.
Joint X-ray/neutron refinement
The joint X-ray/neutron refinement of ligand-free 3CL Mpro was performed using nCNS (40, 59), and the structure was manipulated in Coot (57). After initial rigid-body refinement, several cycles of positional, atomic displacement parameter, and occupancy refinement were performed. The structure was checked for the correctness of side-chain conformations, hydrogen bonding, and orientations of D2O water molecules built based on the mFo − Fc difference neutron-scattering length density maps. The 2mFo − DFc and mFo − DFc neutron-scattering length density maps were then examined to determine the correct orientations of hydroxyl (Ser, Thr, and Tyr), thiol (Cys), and ammonium (Lys) groups and protonation states of the enzyme residues. The protonation states of some disordered side chains could not be obtained directly and remained ambiguous. All water molecules were refined as D2O. Initially, water oxygen atoms were positioned according to their electron density peaks and then were shifted slightly in accordance with the neutron-scattering length density maps. All labile hydrogen positions in 3CL Mpro were modeled as deuterium atoms, and then the occupancies of those atoms were refined individually within the range of −0.56 (pure hydrogen) to 1.00 (pure deuterium). Before depositing the neutron structure to the PDB, a script was run that converts a record for the coordinates of a deuterium atom into two records corresponding to a hydrogen and a deuterium partially occupying the same site, both with positive partial occupancies that add up to unity. The percentage of deuterium at a specific site is calculated according to the following formula: % deuterium = {Occupancy(deuterium) + 0.56}/1.56.
Data availability
The coordinates and structure factors for ligand-free SARS–CoV-2 3CL Mpro have been deposited in the PDB with accession code 7JUN. Any other relevant data are available from the corresponding authors upon reasonable request.
Supplementary Material
This article contains supporting information.
Author contributions—D. W. K., Q. Z., H. M. O., and L. C. formal analysis; D. W. K., K. L. W., S. P., Q. Z., and L. C. validation; D. W. K., G. P., K. L. W., S. P., and Q. Z. investigation; D. W. K. and L. C. visualization; D. W. K. and A. K. writing-original draft; G. P., K. L. W., S. P., and Q. Z. methodology; K. L. W., Q. Z., and H. M. O. resources; H. M. O. funding acquisition; H. M. O., L. C., and A. K. writing-review and editing; L. C. and A. K. conceptualization; L. C. data curation; A. K. supervision; A. K. project administration.
Funding and additional information—This work was supported by the Department of Energy Office of Science through the National Virtual Biotechnology Laboratory, a consortium of Department of Energy national laboratories focused on response to COVID-19, with funding provided by the Coronavirus CARES Act. This work used resources at the Spallation Neutron Source and the High Flux Isotope Reactor, which are Department of Energy Office of Science User Facilities operated by the Oak Ridge National Laboratory. The Office of Biological and Environmental Research supported research at the Oak Ridge National Laboratory Center for Structural Molecular Biology, a Department of Energy Office of Science user facility. This work also used resources at the Second Target Station, which is a Department of Energy Office of Science user facilities construction project at Oak Ridge National Laboratory.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- SARS
- severe acute respiratory syndrome
- CoV-2
- coronavirus 2
- PDB
- Protein Data Bank.
References
- 1. Liu, C., Zhou, Q., Li, Y., Garner, L. V., Watkins, S. P., Carter, L. J., Smoot, J., Gregg, A. C., Daniels, A. D., Jervey, S., and Albaiu, D. (2020) Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Central Sci. 6, 315–331 10.1021/acscentsci.0c00272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hussain, A., Yadav, S., Hadda, V., Suri, T. M., Tiwari, P., Mittal, S., Madan, K., and Mohan, A. (2020) Covid-19: a comprehensive review of a formidable foe and the road ahead. Expert Rev. Respir. Med. 14, 869–879 10.1080/17476348.2020.1782198 [DOI] [PubMed] [Google Scholar]
- 3. Gil, C., Ginex, T., Maestro, I., Nozal, V., Barrado-Gil, L., Cuesta-Geijo, M. A., Urquiza, J., Ramirez, D., Alonso, C., Campillo, N. E., and Martinez, A. (2020) COVID-19: drug tragets and potential treatments. J. Med. Chem., in press 10.1021/acs.jmedchem.0c00606 [DOI] [PubMed] [Google Scholar]
- 4. Sempowski, G. D., Saunders, K. O., Acharya, P., Wiehe, K. J., and Haynes, B. F. (2020) Pandemic preparedness: developing vaccines and therapeutic antibodies for COVID-19. Cell 181, 1458–1463 10.1016/j.cell.2020.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(2020) Osman, E. E. A., Toogood, P. L., Neamati, N. COVID-19: living through another pandemic. ACS Infect. Dis. 6, 1548–1552 10.1021/acsinfecdis.0c00224 [DOI] [PubMed] [Google Scholar]
- 6. Rastogi, Y. R., Sharma, A., Nagraik, R., Aygün, A., and Şen, F. (2020) The novel coronavirus 2019-nCoV: its evolution and transmission into humans causing global COVID-19 pandemic. Int. J. Environ. Sci. Technol. 17, 4381–4388 10.1007/s13762-020-02781-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu, F., Zhao, S., Yu, B., Chen, Y. M., Wang, W., Song, Z. G., Hu, Y., Tao, Z. W., Tian, J. H., Pei, Y. Y., Yuan, M. L., Zhang, Y. L., Dai, F. H., Liu, Y., Wang, Q. M., et al. (2020) A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269; Correction (2020) Nature580, E7 10.1038/s41586-020-2202-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu, J., Zhao, S., Teng, T., Abdalla, A. E., Zhu, W., Xie, L., Wang, Y., and Guo, X. (2020) Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS–CoV-2 and SARS–CoV. Viruses 12, 244 10.3390/v12020244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dai, W., Zhang, B., Jiang, X.-M., Su, H., Li, J., Zhao, Y., Xie, X., Jin, Z., Peng, J., Liu, F., Li, C., Li, Y., Bai, F., Wang, H., Cheng, X., et al. (2020) Structure-based design of antiviral drug candidates targeting the SARS–CoV-2 main protease. Science 368, 1331–1335 10.1126/science.abb4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., Becker, S., Rox, K., and Hilgenfeld, R. (2020) Crystal structure of SARS–CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 368, 409–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., et al. (2020) Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 582, 289–293 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- 12. Rathnayake, A. D., Zheng, J., Kim, Y., Perera, K. D., Mackin, S., Meyerholz, D. K., Kashipathy, M. M., Battaile, K. P., Lovell, S., Perlman, S., Groutas, W. C., and Chang, K.-O. (2020) 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl. Med. 12, eabc5332 10.1126/scitranslmed.abc5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jin, Z., Zhao, Y., Sun, Y., Zhang, B., Wang, H., Wu, Y., Zhu, Y., Zhu, C., Hu, T., Du, X., Duan, Y., Yu, J., Yang, X., Yang, X., Yang, K., et al. (2020) Structural basis for the inhibition of SARS–CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 27, 529–532 10.1038/s41594-020-0440-6 [DOI] [PubMed] [Google Scholar]
- 14. Ma, C., Sacco, M. D., Hurst, B., Townsend, J. A., Hu, Y., Szeto, T., Zhang, X., Tarbet, B., Marty, M. T., Chen, Y., and Wang, J. (2020) Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS–CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692 10.1038/s41422-020-0356-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Riva, L., Yuan, S., Yin, X., Martin-Sancho, L., Matsunaga, N., Pache, L., Burgstaller-Muehlbacher, S., De Jesus, P. D., Teriete, P., Hull, M. V., Chang, M. W., Chan, J. F.-W., Cao, J., Poon, V. K.-M., Herbert, K. M., et al. (2020) Discovery of SARS–CoV-2 antiviral drugs through large-scale compound repurposing. Nature 586, 113–119 10.1038/s41586-020-2577-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gorbalenya, A. E., and Snijder, E. J. (1996) Viral cysteine proteases. Perspect. Drug Discov. Des. 6, 64–86 10.1007/BF02174046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tong, L. (2002) Viral proteases. Chem. Rev. 102, 4609–4626 10.1021/cr010184f [DOI] [PubMed] [Google Scholar]
- 18. Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R., and Hilgenfeld, R. (2003) Coronavirus main protease (3CLpro) structure: basis for design of anti-SARS drugs. Science 300, 1763–1767 10.1126/science.1085658 [DOI] [PubMed] [Google Scholar]
- 19. Huang, C., Wei, P., Fan, K., Liu, Y., and Lai, L. (2004) 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 43, 4568–4574 10.1021/bi036022q [DOI] [PubMed] [Google Scholar]
- 20. Solowiej, J., Thomson, J. A., Ryan, K., Luo, C., He, M., Lou, J., and Murray, B. W. (2008) Steady-state and pre-steady-state kinetic evaluation of severe acute respiratory syndrome coronavirus (SARS–CoV) 3CLpro cysteine protease: development of an ion-pair model for catalysis. Biochemistry 47, 2617–2630 10.1021/bi702107v [DOI] [PubMed] [Google Scholar]
- 21. Fan, K., Wei, P., Feng, Q., Chen, S., Huang, C., Ma, L., Lai, B., Pei, J., Liu, Y., Chen, J., and Lai, L. (2004) Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 279, 1637–1642 10.1074/jbc.M310875200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsu, W.-C., Chang, H.-C., Chou, C.-Y., Tsai, P.-J., Lin, P.-I., and Chang, G.-G. (2005) Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J. Biol. Chem. 280, 22741–22748 10.1074/jbc.M502556200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin, P.-Y., Chou, C.-Y., Chang, H.-C., Hsu, W.-C., and Chang, G.-G. (2008) Correlation between dissociation and catalysis of SARS–CoV main protease. Arch. Biochem. Biophys. 472, 34–42 10.1016/j.abb.2008.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia, B., and Kang, X. (2011) Activation and maturation of SARS–CoV main protease. Protein Cell 2, 282–290 10.1007/s13238-011-1034-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kneller, D. W., Phillips, G., O'Neill, H. M., Jedrzejczak, R., Stols, L., Langan, P., Joachimiak, A., Coates, L., and Kovalevsky, A. (2020) Structural plasticity of SARS–CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 11, 3202 10.1038/s41467-020-16954-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paasche, A., Zipper, A., Schäfer, S., Ziebuhr, J., Schirmeister, T., and Engels, B. (2014) Evidence for substrate binding–induced zwitterion formation in the catalytic Cys-His dyad of the SARS-Co-V main protease. Biochemistry 53, 5930–5946 10.1021/bi400604t [DOI] [PubMed] [Google Scholar]
- 27. Barrila, J., Bacha, U., and Freire, E. (2006) Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry 45, 14908–14916 10.1021/bi0616302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goyal, B., and Goyal, D. (2020) Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 22, 297–305 10.1021/acscombsci.0c00058 [DOI] [PubMed] [Google Scholar]
- 29. Bax, B., Chung, C-W., and Edge, C. (2017) Getting the chemistry right: protonation, tautomers and the importance of H atoms in biological chemistry. Acta Crystallogr. D 73, 131–140 10.1107/S2059798316020283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gardberg, A. S., Del Castillo, A. R., Weiss, K. L., Meilleur, F., Blakeley, M. P., and Myles, D. A. A. (2010) Unambiguous determination of H-atom positions: comparing results from neutron and high-resolution X-ray crystallography. Acta Crystallogr. D 66, 558–567 10.1107/S0907444910005494 [DOI] [PubMed] [Google Scholar]
- 31. Blakeley, M. P., Mitschler, A., Hazemann, I., Meilleur, F., Myles, D. A. A., and Podjarny, A. (2006) Comparison of hydrogen determination with X-ray and neutron crystallography in a human aldose reductase-inhibitor complex. Eur. Biophys. J. 35, 577–583 10.1007/s00249-006-0064-8 [DOI] [PubMed] [Google Scholar]
- 32. Lin, J., Pozharski, E., and Wilson, M. A. (2017) Short carboxylic acid-carboxylate hydrogen bonds can have fully localized protons. Biochemistry 56, 391–402 10.1021/acs.biochem.6b00906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Niimura, N., and Podjarny, A. (2011) Neutron Protein Crystallography, Oxford University Press, Oxford [Google Scholar]
- 34. Golden, E. A., and Vrielink, A. (2014) Looking for hydrogen atoms: neutron crystallography provides novel insights into protein structure and function. Aust. J. Chem. 67, 1751–1762 10.1071/CH14337 [DOI] [Google Scholar]
- 35. Oksanen, E., Chen, J. C.-H., and Fisher, S. Z. (2017) Neutron crystallography for the study of hydrogen bonds in macromolecules. Molecules 22, 596 10.3390/molecules22040596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blakeley, M. P., and Podjarny, A. D. (2018) Neutron macromolecular crystallography. Emerg. Topics Life Sci. 2, 39–55 10.1042/ETLS20170083 [DOI] [PubMed] [Google Scholar]
- 37. Garman, E. F. (2010) Radiation damage in macromolecular crystallography: what is it and why should we care? Acta Crystallogr. D 66, 339–351 10.1107/S0907444910008656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gerlits, O., Weiss, K. L., Blakeley, M. P., Veglia, G., Taylor, S. S., and Kovalevsky, A. (2019) Zooming in on protons: neutron structure of protein kinase A trapped in a product complex. Science Adv. 5, eaav0482 10.1126/sciadv.aav0482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Banco, M. T., Mishra, V., Ostermann, A., Schrader, T. E., Evans, G. B., Kovalevsky, A. Y., and Ronning, D. R. (2016) Neutron structures of the Helicobacter pylori 5´-methylthioadenosine nucleosidase highlight proton sharing and protonation states. Proc. Natl. Acad. Sci. U.S.A. 113, 13756–13761 10.1073/pnas.1609718113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adams, P. D., Mustyakimov, M., Afonine, P. V., and Langan, P. (2009) Generalized X-ray and neutron crystallographic analysis: more accurate and complete structures for biological macromolecules. Acta Crystallogr. D 65, 567–573 10.1107/S0907444909011548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kneller, D. W., Phillips, G., O'Neill, H. M., Tan, K., Joachimiak, A., Coates, L., and Kovalevsky, A. (2020) Room-temperature X-ray crystallography reveals the oxidation and reactivity of cysteine residues in SARS–CoV-2 3CL Mpro: insights into enzyme mechanism and drug design. IUCrJ. 7, 1028–1035 10.1107/S2052252520012634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ding, L., Zhang, X.-X., Wei, P., Fan, K., and Lai, L. (2005) The interactions between severe acute respiratory syndrome coronavirus 3C–like proteinase and a dimeric inhibitor by capillary electrophoresis. Anal. Biochem. 343, 159–165 10.1016/j.ab.2005.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wei, P., Fan, K., Chen, H., Ma, L., Huang, C., Tan, L., Xi, D., Li, C., Liu, Y., Cao, A., and Luhua, L. (2006) The N-terminal octapeptide acts as a dimerization inhibitor of SARS coronavirus 3C-like proteinase. Biochem. Biophys. Res. Commun 339, 865–872 10.1016/j.bbrc.2005.11.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kneller, D. W., Phillips, G., Kovalevsky, A., and Coates, L. (2020) Room-temperature neutron and X-ray data collection of 3CL Mpro from SARS–CoV-2. Acta Crystallogr. 76, 483–487 10.1107/S2053230X20011814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Luft, J. R., Collins, R. J., Fehrman, N. A., Lauricella, A. M., Veatch, C. K., and DeTitta, G. T. (2003) A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J. Struct. Biol. 142, 170–179 10.1016/S1047-8477(03)00048-0 [DOI] [PubMed] [Google Scholar]
- 46. Meilleur, F., Kovalevsky, A., and Myles, D. A. A. (2020) IMAGINE: the neutron protein crystallography beamline at the high flux isotope reactor. Methods Enzymol. 634, 69–85 10.1016/bs.mie.2019.11.016 [DOI] [PubMed] [Google Scholar]
- 47. Meilleur, F., Coates, L., Cuneo, M. J., Kovalevsky, A., and Myles, D. A. A. (2018) The neutron macromolecular crystallography instruments at Oak Ridge National Laboratory: Advances, challenges, and opportunities. Crystals 8, 388 10.3390/cryst8100388 [DOI] [Google Scholar]
- 48. Meilleur, F., Munshi, P., Robertson, L., Stoica, A. D., Crow, L., Kovalevsky, A., Koritsanszky, T., Chakoumakos, B. C., Blessing, R., and Myles, D. A. A. (2013) The IMAGINE instrument: first neutron protein structure and new capabilities for neutron macromolecular crystallography. Acta Crystallogr. 69, 2157–2160 10.1107/S0907444913019604 [DOI] [PubMed] [Google Scholar]
- 49. Coates, L., and Sullivan, B. (2020) The macromolecular neutron diffractometer at the spallation neutron source. Methods Enzymol. 634, 87–99 10.1016/bs.mie.2019.11.020 [DOI] [PubMed] [Google Scholar]
- 50. Coates, L., Cao, H. B., Chakoumakos, B. C., Frontzek, M. D., Hoffmann, C., Kovalevsky, A. Y., Liu, Y., Meilleur, F., dos Santos, A. M., Myles, D. A. A., Wang, X. P., and Ye, F. (2018) A suite-level review of the neutron single-crystal diffraction instruments at Oak Ridge National Laboratory. Rev. Sci. Instrum. 89, 092802 10.1063/1.5030896 [DOI] [PubMed] [Google Scholar]
- 51. Coates, L., Cuneo, M. J., Frost, M. J., He, J., Weiss, K. L., Tomanicek, S. J., McFeeters, H., Vandavasi, V. G., Langan, P., and Iverson, E. B. (2015) The macromolecular neutron diffractometer MaNDi at the Spallation Neutron Source. J. Appl. Crystallogr. 48, 1302–1306 10.1107/S1600576715011243 [DOI] [Google Scholar]
- 52. Arnold, O., Bilheux, J. C., Borreguero, J. M., Buts, A., Campbell, S. I., Chapon, L., Doucet, M., Draper, N., Ferraz Leal, R., Gigg, M. A., Lynch, V. E., Markvardsen, A., Mikkelson, D. J., Mikkelson, R. L., Miller, R., et al. (2014) Mantid: data analysis and visualization package for neutron scattering and μ SR experiments. Nucl. Instrum. Meth. A 764, 156–166 10.1016/j.nima.2014.07.029 [DOI] [Google Scholar]
- 53. Campbell, J. W., Hao, Q., Harding, M. M., Nguti, N. D., and Wilkinson, C. (1998) LAUEGEN version 6.0 and INTLDM. J. Appl. Crystallogr. 31, 496–502 10.1107/S0021889897016683 [DOI] [Google Scholar]
- 54. Helliwell, J. R., Habash, J., Cruickshank, D. W. J., Harding, M. M., Greenhough, T. J., Campbell, J. W., Clifton, I. J., Elder, M., Machin, P. A., Papiz, M. Z., and Zurek, S. (1989) The recording and analysis of synchrotron X-radiation Laue diffraction photographs. J. Appl. Crystallogr. 22, 483–497 10.1107/S0021889889006564 [DOI] [Google Scholar]
- 55. Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L. W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S., and Richardson, D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mustyakimov, M., and Langan, P. (2007) nCNS; an open source distribution patch for CNS for macromolecular structure refinement, Los Alamos National Security, Los Alamos, NM, USA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factors for ligand-free SARS–CoV-2 3CL Mpro have been deposited in the PDB with accession code 7JUN. Any other relevant data are available from the corresponding authors upon reasonable request.