

ABSTRACT
Plant abiotic stresses negative affects growth and development, causing a massive reduction in global agricultural production. Rapeseed (Brassica napus L.) is a major oilseed crop because of its economic value and oilseed production. However, its productivity has been reduced by many environmental adversities. Therefore, it is a prime need to grow rapeseed cultivars, which can withstand numerous abiotic stresses. To understand the various molecular and cellular mechanisms underlying the abiotic stress tolerance and improvement in rapeseed, omics approaches have been extensively employed in recent years. This review summarized the recent advancement in genomics, transcriptomics, proteomics, metabolomics, and their imploration in abiotic stress regulation in rapeseed. Some persisting bottlenecks have been highlighted, demanding proper attention to fully explore the omics tools. Further, the potential prospects of the CRISPR/Cas9 system for genome editing to assist molecular breeding in developing abiotic stress-tolerant rapeseed genotypes have also been explained. In short, the combination of integrated omics, genome editing, and speed breeding can alter rapeseed production worldwide.
KEYWORDS: CRISPR/Cas9, climate-resilient rapeseed, drought, metabolomics, marker-assisted selection, systems biology, salinity, transcriptomics
1. Introduction
As agriculture greatly depends on the prevailing atmosphere, the crop plants often suffer from various environmental or abiotic stress due to climate change. There are many abiotic stresses, viz. salinity, drought, cold, waterlogging, and temperature fluctuations, which considerably hinder the growth rate and reduce agricultural yield globally.1,2 As reported by USDA-FAO, drought and salt stress are important limiting components affecting about 26% and 20% of the agricultural land, respectively (American Geophysical Union; https://sites.agu.org). Soil salinity is the second major limiting factor of agriculture. Salt stress mostly affects the agricultural land of arid and semi-arid regions worldwide.3 Plants under dehydration-inducing conditions ultimately hinder the growth rate, decrease crop production, and encourage adverse effects on plant physiological, metabolic, and biochemical processes.2–4 Moreover, the effect of several other abiotic stresses such as waterlogging,5,6 extreme temperature,7 and metals/metalloids toxicity on crop productivity have also been reported in different crops, including rapeseed.8
Brassica napus L., also called rapeseed or canola, has emerged as an important crop globally through rigorous breeding programs during the past few decades. Among oilseed crops, rapeseed is second in world oilseed production only to soybean. It belongs to the Brassicaceae family, that have 419 genera and 4130 species worldwide. Rapeseed genome is 2 n = 38 (AACC), which arises from the genome hybridization of B. oleracea and B. rapa (Fig. 1; See refs. 14, 15) Rapeseed is now primarily grown in China, Canada, Europe as a cause of edible oil, livestock ration, and industrial derivatives.16,17 Rapeseed oil content is about 30.6–48.3% of the dry weight. The oil profile of rapeseed includes vital fatty acids such as oleic (56.80–64.92%), palmitic (4.18–5.01%), linoleic (17.11–20.92%) acids. The quantity of α-tocopherol is about 13.22–40.01% of the total oil contents.18 Due to climate change, urbanization and industrialization, abiotic stresses have become the main threat to crop production. Rapeseed, along with major crops worldwide, is frequently subjected to stresses which have significant effects on the physiological, biochemical, and molecular functions of plants, which ultimately impacting crop growth and production.8 Improved abiotic stress tolerance of rapeseed is a fundamental approach to increase oilseed production.19,20 General stress signaling pathways are described in Fig. 2.
Figure 1.
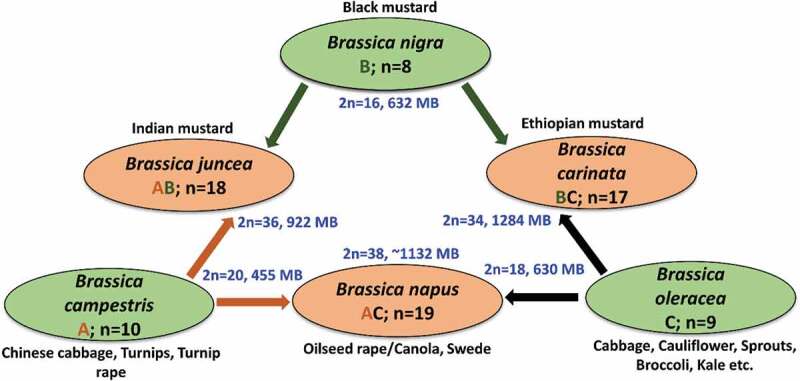
Systematic triangle of cross-breeding among economically important six Brassica species comprising three diploids (green) and three allotetraploids (orange) along with their estimated genome size. Letters (ABC) within the ovals denote the genomic symbols. N means the number of chromosomes. U’s triangle was created according to Nagaharu (1935).9 Genome size was retrieved from refs.10–14
Figure 2.
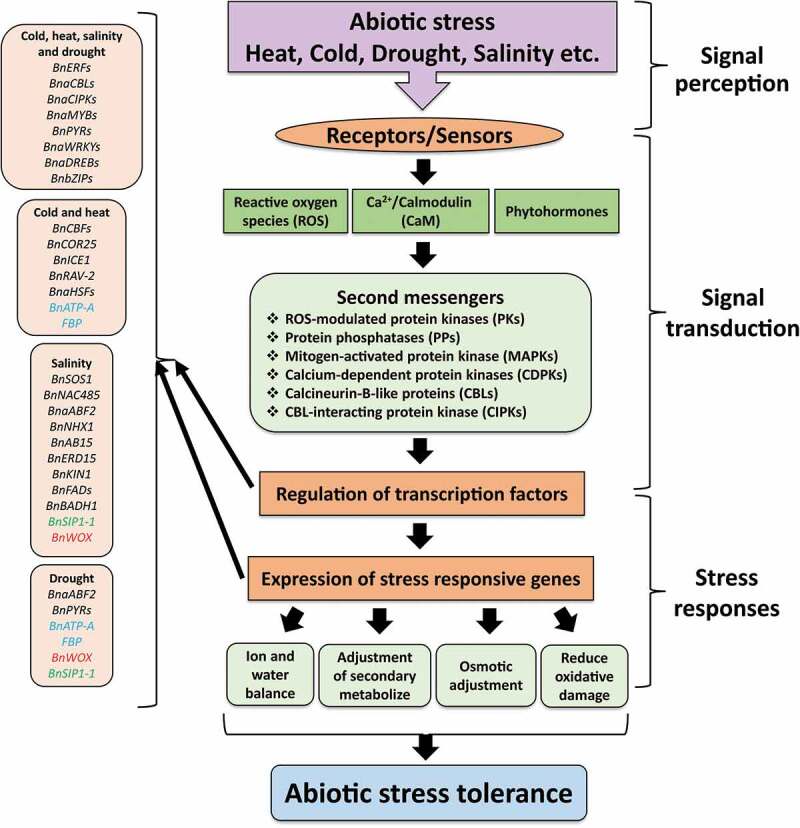
General abiotic stress signaling pathways in plants, starting from signal perception and leads toward the stress responses. Early prevalence of stress sensing via receptors/sensors cascades the downstream stress response by ROS, CaM, and phytohormones. Signal extension and transduction by secondary messengers such as PKs, PPs, MAPKs, CDPKs, CBLs, and CIPKs signaling cause differential regulation of transcription factors (TFs) and stress-responsive genes. The regulation of such TFs and genes leads to the adjustment of physiological, biochemical, and molecular responses, thus, improving stress tolerance. Boxes on the left side, indicating the essential genes and TFs that act and protect plants under extreme environmental stresses. Blue, red and green text color in the gene-boxes showing that these genes or TFs works in different stresses. Adapted and slightly modified from Raza (2020a),8 with permission from Springer Nature
Over the past decade, several reports indicated cloning and overexpression of the genes in rapeseed which enables plants to withstand several abiotic stresses (Table 1). Earlier, the BnNAC485 gene related to the NAC family was cloned, and results show that BnNAC485 was significantly expressed in 21-day-old seedling and cotyledon that was prompted by abiotic stresses.28 In another study, the overexpression of BnNCED3 increases abscisic acid (ABA) accumulation, nitric oxides (NO), and generation of reactive oxygen species (ROS) in transgenic rapeseed. Furthermore, transgenic lines displayed restricted seed growth, lateral root initiation, and improvement of ABA-mediated leaf senescence by modulating ABA synthesis.43 Cloning and expression scrutiny of BnICE1 revealed that BnICE1 is vigorously linked with cold stress tolerance, particularly at low temperatures.21 Regulation and molecular characterization of the C-repeat-binding factor (CBF) exposed that CBF was closely connected with cold stress.44 Recently, the identification and expression analysis of BnBADH1 increases salinity and drought stress tolerance,37 and so on. Further examples of rapeseed abiotic stress-related genes are presented in Table 1.
Table 1.
The overview of abiotic stress-responsive genes or/and transcription factors of rapeseed along with their key responses under stress environment
| Genes | Key stress responses | References |
|---|---|---|
| BnICE1 | Increase cold tolerance and highly expressed in different tissues, like stem, leaf, and hypocotyls | 21 |
| BnCBFs | Increase cold tolerance via ABA-independent pathways | 22 |
| BnCOR25 | Improve cold tolerance via the ABA-dependent pathway | 23 |
| BnSOS1 | High expression of BnSOS1 cause the exclusion of harmful Na+ into the apoplast area from the cellular environment and ultimately increase the salinity tolerance | 15, 24 |
| BnaCBLs and BnaCIPKs | Overexpression and mutant analysis improves tolerance to salinity, cold, heat, drought, ABA signaling, and low potassium | 25–27 |
| BnNAC485 | Transgenic plants show resistance to salinity via ABA-mediated pathway, and it also improves the early flowering in transgenic plants | 28 |
| BnATP-A and FBP | Responsible for cold and drought tolerance | 15 |
| BnERFs and BnRAV-2 | Transcriptome analysis reveals that these genes involved in cold tolerance | 7 |
| BnERF2-like [ERF2.4] | Improve the potential of antioxidant systems which improve resistance to submergence and oxidative stresses | 5 |
| BnaABF2 | Improve drought and salinity tolerance via the ABA-dependent pathway | 29 |
| BnaMYBs | Improves tolerance to cold, heat, drought, and salinity through the regulation of ROS defense genes | 30, 31 |
| BnSIP1-1 | Overexpression improve the growth rate under salinity, osmotic, and ABA stresses; mainly involved in osmotic tolerance | 19 |
| BnNHX1, BnAB15, BnRD29A, BnERD15, and BnKIN1 | Increased salinity tolerance in transgenic plants | 19 |
|
BnbZIP13, BnbZIP28, BnbZIP41, BnbZIP2, BnbZIP53, and BnbZIP60 |
Expression analysis indicated that these genes enhance resistance to heat and salt stress | 32 |
|
BnWOX10, BnWOX50, BnWOX44, and BnWOX18 |
Increase resistance to drought, and salinity by the regulation of plant hormones | 33 |
| BnPYR1-3, BnPYL1-2, and BnPYL7-2 | Expression analysis indicated that these genes enhance resistance to drought, salinity, and elevated temperature | 4 |
| BnaPLDα1, BnaPLDδs and BnaPLDδ | Increase tolerance to salinity, cold, dehydration, and ABA response | 34 |
| BnWRI1 | Regulating the impact of heat stress on fatty acid biosynthesis | 35 |
| BnFADs | Increase resistance to Cd and salt stress | 36 |
| BnBADH1 | Increase resistance to salinity and drought stress | 37 |
| BnKCS1-1, BnKCS1-2, and BnCER1-2 | Upgrade cuticular wax in transgenic plants and increase drought tolerance | 38 |
| BnaNHXs | Differential expression of BnaNHXs improves salinity tolerance [nitrogen and low phosphate] | 39 |
| BnNRT2s and BnNRT2.5 s | Increase tolerance to nutrient deficiency [phosphorus and potassium], waterlogging, and drought stress | 40 |
| BnCOL2 | Regulating the plant response to drought stress | 41 |
| BnNF-YA3 | Regulating the plants’ response to salinity, drought, and ABA | 42 |
The improvement of Brassica spp. for abiotic stress tolerance has been addressed since a few decades, and several tools have been employed, such as identifying and manipulating crops for abiotic stress tolerance, applications of several omics tools for the elucidation of genes associated with abiotic stress tolerance.45 However, the major discoveries in this arena are gene discovery in response to different stress, marker development, gene mapping, transcriptomic analysis, and regulation of physiological and biosynthetic mechanisms.
The advancement of omics tools such as genomics, transcriptomics, proteomics, metabolomics, and phenomics has modernized molecular biology research. These allowed meticulous examination of connections between molecular machinery to assimilate genes, proteins, and many essential regulatory components, collectively organizing systematic molecular strategies.46 The genome sequencing of Brassica crops has allowed more understanding of crop genetics and opens new avenues for crop improvement. Moreover, it has transformed molecular and functional genomics in Brassica cultivars.47 Various transgenic rapeseed cultivars have been developed that allow more tolerance than conventional varieties. Advancement in emerging approaches for genome editing (GE) like CRISPR/Cas9 has helped explore plants’ mechanism against stress responses.48 This review described the integrated omics approaches that could efficiently enhance rapeseed production for global requirements and sustainable agriculture production.
2. The Interplay of Omics Approaches to Address Abiotic Stresses
Omics approaches have been applied to gain insight in all biological mechanisms occurring during abiotic stresses in crops (Fig. 3). Over the past few decades, omics strategies have been improved to develop stress-resistance cultivars. Hence, a paradigm shift has been brought in research by these omics strategies to tackle abiotic factors and open up new horizons for enhanced understanding of numerous features responsible for crop resistance. To combat abiotic stresses, plants adjust their omics profiles accordingly.46 Therefore, the association of omics techniques will help to uncover candidate genes and their biosynthesis pathways in the future. Deciphering the association among omics approaches will potentially help us to illuminate molecular pathways that are actively controlled by abiotic stresses. Additionally, these combined tools will provide a comprehensive data set for biological system analysis.
Figure 3.
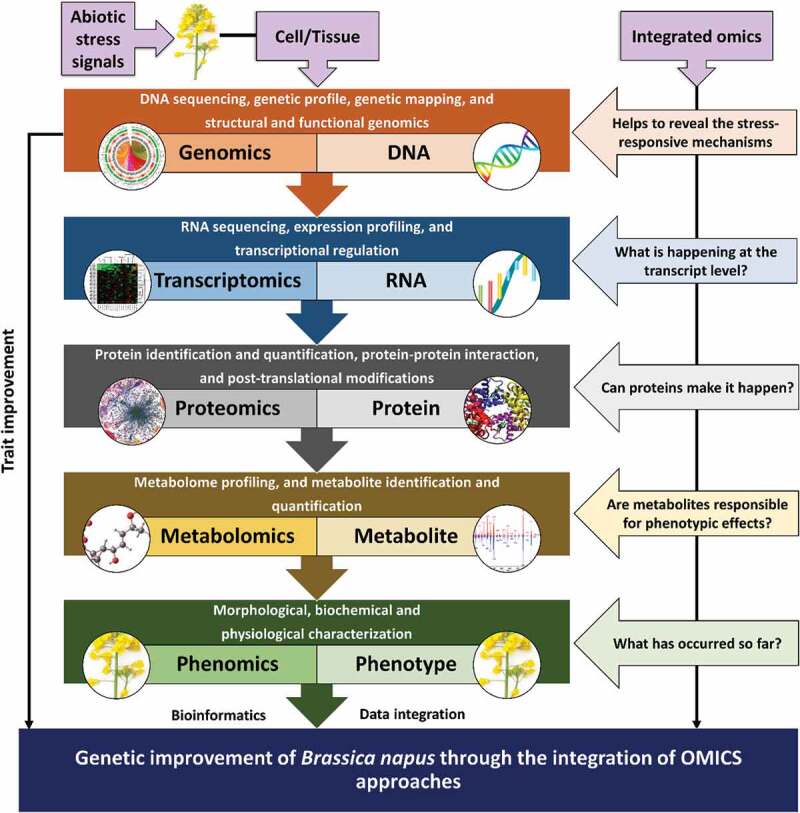
The central dogma of systems biology showing the flow of information from DNA to phenotype. Step-wise presentation of OMICS approaches for studying abiotic stress responses. Ultimately, the integration of omics tools, primarily genomics (mainly single-cell/tissue-specific) leads toward the genetic improvement of rapeseed by modulating several agronomic traits such as environmental stress tolerance, yield, plant height, seed rate, flowering, photosynthesis, and respiration rate, root and shoot length, grain quality and yield, biomass production, etc
2.1. Genomics Approaches: Provide Insights into Stress-associated Mechanisms
Genomics tools have gained enormous importance for the elucidation of desired genes related to abiotic stress tolerance. During the last few years, significant advancement has been achieved by the researchers in the functional genomics era to scan the genomes of many crop species.49 Several genomics tools have been implemented in plant biology to explore the genomes of many crops for abiotic stress-tolerance, such as quantitative trait loci (QTL) mapping, which has emerged as a valuable technique to investigate the genetic diversity of crops. Moreover, up-gradation of QTL mapping and marker technology has allowed deciphering modern breeding approaches like marker-assisted selection (MAS) and single nucleotide polymorphism (SNPs).47,50
High-throughput sequencing techniques, such as next-generation sequencing (NGS), have made it feasible to sequence an organism’s whole genome. The integrated approach of genome-wide association studies (GWAS) and NGS technologies have provided remarkable opportunities to predict stress-related genes.51 Nevertheless, the whole genome comprising genic and intergenic regions can form a particular scheme for plant development. The relationship between functional, comparative, and structural genomics is incredibly desirable for organic production.49 New opportunities have been created by the recent improvements in crop genomics to develop abiotic stress-tolerant crops. Moreover, the availability of genomics platforms for gene identification and trait examination in rapeseed, which have enormous capabilities for molecular breeding integrated with modern tools of breeding related to GE techniques. The accessibility of a reference genome to rapeseed permits the application of such technologies in rapeseed could be predicted to attain significant progress in the future. In the subsequent sections, we have discussed the recent progress in major genomics tools.
2.1.1. Marker-assisted Breeding
For breeding and crop improvement, powerful approaches like marker‐assisted breeding (MAB) and MAS have been comprehensively used to map numerous desired genes in rapeseed.47,50,52 Molecular markers linked with stress-responsive genes or QTLs can be recognized, which can be employed as a secondary choice principle to advance breeding proficiency via MAS. However, the practice of MAS for operating modest traits has been modernized in numerous rapeseed breeding programs. Thus, MAS for enlightening multifaceted traits appears to be at initial stages.47,50,53 Furthermore, to exploit the genetic diversity in different crop plants, a high throughput strategy of genomic tools is used. Rapeseed, which has a comparatively low level of genetic diversity, several studies based on genome-wide MAS have been conducted to describe the genetic variability in rapeseed.47,50,52
Nucleotide sequences of genes have been established using DNA markers, which offer fundamental knowledge to understand the genetic divergence of a species from primitive families and leading landraces that hold their genetic materials well preserved in gene databank for future applications.47,52,54 DNA markers are applied to probe the genome to screen desired loci and transgenes by repeated selective events. Certain important traits were identified, where DNA markers have been applied for genetic mapping in rapeseed.52 Numerous SNPs have been discovered in rapeseed using QTL mapping for desired traits.55 Likewise, RAPD, AFLP, ISSR, and SSR have been used to find the genetic variability in rapeseed.56–59 Detailed information will be presented in the subsequent sections (2.1.1.1-2.1.1.7) to highlight the efforts in rapeseed mapping using different approaches.
2.1.1.1. The contribution of RAPD markers
PCR-based random amplified polymorphic DNA (RAPD) markers primarily comprised on random amplification of segments of polymorphic DNA.60 Previously, several studies have been carried out using RAPD markers for rapeseed mapping for different traits. For example, a double haploid (DH) population resulting from the F1 generation of the cross Apollo (black-seeded) x YN90-1016 (yellow-seeded) rapeseed was examined to detect molecular markers related to the yellow-seed trait.61 An essential gene (pigment 1) lined by 8 RAPD markers was linked with the yellow seed coat color trait in the F1 generation. This gene described 72% phenotypic difference in seed coat color.61 Likewise,62 examined the F2, BC1, and F1-derived DH population for seed color using 810 RAPD and 512 AFLP markers. Out of these, 240 RAPD markers showed polymorphisms among the parents. Additionally, 4 RAPDs and 16 AFLP primer pairs displayed polymorphisms among the bulks, whereas only 2 RAPD and 8 AFLP markers were detected in the locality of the seed coat color.62
Furthermore, a polymorphic marker linkage map conveying nine linkage groups (LGs) was created from F2:3 populations of rapeseed [SLMO46 (winter type and cold-resistant) x Quantum (spring type and cold susceptible)] using 47 RAPD markers.63 They identified one presumed QTL for winter survival elucidating only a 5% phenotypic difference. The QTL was located on LG6 is amid 670 and 650 bp sized RAPD markers with 4 cM distance from the 650 bp marker and had adverse conserving consequence.63 In another study, a similar rapeseed population by 47 RAPD and 32 SSR markers, a linkage map conveying 14 LGs was prepared and presumed QTLs were identified on 3, 8, 9, and 10 LGs. These QTLs explained a 24% phenotypic difference in the freezing tolerance in the studied rapeseed population.64 Besides, RAPD markers have been extensively used to evaluate genetic diversity and polymorphism between different rapeseed genotypes/lines/populations.58–59–65–68
2.1.1.2. The contribution of AFLP markers
Amplified fragment length polymorphism (AFLP) is a PCR-based technique in which a subgroup of constraint remains are selectively amplified with oligonucleotide primers opposite to sequences. It can also be applied to fresh and partly decade-old DNA.69 Previously, the separation of the rapeseed Bnms3 gene (responsible for recessive genic male sterility) for evolving precise PCR markers for MAS and understanding this gene’s crucial function has been investigated. Notably, the Bnms3 gene was labeled with seven AFLP markers; out of these, six AFLPs were recognized to be co-segregated with the target gene in a population of 115 individuals.70 In another study, Brassica 60 K Infinium SNP array, AFLP, and SSR markers were used to make a high-density genetic map in the DH rapeseed population (Zhang et al. 2014c). Using the genetic map, a novel QTL (qBEC-A3a) for boron deficiency was identified, and a similar map was then employed to identify different QTLs related to plant growth (36 QTLs with 6.14–46.27% phenotypic difference) and boron uptake (12 QTLs with >10% phenotypic difference). These findings provided vital QTLs suited for fine mapping and the actual markers for improving breeding productivity in rapeseed.71
Moreover, Fan et al. (2015)72 described the QTLs’ mapping related to waterlogging and drought resistance in the DH rapeseed population’s seedling stage via AFLP and SSR markers. Significant genetic diversity was detected in resynthesized rapeseed lines using AFLP markers compared to the gene pools of conventional spring and winter rapeseed genotypes.73 In the next experiment, these authors correlated the genetic diversity with the heterotic yield in trial hybrids.74 Later, the yield characters of 184 F2:3 lines were accessed via AFLP and SSR markers. Results show that 73 markers were linked with 12 yield characters with an involvement rate of 3.54–15.88% and an average of 6.60%. The genetic distance was expressively associated to heterosis of F1 trials via 66 markers related to yield character and suggesting that the correctness of yield and heterosis estimate reliant on QTLs linked with yield characters in rapeseed required to be upgraded.75,76 On the other hand, AFLP markers have been extensively used to access the genetic diversity among different rapeseed genotypes/lines/populations.56,58,77
2.1.1.3. The contribution of SSR markers
Simple sequence repeats (SSRs) or microsatellites are short tandem repeated motifs of 1–6 nucleotides present in huge amounts and may fluctuate in the number of repetitions at a specified locus. These markers have numerous benefits over other markers, such as hereditary co-dominance.78 The sum of freely accessible Brassica SSR primers is growing because of openly sponsored international enterprises (www.brassica.info). Nevertheless, compared to other imperative crop plant species, limited SSR markers are easily accessible, meaning few consensus maps have been described in DH rapeseed populations.79 The publicly available set of vigorous, extremely polymorphic, mapped SSR markers covering the entire rapeseed genome have helped rapeseed genetics researchers in mapping and genome amalgamation.80
During the past few years, significant progress has been made using SSR markers in integrating available and new genetic maps. Further, the QTL alignment, playing a crucial role in correlation of candidate genes with QTLs. For instance, using F2:3 rapeseed populations,64several major QTLs were detected on 3, 8, 9, and 10 LGs with a phenotypic difference of 24% in freezing tolerance. SSR markers, together with Brassica 60 K Infinium SNP array, were used to make a high-density genetic map in the DH rapeseed population under boron deficiency. This resulted in 12 QTLs with >10% phenotypic difference for boron uptake and a novel QTL linked with the growth and boron deficiency (Zhang et al. 2014c). In rapeseed, 53 SSR markers were expressively linked with 3 phenolic sections, and 11 markers were connected with total phenolic acid contents. Out of these, only 4 SSR markers result from QTL for seed color by association mapping.81 Additionally, 25 and 11 SSR markers are related to seed coat color and oil content, respectively. In contrast, only six SSR markers are connected to both oil content and coat color.82 In a recent study, 36 SSR markers have been used to examine the variability between 41 rapeseed lines linked with drought tolerance QTLs.83 Apart from the above-mentioned traits, SSR markers have shown great potential in deciphering genetic diversity between rapeseed genotypes/lines/populations [58, 84, 85, 86].
2.1.1.4. The contribution of ISSR markers
The commonly used PCR-based markers such as RAPDs, AFLPs, and SSRs have some limitations like low reproducibility, high cost, and need flanking sequences. Therefore, inter simple sequence repeat (ISSR) is an updated method that incapacitates these limitations. This method combines numerous advantages of AFLP- and SSR-based investigations with the universality of RAPD.86–88 Water-stressed rapeseed cultivars showed maximum discrepancy subsidized by ISSR markers went to relative water content (78%) at normal state, to root/shoot index (66%) at modest stress state and to root length (53%) at severe stress environment.89 Recently, while studying drought tolerance signals and their relationship with ISSR markers in rapeseed genotypes, most of the ISSR markers were found to be linked with some of the drought tolerance indices.90 ISSRs have been effectively employed in the genetic diversity studies at inter and intra definite levels in numerous plant species, including rapeseed.89,91,92 Molecular mapping using ISSRs linked with QTLs or candidate genes is yet to be reported in rapeseed, while this has been achieved in some other crop plants.
2.1.1.5. The contribution of SCAR markers
To lessen the difficulties that rise when consuming conventional markers such as RAPD, ISSR, IRAP, AFLP, SSR, etc., the sequence characterized amplified region (SCAR) marker was established. These markers can be derived from ISSR, IRAP, and RAPD markers. It signifies a particular, distinct genomic DNA part that was noticed in PCR extension with a pair of definite primers.93–95 These markers have great potential in plant identification at inter- and intra-specific species or population level.93,96,97 Previously, the linkage mapping using SCAR markers has been linked with 18-carbon fatty acids in rapeseed. Developed SCAR marker (L1L9) shows about 25% genetic difference for 18-carbon fatty acids in rapeseed.98 Later, using AFLP, SCAR markers have been developed for the trait-specific gene. For instance, SCAR markers have been characterized to be linked with a dominant genic male sterility [suppressor gene (Rf)] in rapeseed by several researchers.99–101 Additionally, SCAR markers have been associated with self-incompatibility in rapeseed and showed significant results at a polymorphic level compared to CAPS markers.102 The above-mentioned markers were handy for improving different traits and accelerating MAS progression in rapeseed breeding plans. For cultivar fingerprinting, transposon insertion-SCAR markers have been characterized in rapeseed. These markers can be considered a vital tool in the reverse genetic scheme for separating novel genes in rapeseed.103
2.1.1.6. The contribution of CAPS markers
Cleaved amplified polymorphic sequences (CAPS) markers are PCR-amplifications of DNA remains with specific primers after the digestion of restriction enzymes and the harvests’ isolation in an agarose gel. Practical CAPS markers can be established on a target gene’s identified sequence to analyze its regulation, structure, purpose, and expression. These markers are closely connected with the target gene and are particularly supportive of MAS.104,105 Earlier, SCAR and CAPS markers were developed and mapped to be associated with seed coat color gene in a resynthesized purely yellow rapeseed line obtained from the F1 DH population using a genome-walking method.106 In a recent study, the SCAR and CAPS markers were linked with Leptosphaeria maculans resistance gene Rlm6 in B. napus x B. juncea interspecific hybrids. Segregation of both markers related to the Rlm6 gene was established by using F2 and F3 progeny. Interestingly, the segregation of CAPS markers and phenotype for blackleg disease cruelty in the F2 population had a Mendelian ratio of 3:1 in resilient vs vulnerable plants, respectively, suggesting that the genetic regulator of resistance was carried by a dominant gene.107
In a MAS analysis of new high oleic and low linolenic winter rapeseed inbred lines, genotyping was completed for the assortment of homozygous lines via allele-specific CAPS markers and SNaPshot assay. Lastly, new high oleic and low linolenic winter rapeseed recombinant lines were obtained, ready to be used as a preliminary material in developing new varieties with high oil value than traditional types.108 In another study, CAPS markers were used for the identification of two mutant alleles (BnaA.FAD2 gene) in rapeseed. Further, CAPS markers also provided relatively good results while detecting the heterozygosity in BnaA.FAD2 gene.109 These examples indicate the importance of CAPS markers in MAS studies to identify and develop new breeding lines.
2.1.1.7. The contribution of SNPs
Genomes of all the organisms consist of single base pairs sequences called SNPs. Rapidly decreasing DNA charges for re-sequencing and accessibility of a more advanced SNP platform for rapeseed open up new horizons for significantly more effective gene mining and genetic mapping.47 More than 75% of detected SNPs were useful and could be exploited in modern molecular breeding schemes.47,110 Likewise, SNP analysis was documented to study the heat stress on different rapeseed varieties under field conditions, and data were recorded from flowering to maturity phase for different agronomic traits. Association mapping (AM) for various agronomic traits was executed to detect heat-related QTLs. For these 37,269 SNP markers were employed and showed that several genes were strongly correlated with abiotic stress-tolerance in rapeseed.111 Numerous examples are presented in Table 2 and the next section.
Table 2.
Events performed for the detection of SNPs in rapeseed
| No. of genotype used | Sequencing method | SNP calling tool | No. of SNPs | References |
|---|---|---|---|---|
| 2 | Solexa sequencing Illumina | MAQ | 23,330–41,593 | 112 |
| 8 | Illumina GAIIx system | RADtags | 20,835 | 113 |
| 84 | Illumina mRNA-Seq | Trinity | 101,644 | 114 |
| 4 | Illumina GoldenGate assay and Illumina Infinium | MIRA | 5764 | 115 |
| 4 | Illumina Infinium™ 6 K | SGSAutoSNP | 5306 | 116 |
| 52 | Illumina HiSeq 2000 platform | FaSD, Freebayes and SAMtools | 4.3 Million | 117 |
| 437 | Brassica 60 K Illumina Infinium array | CLC Genomics Server | 52,157 | 118 |
2.1.2. Evidence of QTL and GWAS Studies
A GWAS is a method employed in genetics investigation to associate definite genetic differences with particular traits in different individuals. It overwhelms numerous restrictions of traditional gene mapping (QTL) by providing advanced resolution, often to the gene level, and consuming trials from formerly investigated populations in which frequently happening genetic differences can be connected with a phenotypic difference. The start of high-density SNP typing permitted whole-genome examinations to recognize often insignificant haplotype hunks that are pointedly associated with quantitative traits.119,120 For instance, Jian et al.121 studied the recombinant inbred lines (RIL) population at the seedling stage under salt and drought stress for GWAS. Notably, 2795 SNPs were detected, and the analysis revealed that numerous minor-effect loci regulate the germination percentage of rapeseed, and the various gene is activated under drought or salinity stress. Furthermore, a GWAS experiment was performed to exploit the QTLs correlated with salinity resistance in rapeseed. Genotyping was carried out through Brassica 60 K Illumina Infinium SNP arrays and detected 75 SNPs found on 14 chromosomes. Moreover, 38 candidate genes were evaluated, triggered under salt stress and belongs to different groups such as enzymes, transporters, aquaporins, and transcription factors (TFs). This experiment permits examining the molecular pathway of salt-resistance and may help MAB for salt tolerance in rapeseed.51 Recently, AM was done at the seed germination stage for 214 rapeseed inbred lines to locate QTLs linked with salinity tolerance. The genotyping was performed using 60 K Brassica Infinium® SNP platform, and 110 SNPs were detected with salt stress regulation. Furthermore, 56 targeted genes were mapped in the close locality of QTL regions. Thus, this result provides substantial proof for evaluating rapeseed salt tolerance’s genetic regulation and may support MAB for salt resistance in rapeseed.122
Genetic mapping of QTLs is the 1st phase for discovering the genetic design of a trait and in executing MAS in plant breeding scheme.123 Stress tolerance results from multiple responses/mechanisms, assessment of these mechanisms and stress-related QTLs has assisted plant breeders in developing elite genotypes with stress-tolerance. Genetic investigations for the development of abiotic stress-tolerance rapeseed have been extensively studied for many stresses, as shown in Table 3.
Table 3.
Experiments performed for the identification of QTLs in rapeseed under stressful environment
| Stress type | No. of lines used | Genotyping/Linkage map | No. of QTLs | Chromosomal location | Key findings | References |
|---|---|---|---|---|---|---|
| Freezing | 199 F2:3 lines | 350 SSR and 250 RAPD | 4 | 8, 6, 15, and 14 based on BBSRC maps | Identified QTLs shows 24% of phenotypic variances and with the help of NGS, these QTLs could use in MAS freezing tolerance |
64 |
| Waterlogging and drought | 150 DH-lines | 183 SSR and 157 AFLP | 28-CK, 26, and 31 for stresses | A1 | A few QTLs for both stresses overlapped each other, suggesting that genetic bases for DH were similar to some extent in rapeseed | 124 |
| Salinity | 85 inbred-lines | Illumina Hiseq-2000 | 62 | A1, A2, A3, A5, A7, C3, and C9 | Identified QTLs strongly associated with ion-homeostasis, shoot biomass, and salt-tolerance via AM, and BnaTSN1 as a candidate gene for salt-tolerance | 125 |
| Drought | 225 DH-lines | Brassica-60 K Illumina Infinium array | 20 | A01, A06, A08, C01, and C03 | Identified QTLs colocalized to 2 main QTLs, and showing that compensation is genetically controlled due to a powerful correlation between the morphology of root, productivity, and time of flowering | 126 |
| Drought | IMC106RR and Wichita | Illumina HiSeq-2000 | 1 | A10 | Candidate gene Bna.FLC.A10 were identified through polymorphism, and the results give a sight of genome-wide variation among rapeseed with enhancing information about the root genetic basis under drought stress | 127 |
| Drought, salinity and low-temperature | ZS11 | Microarray and EST-based analysis | 31 | A02, A03, A06, A08, A09, C01- C07, and C09 | 26 BnaWRKY genes were triggered by multiple stresses demonstrating that WRKY genes have a pivotal role in regulating multiple stresses | 128 |
| Salinity | 196 F2:3 lines | 532 molecular markers | 45 | A1–A10 and C1–C9 | Bna003640 was found to be the trigger in salinity and regulate the salt-tolerance mechanism | 129 |
| Salinity | 368 | Brassica 60 K Illumina Infinium SNP array | 25 | A3, A9, C5, C6, and C7 | 38 candidate genes were evaluated, which triggered under salinity and examined the molecular pathway which may help MAB for salt-tolerance | 51 |
| Salinity | 196 F2:3 lines | IP and SCAR markers | 1 | A10 | Lobed leaf gene (Bra009510) gene was mapped, and results presented that this gene might regulate the salt stress -tolerance | 130 |
| Cold | 147 F2:3 lines | 333 SSR markers | 11 | A08 | Identified QTLs showed the phenotypic variation of 42.50% and 1.09%, and two genes BnaA08g15470D and BnaA08g05330D have been strongly linked with cold-tolerance | 123 |
2.2. Transcriptomic Approaches: What Appears to Be Happening at TheTranscript Level?
Transcriptomic is a very promising technique to understand abiotic stress regulation in plants to decipher novel genes and various regulatory networks through transcriptome profiling.131 Transcriptomic or/and gene expression investigation by next-generation sequencing (NGS), RNA-seq profiling, subtractive libraries, expressed sequence tags (ESTs), serial analysis of gene expression (SAGE), and microarray have great potential to improve genomic resources of plants such as gene discovery and functional analysis.132,133 Recently, numerous rapeseed genome sequencing and re-sequencing projects have been completed (Supplementary Table 1). Based on the NGS technologies, a Brassica napus pan-genome information resource (BnPIR) database http://cbi.hzau.edu.cn/bnapus/index.php) has been developed to facilitate the B. napus researchers in the post-genomic era. Global transcriptome profiling provides an excellent opportunity for comprehensive knowledge about gene expression and could help identify numerous regulatory mechanisms. In the subsequent segments, we have explained the potential of widely used approaches for studying gene expression at the transcript level. Table 4 shows a summary of transcriptome-based experiments performed in stressful conditions.
Table 4.
Summary of some transcriptomic studies conducted under stressful environment in rapeseed
| Approach | Stress | Objective | Genotype | Tissue condition or stages | No. of DEGs and TFs, respectively | Key findings | References |
|---|---|---|---|---|---|---|---|
| RNA-seq | Waterlogging | To identify the mechanism of waterlogging tolerance | ZS9 | Roots at 0 h and 12 h of stress | 4432 | Protein degradation is associated with the negative regulation of waterlogging | 134 |
| RNA-seq | Salinity | Regulation in leaves and roots in response to salinity | N119 | Leaves and roots 1 h and 2 h after stress | 14,719, and 582 | Identified genes influenced by salinity in roots and leaves, a novel TFs S1Fa-like reported | 135 |
| RNA-seq | Drought | To decipher the candidate genes for drought tolerance | ZY821 | Six-leaf stage | 3657 | DEGs were triggered under drought stress and providing new genes | 136 |
| RNA-seq | Salinity | To elucidate the relationship between salinity and Strigolactones | ZS11 | Root and shoot after 7-days of treatment | 2162-roots, 5935-shoots DEGs | Strigolactones improves salt-tolerance and results provided stress associated novel genes | 137 |
| RNA-seq | Drought, cold, salinity, heat and ABA | To decipher the function of TF families under stresses | ZS11 | 7-days old seedlings at 12 h after treatments | 315, and 2167 | About 80% DEGs of the identified 5 TFs families BnAP2/ERF, BnbZIP, BnMYB, BnNAC, and BnWRKY triggers abiotic stresses |
133 |
| RNA-seq | Low temperature | To identify candidate genes responsible for fast germination | Ganyouza No. 5 and Huawanyou No. 4 | 1, 2, and 3-days after treatment | 9111-down and 10,233-up-regulated DEGs | Many TFs such as ERF, NAC, DREB, B3, MYB, EFR, bZIP, and WRKY were also found to regulate the low-temperature tolerance in rapid germination cultivar | 138 |
| RNA-seq | Cold | To revealed conserved and novel cold-responsive genes | HX17 and HX58 | After 14-days of treatment, third leafs | 47,328 | Two conserved (the primary metabolism and plant hormone signal transduction) and two novels (plant-pathogen interaction pathway and circadian rhythms pathway) pathways were significantly enriched with DEGs | 139 |
| RNA-seq | Cold | To explore the molecular mechanisms in different rapeseed ecotypes | Five winters and five spring ecotypes | Leaves were harvested at 0 and 12 h after treatment | 25,460 and 28,512 DEGs in spring and winter oilseed ecotype, respectively | Lipid, ABA, secondary metabolism, signal transduction, and transcription factors may play vital roles in both ecotypes under cold stress | 140 |
| EST | Drought | To identify stress-responsive regulatory network | DH-12,075 | Four-leaf stage | 17 TFs | One protein phosphate, eight kinases, 26 regulatory genes, and 17 TFs were investigated for increased transcript level; new miRNAs and regulatory genes modulating drought stress | 141 |
2.2.1. Modern Transcriptomic Analysis by RNA-seq
RNA-seq is an emerging approach that exploits or/and offers a different outlook to examine the transcriptome sequence by granting complete access to transcripts. Therefore, RNA-seq can be conducted as a substitute strategy for many diverse techniques for quantification of the transcript with the advantage of higher sensitivity and ability to differentiate among related gene paralogs, which are different by just a small number of nucleotides.133,135 For instance, five TFs families have been characterized and described under various abiotic stresses in rapeseed. Totally 2167 TFs from 5 different families were documented in rapeseed; i.e., 518 BnAP2/ERF, 252 BnbZIP, 721 BnMYB, 398 BnNAC, and 278 BnWRKY, including some unique transcripts as compared to actual results. Many vital representatives of these TFs and regulatory systems associated with various abiotic stress conditions have been discovered. The investigations in rapeseed against stress tolerance have extended our knowledge toward TFs. RNA-seq represents that more than 80% of TFs triggers abiotic stress, and 315 TFs (DEGs) have been observed, and these 315 DEGs were highly expressed.133
Yong and colleagues performed RNA-seq for the comparative transcriptomic study of rapeseed under increased salt stress. Roots and leaves were collected after 1 hour, and 12 hours of salt stress and de novo transcriptome sequencing yielded a total of 14,719 DEGs. Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment analysis found that 438 transporter genes with 582 TFs were influenced by salinity in roots and leaves. DEGs under salinity provide full knowledge for rapeseed improvement.135 RNA-seq was achieved to elucidate the candidate genes and decipher the molecular pathways that govern the tolerance mechanism under drought stress in rapeseed. Illumina Hiseq 2000 was used for RNA-seq analysis and assemble 28,378,899 and 26,192,312 high-resolution reads. RNA-seq analysis identified 3,657 transcripts triggered by exposure to drought stress.136
Recently, RNA-seq was executed to elucidate the transcriptional modulation process for the fast growth of rapeseed. Various transcriptomic assemblies were established under cold stress and normal temperature with a slow and rapid germination speed rate. Transcriptome analysis showed 9111 and 10,233 DEGs, which were down and up-regulated under cold stress. Moreover, many TFs such as ERF, NAC, DREB, B3, MYB, EFR, bZIP, and WRKY were also found to regulate the low-temperature tolerance in rapid germination cultivars. These results will help future breeding programs to develop abiotic stress tolerance rapeseed genotypes.138 In another study, under cold stress, RNA-seq was used to explore the molecular mechanisms in different rapeseed ecotypes. They identified 25,460 and 28,512 DEGs in spring and winter oilseed ecotype, respectively. Identified DEGs mainly fitted to lipid, ABA, secondary metabolism, signal transduction pathways, and these signaling pathways may play dynamic roles in both ecotypes under cold stress.140 See Table 4 for more examples.
2.2.2. Expressed Sequence Tags (Ests)
ESTs signify a foundation for discovering unique gene structures that provide an outline for further significant studies, like expression system, gene maps, and cDNA sequencing projects. ESTs have been considered the precise technique to unveil sequences-related knowledge under abiotic stress conditions.142,143 The ocsESTdb database provides information about seed ESTs and full-length CDS sequences of oilcrops seeds. The relative investigations of the oilseed databank showed that ESTs are accessible at the database of OCRI-CAAS, Wuhan, China (http://www.ocri-genomics.org/ocsESTdb/).144 Similarly, overtly available Brassica ESTs contigs equivalent to a variety of gene functions (http://brassica.nbi.ac.uk/array_info.html).
Nevertheless, 343 WRKY domains with 287 genes were documented in response to several abiotic stresses in rapeseed. Microarray and EST-based analysis showed that 74 WRKY genes had been expressed beneath stress conditions. After QTL mapping, 77 WRKY genes have been reported in 31 QTL loci associated with many stress resistance. Twenty-six BnaWRKY genes which are triggered by drought, salinity and low-temperature stress.128 Recently, available data of ESTs were examined and found 25 creatine phosphokinase (CPK) genes. A DH line of rapeseed was used to clone the cDNA sequences of 23 genes. Phylogenetic analysis was performed, and green fluorescence protein was used as a reporter gene to detect five candidates in BnaCPKs. Additionally, the expression of 21 BnaCPKs was investigated for oxidative, low potassium, abscisic acid, heat, cold, drought, and salinity stress regulation via qRT-PCR. The results suggested that multiple signaling pathways govern CPKs in response to several stresses in rapeseed.26 Rapeseed EST profiling was carried out to find the function of typical AP2/ERF TFs under biotic and several abiotic stress regulations.145 Table 1 documents the other TFs in response to abiotic stresses in rapeseed.
2.2.3. Serial Analysis of Gene Expression (SAGE)
SAGE has been extensively used to investigate gene expression. SAGE method calculates a tag, which shows a product of gene transcriptome. SAGE is considered the most economical and high-throughput approach, including mRNA isolation, cloning, and sequencing. In the SAGE technique, `tag` is a short length nucleotide sequence with a pointed head-to-head specific restriction enzyme. Therefore, SAGE represents gene expression in digital form. There are many SAGE applications in plants, such as the interaction between host and pathogens, plants’ responses in several abiotic stresses, transcriptome profiling, and metabolism of many hazardous compounds.146,147 About functional genomics investigations of oilseed plants with the importance of seed production and metabolism mechanism of fatty acids, a database was established named Shanghai RAPESEED database (RAPESEED, http://rapeseed.plantsignal.cn/). The rapeseed has 8462 ESTs, including 2526 full-length cDNA; 8095 expressed genes, and 23,895 SAGE tags during seed growth. In RAPESEED database, all the relevant data is stored in the form of nucleotides and protein sequences.148 This technique has been used in several crops but needs to be considered for rapeseed in future work.
2.2.4. Microarray
cDNA microarrays are used for observing gene expression and offer a proficient strategy to evaluate the potential functions of numerous genes.149 Microarray cDNA investigations of gene expression in various plants beneath several abiotic factors have been widely documented.150,151 Gene databank accessibility gave plant gene expression from microarray investigations like Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) and ArrayExpress-functional genomics data (http://www.ebi.ac.uk/arrayexpress).152
Microarray analysis was demonstrated in rapeseed to decipher the function of CBF/DREB1-like TFs under cold stress. Microarray and northern blot analysis confirm that the accumulation of cold-responsive genes that have a function in photosynthesis. Additionally, some other genes, such as GLK1 and GLK2-like TFs, responsible for chloroplast development, trips-p/pi translocator, and β-amylase, were also produced. Hence, increased exchange rate of malate/oxaloacetate, enhancement in glycolysis, more sucrose and starch production, improved Calvin cycle enzymes, and rectification of photosynthetic efficacy was also detected, indicating that BnCBF hyper-expression has moderately imitated freezing-induced tolerance. Resulting in the overexpression of DREB1/BNCBF has shown improved not only cold tolerance but also controlled chloroplast development in rapeseed.153 Therefore, it is essential from an economic point of view to increase rapeseed varieties with freezing tolerance (3–5°C) using CBF genes by means of the addition of millions of acres of lands to rapeseed-production areas worldwide.154 A comprehensive analysis of genes triggered by drought and salinity was performed by microarray to investigate the genes that govern abiotic stress reaction in rapeseed. The results indicated that out of 536 clones, 189 and 141 clones were responsible for gene supersession under salt and drought stress, while 288 and 172 were identified for drought and salinity resistance genes, respectively. The functional examination revealed that these genes play a vital part in the growth, abiotic stress-tolerance, hormone response, signal transduction, regulatory factors, and various metabolic activities in rapeseed.155
2.3. Proteomics Approaches: What Makes It Happen?
Protein components of any living organism at a specific time are termed as proteome of that organism. At the beginning of 2000, due to the accessibility of whole-genome sequencing and mass spectrometry (MS) approaches, the proteomics strategy has been established successfully. To elaborate on the relationship between plant proteome with surrounding environmental stimuli, relative expression at the proteomic level can be studied by subjecting the plants to normal and abiotic stress conditions.156,157 During the last decade, yeast two-hybrid (Y2H), matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI)-MS, and one/two-dimensional gel electrophoresis (2-DE) have gained much attention as excellent tools for separation and as analytical approaches in proteomics analysis. In the sub-sections, we have explained the potential of these approaches.
During abiotic stresses, the protein profiling of drought and salt stress of rapeseed has been conducted to detect the cross-talk between the cells through multiple reaction monitoring. Proteins related to phosphorylation mechanisms, such as CTRI, CDPK21, and TPR, have been identified under salt stress conditions. Similarly, BSL and STN7 proteins were also recognized, which have a role in the phosphorylation process under drought stress.20 Nevertheless, an iTRAQ-proteomic analysis was carried out for rapeseed roots under waterlogging stress, and 7736 proteins were identified. These proteins function differently in response to stress and provide insight knowledge for adaptive mechanisms.158 An experiment was performed on rapeseed for quantitative proteomic profiling under salt stress self-compatibility. Self-compatibility induced by salt stress identified some unique proteins, which elucidate the molecular mechanisms underlying the breeding of rapeseed cultivars.159
2.3.1. MALDI-TOF-MS and ESI
Nowadays, proteomics and metabolomics analysis of endogenous plant components have become very popular, and to carry out these investigations, a powerful tool such as MS imaging has been applied widely. Matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI) are mass spectrometry methods that have been extensively implemented for protein analysis.160,161 MALDI-TOF-MS and ESI are well-established approaches to analyze the cell lysate of protein contents, separated by m/c ratio.162 Ionization in MALDI can also be coupled with time of flight (TOF) analyzer.164
Recently, a study has been reported on rapeseed leaves, in which proteomic matrix, physiological and biochemical variations were evaluated under salt and lipoid acid stress. Comparative proteomic investigations were carried out for control-grown leaves as well as leaves from plants subjected to salinity combined with exogenous lipoic acid (LA) application. The proteins of various sizes were dispersed using 2-DGE and found that 28 proteins have been expressed under stress conditions. Furthermore, for the exact confirmation of these 28 proteins, MALDI-TOF/TOF MS has been used and recognized 21 proteins.163 2-DGE was used for comparative proteomic analysis of thermos-sensitive genic male sterility genotypes of rapeseed during microspore and microsporocyte formation. MALDI-TOF MS has identified 28 protein, from which ten protein expressed during microsporocyte while the remaining 18 protein expressed in the microspore phase.165
2.3.2. Two-dimensional Gel Electrophoresis (2-DE)
In modern proteomic studies, 2-DE is regarded as the driving force. Despite the dominance of MS, this strategy still holds prime importance for proteomic analysis. In various protein separation methods, 2-DE is most promising and accepted as a separation method and an analytical approach because of its powerful separation ability.166 For example, 2-DE was used to decipher the molecular networks regulating the salt stress in rapeseed. A salinity susceptible cultivar of rapeseed was subjected to different salt treatments and found that Na+ content and proline concentration were enhanced in leaf. Therefore, K/Na+ percentage, plant height, and shoot dry weight were reduced. 2-DE detected 110 spots on gels, and 37 of them indicated significantly abundant variations. Proteins were analyzed by employing LC-MS that has a function in photosynthesis and energy production. The results unveiled the reduction in energy production enzymes due to salt stress, whereas the increased accumulation of phosphoribulokinase under salinity in rapeseed.167
2-DE was run to examine the physiological and proteomic regulations of rapeseed under salinity. Seedlings were subjected to different salt stress concentrations that result in reduced photosynthesis activity and growth arrest, and 44 proteins were detected by the spots which contained many salt-resistance proteins. The results showed that the highly expressed proteins, which regulated tolerance, damage repair, and protein metabolism, might mitigate damaging impacts of salinity on respiration, energy production, photosynthesis, and chlorophyll synthesis in the leaves of rapeseed. This will help improve the understanding of the various molecular mechanisms operating under salinity in rapeseed and aid in developing GE rapeseed with increased salinity tolerance.168 Recently, 2-DE was used for the separation of root proteins, while MALDI-TOF-MS was employed for protein identification under salinity. Different protein spots were detected and their abundance was significantly influenced by salinity. Functional studies demonstrated the nine categories of DEPs, while 14 protein categories were detected in tolerant genotypes. The most important findings are the DEPs associated with energy metabolism, redox regulation, heat shock proteins, fructose synthesis, and glycolysis mechanism was enhanced only in the salt-tolerant genotype rapeseed.169 There are few databases available for proteomic analysis by using 2-DE, including rapeseed, rice, banana, tobacco, and Arabidopsis having protein profiles present at SWISS-2D-PAGE (https://world-2dpage.expasy.org/swiss-2dpage/) and WORLD-2D-PAGE (https://world-2dpage.expasy.org/list/).170
2.3.3. Yeast Two-hybrid (Y2H)
Y2H system is a handy technique for genetic mapping and to exploit the various mechanism basal in protein–protein interactions by activating the reporter genes.171 Y2H assay was conducted to identify the various amino acids having significant potential in modulating drought stress resistance factors like DREBs in rapeseed.172 Y2H and bimolecular fluorescence complementation (BiFC) were executed for identification and cloning of protein phosphatase type 2 C (PP2C) and basic leucine zipper (bZIP) from rapeseed. It showed the interaction between BnaPP2Cs and BnaCPKs and unveiled the novel interaction among BnaCPK.26 Y2H system was used to study the interaction among various CBLs and CIPKs proteins under several rapeseed abiotic stresses. Results show that 23 CIPK and 7 CBL genes were detected from rapeseed database research and cloning of 17 CIPKs and 6 CBLs cDNA sequences. Green fluorescence protein (GFP) was used to determine the subcellular detection of 2 BnaCIPKs and 3 BnaCBLs genes in rapeseed. Y2H assay was performed for protein interactions among 17 BnaCIPKs and 6 BnaCIPKs. Besides, the expression level of selected 12 BnaCIPKs and 6 BnaCBLs genes were analyzed under ABA, heat, cold, drought, and salinity stress via qRT-PCR. The findings revealed that CIPK and CBL families established a complicated signaling framework under several rapeseed abiotic factors.27
Another experiment was conducted to elucidate the function of MPK and MKK protein families in multiple stresses. The amino acid sequence was predicted for both MPK and MKK through sequencing and phylogenetic analysis. To unveil the cellular localization reporter gene, GFP was used. For protein–protein interactions among MPK and MKK families, the Y2H assay was run, and the results were further assessed through BiFC assay. Moreover, the expression of selected MPKs and MKKs under multiple stresses was evaluated by using RT-PCR. The result showed that 12 MPK and 7 MKK genes had been identified, which can be employed as a marker to generate stress-tolerance rapeseed.173 Genetic studies documented that ds-3 is responsible for dwarfism in rapeseed. The proteomic evaluation was performed for rapeseed to elucidate the role of genes, BnaC07.RGA, BnaA06.RGA, BnaC09.RGA and BnaA09.RGA. qRT-PCR and Y2H strategies were used for BnaC09.RGA and BnaA09.RGA gene expression and identified that both these genes have a considerable impact on the production of semi-dwarf rapeseed.174
2.4. Metabolomics: Are Metabolites Associated with the Closest Link to Phenotype?
To understand plant biochemistry at the organism and cellular level, metabolomics arising as a new era strategy to examine the whole metabolome of any crop plant. Due to its wide applications since the 1990s, metabolomics has been magnificently applied to plants to detect novel metabolites, genes and their metabolic pathways.175 Using metabolomics, GM crops can be evaluated in terms of their improved agronomic characters. Metabolomics consists of different signaling pathways, the interaction between proteins, the involvement of plant primary and secondary metabolites, and the epigenetic regulation process. Primary and secondary metabolites of plants are crucial to regulating biological and biochemical mechanisms.175–177
Metabolome analysis of high expression of the phospholipase C2 gene was studied in GE rapeseed and revealed that it has resistance against low temperature at a metabolomic level. GE plants show a remarkable increase in maltose and considerable enhancement in some free fatty acids, glycerol 3-phosphate and glycerol, stachyose, raffinose, and some flavonoids.179 For metabolome profiling gas chromatography-mass spectrometry (GC-MS) approach has been used for various rapeseed cultivars. Out of 162 compounds, metabolic profiling of 52 compounds has been successfully achieved. Different multivariate tools were applied, which showed remarkable variance among different rapeseed varieties.176 With the emergence of omics approaches, various researches have concentrated on elucidating the expression and function of several genes, proteins, and metabolites in rapeseed. Because of its commercial importance, oil-producing rapeseed has been subjected to MS tools to quantify and characterize rapeseed metabolome. Therefore, profiling of rapeseed metabolome aids in moving more deepen in plant biology and physiology to exploit an abiotic factor regulation.179 Recently,140 identified 41 differentially accumulated metabolites in the spring and 47 in the winter rapeseed ecotypes under cold stress. Notably, 81 metabolites primarily went to primary metabolites, including amino acids, organic acids, and sugars. They are suggesting that carbohydrates and amino acid compounds play a vital role in improving cold tolerance. Metabolomics is yet to be reported for several abiotic stresses in rapeseed.
2.4.1. Metabolic QTL for Abiotic Stress Tolerance
For phenotypical and morphological determination, various biomarkers have been developed. Thus, metabolic QTL (mQTL) and metabolic GWAS (mGWAS) approaches have been used to predict phenotype and establishment of metabotypes.180 For gene expression, protein and metabolome profiling are coupled with QTL mapping, as shown in Fig. 4. Genomic and metabolic markers are frequently correlated in mQTL mapping. In mQTL, a major difficulty arises in the detection of desired genes and metabolites. Therefore, a precise, measurable mapping of metabotypes is permitted to detect candidate gene.176,179 Metabolic levels of fatty acids, G3P, glycerol, and maltose significantly rose in GE plants in cold stresses by elevated concentrations of flavonoids, raffinose, and other sugars.181 Nuclear magnetic resonance (NMR)-based seed metabolites identification was carried out between two species, rapeseed and turnip. It was concluded that unsaturated fatty acids, sucrose, and sinapine are the most perspicacious metabolites influencing by changing the environment.182 The LC-TOFMS-based metabolomic and allopathic analysis was performed to understand the chemistry of rapeseed allelochemicals and their ability to hinder root and shoot growth.183
Figure 4.
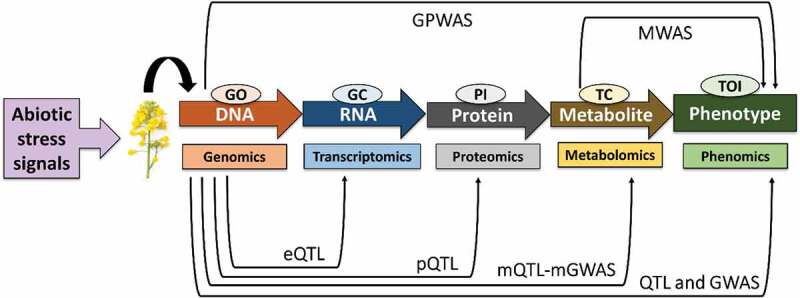
Systematic sketch of QTL mapping for gene expression together with molecular phenotype. The movement of particular knowledge is described from DNA-phenotype under abiotic stress signal. Notably, black arrows show that each molecular phenotype can be mapped onto the genome by using QTL mapping and GWAS techniques. However, MWAS does not demand particular knowledge to exploit the impacts of genetic deviant on metabolites. eQTL means epigenomic QTL, pQTL means proteomic QTL, mQTL means metabolomics QTL, mGWAS means metabolomic GWAS, MWAS means metabolome-wide association studies, GPWAS means genome-phenome wide association studies, GO means gene ontology, GC means gene co-expression, PI means protein interaction, TC means trait correlation, TOI means trait of interest. Modified from Razzaq et al. (2019).176
2.5. Phenomics: What Has Occurred?
The relationship between genotypes and phenotypes is very crucial for breeding programs. Plant phenomics is considered the phenotype of a plant or genotypic expression in a specific environmental condition.184 Additionally, a plant’s phenotype included several parameters that can be assessed through direct examination or by applying numerous analytical techniques and can also be defined by the association between environment and plant genotype.185 Phenotyping is still a massive task under abiotic stress conditions due to complex biosynthesis processes that govern several abiotic stress tolerance in plants.185 The significance of phenotyping has become apparent in the postgenomic era because the approaches used for crop improvement such as GWAS, GS, MAS, QTL mapping heavily depend upon the high-throughput phenotyping (HTP) in crops.186 Nonetheless, HTP technologies have been getting tremendous advancement. Data mining, interpretation, and storage strategies are automated, precise, accurate, and economical. Genetic dissection of various characteristics and detailed examination of plant structure and function permits studying the plant phenotypic expression.188
Recently, advanced HTP tools have been designed with multifunctional software coordination, consisting of X-ray tomography, hyperspectral imaging, and visible light imaging. This HTP allows the imaging of thousands of plants automatically at the same time. Many phenotyping centers have been set up in many countries (Table 5) and, notably, some unique QTLs have been documented in several plants.187 Recently, a few studies have been conducted using HTP in rapeseed. For instance, to speed-up the dissection of the dynamic genetic architecture of rapeseed growth and yield under different developmental stages.189 The phenotyping and genome-wide association mapping for the improvement of root architectural traits.190 Moreover, it has been summarized that the integration of holistic phenotyping tools can elucidate the functional gene polymorphism and explain the complex mechanisms responsible for abiotic stress tolerance in crop plants. Notably, in rapeseed, the utilization of different HTP tools under environmental stresses is still lacking.
Table 5.
Globally available phenomics and phenotyping center to study the genotypic expression of plants under specific environmental conditions
| Center name | URL | Country |
|---|---|---|
| PHENOPSIS- INRA | http://bioweb.supagro.inra.fr/phenopsis/ | France |
| German Plant Phenotyping Network (DPPN) | https://www.ipk-gatersleben.de/en/phenotyping/ | Germany |
| High-Resolution Plant Phenomics Center (HRPPC) | http://www.plantphenomics.org.au/HRPPC | Australia |
| National Plant Phenomics Center (NPPC) | https://www.plant-phenomics.ac.uk/ | UK |
| European Plant Phenotyping Infrastructure (EMPHASIS) | https://emphasis.plant-phenotyping.eu/ | Europe |
| Nordic Plant Phenotyping Network (NPPN) | https://nordicphenotyping.org/ | Denmark |
| European Plant Phenotyping Network (EPPN) | https://eppn2020.plant-phenotyping.eu/EPPN2020 | Europe |
| Phenome UK | https://www.phenomuk.net/ | UK |
| PHEN-ITALY | http://www.phen-italy.it/index.php | Italy |
3. An Overview of Modern Technologies for Developing Climate-resilient Rapeseed Plants
3.1. Transgenic Approaches
In the past few decades, transgenic methods have been widely used for developing abiotic stress tolerance plants. Genetic engineering for the development of stress resistance plants relies on the manipulation of genes responsible for stress regulation, which might be a way forward for enhancing plant health under stressful conditions. Assessment of the transgenic plants under stressful environments and regulation of physiological, biochemical, and cellular effects of the manipulated genes at the whole plant extent is still considered a persisting challenge to overwhelmed in rapeseed. So far, several transgenic rapeseed lines have been reported with improved abiotic stress tolerance (Table 6). For instance, transgenic rapeseed lines overexpressing Arabidopsis CBF1/2/3 genes exhibited improved freezing tolerance due to the introduction of CBF-targeted orthologous rapeseed Bn115 gene.200 Likewise, homologous overexpression of BnCBF5 and BnCBF17 enhanced freezing tolerance in transgenic lines.153 Whereas transgenic lines showed improved performance with BnCBF17 as compared to BnCBF5, possibly due to higher expression of cor genes under cold stress.153 The transgenic lines expressing the AtPLD-α-1 gene from Arabidopsis thaliana show it improves tolerance to drought and salinity stresses. It also reduced the H2O loss, improve biomass, and yield production under stress conditions.192 The hyperactive expression of the Arabidopsis AtDWF4 gene elevated seed productivity and tolerance to heat, drought, dehydration stresses and enhances the seed yield in transgenic lines.195 The cloning of tobacco serine acetyltransferase (SAT; a rate-limiting enzyme for cysteine biosynthesis)-encoding gene (NtSAT4) generated high levels of glutathione and cysteine in transgenic rapeseed lines for improved heavy metals tolerance.197 In another study, the Arabidopsis thaliana AtTrx-h2 gene improves the salinity tolerance by modulating antioxidant defense systems in transgenic lines.198 Several other examples are presented in Table 6.
Table 6.
Stories of successfully developed transgenic rapeseed and transferred genes related to different abiotic stresses
| Source plant | Gene | Coding protein or action mechanisms | Stress resistance | References |
|---|---|---|---|---|
| Oryza sativa | OsNASI | POD, dehydrogenase, GST, PSMB5 and RuBPco | Salinity | 191 |
| Arabidopsis thaliana | AtPLD-α-1 | Phospholipase D-α-1 | Reduced H2O loss, enhance biomass amassing and yield beneath drought and salinity conditions | 191 |
| N/A | Cyp-11A1 | Bovine side-chain cleavage cytochrome P450scc | High temperature | 194 |
| Transgenic Brassica napus | YHem1 | Promote the metabolism of endogenetic 5-ALA | Salinity | 193 |
| Transgenic Brassica napus | BnPLC2 | Phosphatidylinositol-specific phospholipase C2 (PLC2] | Low temperature | 178 |
| Transgenic Brassica napus | DREB | DREB factor and improves the expression of several stress-related transcripts | Salinity and drought | 196 |
| Arabidopsis thaliana | AtDWF4 | Brassinosteroid insensitive1 (BRI1)-EMS suppressor1 (BES1) and Brassinazole-resistant1 (BZR1) | Heat, drought, dehydration, enhance seed yield and also involved in biotic stress | 195 |
| Arabidopsis thaliana | AtDFR | dihydroflavonol-4-reductase | Improve salinity and drought tolerance by the accumulation of anthocyanins | 197 |
| Nicotiana tabacum | NtSAT4 | Serine acetyl transferase (SAT) | Heavy metals | 197 |
| Arabidopsis thaliana | AtTrx-h2 | h-type thioredoxins | Improves salt-tolerance and antioxidant systems | 199 |
| Nicotiana tabacum | NtHSP17.6 | Heat shock proteins | Heat, drought, and salinity | 201 |
| Brassica napus | BnKCS1-1, BnKCS1-2, and BnCER1-2 | – | Improve drought tolerance and cuticular wax | 38 |
3.2. CRISPR/Cas System: The Promising Genome Editing Tool
Genome editing is an efficient tool for crop improvement either by the loss-of-function, the gain-of-function, or a multiplex genome editing methods. Thus, clustered regularly interspaced short palindromic repeats/-associated proteins (CRISPR/Cas9) have come out as a robust GE strategy and are considered a fast, modest, and multipurpose procedure for GE and development of a transgene-free end-product.48,202 The step-wise presentation of CRISPR/Cas9 based GE for the evolution of different stress-tolerance varieties of rapeseed is described in Fig. 5. Nevertheless, CRISPR/Cas9 mediated GE’s mutation efficiency was observed and examined the heritability and pattern of gene manipulations in rapeseed. Interestingly, no off-target mutation was produced in mutated lines, and this investigation has opened new horizons for biotechnological applications in rapeseed for the development of stress-tolerance cultivars.48 The difficulty of the rapeseed genome and gene redundancy is the main limiting factor for simultaneous mutagenesis of several homologs in the first progeny. Furthermore, the widespread identification of mutant plants is time-consuming through conventional approaches.203 Recently, an expression cassette for sgRNA targeted five homologous genes, such as BnSPL3-Cnn, BnSPL3-C4, BnSPL3-C3, BnSPL3-A4, and BnSPL3-A5 have been developed. High-throughput sequencing investigation disclosed a very high knock out the efficiency of about 96.8% to 100% by CRISPR/Cas9.203
Figure 5.
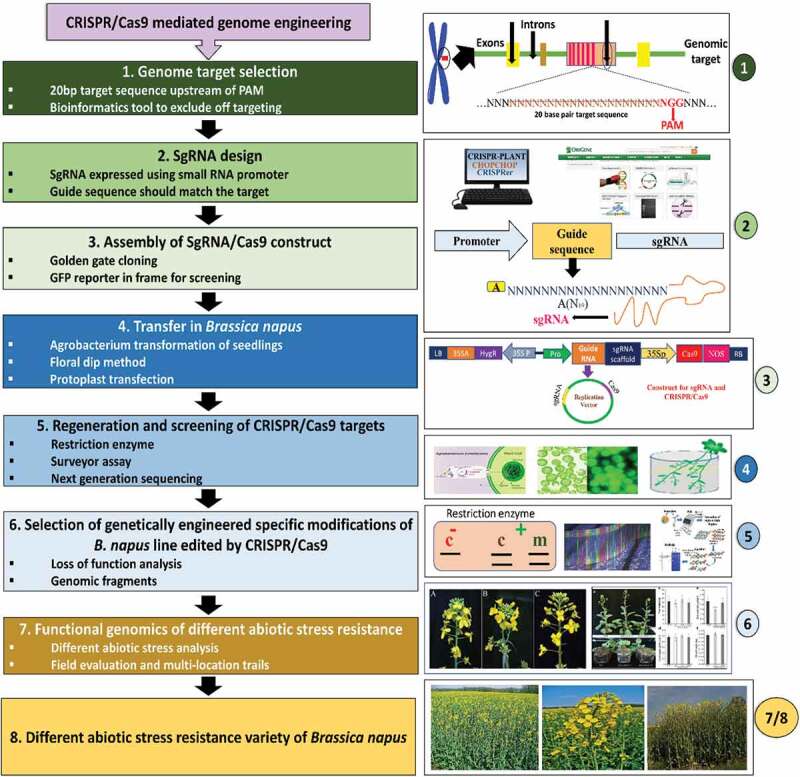
Step-by-step presentation of CRISPR/Cas9 mediated genome editing for the development of different abiotic stress resistance varieties of rapeseed. Each step (1–8) shows some basic sub-steps within the box (left side). In contrast, the right side indicates the practically on-table illustrations of the steps involved in the CRISPR/Cas9 mediated genome editing in rapeseed
CRISPR/Cas9 based GE has excellent power in breeding programs to develop improved varieties for agronomic traits in many crops. However, the early exploitation of this tool in enlightening agronomic traits of rapeseed, e.g., the FAD2 gene, which controls the enzyme that catalyzes oleic acid desaturation in rapeseed,204 the BnLLA10 gene which controls the lobed-leaf shape and regulates salt tolerance in rapeseed.205 Moreover, the knockout of two BnaMAX1 homologs advances plant architecture and upsurges yield,206 precise editing of CLAVATA genes normalizes silique development,207 pod shatter resistance by the multiplex editing of INDEHISCENT homologues,208 and JAGGED genes,209 flowering time and plant architecture by knockout of BnaTFL1 gene.210 In a recent study, the gain-of-function mutant (bnaa6.rga-D) showed boosted drought tolerance, and its stomata were oversensitive to ABA signaling, whereas bnarga (loss-of-function mutant) owned compact drought tolerance and less sensitivity to ABA signaling.211, 212 Notably, in rapeseed, the CRISPR/Cas9 system’s utilization for the development of abiotic stress-tolerant rapeseed is still lacking and yet to be documented under several stress conditions.
4. Persisting Bottlenecks in the Production of Climate-resilient Rapeseed
Although marvelous advancement has been achieved in the biotechnological era, many questions and bottlenecks presently restrict the implementation of omics approaches for abiotic stress tolerance studies in rapeseed. Therefore, few bottlenecks demand to be abundantly handled to decipher the potential of omics tools for rapeseed (Fig. 6). Based on the available literature, there are very few abiotic stress tolerance rapeseed genotypes developed via omics tools. However, after the rapeseed genome sequence, researchers pay attention to identify candidate genes and regulators to improve abiotic stress tolerance in rapeseed. Still, few genes have been reported for abiotic stresses.
Figure 6.
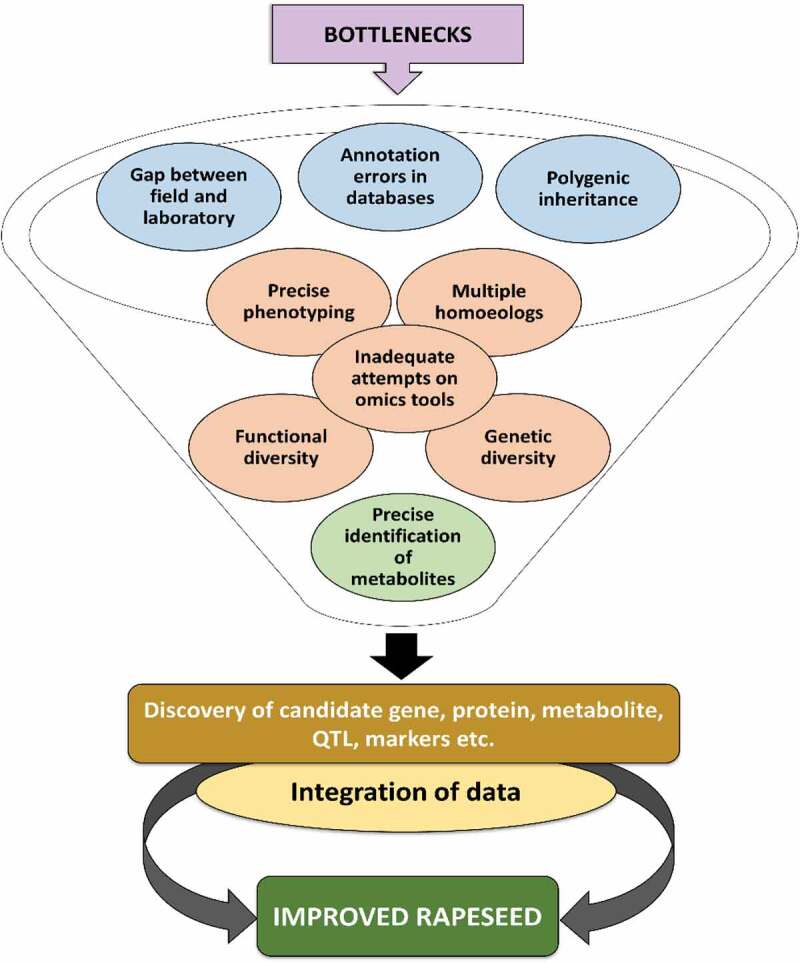
Persisting bottlenecks in the utilization of omics approaches to develop climate-resilient rapeseed. The dismissal of these bottlenecks using different molecular tools will help us to exploit the new manifesto to develop abiotic stress tolerance rapeseed; and successively will assure global food security
Moreover, polygenic inheritance in abiotic stress tolerance rapeseed is still acting as a major bottleneck in conventional breeding programs. The misplacement of genetic divergence during the domestication of rapeseed is still considered a significant bottleneck among the existing ones. There is still a noticeable gap between the field and laboratory. However, less genetic diversity directly leads to less functional diversity. The biggest question is to utilize wild diversity more quickly, which can be utilized to explore genomic parts of loci related to essential QTL for abiotic stress tolerance in rapeseed.
Even though it is viable to identify thousands of metabolites, however, the precise identification of leftovers as the utmost bottleneck in plant metabolomics investigations. Insufficient knowledge and inadequate attempts on omics tools, mainly metabolomics, proteomics, and annotation mistakes in databases, are a significant barrier for functional characterization, and still act as significant bottlenecks in genome analysis.
However, the integration of newly emerged molecular platforms for identifying complex QTLs has remained a key restriction due to restricted potential to phenotype the crops precisely. Together, precise phenotyping concealed by native habitat is still a big phenotyping-bottleneck in several diverse development stages, including translation of genotype to phenotype. Another significant bottleneck is multiple homoeologs of a single gene in the rapeseed genome; in this regard, more homoeologs need to be targeted using GE tools. Thus, the dismissal of these bottlenecks using molecular tools will help us to exploit the new manifesto to develop stress resistance rapeseed; and successively will assure global food security.
5. Concluding Remarks
Rapidly changing climate is the main reason for several environmental stresses in the current era. Among them, abiotic stresses immensely affect rapeseed growth and production globally. These stresses may cause irregularities and a decrease in crop yield parameters. They can execute physiological, morphological, and molecular variations that deleteriously disturb production, growth, and final rapeseed yield. Therefore, it is vital to explore the stress-responsive mechanisms to enhance rapeseed production and quality under harsh environmental conditions. To solve this persisting issue, omics tools have demonstrated the most outstanding biotechnological applications to develop abiotic stress-tolerance crops.
Omics, GE tools, and molecular breeding strategies have been extensively used to elucidate the complex biological mechanism and pathways regulating abiotic stress-tolerance in rapeseed. The multi-dimensional omics studies have gathered numerous datasets at the transcriptome, proteome, and metabolome level to unveil various molecular, physiological, and metabolic pathways related to abiotic stress-tolerance in rapeseed, which open ups new horizons for future investigations. However, few bottlenecks demand to be abundantly handled to decipher the potential of omics tools for rapeseed and other crops (Fig. 6). However, after the rapeseed genome sequence, researchers pay attention to identify candidate genes and regulators to improve stress tolerance in rapeseed. Still, few genes have been reported for multiple abiotic stresses. More work needs to be carried out to fully explore the beneficial role of omics in abiotic stress tolerance.
Molecular markers are the most powerful technology in plant molecular biology to determine the genetic variations in rapeseed at the polyploidy level. Additionally, with the accessibility of the rapeseed genome sequence, we can detect candidate genes related to several abiotic stresses in modern breeding programs. High-throughput sequencing technologies and EST construction for rapeseed are very promising for identifying SNPs for various abiotic stress-related QTLs. The applications of SNPs in rapeseed are very diverse due to its complex genome. Continuous improvements in NGS and genotyping techniques permit genotyping by sequencing, and SNPs will offer valuable information for rapeseed scientists. Moreover, genomic selection tools, along with HTP stages for selecting huge populations, permit more insights.
6. Future Directions
Genome editing via the CRISPR/Cas system and engineering stress-associated genes should be considered one of the most promising research directions to boost stress tolerance. Likewise, the engineering of metabolic pathways can open novel gaps for climate-resilient development, ready to grow rapeseed plants. Speed breeding has developed as a time-saving tool to expand plant growth and development under preferred conditions with enhanced stress tolerance. In the future, speed breeding will boost the genetic understanding and allow abiotic stress-tolerance rapeseed as established in other crops. Nevertheless, the CRISPR/Cas9 system can be integrated with speed breeding to modernize world rapeseed production. Shortly, the combination of omics, genome editing, and speed breeding can speed up the rapeseed production with improved traits and increased abiotic stress tolerance. On the other hand, another emerging approach, “synthetic biology” can be mainly applied to plant biology and engineering methods to develop climate-smart rapeseed plants.
Supplementary Material
Acknowledgments
We are thankful to the researchers whose contributions have been cited in this review, which have helped us prepare this review paper. Further, the author also apologizes to all researchers whose relevant work could not be cited due to space limitations.
Funding Statement
This work was supported by the Agricultural Science and Technology Innovation Program (ASTIP) and The Hubei Agricultural Science and Technology Innovation Center (2016-620-000-001-048).
Disclosure statement
The authors declare no conflict of interest.
Supplementary Material:
Supplemental data for this article can be accessed on the publisher’s website.
Author Contributions
Ali Raza (AR) conceived the idea and wrote the manuscript. Ali Razzaq (ARa) and SSM wrote some parts in the initial draft. SSM, MAH, SW, HH, and QZ helped in the literature. AR prepared the tables and figures. AR, ZX, CY, and MH proofread and edited the manuscript. ZX and CY supervised the work. All authors have read and approved the final version of the manuscript.
References
- 1.Raza A, Ashraf F, Zou X, Zhang X, Tosif H.. Plant adaptation and tolerance to environmental stresses: mechanisms and perspectives. In: Hasanuzzaman M, editor. Plant ecophysiology and adaptation under climate change: mechanisms and perspectives I. Springer, Singapore; 2020a. p. 117–45. [Google Scholar]
- 2.Raza A, Razzaq A, Mehmood SS, Zou X, Zhang X, Lv Y, Xu J. Impact of climate change on crops adaptation and strategies to tackle its outcome: A review. Plants. 2019a;8:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabagh AE, Hossain A, Barutçular C, Islam MS, Ratnasekera D, Kumar N, Meena RS, Gharib HS, Saneoka H, da Silva JAT. Drought and salinity stress management for higher and sustainable canola (‘Brassica napus’ L.) production: A critical review. Aust J Crop Sci. 2019;13:88. [Google Scholar]
- 4.Di F, Jian H, Wang T, Chen X, Ding Y, Du H, Lu K, Li J, Liu L. Genome-wide analysis of the PYL gene family and identification of PYL genes that respond to abiotic stress in Brassica napus. Genes. 2018;9(3):156. doi: 10.3390/genes9030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lv Y, Fu S, Chen S, Zhang W, Qi C. Ethylene response factor BnERF2-like (ERF2.4) from Brassica napus L. enhances submergence tolerance and alleviates oxidative damage caused by submergence in Arabidopsis thaliana. Crop J. 2016;4(3):199–211. doi: 10.1016/j.cj.2016.01.004. [DOI] [Google Scholar]
- 6.Xu M, Ma H, Zeng L, Cheng Y, Lu G, Xu J, Zhang X, Zou X. The effect of waterlogging on yield and seed quality at the early flowering stage in Brassica napus L. Field Crop Res. 2015;180:238–45. [Google Scholar]
- 7.Du C, Hu K, Xian S, Liu C, Fan J, Tu J, Fu T. Dynamic transcriptome analysis reveals AP2/ERF transcription factors responsible for cold stress in rapeseed (Brassica napus L.). Mol Genet Genomics. 2016;291(3):1053–1067. doi: 10.1007/s00438-015-1161-0. [DOI] [PubMed] [Google Scholar]
- 8.Raza A. Eco-physiological and biochemical responses of rapeseed (Brassica napus L.) to abiotic stresses: consequences and mitigation strategies. J Plant Growth Regul. 2020a. doi: 10.1007/s00344-020-10231-z. [DOI] [Google Scholar]
- 9.Nagaharu U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn J Bot. 1935;7:389–452. [Google Scholar]
- 10.Liu S, Liu Y, Yang X, Tong C, Edwards D, Parkin IA, Zhao M, Ma J, Yu J, Huang S. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun. 2014;5:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Liu D, Wang X, Ji C, Cheng F, Liu B, Hu Z, Chen S, Pental D, Ju Y. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat Genet. 2016;48:1225–32. [DOI] [PubMed] [Google Scholar]
- 12.Waminal NE, Perumal S, Lee J, Kim HH, Yang T-J. Repeat Evolution in Brassica rapa (AA), B. oleracea (CC), and B. napus (AACC) Genomes. Plant Breed Biotechnol. 2016;4:107–22. [Google Scholar]
- 13.Zhang L, Cai X, Wu J, Liu M, Grob S, Cheng F, Liang J, Cai C, Liu Z, Liu B. Improved Brassica rapa reference genome by single-molecule sequencing and chromosome conformation capture technologies. Hortic Res. 2018a;5:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song J-M, Guan Z, Hu J, Guo C, Yang Z, Wang S, Liu D, Wang B, Lu S, Zhou R. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat Plant. 2020;6:34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, Wang X, Chiquet J, Belcram H, Tong C, Samans B. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345(6199):950–53. doi: 10.1126/science.1253435. [DOI] [PubMed] [Google Scholar]
- 16.OECD . Organisation for Economic Co-operation and Development. Consensus document on the biology of the Brassica crops (Brassica spp.). Series on Harmonisation of Regulatory oversight of Biotechnology, No 54, OECD, Paris, 2012. p. 142. [Google Scholar]
- 17.USDA-ARS . Germplasm resources information network (GRIN) taxonomy for plants. Taxon: Brassica napus L. United States Department of Agriculture. Agricultural Research Service, Beltsville Area, MD, USA. 2017. https://www.ars-grin.gov/cgi-bin/npgs/html/taxon.pl?7661. [Google Scholar]
- 18.Matthaus B, Özcan MM, Al Juhaimi F. Some rape/canola seed oils: fatty acid composition and tocopherols. Zeitschrift für Naturforschung C. 2016;71(3–4):73–77. doi: 10.1515/znc-2016-0003. [DOI] [PubMed] [Google Scholar]
- 19.Luo J, Tang S, Mei F, Peng X, Li J, Li X, Yan X, Zeng X, Liu F, Wu Y. BnSIP1-1, a trihelix family gene, mediates abiotic stress tolerance and ABA signaling in Brassica napus. Front Plant Sci. 2017;8:44. doi: 10.3389/fpls.2017.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo J, Tang S, Peng X, Yan X, Zeng X, Li J, Li X, Wu G, Yang D. Elucidation of cross-talk and specificity of early response mechanisms to salt and PEG-simulated drought stresses in Brassica napus using comparative proteomic analysis. PloS One. 2015;10(10):e0138974. doi: 10.1371/journal.pone.0138974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang T, Chang Y, Wang J, Wang N, Wang Y, Chen Q, Sun W. Cloning and expression analysis of a BnICE1 from Brassica napus L. Sci Agric Sin. 2013;1:205–14. [Google Scholar]
- 22.Zhou M, Shen C, Wu L, Tang K, Lin J. CBF-dependent signaling pathway: a key responder to low temperature stress in plants. Crit Rev Biotechnol. 2011;31:186–92. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Zhong H, Ren F, Guo -Q-Q, Hu X-P, Li X-B. A novel cold-regulated gene, COR25, of Brassica napus is involved in plant response and tolerance to cold stress. Plant Cell Rep. 2011;30(4):463–71. doi: 10.1007/s00299-010-0952-3. [DOI] [PubMed] [Google Scholar]
- 24.Chakraborty K, Sairam RK, Bhattacharya R. Differential expression of salt overly sensitive pathway genes determines salinity stress tolerance in Brassica genotypes. Plant Physiol Biochem. 2012;51:90–101. doi: 10.1016/j.plaphy.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Chen L, Ren F, Zhou L, Wang -Q-Q, Zhong H, Li X-B. The Brassica napus calcineurin B-Like 1/CBL-interacting protein kinase 6 (CBL1/CIPK6) component is involved in the plant response to abiotic stress and ABA signalling. J Exp Bot. 2012;63(17):6211–6222. doi: 10.1093/jxb/ers273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H, Liu W-Z, Zhang Y, Deng M, Niu F, Yang B, Wang X, Wang B, Liang W, Deyholos MK. Identification, expression and interaction analyses of calcium-dependent protein kinase (CPK) genes in canola (Brassica napus L.). BMC Genomics. 2014a;15:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Yang B, Liu W-Z, Li H, Wang L, Wang B, Deng M, Liang W, Deyholos MK, Jiang Y-Q. Identification and characterization of CBL and CIPK gene families in canola (Brassica napus L.). BMC Plant Biol. 2014b;14:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ying L, Chen H, Cai W. BnNAC485 is involved in abiotic stress responses and flowering time in Brassica napus. Plant Physiol Biochem. 2014;79:77–87. [DOI] [PubMed] [Google Scholar]
- 29.Zhao B-Y, Hu Y-F, Li J-J, Yao X. BnaABF2, a bZIP transcription factor from rapeseed (Brassica napus L.), enhances drought and salt tolerance in transgenic Arabidopsis. Bot Stud. 2016;57:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen B, Niu F, Liu W-Z, Yang B, Zhang J, Ma J, Cheng H, Han F, Jiang Y-Q. Identification, cloning and characterization of R2R3-MYB gene family in canola (Brassica napus L.) identify a novel member modulating ROS accumulation and hypersensitive-like cell death. DNA Res. 2016;23(2):101–14. doi: 10.1093/dnares/dsv040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hajiebrahimi A, Owji H, Hemmati S, Cloutier S. Genome-wide identification, functional prediction, and evolutionary analysis of the R2R3-MYB superfamily in Brassica napus. Genome. 2017;60(10):797–814. doi: 10.1139/gen-2017-0059. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y, Xu D, Jia L, Huang X, Ma G, Wang S, Zhu M, Zhang A, Guan M, Lu K. Genome-wide identification and structural analysis of bZIP transcription factor genes in Brassica napus. Genes. 2017;8:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang -M-M, Liu -M-M, Ran F, Guo P-C, Ke Y-Z, Wu Y-W, Wen J, Li P-F, Li J-N, Du H. Global analysis of WOX transcription factor gene family in Brassica napus reveals their stress-and hormone-responsive patterns. Int J Mol Sci. 2018a;19:3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu S, Fadlalla T, Tang S, Li L, Ali U, Li Q, Guo L. Genome-wide analysis of Phospholipase D gene family and profiling of phospholipids under abiotic stresses in Brassica napus. Plant Cell Physiol. 2019;60(7):1556–66. doi: 10.1093/pcp/pcz071. [DOI] [PubMed] [Google Scholar]
- 35.Huang R, Liu Z, Xing M, Yang Y, Wu X, Liu H, Liang W. Heat stress suppresses Brassica napus seed oil accumulation by inhibition of photosynthesis and BnWRI1 pathway. Plant Cell Physiol. 2019;60(7):1457–70. doi: 10.1093/pcp/pcz052. [DOI] [PubMed] [Google Scholar]
- 36.Xu L, Zeng W, Li J, Liu H, Yan G, Si P, Yang C, Shi Y, He Q, Zhou W. Characteristics of membrane-bound fatty acid desaturase (FAD) genes in Brassica napus L. and their expressions under different cadmium and salinity stresses. Environ Exp Bot. 2019;162:144–56. [Google Scholar]
- 37.Chai L, Li H, Yang L, Zheng B, Cui C, Jiang J, Zhang J, Jiang L. Rapid identification of enhanced drought and salt tolerances in Arabidopsis conferred by BnBADH1 gene. Int J Agric Biol. 2019;22:633–38. [Google Scholar]
- 38.Wang Y, Jin S, Xu Y, Li S, Zhang S, Yuan Z, Li J, Ni Y. Overexpression of BnKCS1-1, BnKCS1-2, and BnCER1-2 promotes cuticular wax production and increases drought tolerance in Brassica napus. Crop J. 2020a;8:26–37. [Google Scholar]
- 39.Cui J-Q, Hua Y-P, Zhou T, Liu Y, Huang J-Y, Yue C-P. Global landscapes of the Na+/H+ antiporter (NHX) family members uncover their potential roles in regulating the rapeseed resistance to salt stress. Int J Mol Sci. 2020;21(10):3429. doi: 10.3390/ijms21103429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong J, Walk TC, Han P, Chen L, Shen X, Li Y, Gu C, Xie L, Hu X, Liao X, et al. Genome-wide identification and analysis of high-affinity nitrate transporter 2 (NRT2) family genes in rapeseed (Brassica napus L.) and their responses to various stresses. BMC Plant Biol. 2020;20:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu L, Ding Q, Liu J, Yang C, Chen H, Zhang S, Zhu J, Wang D. Brassica napus COL transcription factor BnCOL2 negatively affects the tolerance of transgenic Arabidopsis to drought stress. Environ Exp Bot. 2020;178:104171. doi: 10.1016/j.envexpbot.2020.104171. [DOI] [Google Scholar]
- 42.Wang J, Jin Z, Zhou M, Yu Y, Liang M. Characterization of NF-Y transcription factor families in industrial rapeseed (Brassica napus L.) and identification of BnNF-YA3, which functions in the abiotic stress response. Indus Crops Prod 1. 2020b;48:112253. [Google Scholar]
- 43.Xu P, Cai W. Functional characterization of the BnNCED3 gene in Brassica napus. Plant Sci. 2017;256:16–24. [DOI] [PubMed] [Google Scholar]
- 44.Gao M-J, Allard G, Byass L, Flanagan AM, Singh J. Regulation and characterization of four CBF transcription factors from Brassica napus. Plant Mol Biol. 2002;49(5):459–71. doi: 10.1023/A:1015570308704. [DOI] [PubMed] [Google Scholar]
- 45.Bancroft I, Morgan C, Fraser F, Higgins J, Wells R, Clissold L, Baker D, Long Y, Meng J, Wang X. Dissecting the genome of the polyploid crop oilseed rape by transcriptome sequencing. Nat Biotechnol. 2011;29(8):762. doi: 10.1038/nbt.1926. [DOI] [PubMed] [Google Scholar]
- 46.Muthamilarasan M, Singh NK, Prasad M. Multi-omics approaches for strategic improvement of stress tolerance in underutilized crop species: A climate change perspective. In: Dhavendra Kumar, editor. Advances in genetics. Vol. 103. Elsevier, USA; 2019. p. 1–38. [DOI] [PubMed] [Google Scholar]
- 47.Snowdon RJ, Iniguez Luy FL. Potential to improve oilseed rape and canola breeding in the genomics era. Plant Breed. 2012;131:351–60. [Google Scholar]
- 48.Yang H, Wu -J-J, Tang T, Liu K-D DC. CRISPR/Cas9-mediated genome editing efficiently creates specific mutations at multiple loci using one sgRNA in Brassica napus. Sci Rep. 2017;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brozynska M, Furtado A, Henry RJ. Genomics of crop wild relatives: expanding the gene pool for crop improvement. Plant Biotechnol J. 2016;14(4):1070–85. doi: 10.1111/pbi.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Werner C, Snowdon R. Genome-facilitated breeding of oilseed rape. In: Liu S, Snowdon R, Chalhoub B, editors. The Brassica napus genome. Springer, Cham; 2018. p. 245–69. [Google Scholar]
- 51.Wan H, Chen L, Guo J, Li Q, Wen J, Yi B, Ma C, Tu J, Fu T, Shen J. Genome-wide association study reveals the genetic architecture underlying salt tolerance-related traits in rapeseed (Brassica napus L.). Front Plant Sci. 2017;8:593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heng S, Chen F, Wei C, Hu K, Yang Z, Wen J, Yi B, Ma C, Tu J, Si P. Identification of different cytoplasms based on newly developed mitotype-specific markers for marker-assisted selection breeding in Brassica napus L. Plant Cell Rep. 2017;36(6):901–09. doi: 10.1007/s00299-017-2121-4. [DOI] [PubMed] [Google Scholar]
- 53.Miladinović D, Miler M, Marjanović-Jeromela A, Imerovski I, Dimitrijevic A, Kovacevic B, Jocic S, Cvejic S, Hladni N, Obreht-Vidakovic D. Evaluation of RAPD markers as a marker-assisted selection tool for variety type and erucic acid content in rapeseed. ABI Genetika. 2018;50(2):421–30. doi: 10.2298/GENSR1802421M. [DOI] [Google Scholar]
- 54.Fu S, Yin L, Xu M, Li Y, Wang M, Yang J, Fu T, Wang J, Shen J, Ali A. Maternal doubled haploid production in interploidy hybridization between Brassica napus and Brassica allooctaploids. Planta. 2018;247(1):113–25. doi: 10.1007/s00425-017-2772-y. [DOI] [PubMed] [Google Scholar]
- 55.Basunanda P, Radoev M, Ecke W, Friedt W, Becker H, Snowdon R. Comparative mapping of quantitative trait loci involved in heterosis for seedling and yield traits in oilseed rape (Brassica napus L.). Theor Appl Genet. 2010;120(2):271. doi: 10.1007/s00122-009-1133-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ecke W. Construction of a high fidelity genetic linkage map using AFLP and SSR markers in rapeseed (Brassica napus L.). SABRAO J Breed Genet. 2016;48:189-199. [Google Scholar]
- 57.Havlíčková L, Jozova E, Rychla A, Klima M, Kučera V, Čurn V. Genetic diversity assessment in winter oilseed rape (Brassica napus L.) collection using AFLP, ISSR and SSR markers. Czech J Genet Plant Breed. 2014;50(3):216–25. doi: 10.17221/220/2013-CJGPB. [DOI] [Google Scholar]
- 58.Moghaieb RE, Mohammed EH, Youssief SS. Genetic diversity among some canola cultivars as revealed by RAPD, SSR and AFLP analyses. 3 Biotech. 2014;4(4):403–10. doi: 10.1007/s13205-013-0165-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raza A, Mehmood SS, Ashraf F, Khan RSA. Genetic diversity analysis of Brassica species using PCR-based SSR markers. Gesunde Pflanz. 2019b;71:1–7. [Google Scholar]
- 60.Williams JG, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucl Acids Res. 1990;18:6531–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Somers DJ, Rakow G, Prabhu VK, Friesen KR. Identification of a major gene and RAPD markers for yellow seed coat colour in Brassica napus. Genome. 2001;44:1077–82. [PubMed] [Google Scholar]
- 62.Zhi-Wen L, Ting-Dong F, Jin-Xing T, Bao-Yuan C. Inheritance of seed colour and identification of RAPD and AFLP markers linked to the seed colour gene in rapeseed (Brassica napus L.). Theor Appl Genet. 2005;110:303–10. [DOI] [PubMed] [Google Scholar]
- 63.Asghari A, Mohammadi S, Moghaddam M, Mohammaddoost H. Identification of QTLS controlling winter survival in Brassica napus using rapd markers. Biotechnol Biotechnol Equip. 2007;21(4):413–16. doi: 10.1080/13102818.2007.10817485. [DOI] [Google Scholar]
- 64.Asghari A, Mohammadi S, Moghaddam M, Toorchi M, Mohammadinasab AD. Analysis of quantitative trait loci associated with freezing tolerance in rapeseed (Brassica napus L.). Biotechnol Biotechnol Equip. 2008;22(1):548–52. doi: 10.1080/13102818.2008.10817509. [DOI] [Google Scholar]
- 65.Özbek Ö, Gidik BU. Genetic diversity in commercial rapeseed (Brassica napus L.) varieties from Turkey as revealed by RAPD. Not Sci Biol. 2013;5(1):114–19. doi: 10.15835/nsb518911. [DOI] [Google Scholar]
- 66.Hedayati MH, Samiezadeh LHA. Genetic diversity assessment of lines and varieties in winter rapeseed (Brassica napus L.) using RAPD and SSR molecular markers. J Crop Breed. 2016;8:31–39. [Google Scholar]
- 67.Raza A, Farooq ABU, Khan WA, Iqbal A, Çelik S, Ali M, Khan RSA. Polymorphic information and genetic diversity in Brassica species revealed by RAPD markers. BIOCELL. 2020b;44:769–776. [Google Scholar]
- 68.Safari S, Mehrabi -A-A, Safari Z. Efficiency of RAPD and ISSR markers in assessment of genetic diversity in Brassica napus genotypes. Int J Agric Crop Sci. 2013;5:273–79. [Google Scholar]
- 69.Vos P, Hogers R, Bleeker M, Reijans M, Lee T, Hornes M, Friters A, Pot J, Paleman J, Kuiper M. AFLP: a new technique for DNA fingerprinting. Nucl Acids Res. 1995;23:4407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ke L, Sun Y, Hong D, Liu P, Yang G. Identification of AFLP markers linked to one recessive genic male sterility gene in oilseed rape, Brassica napus. Plant Breed. 2005;124(4):367–370. doi: 10.1111/j.1439-0523.2005.01115.x. [DOI] [Google Scholar]
- 71.Zhang D, Hua Y, Wang X, Zhao H, Shi L, Xu F. A high-density genetic map identifies a novel major QTL for boron efficiency in oilseed rape (Brassica napus L.). PloS One. 2014c;9:e112089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fan Y, Shabala S, Ma Y, Xu R, Zhou M. Using QTL mapping to investigate the relationships between abiotic stress tolerance (drought and salinity) and agronomic and physiological traits. BMC Genomics. 2015;16(1):1–11. doi: 10.1186/s12864-015-1243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seyis F, Snowdon R, Luhs W, Friedt W. Molecular characterization of novel resynthesized rapeseed (Brassica napus) lines and analysis of their genetic diversity in comparison with spring rapeseed cultivars. Plant Breed. 2003b;122:473–78. [Google Scholar]
- 74.Seyis F, Friedt W, Lühs W. Resynthesised Brassica napus as a genetic resource in rapeseed improvement for quality and agronomic performance. Schriften zu Genetischen Ressourcen. 2003a;19:334–38. [Google Scholar]
- 75.Shen J-X, Fu T-D, Yang G-S. Heterosis of double low self-incompatibility in oilseed rape (Brassica napus L.). Agric Sci China. 2002;1:732–37. [Google Scholar]
- 76.Shen J-X, Fu T-D, Yang G-S, Tu J-X, Ma C-Z. Prediction of heterosis using QTLs for yield traits in rapeseed (Brassica napus L.). Euphytica. 2006;151:165–71. [Google Scholar]
- 77.Guanghuan Y, Jibin N, Suping G, Yan Y, Ba D. Research progress of molecular markers in genetic diversity of rapeseed. Asian Agric Res. 2019;11:54–58. [Google Scholar]
- 78.Tautz D. Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucl Acids Res. 1989;17:6463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lombard V, Delourme R. A consensus linkage map for rapeseed (Brassica napus L.): construction and integration of three individual maps from DH populations. Theor Appl Genet. 2001;103(4):491–507. doi: 10.1007/s001220100560. [DOI] [Google Scholar]
- 80.Lydiate D, Sharpe A. Aligning genetic maps of Brassica napus using microsatellite markers. In: Plant & animal genomes XI conference, San Diego, USA; 2003. [Google Scholar]
- 81.Rezaeizad A, Wittkop B, Snowdon R, Hasan M, Mohammadi V, Zali A, Friedt W. Identification of QTLs for phenolic compounds in oilseed rape (Brassica napus L.) by association mapping using SSR markers. Euphytica. 2011;177:335–42. [Google Scholar]
- 82.Qu C, Hasan M, Lu K, Liu L, Zhang K, Fu F, Wang M, Liu S, Bu H, Wang R. Identification of QTL for seed coat colour and oil content in Brassica napus by association mapping using SSR markers. Canadian J Plant Sci. 2015;95(2):387–95. doi: 10.4141/cjps2013-411. [DOI] [Google Scholar]
- 83.Zali H, Sofalian O, Zeinalabedini M, Alizadeh B. Assessment of variability and genetic structure of canola cultivars and lines using SSR markers related on drought tolerance QTLs. J Crop Breed. 2018a;10:65–75. [Google Scholar]
- 84.Zali H, Sofalian O, Zeinalabedini M, Hasanloo T, Asghari A, Alizadeh B. Assessment of haplotype and allelic diversity of SSR markers in canola. MGj. 2018b;13:63–77. [Google Scholar]
- 85.Klyachenko O, Prysiazhniuk L, Shofolova N, Piskova O. Polymorphism in spring and winter rapeseed varieties (Brassica napus L.) identified by ssr markers. Plant Varieties Stud Prot. 2018;14:366–374. [Google Scholar]
- 86.Qamar H, Shabbir G, Ilyas M, Arshad A, Imran S, Malik T, Mustafa Hsb. Studies on genetic divergence of rapeseed genotypes using SSR markers. Pak J Bot. 2020;52:197–204. [Google Scholar]
- 87.Gupta M, Chyi Y-S, Romero-Severson J, Owen J. Amplification of DNA markers from evolutionarily diverse genomes using single primers of simple-sequence repeats. Theor Appl Genet. 1994;89(7–8):998–1006. doi: 10.1007/BF00224530. [DOI] [PubMed] [Google Scholar]
- 88.Liu B, Wendel JF. Intersimple sequence repeat (ISSR) polymorphisms as a genetic marker system in cotton. Mol Ecol Notes. 2001;1(3):205–08. doi: 10.1046/j.1471-8278.2001.00073.x. [DOI] [Google Scholar]
- 89.Tamura K, Nishioka M, Hayashi M, Zhang Z, Lian C, Hougetsu T, Harada K. Development of microsatellite markers by ISSR-suppression-PCR method in Brassica rapa. Breed Sci. 2005;55:247–52. [Google Scholar]
- 90.Nemati M, Asghari A, Sofalian O, Rasoulzadeh A, Mohamaddoust Chamanabad H. Effect of water stress on rapeseed cultivars using morpho-physiological traits and their relations with ISSR markers. J Plant Physiol Breed. 2012;2:55–66. [Google Scholar]
- 91.Motallebinia S, Sofalian O, Asghari A, Rasoulzadeh A, Fathi B. Study of drought tolerance indices and their relationship with ISSR markers in some canola (Brassica napus L.) Cultivars. J Plant Genet Res. 2019;6(1):99–114. doi: 10.29252/pgr.6.1.99. [DOI] [Google Scholar]
- 92.AbdElsalam AE, Attaya AS, Mekki BE, ElSarag EI. Assessment of genetic diversity of some canola genotypes. Sinai J Appl Sci. 2017;6(3):241–48. doi: 10.21608/sinjas.2017.78829. [DOI] [Google Scholar]
- 93.Paul M, Islam T, Hoque M, Sarker R. Analysis of genetic diversity in oilseed Brassica germplasm through ISSR markers and isozyme profiling. Bangladesh J Bot. 2020;49(1):147–58. doi: 10.3329/bjb.v49i1.49123. [DOI] [Google Scholar]
- 94.Basha S, Sujatha M. Inter and intra-population variability of Jatropha curcas (L.) characterized by RAPD and ISSR markers and development of population-specific SCAR markers. Euphytica. 2007;156(3):375–86. doi: 10.1007/s10681-007-9387-5. [DOI] [Google Scholar]
- 95.Julio E, Verrier J, De Borne FD. Development of SCAR markers linked to three disease resistances based on AFLP within Nicotiana tabacum L. Theor Appl Genet. 2006;112(2):335–46. doi: 10.1007/s00122-005-0132-y. [DOI] [PubMed] [Google Scholar]
- 96.Scheef EA, Casler MD, Jung G. Development of species‐specific SCAR markers in bentgrass. Crop Sci. 2003;43:345–49. [Google Scholar]
- 97.Bautista R, Crespillo R, Cánovas FM, Claros MG. Identification of olive-tree cultivars with SCAR markers. Euphytica. 2003;129(1):33–41. doi: 10.1023/A:1021528122049. [DOI] [Google Scholar]
- 98.Marieschi M, Torelli A, Beghé D, Bruni R. Authentication of Punica granatum L.: development of SCAR markers for the detection of 10 fruits potentially used in economically motivated adulteration. Food Chem. 2016;202:438–44. doi: 10.1016/j.foodchem.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 99.Hu J, Li G, Struss D, Quiros C. SCAR and RAPD markers associated with 18-carbon fatty acids in rapeseed, Brassica napus. Plant Breed. 1999;118(2):145–50. doi: 10.1046/j.1439-0523.1999.118002145.x. [DOI] [Google Scholar]
- 100.Hong D, Wan L, Liu P, Yang G, He Q. AFLP and SCAR markers linked to the suppressor gene (Rf) of a dominant genetic male sterility in rapeseed (Brassica napus L.). Euphytica. 2006;151(3):401–09. doi: 10.1007/s10681-006-9162-z. [DOI] [Google Scholar]
- 101.Xie Y, Hong D, Xu Z, Liu P, Yang G. Identification of AFLP markers linked to the epistatic suppressor gene of a recessive genic male sterility in rapeseed and conversion to SCAR markers. Plant Breed. 2008;127:145–49. [Google Scholar]
- 102.Hong D, Liu J, Yang G, He Q. Development and characterization of SCAR markers associated with a dominant genic male sterility in rapeseed. Plant Breed. 2008;127:69–73. [Google Scholar]
- 103.Zhang X, Ma C, Fu T, Li Y, Wang T, Chen Q, Tu J, Shen J. Development of SCAR markers linked to self-incompatibility in Brassica napus L. Molecular Breed. 2008;21:305–15. [Google Scholar]
- 104.Lee S-I, Park K-C, Ha M-W, Kim K-S, Jang Y-S, Kim N-S. CACTA transposon-derived Ti-SCARs for cultivar fingerprinting in rapeseed. Genes Genomics. 2012;34(5):575–79. doi: 10.1007/s13258-012-0190-x. [DOI] [Google Scholar]
- 105.Konieczny A, Ausubel FM. A procedure for mapping Arabidopsis mutations using co‐dominant ecotype‐specific PCR‐based markers. Plant J. 1993;4(2):403–10. doi: 10.1046/j.1365-313X.1993.04020403.x. [DOI] [PubMed] [Google Scholar]
- 106.Shavrukov Y. CAPS markers in plant biology. Russian J Genet Appl Res. 2016;6:279–87. [Google Scholar]
- 107.Xiao S, Xu J, Li Y, Zhang L, Shi S, Shi S, Wu J, Liu K. Generation and mapping of SCAR and CAPS markers linked to the seed coat color gene in Brassica napus using a genome-walking technique. Genome. 2007;50:611–18. [DOI] [PubMed] [Google Scholar]
- 108.Rashid MH, Zou Z, Fernando WD. Development of molecular markers linked to the Leptosphaeria maculans resistance gene Rlm6 and inheritance of SCAR and CAPS markers in Brassica napus× Brassica juncea interspecific hybrids. Plant Breed. 2018;137:402–11. [Google Scholar]
- 109.Spasibionek S, Mikołajczyk K, Ćwiek–Kupczyńska H, Piętka T, Krótka K, Matuszczak M, Nowakowska J, Michalski K, Bartkowiak-Broda I. Marker assisted selection of new high oleic and low linolenic winter oilseed rape (Brassica napus L.) inbred lines revealing good agricultural value. PloS One. 2020;15:e0233959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Matuszczak M, Spasibionek S, Gacek K, Bartkowiak-Broda I. Cleaved amplified polymorphic sequences (CAPS) marker for identification of two mutant alleles of the rapeseed BnaA.FAD2 gene. Mol Biol Rep. 2020;47(10):7607–21. doi: 10.1007/s11033-020-05828-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang X, Long Y, Wang N, Zou J, Ding G, Broadley MR, White PJ, Yuan P, Zhang Q, Luo Z. Breeding histories and selection criteria for oilseed rape in Europe and China identified by genome wide pedigree dissection. Sci Rep. 2017;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rahaman M, Mamidi S, Rahman M. Association mapping of agronomic traits of canola (Brassica napus L.) subject to heat stress under field conditions. Aust J Crop Sci. 2017;11(09):1094. doi: 10.21475/ajcs.17.11.09.pne512. [DOI] [Google Scholar]
- 113.Trick M, Long Y, Meng J, Bancroft I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol J. 2009;7:334–46. [DOI] [PubMed] [Google Scholar]
- 114.Bus A, Hecht J, Huettel B, Reinhardt R, Stich B. High-throughput polymorphism detection and genotyping in Brassica napus using next-generation RAD sequencing. BMC Genomics. 2012;13(1):1–11. doi: 10.1186/1471-2164-13-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Harper AL, Trick M, Higgins J, Fraser F, Clissold L, Wells R, Hattori C, Werner P, Bancroft I. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat Biotechnol. 2012;30(8):798–802. doi: 10.1038/nbt.2302. [DOI] [PubMed] [Google Scholar]
- 116.Delourme R, Falentin C, Fomeju BF, Boillot M, Lassalle G, André I, Duarte J, Gauthier V, Lucante N, Marty A. High-density SNP-based genetic map development and linkage disequilibrium assessment in Brassica napus L. BMC Genomics. 2013;14(1):120. doi: 10.1186/1471-2164-14-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dalton-Morgan J, Hayward A, Alamery S, Tollenaere R, Mason AS, Campbell E, Patel D, Lorenc MT, Yi B, Long Y. A high-throughput SNP array in the amphidiploid species Brassica napus shows diversity in resistance genes. Funct Integr Genomics. 2014;14(4):643–55. doi: 10.1007/s10142-014-0391-2. [DOI] [PubMed] [Google Scholar]
- 118.Schmutzer T, Samans B, Dyrszka E, Ulpinnis C, Weise S, Stengel D, Colmsee C, Lespinasse D, Micic Z, Abel S. Species-wide genome sequence and nucleotide polymorphisms from the model allopolyploid plant Brassica napus. Sci Data. 2015;2:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Clarke WE, Higgins EE, Plieske J, Wieseke R, Sidebottom C, Khedikar Y, Batley J, Edwards D, Meng J, Li R. A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor Appl Genet. 2016;129(10):1887–99. doi: 10.1007/s00122-016-2746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brachi B, Morris GP, Borevitz JO. Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol. 2011;12(10):1–8. doi: 10.1186/gb-2011-12-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang X, Han B. Natural variations and genome-wide association studies in crop plants. Ann Rev Plant Biol. 2014;65(1):531–51. doi: 10.1146/annurev-arplant-050213-035715. [DOI] [PubMed] [Google Scholar]
- 122.Jian H, Xiao Y, Li J, Ma Z, Wei L, Liu L. QTL mapping for germination percentage under salinity and drought stresses in Brassica napus L. using a SNP genetic map. Acta Agron Sin. 2014;40(4):629–35. doi: 10.3724/SP.J.1006.2014.00629. [DOI] [Google Scholar]
- 123.Wan H, Wei Y, Qian J, Gao Y, Wen J, Yi B, Ma C, Tu J, Fu T, Shen J. Association mapping of salt tolerance traits at germination stage of rapeseed (Brassica napus L.). Euphytica. 2018;214:190. [Google Scholar]
- 124.Huang Z, Zhao N, Qin M, Xu A. Mapping of quantitative trait loci related to cold resistance in Brassica napus L. J Plant Physiol. 2018;231:147–54. doi: 10.1016/j.jplph.2018.09.012. [DOI] [PubMed] [Google Scholar]
- 125.Li Z, Mei S, Mei Z, Liu X, Fu T, Zhou G, Tu J. Mapping of QTL associated with waterlogging tolerance and drought resistance during the seedling stage in oilseed rape (Brassica napus). Euphytica. 2014;197(3):341–53. doi: 10.1007/s10681-014-1070-z. [DOI] [Google Scholar]
- 126.Yong H-Y, Wang C, Bancroft I, Li F, Wu X, Kitashiba H, Nishio T. Identification of a gene controlling variation in the salt tolerance of rapeseed (Brassica napus L.). Planta. 2015;242:313–26. [DOI] [PubMed] [Google Scholar]
- 127.Fletcher RS, Mullen JL, Heiliger A, McKay JK. QTL analysis of root morphology, flowering time, and yield reveals trade-offs in response to drought in Brassica napus. J Exp Bot. 2015;66(1):245–56. doi: 10.1093/jxb/eru423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fletcher RS, Herrmann D, Mullen JL, Li Q, Schrider DR, Price N, Lin J, Grogan K, Kern A, McKay JK. Identification of polymorphisms associated with drought adaptation QTL in Brassica napus by resequencing. Genes Genomes Genet. 2016;6:793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.He Y, Mao S, Gao Y, Zhu L, Wu D, Cui Y, Li J, Qian W, Li M. Genome-wide identification and expression analysis of WRKY transcription factors under multiple stresses in Brassica napus. PloS One. 2016;11(6):e0157558. doi: 10.1371/journal.pone.0157558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lang L, Xu A, Ding J, Zhang Y, Zhao N, Tian Z, Liu Y, Wang Y, Liu X, Liang F. Quantitative trait locus mapping of salt tolerance and identification of salt-tolerant genes in Brassica napus L. Front Plant Sci. 2017;8:1000. doi: 10.3389/fpls.2017.01000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang Y, Xu A, Lang L, Wang Y, Liu X, Liang F, Zhang B, Qin M, Dalelhan J, Huang Z. Genetic mapping of a lobed-leaf gene associated with salt tolerance in Brassica napus L. Plant Sci. 2018b;269:75–84. [DOI] [PubMed] [Google Scholar]
- 132.Yang H, Liu J, Huang S, Guo T, Deng L, Hua W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene. 2014;538:113–22. [DOI] [PubMed] [Google Scholar]
- 133.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang P, Yang C, Chen H, Luo L, Leng Q, Li S, Han Z, Li X, Song C, Zhang X. Exploring transcription factors reveals crucial members and regulatory networks involved in different abiotic stresses in Brassica napus L. BMC Plant Biol. 2018b;18:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zou X, Tan X, Hu C, Zeng L, Lu G, Fu G, Cheng Y, Zhang X. The transcriptome of Brassica napus L. roots under waterlogging at the seedling stage. Int J Mol Sci. 2013;14:2637–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yong H-Y, Zou Z, Kok E-P, Kwan B-H, Chow K, Nasu S, Nanzyo M, Kitashiba H, Nishio T. Comparative transcriptome analysis of leaves and roots in response to sudden increase in salinity in Brassica napus by RNA-seq. Biomed Res Int. 2014;2014:467395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lu K, Zhang L, Qu C-M, Liang Y, Tang Z-L, Li J-N. Identification of drought stress-responsive genes in leaves of Brassica napus by RNA sequencing. Sci Agric Sin. 2015;48:630–45. [Google Scholar]
- 138.Ma N, Hu C, Wan L, Hu Q, Xiong J, Zhang C. Strigolactones improve plant growth, photosynthesis, and alleviate oxidative stress under salinity in rapeseed (Brassica napus L.) by regulating gene expression. Front Plant Sci. 2017;8:1671. doi: 10.3389/fpls.2017.01671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Luo T, Xian M, Zhang C, Hu L, Xu Z. Associating transcriptional regulation for rapid germination of rapeseed (Brassica napus L.) under low temperature stress through weighted gene co-expression network analysis. Sci Rep. 2019;9:1–16. doi: 10.1038/s41598-018-37186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xin H, Xianchao N, Pan X, Wei L, Min Y, Yu K, Lunwen Q, Wei H. Comparative transcriptome analyses revealed conserved and novel responses to cold and freezing stress in Brassica napus L. G3: Genes Genomes Genet. 2019;9:2723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jian H, Xie L, Wang Y, Cao Y, Wan M, Lv D, Li J, Lu K, Xu X, Liu L. Characterization of cold stress responses in different rapeseed ecotypes based on metabolomics and transcriptomics analyses. PeerJ. 2020;8:e8704. doi: 10.7717/peerj.8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shamloo-Dashtpagerdi R, Razi H, Ebrahimie E. Mining expressed sequence tags of rapeseed (Brassica napus L.) to predict the drought responsive regulatory network. Physiol Mol Biol Plants. 2015;21:329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Saini P, Kamboj D, Yadav R, Yadav NR. SRAPs and EST-SSRs provide useful molecular diversity for targeting drought and salinity tolerance in Indian mustard. Mol Biol Rep. 2019;46:1213–25. [DOI] [PubMed] [Google Scholar]
- 144.Sathasivam R, Guo R, Wang H, Lim W-A, Ki J-S. Expressed sequence tag library of the marine green alga Tetraselmis suecica: a focus on stress-related genes for marine pollution. J Appl Phycol. 2018;30:2387–402. [Google Scholar]
- 145.Ke T, Yu J, Dong C, Mao H, Hua W, Liu S. ocsESTdb: a database of oil crop seed EST sequences for comparative analysis and investigation of a global metabolic network and oil accumulation metabolism. BMC Plant Biol. 2015;15(1):19. doi: 10.1186/s12870-014-0399-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhuang J, Zhu B. Analysis of Brassica napus ESTs: gene discovery and expression patterns of AP2/ERF-family transcription factors. Mol Biol Rep. 2014;41:45–56. [DOI] [PubMed] [Google Scholar]
- 147.Cheng CK, Au CH, Wilke SK, Stajich JE, Zolan ME, Pukkila PJ, Kwan HS. 5’-serial analysis of gene expression studies reveal a transcriptomic switch during fruiting body development in Coprinopsis cinerea. BMC Genomics. 2013;14(1):195. doi: 10.1186/1471-2164-14-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vega-Sánchez ME, Gowda M, Wang G-L. Tag-based approaches for deep transcriptome analysis in plants. Plant Sci. 2007;173:371–80. [Google Scholar]
- 149.Wu G-Z, Shi Q-M, Niu Y, Xing M-Q, Xue H-W. Shanghai RAPESEED database: a resource for functional genomics studies of seed development and fatty acid metabolism of Brassica. Nucleic Acids Res. 2007;36:D1044–D1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.He M, Zhu C, Dong K, Zhang T, Cheng Z, Li J, Yan Y. Comparative proteome analysis of embryo and endosperm reveals central differential expression proteins involved in wheat seed germination. BMC Plant Biol. 2015;15(1):1–17. doi: 10.1186/s12870-015-0471-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Erice G, Ruíz-Lozano JM, ÁM ÁM, García-Mina JM, Aroca R. Transcriptomic analysis reveals the importance of JA-Ile turnover in the response of Arabidopsis plants to plant growth promoting rhizobacteria and salinity. Environ Exp Bot. 2017;143:10–19. doi: 10.1016/j.envexpbot.2017.08.006. [DOI] [Google Scholar]
- 152.Huang Y, Shi J, Tao Z, Zhang L, Liu Q, Wang X, Yang Q, Liu G, Wang H . Microarray expression analysis of the main inflorescence in Brassica napus. PloS One. 2014;9:e102024. Biology. 2016;5(2):20. doi: 10.3390/biology5020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Moustafa K, Cross JM. Genetic approaches to study plant responses to environmental stresses: an overview. Biology. 2016;5(2):20. doi: 10.3390/biology5020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Savitch LV, Allard G, Seki M, Robert LS, Tinker NA, Huner NP, Shinozaki K, Singh J. The effect of overexpression of two Brassica CBF/DREB1-like transcription factors on photosynthetic capacity and freezing tolerance in Brassica napus. Plant Cell Physiol. 2005;46:1525–39. [DOI] [PubMed] [Google Scholar]
- 155.Thomashow MF. Molecular basis of plant cold acclimation: insights gained from studying the CBF cold response pathway. Plant Physiol. 2010;154:571–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen L, Ren F, Zhong H, Jiang W, Li X. Identification and expression analysis of genes in response to high-salinity and drought stresses in Brassica napus. Acta Biochim Biophys Sin. 2010;42(2):154–164. doi: 10.1093/abbs/gmp113. [DOI] [PubMed] [Google Scholar]
- 157.Jacq A, Burlat V, Jamet E. Plant cell wall proteomics as a strategy to reveal candidate proteins involved in extracellular lipid metabolism. Curr Protein Pept Sci. 2018;19(2):190–99. doi: 10.2174/1389203718666170918152859. [DOI] [PubMed] [Google Scholar]
- 158.Kosová K, Vítámvás P, Urban MO, Prášil IT, Renaut J. Plant abiotic stress proteomics: the major factors determining alterations in cellular proteome. Front Plant Sci. 2018;9:122. doi: 10.3389/fpls.2018.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xu J, Qiao X, Tian Z, Zhang X, Zou X, Cheng Y, Lu G, Zeng L, Fu G, Ding X. Proteomic analysis of rapeseed root response to waterlogging stress. Plants. 2018;7:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yang Y, Liu Z, Zhang T, Zhou G, Duan Z, Li B, Dou S, Liang X, Tu J, Shen J. Mechanism of salt-induced self-compatibility dissected by comparative proteomic analysis in Brassica napus L. Int J Mol Sci. 2018a;19:1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cheng D, Qiao L, Horvatovich P. Toward spectral library-free matrix-assisted laser desorption/ionization time-of-flight mass spectrometry bacterial identification. J Proteome Res. 2018;17(6):2124–30. doi: 10.1021/acs.jproteome.8b00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Qin L, Zhang Y, Liu Y, He H, Han M, Li Y, Zeng M, Wang X. Recent advances in matrix-assisted laser desorption/ionisation mass spectrometry imaging (MALDI-MSI) for in situ analysis of endogenous molecules in plants. Phytochem Anal. 2018;29(4):351–64. doi: 10.1002/pca.2759. [DOI] [PubMed] [Google Scholar]
- 163.Freitas JR, Vendramini PH, Melo JO, Eberlin MN, Augusti R. An appraisal on the source-to-sink relationship in plants: an application of desorption electrospray ionization mass spectrometry imaging. J Brazil Chem Soc. 2018;29:17–23. [Google Scholar]
- 164.Signor L, Erba EB. Matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometric analysis of intact proteins larger than 100 kDa. J Visual Exp. 2013;108:e50635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yıldız M, Akçalı N, Terzi H. Proteomic and biochemical responses of canola (Brassica napus L.) exposed to salinity stress and exogenous lipoic acid. J Plant Physiol. 2015;179:90–99. [DOI] [PubMed] [Google Scholar]
- 166.Zhang J, Liu Z, Liu X, Dong J, Pang H, Yu C. Proteomic alteration of a thermo‐sensitive male sterility SP2S in rapeseed (Brassica napus) in response to mild temperature stress. Plant Breed. 2016;135:191–99. [Google Scholar]
- 167.Naryzhny S. Inventory of proteoforms as a current challenge of proteomics: some technical aspects. J Proteom. 2019;191:22–28. doi: 10.1016/j.jprot.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 168.Dolatabadi N, Toorchi M, Valizadeh M, Bandehagh A. The proteome response of salt-sensitive rapeseed (Brassica napus L.) genotype to salt stress. Not Bot Horti Agrobot Cluj-Napoca. 2019;47(1):17–23. doi: 10.15835/nbha47111133. [DOI] [Google Scholar]
- 169.Jia H, Shao M, He Y, Guan R, Chu P, Jiang H, Dai SJ. Proteome dynamics and physiological responses to short-term salt stress in Brassica napus leaves. PloS One. 2015;10(12):e0144808. doi: 10.1371/journal.pone.0144808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kholghi M, Toorchi M, Bandehagh A, Ostendorp A, Ostendorp S, Hanhart P, Kehr J. Comparative proteomic analysis of salt-responsive proteins in canola roots by 2-DE and MALDI-TOF MS. Biochim Biophys Acta Proteins Proteom. 2019;1867(3):227–36. doi: 10.1016/j.bbapap.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 171.Jorrin-Novo JV, Komatsu S, Sanchez-Lucas R, de Francisco LER. Gel electrophoresis-based plant proteomics: past, present, and future. Happy 10th Anniversary Journal of Proteomics! J Proteom. 2019;198:1–10. [DOI] [PubMed] [Google Scholar]
- 172.Voisin M, Vanrobays E, Tatout C. Investigation of nuclear periphery protein interactions in plants using the membrane yeast two-hybrid (MbY2H) system. In: Gundersen G, Worman H, editors The LINC complex. Humana Press, NY:Springer; 2018. p. 221–35. [DOI] [PubMed] [Google Scholar]
- 173.Zhao T-J, Liu Y, Yan Y-B, Feng F, Liu W-Q, Zhou H-M. Identification of the amino acids crucial for the activities of drought responsive element binding factors (DREBs) of Brassica napus. FEBS Lett. 2007;581:3044–50. [DOI] [PubMed] [Google Scholar]
- 174.Liang W, Yang B, Yu B-J, Zhou Z, Li C, Jia M, Sun Y, Zhang Y, Wu F, Zhang H. Identification and analysis of MKK and MPK gene families in canola (Brassica napus L.). BMC Genomics. 2013;14(1):1–24. doi: 10.1186/1471-2164-14-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhao B, Li H, Li J, Wang B, Dai C, Wang J, Liu K. Brassica napus DS-3, encoding a DELLA protein, negatively regulates stem elongation through gibberellin signaling pathway. Theor Appl Genet. 2017;130:727–41. [DOI] [PubMed] [Google Scholar]
- 176.Raza A. Metabolomics: a systems biology approach for enhancing heat stress tolerance in plants. Plant Cell Rep. 2020b. doi: 10.1007/s00299-020-02635-8. [DOI] [PubMed] [Google Scholar]
- 177.Razzaq A, Sadia B, Raza A, Khalid Hameed M, Saleem F. Metabolomics: A Way Forward for Crop Improvement. Metabolites. 2019;9:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Smolikova G, Shavarda A, Alekseichuk I, Chantseva V, Medvedev S. The metabolomic approach to the assessment of cultivar specificity of Brassica napus L. Seeds Russ J Genet Appl Res. 2016;6:78–83. [Google Scholar]
- 179.Nokhrina K, Ray H, Bock C, Georges F. Metabolomic shifts in Brassica napus lines with enhanced BnPLC2 expression impact their response to low temperature stress and plant pathogens. GM Crops Food. 2014;5(2):120–31. doi: 10.4161/gmcr.28942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Misra BB. Cataloging the Brassica napus seed metabolome. Cogent Food Agric. 2016;2:1254420. [Google Scholar]
- 181.Dumas M-E. Metabolome 2.0: quantitative genetics and network biology of metabolic phenotypes. Mol BioSys. 2012;8(10):2494–502. doi: 10.1039/c2mb25167a. [DOI] [PubMed] [Google Scholar]
- 182.Georges F, Das S, Ray H, Bock C, Nokhrina K, Kolla VA, Keller W. Over‐expression of Brassica napus phosphatidylinositol‐phospholipase C2 in canola induces significant changes in gene expression and phytohormone distribution patterns, enhances drought tolerance and promotes early flowering and maturation. Plant Cell Environ. 2009;32(12):1664–81. doi: 10.1111/j.1365-3040.2009.02027.x. [DOI] [PubMed] [Google Scholar]
- 183.Kortesniemi M, Vuorinen AL, Sinkkonen J, Yang B, Rajala A, Kallio H. NMR metabolomics of ripened and developing oilseed rape (Brassica napus) and turnip rape (Brassica rapa). Food Chem. 2015;172:63–70. doi: 10.1016/j.foodchem.2014.09.040. [DOI] [PubMed] [Google Scholar]
- 184.Asaduzzaman M, Pratley JE, An M, Luckett DJ, Lemerle D. Metabolomics differentiation of canola genotypes: toward an understanding of canola allelochemicals. Front Plant Sci. 2015;5:765. doi: 10.3389/fpls.2014.00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pratap A, Gupta S, Nair RM, Gupta S, Schafleitner R, Basu P, Singh CM, Prajapati U, Gupta AK, Nayyar H. Using plant phenomics to exploit the gains of genomics. Agronomy. 2019;9:126. [Google Scholar]
- 186.Walter A, Liebisch F, Hund A. Plant phenotyping: from bean weighing to image analysis. Plant Methods. 2015;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mickelbart MV, Hasegawa PM, Bailey-Serres J. Genetic mechanisms of abiotic stress tolerance that translate to crop yield stability. Nat Rev Genet. 2015;16(4):237–51. doi: 10.1038/nrg3901. [DOI] [PubMed] [Google Scholar]
- 188.Cabrera‐Bosquet L, Crossa J, von Zitzewitz J, Serret MD, Luis Araus J. High-throughput phenotyping and genomic selection : the frontiers of crop breeding converge F. J Integr Plant Biol. 2012;54(5):312–320. doi: 10.1111/j.1744-7909.2012.01116.x. [DOI] [PubMed] [Google Scholar]
- 189.Zhang X, Huang C, Wu D, Qiao F, Li W, Duan L, Wang K, Xiao Y, Chen G, Liu Q. High-throughput phenotyping and QTL mapping reveals the genetic architecture of maize plant growth. Plant Physiol. 2017;173:1554–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li H, Feng H, Guo C, Yang S, Huang W, Xiong X, Liu J, Chen G, Liu Q, Xiong L, et al. High‐throughput phenotyping accelerates the dissection of the dynamic genetic architecture of plant growth and yield improvement in rapeseed. Plant Biotechnol J. 2020;18(11):2345–53. doi: 10.1111/pbi.13396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Arifuzzaman M, Oladzadabbasabadi A, McClean P, Rahman M. Shovelomics for phenotyping root architectural traits of rapeseed/canola (Brassica napus L.) and genome-wide association mapping. Mol Genet Genomics. 2019;294(4):985–1000. doi: 10.1007/s00438-019-01563-x. [DOI] [PubMed] [Google Scholar]
- 192.Kong F, Mao S, Du K, Wu M, Zhou X, Chu C, Wang Y. Comparative proteomics analysis of OsNAS1 transgenic Brassica napus under salt stress. Chin Sci Bull. 2011;56(22):2343–50. doi: 10.1007/s11434-011-4585-x. [DOI] [Google Scholar]
- 193.Lu S, Bahn SC, Qu G, Qin H, Hong Y, Xu Q, Zhou Y, Hong Y, Wang X. Increased expression of phospholipase Dα1 in guard cells decreases water loss with improved seed production under drought in Brassica napus. Plant Biotechnol J. 2013;11(3):380–89. doi: 10.1111/pbi.12028. [DOI] [PubMed] [Google Scholar]
- 194.Sakhno L, Slyvets M, Kuchuk M. cyp11A1 Canola plants under short time heat stress conditions. Cytol Genet. 2014;48:279–84. [PubMed] [Google Scholar]
- 195.Sun X, Feng X, Li C, Zhang Z, Wang L. Study on salt tolerance with YHem1 transgenic canola (Brassica napus). Physiol Plant. 2015;154:223–42. [DOI] [PubMed] [Google Scholar]
- 196.Qamarunnisa S, Jamil I, Raza S, Azhar A, Naqvi SM. Genetic improvement of canola against abiotic stress through incorporation of DREB gene. Asian J Agric Biol. 2015;3:77–104. [Google Scholar]
- 197.Sahni S, Prasad BD, Liu Q, Grbic V, Sharpe A, Singh SP, Krishna P. Overexpression of the brassinosteroid biosynthetic gene DWF4 in Brassica napus simultaneously increases seed yield and stress tolerance. Sci Rep. 2016;6:28298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kim J, Lee WJ, Vu TT, Jeong CY, Hong S-W S-W, Lee H. High accumulation of anthocyanins via the ectopic expression of AtDFR confers significant salt stress tolerance in Brassica napus L. Plant Cell Rep. 2017;36(8):1215–24. doi: 10.1007/s00299-017-2147-7. [DOI] [PubMed] [Google Scholar]
- 199.Rajab H, Khan MS, Shah SH, Shah SMA. Genetic transformation of tobacco serine acetyltransferase 4 (NtSAT4) gene in Brassica napus L. Sarhad J Agric. 2019;35:1224–33. [Google Scholar]
- 200.Ji MG, Park HJ, Cha J-Y, Kim JA, Shin G-I, Jeong SY, Lee ES, Yun D-J, Lee SY, Kim W-Y. Expression of Arabidopsis thaliana Thioredoxin-h2 in Brassica napus enhances antioxidant defenses and improves salt tolerance. Plant Physiol Biochem. 2020;147:313–21. doi: 10.1016/j.plaphy.2019.12.032. [DOI] [PubMed] [Google Scholar]
- 201.Zafar S, Yu Y, Zhu K, Wang W, Tan X. Overexpression of Nicotiana tabacum HSP17.6 enhances abiotic stress tolerance in Brassica napus. Int J Agric Biol. 2020;23:164–70. [Google Scholar]
- 202.Jaglo KR, Kleff S, Amundsen KL, Zhang X, Haake V, Zhang JZ, Deits T, Thomashow MF. Components of the arabidopsis C-repeat/dehydration-responsive element binding factor cold-response pathway are conserved in Brassica napus and other plant species. Plant Physiol. 2001;127(3):910–17. doi: 10.1104/pp.010548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zaman QU, Li C, Cheng H, Hu Q. Genome editing opens a new era of genetic improvement in polyploid crops. Crop J. 2019a;7:141–50. [Google Scholar]
- 204.Li C, Hao M, Wang W, Wang H, Chen F, Chu W, Zhang B, Mei D, Cheng H, Hu Q. An efficient CRISPR/Cas9 platform for rapidly generating simultaneous mutagenesis of multiple gene homoeologs in allotetraploid oilseed rape. Front Plant Sci. 2018;9:442. doi: 10.3389/fpls.2018.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Okuzaki A, Ogawa T, Koizuka C, Kaneko K, Inaba M, Imamura J, Koizuka N. CRISPR/Cas9-mediated genome editing of the fatty acid desaturase 2 gene in Brassica napus. Plant Physiol Biochem. 2018;131:63–69. doi: 10.1016/j.plaphy.2018.04.025. [DOI] [PubMed] [Google Scholar]
- 206.Hu L, Zhang H, Yang Q, Meng Q, Han S, Nwafor CC, Khan MHU, Fan C, Zhou Y. Promoter variations in a homeobox gene, BnA10.LMI1, determine lobed leaves in rapeseed (Brassica napus L.). Theor Appl Genet. 2018;131(12):2699–708. doi: 10.1007/s00122-018-3184-5. [DOI] [PubMed] [Google Scholar]
- 207.Zheng M, Zhang L, Tang M, Liu J, Liu H, Yang H, Fan S. Knockout of two BnaMAX1 homologs by CRISPR/Cas9-targeted mutagenesis improves plant architecture and increases yield in rapeseed (Brassica napus L.). Plant Biotechnol J. 2020;18:644–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yang Y, Zhu K, Li H, Han S, Meng Q. Precise editing of CLAVATA genes in Brassica napus L. regulates multilocular silique development. Plant Biotechnol J. 2018b;16:1322–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zhai Y, Cai S, Hu L, Yang Y, Amoo O, Fan C, Zhou Y. CRISPR/Cas9-mediated genome editing reveals differences in the contribution of INDEHISCENT homologues to pod shatter resistance in Brassica napus L. Theor Appl Genet. 2019;132:2111–23. [DOI] [PubMed] [Google Scholar]
- 210.Zaman QU, Chu W, Hao M, Shi Y, Sun M, Sang SF, Mei D, Cheng H, Liu J, Li C, et al. CRISPR/Cas9-mediated multiplex genome editing of JAGGED gene in Brassica napus L. Biomolecules. 2019b;9:725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sriboon S, Li H, Guo C, Senkhamwong T, Dai C, Liu K. Knock-out of TERMINAL FLOWER 1 genes altered flowering time and plant architecture in Brassica napus. BMC Genet. 2020;21:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wu J, Yan G, Duan Z, Wang Z, Kang C, Guo L, Liu K, Tu J, Shen J, Yi B, et al. Roles of the Brassica napus DELLA protein BnaA6.RGA, in modulating drought tolerance by interacting with the ABA signaling component BnaA10.ABF2. Front Plant Sci. 2020;11:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.