

ABSTRACT
We report here the discovery and optimization of a novel T cell retargeting anti-GUCY2C x anti-CD3ε bispecific antibody for the treatment of solid tumors. Using a combination of hybridoma, phage display and rational design protein engineering, we have developed a fully humanized and manufacturable CD3 bispecific antibody that demonstrates favorable pharmacokinetic properties and potent in vivo efficacy. Anti-GUCY2C and anti-CD3ε antibodies derived from mouse hybridomas were first humanized into well-behaved human variable region frameworks with full retention of binding and T-cell mediated cytotoxic activity. To address potential manufacturability concerns, multiple approaches were taken in parallel to optimize and de-risk the two antibody variable regions. These approaches included structure-guided rational mutagenesis and phage display-based optimization, focusing on improving stability, reducing polyreactivity and self-association potential, removing chemical liabilities and proteolytic cleavage sites, and de-risking immunogenicity. Employing rapid library construction methods as well as automated phage display and high-throughput protein production workflows enabled efficient generation of an optimized bispecific antibody with desirable manufacturability properties, high stability, and low nonspecific binding. Proteolytic cleavage and deamidation in complementarity-determining regions were also successfully addressed. Collectively, these improvements translated to a molecule with potent single-agent in vivo efficacy in a tumor cell line adoptive transfer model and a cynomolgus monkey pharmacokinetic profile (half-life>4.5 days) suitable for clinical development. Clinical evaluation of PF-07062119 is ongoing.
KEYWORDS: T cell bispecific, T cell retargeting, T-BsAb, immuno-oncology, GUCY2C, Guanlyate cyclase 2c (GCC), PF-07062119, antibody engineering, antibody optimization, developability, high-throughput protein production
Introduction
Bispecific antibodies (BsAb) that recruit and direct immune effector cells to kill tumor cells represent an exciting and rapidly expanding area of cancer research. While there are numerous immune cell-engaging BsAb formats, they share the fundamental aspects of pairing one antibody that targets a tumor-associated antigen with another antibody targeting cytotoxic lymphocytes, typically T cells via CD3 epsilon (CD3ε).1 T cell bispecifics (T-BsAb) function by driving the formation of a synapse between T cells and tumor cells, where the synapse is independent of the normal T cell receptor (TCR) and peptide-major histocompatibility complex (pMHC) formation. Instead, the T-BsAb mediates the cytotoxic synapse through simultaneous binding of both targets, leading to T cell activation through CD3ε signaling alone, independent of TCR sequence. T-BsAb-directed cytotoxicity of cancer cells occurs primarily through membrane perforation by perforin, followed by transfer of Granzyme B and subsequent apoptotic signaling through caspases; and secondarily through cytokine release and further immune cell activation and recruitment.1,2 To date, only two T-BsAbs, catumaxomab (withdrawn) and blinatumomab have been granted marketing approvals, but at least 47 new T-BsAbs are in clinical development.1,3
Advances in protein engineering have led to an upsurge of preclinical and clinical studies harnessing BsAb technology.4 This is evident by more than 85 ongoing clinical trials involving BsAbs, consisting of over 20 different BsAb formats.3 T cell engaging bispecific formats include fragment-based formats such as the Bispecific T cell Engager (BiTE), as well as symmetric and asymmetric IgG-like formats that may be designed to have a 1-to-1, 2-to-1 or 2-to-2 tumor cell-to-T cell binding domain orientation.1,3 Optimal manufacturability properties of these novel BsAb formats are critical for their successful development and clinical administration.5 While many BsAb formats leverage the inherent properties of regular antibodies, some still require extensive optimization to meet manufacturability criteria.6 Other BsAb formats incorporate antibody fragments appended to Fc scaffolds in non-natural configurations, and therefore may also require further optimization of individual antibody domains to achieve a desired manufacturability profile.7 These ideal “developability” characteristics include high conformational stability, high solubility/low self-association potential, low polyreactivity, low risk of post-translational modifications and low immunogenicity.8 Combining the requirements for good developability with a complex format can lead to a need for extensive optimization and multiple design-build-test cycles. In this report, we describe the application of a suite of high-throughput technologies to achieve multi-parameter optimization of a T-BsAb in the format of a diabody-Fc fusion.9–11
Colorectal cancer (CRC) is among the most frequently diagnosed cancers and a leading cause of cancer deaths worldwide.12,13 Guanylyl cyclase 2 C (GUCY2C or GCC) belongs to the guanylyl cyclase family of receptors and has been reported to be expressed in several gastrointestinal cancers including more than 90% of CRC across all stages.14,15 GUCY2C is a brush border membrane receptor for the hormones guanylin and uroguanylin as well as the heat-stable enterotoxin (STa) derived from enteric Escherichia coli.16–18 Expression of GUCY2C is confined to the luminal surfaces of healthy intestinal epithelium, and because tumors have disrupted tight junction architecture, GUCY2C may offer a promising target antigen for tumor-specific activation of systemically administered T cell redirection therapies.19–21 GUCY2C cell-surface expression is reported to be higher on moderately to well-differentiated tumors compared to more poorly differentiated tumors.21 We therefore sought to develop an anti-GUCY2C/anti-CD3ε T-BsAb that demonstrated potent in vitro cytotoxicity (EC50 below 1 nM) and in vivo single-agent efficacy below 1 mg/kg in preclinical models, good tolerability in cynomolgus monkeys at the predicted human efficacious dose, a pharmacokinetic (PK) profile compatible with dosing every three weeks, and developability characteristics supporting clinical development. Here we report on the discovery and optimization of the anti-GUCY2C/anti-CD3ε T-BsAb PF-07062119 using multiple complementarity-determining region (CDR)-based approaches.22,23 Clinical evaluation of PF-07062119 is ongoing (NCT04171141).
Results
Multifaceted discovery and optimization of diabody-Fc
We and others have shown that cytotoxic potency of CD3-based bispecifics can be strongly influenced by a combination of epitope, affinity, and geometry,10,24,25 and therefore the choice of a molecular format for the initial screen can dictate which tumor-associated antigen binding domains are identified as effective components of the bispecific molecule. In order to develop GUCY2C-targeted cytotoxic molecules, we selected the diabody-Fc format based on our successful development of a clinical-stage molecule using this design.10 The diabody-Fc format (Figure 1(a,b)), in which single-chain variable fragment (scFv) binding to CD3ε and a tumor-associated antigen are forced by short linkers to adopt a compact structure containing one VH-VL pair of each specificity, achieves efficient formation of the desired product using human IgG1 Fc containing knobs-into-holes mutations,26,27 as well as mutations to minimize effector function.28–30 Diabody-Fcs of this type have shown potent T cell-dependent cytotoxicity and clinical utility.9–11,25 Furthermore, molecules using this format can be produced in a single cell line with a simpler purification process than alternative IgG-like formats we have explored, which require expression of components in two cell lines and subsequent assembly and purification.31
Figure 1.
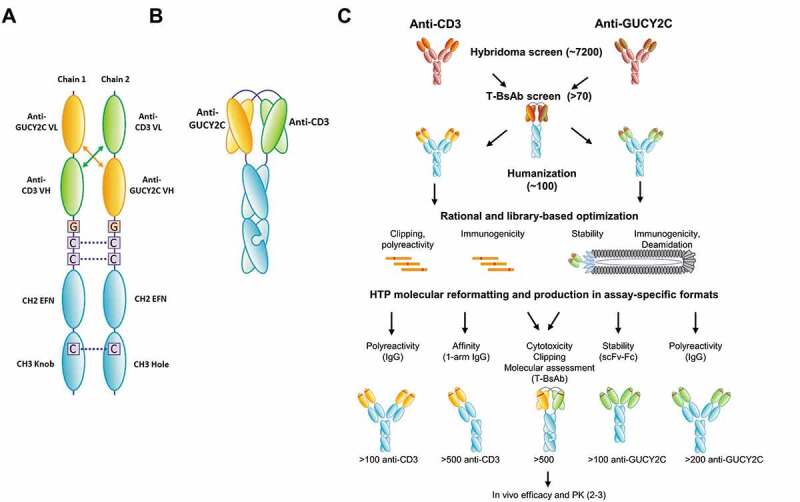
(a) Design and format of the GUCY2C T-BsAb. (A) Schematic of individual chains. For the GUCY2C T-BsAb, we applied the diabody Fc format. Heterodimerization is driven using knobs-into-holes. The Fc-knob (containing Y349C and T366W, numbered according to the EU index) and Fc-hole (containing S354C, T366S, L368A, and Y407V) are derived from human IgG126,27 and contain mutations L234A, L235A, and G237A in the lower hinge to reduce Fcγ receptor binding, antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity.28–30 EFN = effector function null. (b) Schematic of paired domains. (c) High-throughput anti-GUCY2C x anti-CD3 bispecific discovery workflow. To support bispecific discovery efforts for the GUCY2C T-BsAb program, automated reformatting, production and screening tools were implemented. This allows for parallel discovery and optimization activities where hundreds-to-thousands of hits can be cloned, produced and characterized in a very short time frame. Multiple formats were used to test for improvement of specific characteristics during optimization. This allows for rapid triaging and lead selection for scale-up as the final bispecific
We identified a panel of anti-GUCY2C hybridomas and determined, by reformatting into diabody format with an anti-CD3 antibody, that a subset of these antibodies supported high-potency T cell-dependent cytotoxicity in vitro (Table S1). Progressing from these initial hits to a therapeutic molecule suitable for clinical development required a multiparameter optimization process, including humanization and adjustments to binding affinity and specificity, thermal stability, and sequences associated with posttranslational modification or potential immunogenicity risk. To conduct this optimization, we addressed parameters in a modular fashion, using formats and methods suitable for their optimization (Figure 1(c)). For example, we applied structure-guided rational design to address nonspecific binding of the CD3 domain and assessed the outcome in IgG format, for which a suite of high-throughput assays has been extensively validated.32 Thermal stability of the GUCY2C domain was addressed using CDR randomization and phage display to identify stable scFv, followed by screening in scFv-Fc format in a sensitive high-throughput assay. High-throughput production of diabody-Fc was a critical component to this process, allowing the results of independent optimization steps to be assessed in combination in the final therapeutic format and enabling both the incorporation of improvements into subsequent optimization steps and the avoidance of variants that showed unexpected poor behavior in the diabody-Fc format. Figure 1(c) illustrates how we produced and characterized hundreds of antibodies against CD3 and GUCY2C in multiple protein formats simultaneously to rapidly address separate functional and biophysical properties, combining independently improved variants in diabody-Fc format to identify molecules with suitable therapeutic properties. Anti-CD3 and anti-GUCY2C variants demonstrating the preferred activity and manufacturability profiles were selected for the final T-BsAb.
Re-humanization and optimization of anti-CD3 domain H2B4
An anti-CD3 antibody, H2B4, has previously been used in an asymmetrical heterodimeric IgG format as part of an anti-FLT3 T-BsAB and has demonstrated potent in vivo efficacy in liquid tumor settings.33 However, H2B4 in the diabody-Fc format with binding domains directed against several other targets showed poor stability, nonspecific binding (polyreactivity) and clipping in CDR H2 during stable cell expression but not transient expression (data not shown), and therefore required optimization for use in this format. To address these issues, the CDRs of the H2B4 variable light (VL) domain were grafted from the original light chain, VK4-01, to VK1-39 germline frameworks. The variable heavy (VH) framework VH3-7 was kept the same. VK1-39 represents one of the most abundant VL germlines in the human repertoire and has shown favorable stability and manufacturability.34,35 Using a flow cytometry binding assay, we observed that the re-humanized molecule (referred to as H2B5) in bispecific format retained most of the H2B4 binding activity to CD3ε on naïve T cells (13.24 nM compared to 8.18 nM), and in vitro T cell-mediated cytotoxicity of target-expressing tumor cells in a cytotoxic T lymphocyte (CTL) assay (0.421 nM compared to 0.176 nM) (Figure S1A and B). Changing the light chain framework region to VK1-39 also improved thermal stability by approximately 5°C, as measured by differential scanning calorimetry (DSC) (Figure S1C).
To address polyreactivity and clipping, a crystal structure of H2B4 was used to explore positive charge patches that might contribute to off-target binding, as well as to model potential mutations in CDR H2 (Fig. S2). Over 100 unique variant IgGs designed to reduce positive charge on the H2B5 surface were produced and tested for retention of human CD3ε binding by a high-throughput Octet binding assay. Clones demonstrating binding (data not shown) were then assessed for specific binding affinity by surface plasmon resonance (SPR) and for polyreactivity using nonspecific DNA and insulin binding enzyme-linked immunosorbent assay (ELISA), an assay for nonspecific binding in IgG format that has been demonstrated to provide a reliable indication of risk of poor PK (Figure 2(a)).32
Figure 2.
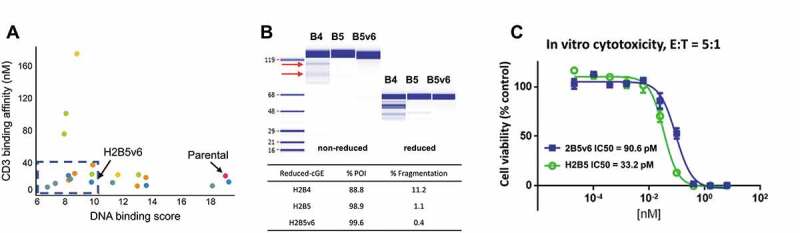
Rational optimization of anti-CD3 H2B4 to reduce polyreactivity and clipping. (a) Rational IgG variants with retained binding demonstrated lower polyreactivity (DNA score) compared to the parental 2B5 while maintaining desired affinity as measured by Biacore. (b) Re-humanization to H2B5 as T-BsAb with a stable control binding arm minimized clipping compared to H2B4, while H2B5v6 showed further clipping reduction as measured by cGE. Red arrows indicate clipped species. (c) H2B5v6 T-BsAb demonstrated reduced but acceptable in vitro CTL activity compared to re-humanized H2B5
To analyze clipping, clones showing favorable binding (KD ≤ 40 nM) and reduced polyreactivity (<10 in DNA ELISA) were paired with a stable control binding domain in diabody-Fc format, stably transfected into a Chinese hamster ovary (CHO) cell line, expressed and purified by protein A, then monitored for clipping by capillary gel electrophoresis (cGE) under reducing and non-reducing conditions. Clone H2B5v6, containing the mutations R52Q in the VH and R29Q in the VL (Figure S2), demonstrated reduced clipping during CHO expression (Figure 2(b)). R52Q resides in the middle of the stretch of amino acids RARNR in CDR H2, and modification to a glutamine appears to disrupt the enzymatic cleavage of this site. The removal of two positively charged, surface-exposed arginine residues is also likely playing a role in reducing polyreactivity from a DNA score of 19 as H2B4 to 9.8 as H2B5v6. When combined into the diabody-Fc format and assessed for in vitro cytotoxicity (Figure 2(c)), H2B5v6 showed a slight reduction of T cell-dependent killing (90.6 pM compared to 33.2 pM for H2B5). This was determined to be acceptable based on PK/pharmacodynamic (PD) modeling,36 and may even reduce the risk associated with cytokine toxicity driven by T cell activation.25 Therefore, H2B5v6 was selected as the lead anti-CD3 antibody for building the anti-GUCY2C T-BsAb.
Discovery and characterization of anti-GUCY2C T-BsAb
In parallel, anti-GUCY2C antibodies were discovered by mouse immunization using GUCY2C over-expressing cells. From a screen of 7200 hybridoma wells, specific human, cynomolgus monkey and mouse GUCY2C cross-reactive antibodies were identified and confirmed by flow cytometry and cell-based ELISA using engineered GUCY2C-over-expressing cell lines, as well as tumor cell lines with and without GUCY2C expression (Figures S3 and S4). Hybridoma supernatants demonstrating specific binding to GUCY2C-positive T84 tumor cells, but not GUCY2C-negative HCT-116 cells, were selected for antibody variable region cloning and further analysis.
We chose to produce these variable regions in both chimeric human IgG1 and chimeric T-BsAb formats (Figure 1). Production of multiple formats simultaneously allowed for multidimensional screening of various properties at once. While the behavior of isolated binding arms may not translate directly to the behavior of the final T-BsAb, assessment in isolation facilitated identification of beneficial changes in comparison to reference compounds using established assays. With the aid of automation, producing and characterizing the proteins in multiple assay-specific formats (IgG, monovalent Fab-Fc, scFv-Fc; Figure 1(c)) was not a rate-limiting process.
Over 70 chimeric IgG and T-BsAb were cloned, expressed, and purified at small scale, generating enough protein for binding, functional (CTL) and biophysical assessments. The high throughput protein production methods used only protein A capture and buffer exchange, meaning that high molecular mass species (HMMS) and homodimer species were not removed prior to initial characterization of the T-BsAbs. Our analytical purity data for these samples showed a range of 5–30% HMMS and 0–20% homodimer (data not shown). Although this material was not purified to homogeneity, it could reliably be used to identify molecules with binding activity to both targets, and more importantly, molecules with potent T cell-dependent cytotoxicity. Precise quantitation of activity with molecules purified to homogeneity later enabled more definitive ranking.
Chimeric GUCY2C T-BsAb were first analyzed for binding to recombinant human GUCY2C, binding to human CD3ε, and in a bispecific sandwich ELISA (Table S1). Clones demonstrating good bispecific binding were then analyzed for binding and T cell-dependent killing (CTL) of T84 tumor cells (Table S1). Here, clones showing effective killing by CTL assay were selected for scale-up expression and purification (with removal of HMMS and homodimer) to confirm T cell-dependent killing activity (Figure 3(a)). These clones were also assessed for stability by DSC (Figure 3(b)), polyreactivity by DNA and insulin ELISAs in the IgG and T-BsAb formats (Figure 3(c)), and binding affinity by SPR to human GUCY2C and human CD3 (Figure 3(d), S5 and Table S2). While most showed good CTL activity, many showed poor thermal stability, with Tm1 < 65°C, and high levels of polyreactivity (>10). Interestingly, GUCY2C-binding antibodies in IgG format showed much lower levels of DNA and insulin binding compared to the T-BsAb, possibly a result of inclusion of the H2B4 domain in the T-BsAb. Clone GUCY2C-0098 in the T-BsAb format, derived from parental hybridoma 9H3 (Figure S3), demonstrated not only the strongest CTL killing activity on T84 cells (EC50 0.4 nM: Figure 3(a)) and no CTL activity on the GUCY2C-negative cell line HCT-116 at concentrations up to 100 nM (data not shown), but also the strongest binding affinity to GUCY2C, and was therefore selected for humanization and optimization.
Figure 3.
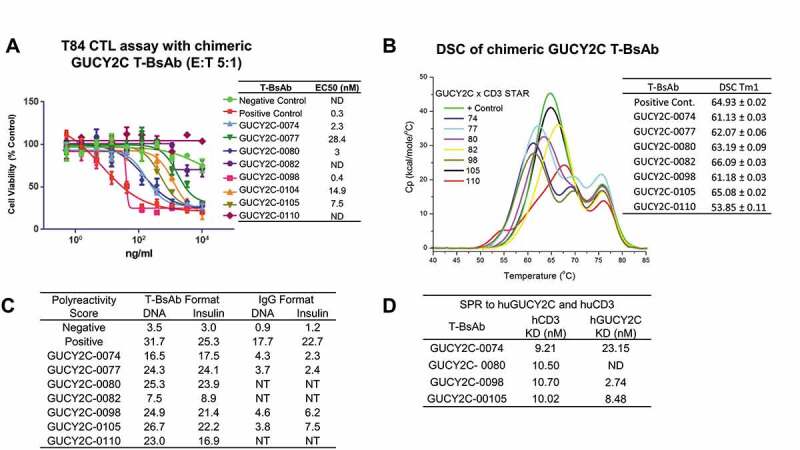
Characterization of chimeric GUCY2C T-BsAbs. (a) Top chimeric GUCY2C T-BsAb demonstrating CTL activity were scaled up and CTL activity against T84 tumor cells was repeated. These chimeric T-BsAbs were also analyzed for stability by DSC (b) and polyreactivity in both bispecific T-BsAb and monospecific anti-GUCY2C IgG format (c), as well as for binding to human CD3 and human GUCY2C recombinant protein by surface plasmon resonance (d)
Humanization of anti-GUCY2C T-BsAb
The variable regions of GUCY2C-0098 were humanized by CDR grafting into commonly used frameworks VH3-7 and VK1-39 using conventional methods.37 Humanization was initially performed in the IgG format to facilitate assessment of affinity and nonspecific binding, and later, humanized V-genes were transferred to the T-BsAb for confirmation of retained cytotoxicity. Over 100 unique antibodies were made through pairing heavy and light chain variants and screened for binding to determine necessary back-mutations. To restore full binding activity, a total of seven back-mutations to the original mouse sequence were required, six to the VH (V48I, A49G, R71V, N73K, L78A and A93T) and one to the VL (M4L). Binding was assessed by direct and competition ELISAs (Table S3). The resulting humanized T-BsAb GUCY2C-0247 (which incorporated the H2B4 anti-CD3 domain, as the anti-CD3 optimization was happening concurrently) showed equivalent binding and T cell-dependent cytotoxicity compared with the parental clone in the IgG and T-BsAb formats, respectively (Table S3). Binding affinity measured by SPR was 4.05 nM for the T-BsAb compared to 2.74 nM for the chimeric T-BsAb GUCY2C-0098. CTL killing activity of T84 cells also remained close to the parental clone at 0.098 nM compared to 0.089 nM. Unexpectedly, nonspecific binding to DNA and insulin were slightly increased in the IgG format upon humanization but reduced in the T-BsAb format, whereas the opposite trend was observed for affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS). This observation suggests that the format affects the contributions of the separate binding domains to non-target-directed binding, thus highlighting the need to screen in multiple formats. Thermal stability of the T-BsAb as measured by DSC did not appear to improve upon humanization, with Tm1 remaining below 65°C (Table S3), thus requiring further attention.
Optimization of humanized GUCY2C-0247 using phage display
Following humanization, several liabilities remained, including poor thermal stability, polyreactivity, high self-association potential, and a putative asparagine deamidation site (NG) in CDR H2. Affinity was determined to be appropriate based on in vitro cytotoxicity and PK/PD modeling, and therefore no efforts were made to increase affinity during the optimization process. To address these other liabilities, a phage display approach was taken in which 8 libraries were built based on GUCY2C-0247 in scFv format (VH-GGGGSGGGGSGGGGSG-VL), intended to allow VH-VL interaction similar to that present in the diabody format. Of these libraries, 6 were designed as individual-CDR-walk libraries made up of overlapping diversified 4- or 5-residue fragments that spanned each CDR. Library 7 was designed by diversifying specific residues (H35, H100e, L34, L89, L91 and L96) at the VH-VL interface. For these 7 libraries, we used a homology model of GUCY2C-0247 (data not shown) to determine the likely surface-exposed and buried residues, as well as residues of the VH-VL interface. Library 8 focused on perturbing the NG deamidation site by forcing 19 specific motifs (at positions H54 and H55) flanked by 4 diversified residues. For all libraries, with the exception of library 8, we used soft-randomization as the diversification method, where at each diversified position, 50% of the clones are expected to encode the wild-type amino acid and the remaining 50% of the clones will be distributed across the other 19 amino acids. During phage selections, thermal pressure was applied to rounds 2 and 3 at 60°C and 65°C, respectively, to select for variants with increased stability (Table S4). Additionally, deselections were performed against uncoated streptavidin beads, DNA, insulin, and a membrane extract reagent to reduce polyreactivity.
Following three rounds of selections, colonies were picked and analyzed for binding as periplasmic extracts (peripreps) to human GUCY2C with and without thermal challenge, using a “cook-and-bind” dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA) assay modified from methods previously described (Figure 4(a)).7,38,39 Output pools showing hits with improved thermal stability were selected for combining into a CDR shuffling library. CDR pools from each library were amplified with framework-specific primers, phosphorylated, and used as templates for additional multi-site directed mutagenesis (Figure 4(b)). The newly created, CDR-shuffled library was selected as before, with thermal challenges increasing to 75°C (Table S4). Again, clones were selected and screened for binding in the periprep cook-and-bind DELFIA assay.
Figure 4.
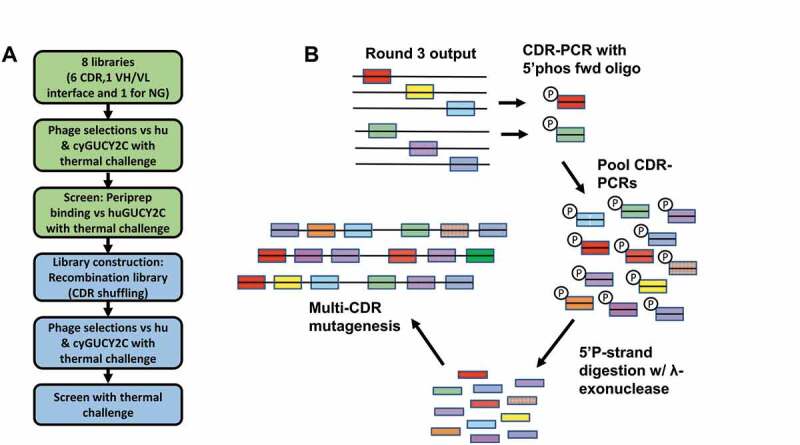
Soft randomization of CDRs coupled with CDR shuffling by phage display improves stability. (a) Phage display was performed on soft-randomization libraries targeting individual CDRs and the VH-VL interface of GUCY2C-0247. Thermal challenge was employed at 60°C and 65°C along with deselection using polyreactivity reagents. Output pools were recombined, creating a CDR shuffling library and additional phage selections were performed with increasing thermal challenge to 75°C to further improve stability. (b) Schematic illustration of the shuffling process to recombine CDRs from hits in the first 8-library selection
For periprep screening, we tested binding following incubation at 60°C, 65°C, 70°C and 75°C (Figure 5 (a b,). Peripreps containing the parental anti-GUCY2C scFv in the same VH-linker-VL format as the library lost binding after incubation at 55°C for 30 min. Hits identified from individual CDR soft-randomization libraries demonstrated modest improvement, with many showing binding following 60°C incubation, but most lost activity following incubation at 65°C. However, clones coming from the combined, CDR shuffled library showed strong binding after 65°C treatment. Many still remained active after incubation at 70°C, but by 75°C, all clones appear to have lost activity (data not shown).
Figure 5.
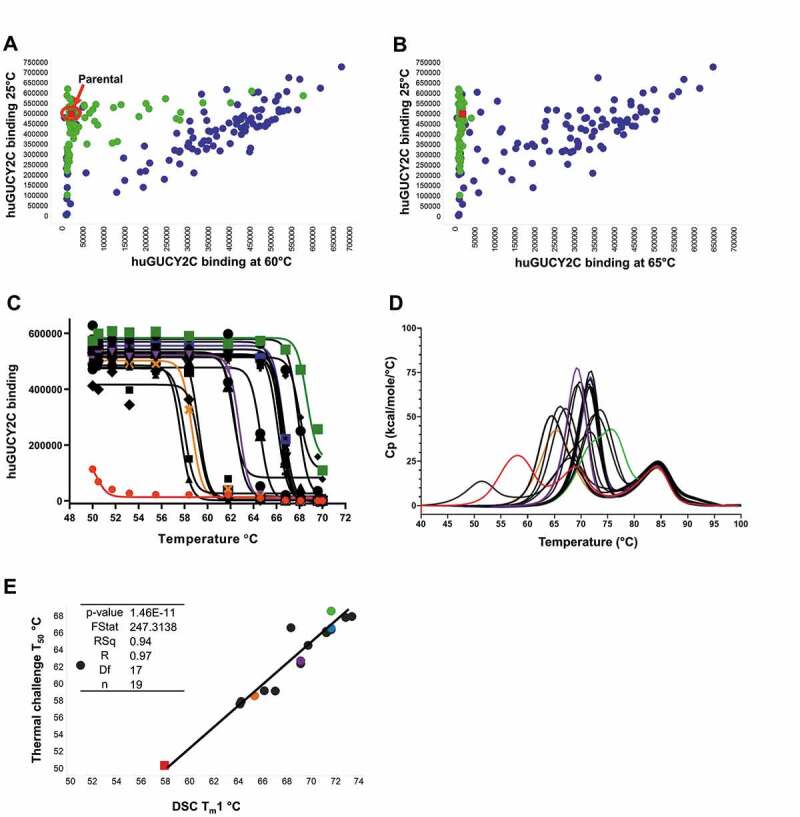
(a and b) Thermal challenge DELFIA assay demonstrates clones with improved thermal stability following phage display optimization. Cook-and-bind DELFIA time resolved fluorescence (TRF) measuring binding to human GUCY2C protein at 60°C, and 65°C, (X-axis) were compared with normal binding with no thermal challenge (Y-axis). Clones from individual CDR soft-randomization libraries are shown in green. Clones from CDR shuffling are shown in blue. The parental clone is shown in red. (c) ScFv-Fc binding activity following incubation at a range of temperatures, from which a T50 can be derived. Parental clone GUCY2C-0295 as scFv-Fc is shown in red and other representative clones with a range of T50 values are shown in orange, purple, blue and green. (d) DSC profiles show a spectrum of unfolding over a range of temperatures, from which a Tm1 can be derived. Representative clones are highlighted with the same coloring. (e) Correlation between T50 and Tm1 using a linear regression model
Following thermal screening of peripreps, hits demonstrating retained activity and improved thermal stability were sequenced and reformatted to IgG, scFv-Fc and T-BsAb formats using automated workflows. The IgG format was used to assess expression level, purity, polyreactivity, retained activity by competition DELFIA assay, and binding affinity by SPR (data not shown). Due to stability provided by the constant light and constant heavy 1 domains of the Fab domain, it can be difficult to assess stability improvements in the variable regions when tested in normal IgG1 format. Therefore, scFv-Fc were produced in parallel and tested by high-throughput cook-and-bind DELFIA assay and by DSC (Figure 5 (c,d)). The temperature at which 50% maximum signal is detected, referred to as the T50,37 was used to rank stability improvement. The most stable scFv-Fc were derived from the combination CDR-shuffling library, consistent with periprep screening results. Plotting the cook-and-bind T50 values against the Tm1 from DSC revealed a strong correlation between these separate stability assessments (Figure 5 (e)). This suggests that the cook-and bind-assay may offer an inexpensive and high-throughput substitute method to triage clones for improved stability. The best performing clones exhibited an increase of over 18°C by cook-and-bind and over 15°C by DSC (Table S5).
The T-BsAb format incorporating the optimized anti-CD3 antibody H2B5v6 was used to assess bispecific binding and T-cell dependent cytotoxicity. Even with the reduced CD3 affinity of H2B5v6 compared to that of H2B4, several clones demonstrated retained CTL killing activity comparable to that of the parental humanized GUCY2C-0247 (Figure S6). However, top clones demonstrating improved stability and binding activity equivalent to GUCY2C-0247 still contained the putative asparagine deamidation motif as well as undesirable in silico immunogenicity scores, therefore requiring further optimization.
Asparagine deamidation removal and immunogenicity de-risking
Asparagine deamidation removal
Screening of the initial soft randomization library that focused on the potential asparagine deamidation site in CDR H2 produced several tolerated mutations that maintained the desired binding activity. However, none of these clones exhibited improved thermal stability. Deamidation at this site in a T-BsAb (GUCY2C-1478), which contained a thermally stabilized GUCY2C binding domain, was confirmed by tandem mass spectrometry (MS-MS; data not shown), and was accompanied by a reduction in binding activity following incubation at 40°C for 4 weeks40 (Figure S7A) and an increase in acidic species by capillary isoelectric focusing (cIEF; data not shown). In order to remove this asparagine-glycine motif, we selected 11 of the top stability-optimized clones from the shuffling library and forced the introduction of the tolerated mutations identified previously into the designs for the immunogenicity de-risking mutations described below. We screened for retained activity following thermal challenge as described above, selecting clones that no longer contained the asparagine deamidation motif. Incorporation of a single mutation (Gly H55 Glu) to the stability-optimized T-BsAb clones demonstrated a reduction in the acidic species by cIEF and showed no significant impact on binding (Figure S7B and C).
Immunogenicity de-risking
During the discovery and optimization of the GUCY2C and CD3 antibodies, potential immunogenicity was assessed and monitored using Epivax and IEDB in silico T cell epitope prediction tools.41–43 An effort was made to minimize immunogenicity risk by introducing mutations that disrupted possible T cell epitopes and by avoiding mutations that increased in silico immunogenicity scoring (Epivax EpiMatrix T-regitope adjusted score; see ref. 41) or non-germline CDR content. To further reduce immunogenicity risk of the stability optimized anti-GUCY2C clones, we used a phage display approach in which we designed mutagenic oligonucleotides to the predicted immunogenic hot spots in the top 11 stability optimized variants. Specifically, we targeted L27, L33, L50, L91 on the VL and H27, H30 and H65 on the VH to disrupt the potentially immunogenic peptides. We introduced these mutations in combination with the asparagine-deamidation mutations above during phage library construction. We performed semi-automated selections with thermal challenge as before and screened for retained activity and stability, selecting clones with reduced in silico immunogenicity scores. Top clones were reformatted to IgG and T-BsAb and screened for activity and stability. We identified several clones with low in silico scores, (<-50) but these clones did not possess the required stability or activity. Mutations designed to modify T cell epitopes in CDR L2 appeared to be destabilizing, leading to a decrease in activity following incubation at 60°C (Table S6). We prioritized clones based on preferred activity and stability profiles possessing the lowest in silico immunogenicity scores.
Simultaneously, we screened immunogenicity de-risking mutations in the anti-CD3 antibody. A panel of over 500 rationally designed variants designed to remove T cell epitopes in H2B5 in CDRL1, L2 and H2, without interfering with residues involved in binding was produced and tested as individual IgG. We were unable to identify further de-risked variants that did not also show an impact on activity (data not shown). Therefore, we kept H2B5v6 as our lead anti-CD3 domain. The combined de-risked and optimized GUCY2C T-BsAb PF-07062119 demonstrated a considerably reduced immunogenicity risk score compared to the parental humanized GUCY2C-0247 and the stability optimized GUCY2C-1478 (Table S6). Using the Epivax scoring method, the final GUCY2C T-BsAb PF-07062119 had a predicted T cell epitope score (−34.8) comparable to those of bevacizumab (−31.62), palivizumab (−28.02) and rovelizumab (−29.72), which have all shown low incidence of anti-drug antibodies (ADA) in the clinic.44,45
Optimized GUCY2C T-BsAB, PF-07062119, demonstrates potent in vitro activity and desirable manufacturability
Manufacturability profile
Following optimization, the lead GUCY2C T-BsAb PF-07062119 sequence was cloned into a site-specific integration (SSI) vector and stably transfected into CHO cells.46 The average expression level of CHO stable pools was measured at 0.7 g/L in a 12-day fed-batch (Table S7). PF-07062119 was 60% pure following Protein A chromatography (with the remaining 40% a mixture of homodimer and high molecular mass species) and was purified to >99% purity using conventional chromatography methods, with a final yield across the entire process of 23% (data not shown). Optimization of both parental antibodies contributed to substantially reduced self-association and polyreactivity potential for the final T-BsAb molecule (Figure 6(b), Table S7). In the T-BsAb format, the thermal stability improvement as measured by DSC increased by 8°C (Figure 6(c)). Analysis by cGE confirmed the removal of clipping in CDRH2 of 2B5v6 (Figure 6(d)). Additionally, long-term stability analysis displayed a desirable profile with no increase in HMMS for over 6 weeks at either 4°C or 25°C (Figure 6(e)) and no loss of binding activity to GUCY2C or CD3 following 4 weeks of challenge at 40°C (Figure S7) Asparagine deamidation and aspartate isomerization were also low (<3%) under forced degradation conditions (data not shown). PF-07062119 also exhibited low viscosity at concentrations over 100 mg/mL (Figure 6(f)). Collectively, these findings demonstrate that PF-07062119 has a good manufacturability profile.
Figure 6.
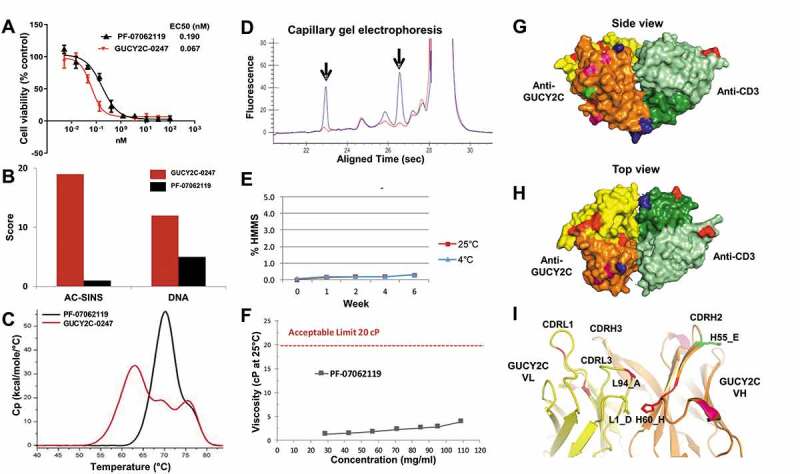
Optimization for manufacturability, efficacy and predicted immunogenicity. (a) CTL cytotoxicity assay (T84 cells, 48 hr, 5:1 E:T) demonstrates potent killing activity of PF-07062119 as compared to the parental GUCY2C-0247. (b) PF-07062119 demonstrates substantially reduced self-association potential and polyreactivity compared with GUCY2C-0247, as measured by AC-SINS and DNA binding assays. (c) Thermal stability in the T-BsAb demonstrated an increase in Tm1 of over 8°C compared to the parental molecule. (d) Clipped species in the parental anti-CD3 antibody H2B4 indicated by arrows (blue curve) were successfully removed in H2B5v6 (red curve) as demonstrated by cGE. (e) Long term stability analysis demonstrated good stability and no increase in HMMS for over 6 weeks at 4°C or at 25°C. (f) PF-07062119 also demonstrated low viscosity at concentrations over 100 mg/mL. Limit of 20 cP indicated by dashed red line. (g) Crystal structure of PF-07062119 (side view) shows the anti-GUCY2C domain in yellow and orange, the anti-CD3 in light and dark green, the linker sequences in blue, the stabilizing mutations in red, the immunogenicity-reducing mutations in pink, the deamidation mutation in bright green, and the disulfide constrained C-terminal domain (at bottom) also in blue. (h) Top view of tightly packed diabody structure. (i) Zoomed in view of GUCY2C VH-VL interface highlighting key stabilizing mutations with same coloring pattern of domains and mutations. Mutations at positions L94 (Ala) and H60 (His) were observed in most stability optimized clones. These mutations appear to help stabilize the VH-VL interface
In vitro cytotoxicity
PF-07062119 also maintained potent in vitro T cell-dependent cytotoxicity of T84 cells, with CTL EC50 measured at 0.19 nM. While the potency of the parental humanized molecule GUCY2C-0247 was ~2.8 fold stronger than that of PF-07062119 in CTL assays (Figure 6(a), Table S7), which was likely a function of slightly higher affinity to human GUCY2C (~2.5-fold) and to CD3 (~7.2-fold), the potency and kinetic profile of PF-07062119 were determined to be suitable for therapeutic use.36 This conclusion is further supported by the potent anti-tumor activity of PF-07062119 in other tumor lines expressing between 875 and 8,067 GUCY2C molecules per cell21 and its in vivo performance (below).
Crystallography of PF-07062119
As part of the optimization effort, we solved the crystal structure of PF-07062119. A non-Fc version of PF-07062119 was expressed and purified to homogeneity using nickel and anti-FLAG chromatography. This structure shows the two antigen binding sites directly opposite each other, spaced apart by approximately 70 Å (Figure 6(g,h)). In total, 13 mutations were introduced during the optimization campaigns for this GUCY2C T-BsAb, 6 in the GUCY2C VH domain, 5 in the GUCY2C VL domain and 1 each in the CD3 VH and VL domains (not including change in frameworks for H2B5 VL or humanization mutations). Two key stabilizing mutations at the GUCY2C VH/VL interface included an isoleucine to histidine mutation at H60 (an asparagine was also stabilizing at this position) and a valine to alanine mutation at position L94 (Figure 6(i)). These appear to stabilize this VH/VL interface by allowing better packing (Val to Ala at L94) and potentially forming stabilizing electrostatic bonds (His at H60 with Asp at L1). Another important stabilizing mutation, glutamic acid to alanine at L55, appears to have stabilized the interface between L2 and H3 of the anti-GUCY2C VH-VL (data not shown).
T-BsAb format comparison
To understand the impact of format on activity, we made the lead GUCY2C antibody in a bispecific IgG format with H2B5v6.31 However, no CTL killing activity was observed (Figure S8). While we did not conduct structural studies of this bispecific IgG, the Fab arms of a typical IgG are expected to be held in a flexible orientation 90–100 Å apart,47 which is significantly different from the rigid arrangement observed in the crystal structure of PF-07062119. This finding was not a surprise, since CTL killing potency of CD3 bispecifics has frequently been observed to depend on both the epitope and the spatial arrangement between CD3 and the tumor antigen binding arm.10,24 Rigid arrangement of two binding sites facing 180 degrees from each other at 70 Å apart compared to the flexible Fab orientation of a typical IgG (90–100 Å apart) may enable more efficient synapse formation with this particular epitope on GUCY2C, and therefore a more potent T cell-dependent cytotoxicity. We estimate that the distance between the membranes of the tumor cell and the T cell would be about 150 Å when spanned by the diabody-Fc bound to GUCY2C and CD3ε, which is consistent with the spacing conferred by other active CD3-bispecifics and by the T cell receptor-MHC complex at the immunological synapse.24 The bispecific IgG containing our GUCY2C and CD3 binding domains would be estimated to increase the separation between cells to a suboptimal distance, about 210 Å.
In vivo characterization of optimized GUCY2C T-BsAb
Pharmacokinetics of GUCY2C T-BsAb in cynomolgus monkey
The PK of GUCY2C-0247 and PF-07062119 were evaluated in cynomolgus monkeys at 30 and 60 µg/kg (Figure 7(a)). PK analysis in both treatment cohorts, assessed by maximum observed concentration (Cmax) and area under the concentration-time curve (AUC), showed near dose-proportional systemic exposure, as well as linear PK profiles (Figure 7(a)) for both T-BsAbs.
Figure 7.
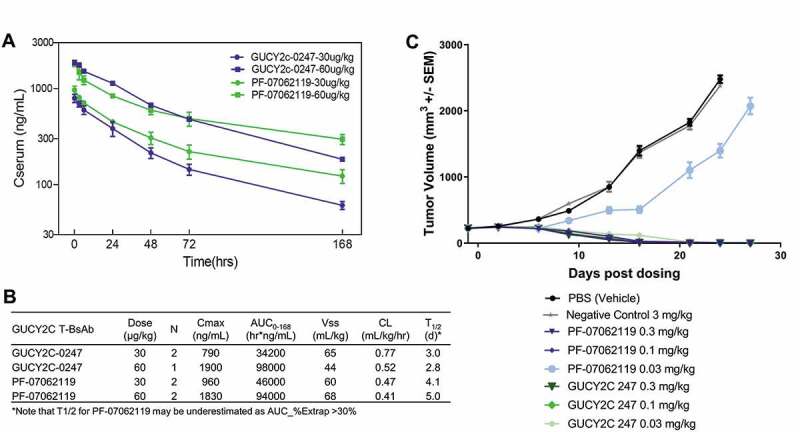
In vivo efficacy and PK of PF-07062119. (a) Mean serum pharmacokinetic profiles of GUCY2C-0247, PF‑06929607 and PF-07062119 in cynomolgus monkeys following single dose IV administration at 30 µg/kg and 60 µg/kg. (b) Results of non-compartmental analysis of the observed PK (0–168 hrs) of GUCY2C-0247 and PF‑07062119. Note that apparent CL estimates may be overestimated, and half-life may be underestimated for PF‑07062119 due to the limited observation period. (c) In vivo efficacy LS1034 adoptive transfer xenograft model demonstrates potent activity down to 0.1 mg/kg for PF-07062119 (n = 10 animals/group). Data shown from one donor. Similar results were obtained from separate experiments with other donor T cells
Notably, although both T-BsAbs exhibited near dose-proportional systemic exposure, PF-07062119 exhibited an improved PK profile (Figure 7(b)). Specifically, based on non-compartmental analysis of 0–168 hr observed PK data, PF-07062119 exhibited a lower apparent CL and higher apparent mean terminal half-life (T1/2 > 4.5 days for PF-07062119 compared to T1/2 ≤ 3 days for GUCY2C-0247). These PK parameters of PF-07062119, unlike GUCY2C-0247, were close to the typical PK profile reported for antibodies in cynomolgus monkeys.48 Although we cannot rule out the possibility that the higher CD3 affinity of GUCY2C-0247 is responsible for its higher apparent clearance via a CD3-mediated drug disposition mechanism, similar to that reported for other CD3 bispecifics, there was no clear indication of non-linear drug disposition for either GUCY2C-CD3 bispecific. Since the combination of linear PK and fast clearance is consistent with a nonspecific binding effect on IgG clearance,32,49 these results suggest that the improved biophysical properties and polyreactivity of PF-07062119 may be responsible for its improved PK.
In vivo efficacy
To assess PF-07062119 activity in vivo, NOD scid gamma (NSG) mice were implanted subcutaneously with the CRC cell line LS1034, which showed GUCY2C expression comparable to that observed in human colorectal tumors with high GUCY2C expression.21 We administered an initial dose of PF-07062119, GUCY2C-0247, a non-targeted-CD3 negative control bispecific, or vehicle to animals with established tumors, along with the adoptive transfer of human T cells, followed by weekly dosing with PF-07062119, GUCY2C-0247, or control agents. In this LS1034 model, PF-07062119 treatment at 0.1 mg/kg led to complete tumor regressions (p-value<0.0001) (Figure 7(c)). The anti-tumor activity was dose-dependent, since partial reduction in tumor volumes was observed at 0.03 mg/kg of PF-07062119. Compared with the parental humanized GUCY2C-0247 in this model, we observed similar activity at all doses except 0.03 mg/kg, where −0247 showed better activity and complete regression, consistent with potency differences observed in vitro and the ~ 7-fold decrease in CD3 affinity between these molecules (H2B4 vs H2B5v6). The decrease in PF-07062119 potency may have been offset by the improvement in the biophysical properties and half-life. Neither the phosphate-buffered saline (PBS) vehicle control nor the non-targeted CD3 bispecific inhibited tumor growth in this model. These data demonstrate potent, GUCY2C-dependent anti-tumor efficacy with PF-07062119. This molecule also showed anti-tumor efficacy in additional cell-line xenograft, patient-derived xenograft, and immunocompetent mouse tumor models, including a cell line representative of lower GUCY2C-expressing human tumors, described in detail by Mathur et al.21
Discussion
The discovery and development of antibody-based biotherapeutics is a challenging, costly, and risky endeavor, often taking over 10 years to achieve commercialization of a product.50,51 Though antibodies are approved by regulatory agencies at twice the rate of small molecule drugs, they are complex biomolecules that require significant effort and resources to address challenges related to safety, efficacy, stability, formulation, and expression and purification.50,52 These challenges are often increased in the context of bispecific antibodies, where two separate antibody domains may not only have their own distinct optimization needs, but also may influence each other’s behavior. Therefore, implementing simultaneous, multidimensional optimization campaigns with early developability screening is essential.6,53 We have shown here the successful use of a multifaceted, high-throughput discovery workflow that allowed for simultaneous identification and optimization of two lead antibodies that make up our bispecific therapeutic candidate.
High thermal stability of an antibody is critical for successful development, as it can affect solubility, aggregation propensity, expression level, and safety related to formation of immunogenic aggregates.8,54–56 The roles of the variable domains and the VH/VL interface in the stability of an antibody have been well documented, particularly in scFv-containing molecules that do not benefit from the compensatory stabilization of the CH1 and CL domains, such as the diabody Fc T-BsAb format.57–59 Many approaches have been described for improving antibody thermal stability, as summarized by McConnell et al.,56 including grafting CDR loops onto stable framework regions,60,61 introducing stabilizing disulfide bonds,62 using in vitro display for random mutagenesis combined with increased thermal pressure,63 and using structure-based computational design.64 More recently, groups have reported combining two or more of these stability optimization approaches, along with optimization of affinity or other properties yielding substantially improved functional and biophysical properties.39,53,56,65,66 Here, we combined structure-guided computational design with framework swapping to optimize the anti-CD3 antibody domains. Transfer of the light chain CDRs of H2B4 from VK4-1 to the VK1-39 framework alone led to an improvement in stability of approximately 5°C as measured by DSC.
For the GUCY2C antibody domains, we combined CDR grafting into stable human frameworks during humanization, computational design based on homology modeling, and random mutagenesis through phage display. These structure-guided phage display libraries were subjected to increased thermal pressure up to 75°C. The key mutations driving improved stability were observed at or near the interface between the VH and VL domains. Interestingly, the VH/VL interface-focused library generated few hits, with most hits coming from the CDR-focused libraries. These individual mutations were identified in the separate CDR soft-randomization libraries, and the CDR shuffling approach allowed us to combine these favorable mutations for superior stability. The resulting anti-GUCY2C domains demonstrated markedly improved thermal stability with an increase in Tm1 as high as 15°C as measured by DSC. We also observed optimal long-term stability at 25°C and low viscosity at concentrations over 100 mg/mL for the final T-BsAb. Improvement in thermal stability may be contributing to these results. Considering that this class of T-BsAb is typically dosed at extremely low concentrations following dilution at time of administration (e.g., 5–28 µg/day for blinatumomab67), the biophysical properties of the GUCY2C T-BsAb are well within the range needed to meet formulation requirements. We demonstrated that applying heat stress during phage selections translated to improved thermal stability of scFv in crude periplasmic extracts, which in turn translated to improved stability of diabody-Fc, and which ultimately translated to a well-behaved final T-BsAb.
As part of the multiparameter optimization, we successfully addressed the manufacturability issue associated with an asparagine deamidation chemical liability. Asparagine deamidation of antibodies in the solvent-exposed CDR regions can lead to reduced function,40 and we demonstrated that deamidation could occur in the CDR H2 of our starting anti-GUCY2C binding domain under stress conditions in vitro. After testing several combinations of mutations targeting the asparagine-glycine motif at H54/H55 in GUCY2C-0247, we identified a glycine to glutamic acid mutation at H55 that appeared to disrupt this deamidation as determined by a reduction in the acidic species and through functional analysis following an accelerated stability study. By removing this liability, lyophilization is not required to minimize the risk of deamidation that may occur during liquid formulation of PF-07062119.
Cleavage of antibodies, particularly in CDR regions, arising during expression can complicate downstream purification processes and lead to reduced activity.68 We identified a low molecular weight degradation product resulting from cleavage of the CDR H2 loop in anti-CD3 H2B4. Changing an arginine residue to a glutamine at H52b (Kabat numbering) resolved this issue, though it remains unclear what mechanism was driving this cleavage.
The efficacy and safety of antibody-based therapeutics are heavily influenced by their PK properties.32 First-generation T-BsAbs such as BiTEs have a short half-life (~ 2 hours) and require administration by constant intravenous (IV) infusion. We have previously demonstrated the correlation of in vitro poly-specificity and self-association assays with the clearance rates of antibodies in human FcRn transgenic mice.32 Using simple, high-throughput DNA and insulin ELISAs69 and AC-SINS assays,70 we were able to monitor the optimized GUCY2C and CD3 hits, selecting for clones demonstrating low polyreactivity and self-association. PF-07062119 showed low scores in both AC-SINS and in polyreactivity ELISAs, significantly below the thresholds correlated with risk of high mAb clearance.32 This correlated with an improved PK profile and increased half-life in cynomolgus monkeys compared with GUCY2C-0247, which had AC-SINS and polyreactivity scores above the clearance threshold, and further suggests that mechanisms connecting nonspecific binding of IgG with increased clearance also apply to the diabody-Fc format. The longer half-life observed in monkeys for PF-07062119 compared to that of BiTEs should enable more convenient dosing regimens in the clinic (once weekly or less).
Immunogenicity of therapeutic proteins results, in part, from intrinsic factors within the protein sequence such as T cell epitopes, which are sequences predicted to be presented on class II MHC for recognition by corresponding TCRs.44 Formation of ADA can negatively impact efficacy and PK, and potentially lead to unwanted adverse events during treatment.44 These safety risks are especially relevant considering the mechanism of action for T-BsAbs and the potential for T cell activation through crosslinking by ADA in the absence of tumor target.71 Here, we used a set of in silico T cell prediction tools to identify and partially remove potential immunogenic epitopes. The immunogenicity score of PF-07062119 was in the range of other antibodies that have shown low incidence of ADA in human clinical trials.45 While this low immunogenicity score may indicate a lower risk of developing ADA, in silico tools provide a risk assessment of only one step in a complex series of events leading to an in vivo immune response, and therefore, the true immunogenicity of PF-07062119 must be determined in clinical trials.
Significant efforts were made to further reduce the remaining putative T cell epitopes. However, mutations beneficial to reducing immunogenicity in CDRs (H1, H2, L3) led to detrimental changes of other properties such as activity and stability. Lead selection required applying appropriate weight to factors influencing key features, such as efficacy, safety, and manufacturability. PK/PD modeling was able to demonstrate that 3- and 7-fold decreases in affinity to the GUCY2C and CD3 binding domains, respectively, were consistent with the targeted efficacy and safety profile for this molecule. Ultimately, maintenance of appropriate affinities along with a good manufacturing profile and suitable predicted PK could be achieved with removal of a substantial subset of predicted T cell epitopes (6 in total), but removal of the remaining sites would have required significant compromises of molecular performance.
The value of the high-throughput protein production system employed in this work was underscored by the need to optimize multiple properties and the observation that optimizing one property can come at the expense of another. For example, removal of predicted T cell epitopes in the anti-CD3 domain led to loss of affinity, and phage selection yielding thermostable GUCY2C domains did not produce hits that altered the H54/H55 asparagine deamidation site despite use of a library designed to do so. The production and testing of over 1600 variants at the 1 mg scale compensated for the relative rarity of designs that achieved a balance among the set of targeted biophysical and functional properties. The availability of protein in formats attuned to specific assays (for example, the rapid thermal stability assay in the scFv-Fc format and affinity measurements in monovalent Fc format) ensured that sequence variants contributing to antibody stability, posttranslational modification, and potential immunogenicity could be evaluated thoroughly. The availability in parallel of proteins in the final therapeutic format – over 500 BsAb were produced for the program – enabled rapid identification of molecules that successfully combined the outputs of multiple strands of optimization. The final result was a well-behaved bispecific antibody suitable for manufacturing and clinical development.
Materials and methods
Identification, characterization and optimization of lead Anti-CD3
A humanized anti-CD3 antibody, H2B4, with specificity to human and cynomolgus monkey CD3ε was generated by mouse immunization followed by humanization into preferred human frameworks as described previously.33 CDR regions of H2B4 VL were grafted into the VK1-39 framework using conventional grafting methods37 and gene synthesis (Blue Heron Bio, Bothell, WA). Following re-grafting of the VL CDRs, mutations were introduced in the CDR regions to improve stability, to reduce proteolytic degradation at or near previously determined cleavage sites, and to reduce potential charge patches within the CDRs.72–74 Mutations were designed and selected using a homology model of an existing x-ray crystal structure of H2B4 Fab complexed to the N-terminal peptide of human CD3ε (data not shown). Discovery Studio 4.0 (Accelrys Inc, San Diego, CA) was used to design high-affinity, stable variants. Additionally, stability calculations were performed using scripts and methods described along with the program FoldX 3.0 Beta 6.1 c (see supplementary methods).75–77
Generation of T-BsAbs with an optimized anti-CD3
Variants of anti-CD3 H2B4, termed H2B5, were synthesized and sub-cloned as before into proprietary mammalian expression vectors containing either anti-tumor or negative control VH or VL domains and “knobs-into-holes” heterodimerization-promoting domains.26,27 H2B5 VH domains were fused in frame with an anti-tumor (or negative control) VL domain, separated by a glycine-serine linker (GGGSGGGG), followed by a cysteine-containing linker (GCPPCP), connecting to the knob Fc chain. Similarly, H2B5 VL domains were fused in frame with a glycine-serine linker (GGGSGGGG or GGGGSGGGG), an anti-tumor (or negative control) VH domain, followed by a cysteine-containing linker, GCPPCP, and a hole Fc chain. The Fc-knob and Fc-hole domains are derived from human IgG1 and contain mutations in the CH2 region to reduce antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity.28–30 The negative control used here was an inert, germline antibody in VH3-23/VK1-39 frameworks. DNA was prepared as above.
AC-SINS assay and DNA and insulin ELISA
To assess self-interaction and polyspecificity potential of anti-CD3 antibodies, two methods were used. The affinity capture self-interaction nanoparticle spectroscopy method was adapted from Liu et al.,70 and samples were characterized in this assay as previously described.32 The DNA and insulin polyspecificity assays were performed as previously described.32
Immunization and hybridoma generation of mouse anti-human GUCY2C antibodies
The 300.19 cells over-expressing hu GUCY2C (300.19/huGUCY2C) were injected as immunogens into eight-week old female Balb/c mice for the generation of hybridomas. Balb/c mice were immunized with 5 × 106 300.19/huGUCY2C cells twice per week for one month without adjuvant through intraperitoneal (IP) injection. To determine the anti-GUCY2C titers, test bleeds were collected from immunized animals and the anti-GUCY2C specific titers were examined by ELISA on huGUCY2C-mIgG2a-Fc recombinant protein and cynomolgus monkey GUCY2C-mIgG2a-Fc. Splenocytes from the animals with the highest anti-GUCY2C titers were fused with P3X myeloma at 1:1 ratio using polyethylene glycol (PEG). Fused cells were selected in the presence of hypoxanthine-aminopterin-thymidine (HAT) media for seven days, after which the hybridomas were maintained in HT-containing media prior to screening. To identify anti-huGUCY2C-specific hybridomas, hybridoma supernatants were screened for anti-huGUCY2C IgG reactivity by ELISA. Hybridomas were analyzed for reactivity against 300.19/hu and cynomolgus GUCY2C cells and primary tumor lines expressing GUCY2C and supernatants with confirmed cell surface binding activity were sub-cloned into mouse IgG1 format (see supplementary methods),
Characterization of GUCY2C T-BsAbs binding to human T cells and tumor cells and cytotoxic T lymphocyte (CTL) assay
Binding activity of GUCY2C T-BsAbs to T cells and GUCY2C expressing tumor cells was analyzed by flow cytometry as previously described.20 T cell-dependent cytotoxicity of GUCY2C T-BsAbs on colorectal tumor cell lines endogenously expressing GUCY2C and engineered to express firefly luciferase was performed as previously described.21
Thermal challenge “cook-and-bind” ELISA for characterizing optimized GUCY2C antibodies
To evaluate improved thermal stability of the optimized variants, either as peripreps or as purified proteins, samples were assessed for binding activity following a thermal challenge at elevated temperatures in a PCR block. Greiner Bio-One 384-well white ELISA plates (Greiner Bio-One, Cat. no. 781074) were coated with 1 µg/mL of target protein in coating buffer (25 mM Na2CO3, 75 mM NaHCO3 pH 9.6) O/N at 4°C. Plates were washed 3 times with PBS + 0.05% Tween 20 and then blocked with blocking buffer (PBS plus 3% milk) for 1 hour at room temperature (RT) with shaking. Separately, dilution plates of periprep (at a single dilution) or purified proteins were prepared at either 1 µg/mL or EC80 of recombinant protein binding at room temperature in blocking buffer, and 100 µL were transferred to a 96-well PCR plate. The PCR plate containing 100 µL diluted protein was then heated at the desired temperatures (generally at 60°, 65°, 70° or 75°C) for 30 minutes. The plates were then allowed to cool for 15 minutes at room temperature and centrifuged at 4000 rpm for 10 minutes. The blocked ELISA plates were emptied and 20 µL of the thermally challenged protein supernatant was added per well, then incubated for 1 hour, shaking at RT. The plates were washed 3 times as before and 20 µL of an anti-human IgG-Europium conjugate reagent protein (Perkin Elmer, Cat. no. 1244–330) diluted 1:1000 in DELFIA assay buffer (Perkin Elmer 4002–0010) or an anti-pentaHis antibody (Qiagen, Cat. no. 34660) diluted 1:1000 in blocking buffer for peripreps was added per well and incubated at RT for 1 hour with shaking. For peripreps, an additional step was required, where 20 µL per well of anti-mouse Europium conjugate (Perkin Elmer, Cat. no. AD0207) diluted 1:500 in DELFIA assay buffer was added. The plates were washed 3x with 1x DELFIA Wash Solution (Perkin Elmer, Cat. no. 1244–114) and 20 µl of Enhancement solution (Perkin Elmer, Cat. no. 1244–105) was added to each well and incubated for 30 minutes. The plates were read on an Envision plate reader for time-resolved fluorescence with excitation at 320 nm and emission at 615 nm following the manufacturer’s methods. For purified proteins, the temperature where 50% of activity is retained is reported as T50.
Humanization of mouse anti-GUCY2C antibody
Humanization of mouse anti-GUCY2C variable regions was performed using a CDR-graft strategy as described previously for the anti-CD3 antibody. Briefly, cDNAs containing human acceptor framework, VH3-7 for heavy chain and VK1-39 for light chain, with anti-GUCY2C CDR donor sequences were synthesized in vectors containing either human IgG1 constant region for the heavy chain, or human kappa constant region for the light chain. To recover binding activity, back-mutations to mouse sequence in the framework regions of both the VH and VL domains were introduced. Proteins were expressed and purified as IgG and diabody-Fc as before, then assessed for binding activity by direct or competition ELISA, and CTL assay as described above.
Optimization of humanized anti-GUCY2C antibody
Structure-guided rational optimization
The humanized GUCY2C antibody and T-BsAb demonstrated poor thermal stability, high polyreactivity scores, a high AC-SINS score and an undesirable in silico immunogenicity score. Additionally, a potential deamidation site (NG) was identified in CDRH2. To address these liabilities, sequence and crystal structure model-guided methods were used to identify and selectively mutate the humanized anti-GUCY2C antibody. Rational variants were identified and mutations were designed using similar methods described above for the anti-CD3 antibody. Rationally designed variants were synthesized as before and proteins in monovalent and bivalent IgG format, and diabody-Fc format were generated using high-throughput methods described above.
Library-based optimization of humanized GUCY2C antibody using phage display
Three structure-guided phage display approaches were also employed to optimize the anti-GUCY2C antibody. In one approach, focusing on improving stability and reducing charge patches that were potentially causing high polyreactivity, individual CDRs were soft-randomized. Here, oligos covering four amino acid stretches within the CDRs were designed such that 50% wild-type sequence and 50% random sampling were allowed at each CDR position. A second approach taken to improve stability focused on introducing mutations at the VH-VL interface, where H35, H100e, L34, L89, L91 and L96 were specifically targeted. Soft-randomization oligos were again designed to allow 50% wild-type and 50% random sequence at these positions. A third phage display approach was taken to remove the deamidation site (NG) in CDRH2 where oligos were designed to introduce specific amino acid changes at positions H54 and H55. Oligos for each approach were synthesized, gel purified and phosphorylated at a vendor (Integrated DNA Technologies, Coralville, IA). Uracil-containing single stranded template DNA of humanized GUCY2C antibody in scFv format (VH-GGGGSGGGGSGGGGSG-VL) in a proprietary phage display vector, pWRIL1,78 was prepared and mutagenesis was performed as previously described.79 In total, eight libraries were prepared, six representing each one of the CDR soft-randomization libraries, and one each for the VH-VL interface library and the deamidation library.
Phage display libraries were transformed into E. coli TG1 cells (Lucigen, Cat. no. 60502–1), and phage was rescued and assessed for library size and sequence diversity as previously described.78,80 Solution-based phage selections of individual libraries were performed over three rounds using biotinylated human (rounds 1 and 3) and cynomolgus (round 2) GUCY2C with decreasing target concentration after each round. As affinity optimization was not required for this selection campaign, target concentration was gently reduced from 10 nM in round 1 to 1 nM in rounds 2 and 3. To improve stability, thermal challenges were performed at 60°C in round 2 and 65°C in round 3 (see supplementary methods). Following initial screening for retained binding activity and improved stability, phage outputs containing clones demonstrating desired properties were used to generate new multi-site directed mutagenesis oligos for a combined CDR shuffling library (see supplementary methods). Clones were picked and screened as described above.
Immunogenicity de-risking of anti-CD3 and anti GUCY2C antibodies
To minimize the potential for immunogenicity arising through T cell-dependent responses, efforts were made to reduce the antigenic potential of the anti-CD3 and anti-GUCY2C antibody sequences using in silico methods described below. Amino acid sequences were analyzed using two methods to identify potential T cell epitopes. Sequences (9-mers) flagged by these methods were considered an epitope. Sequences were analyzed for EpiMatrix analysis using ISPRI software (ISPRI v 1.8.0, EpiVax Inc, Providence, RI; see ref. 41), which provides rankings of likelihood of binding of each 9-mer amino acid fragment against 8 different HLA types. A second method analyzed sequences using the MHC-II binding Consensus method42,43 in IEDB (IEDB MHC-II Binding Predictions), which provides ranking of likelihood of binding of 9-mers and 15-mers against 8 HLA types. Each epitope determined by these methods was classified as a germline or non-germline epitope, then further classified based on its location within the antibody (CDR or non-CDR). Non-germline epitopes were targeted for either rational mutagenesis or soft-randomization mutagenesis methods described previously to replace residues that are predicted to lead to high HLA binding with residues predicted to reduce HLA binding. Since these potential mutations may also affect binding and stability, care was taken to consider affinity and stability data predictions described above to identify and select tolerated mutations.
Additionally, for humanized GUCY2C antibody, a small, focused phage display library was generated using 11 stability-optimized variants from the previous selections as templates. Mutagenic oligos introducing amenable amino acid changes in potential immunogenic hotspots in FW L1 and CDRs L1, L2, L3 and H2 were generated using structural modeling and in silico immunogenicity methods described above. Some stability optimized templates still retained the putative deamidation site in H2 following initial optimization, so mutagenic oligos targeting this position were included in the library build. Phage display libraries were prepared and selections were performed as described previously. Hits demonstrating retained activity and thermal stability were selected for conversion to diabody-Fc format and expressed and purified as before.
Pharmacokinetic measurements in cynomolgus monkeys
The PK of PF-07062119 and GUCY2C-0247 were assessed in cynomolgus monkeys as part of studies approved by Pfizer Institutional Animal Care and Use Committees, conducted in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited institution, and conducted in accordance with the current guidelines for animal welfare.81,82 In each study, cynomolgus monkeys were administered PF-07062119 or GUCY2C-0247 at 30 or 60 ug/kg by IV bolus injection.
A Meso-Scale Discovery (MSD; Meso Scale Diagnostics, Rockville, MD) assay was used to measure the PK of PF-07062119 and GUCY2C-0247 in cynomolgus monkey serum. Biotinylated goat anti-human IgG was bound to pre-blocked streptavidin-coated plates. After washing, samples containing drug were added. After 1 hr incubation and washing, a ruthenylated mouse anti-human IgG Fc was added. After 1 hr incubation and a final wash, tripropylamine was added to each well and the amount of bound bispecific was measured based on the amount electro-chemiluminescent signal within the MSD instrument. Sample concentrations were determined by interpolation from a standard curve fit using a 4-parameter logistic equation weighted by 1/y^2. The dynamic range of quantification in 100% serum was 20 ng/mL to 2560 ng/mL for both drugs. The PK parameters were determined from individual animal data using non-compartmental analysis in Watson LIMS (Version 7.5, Thermo, Inc. Philadelphia, PA). Concentrations below the limit of quantitation (BLQ) were not used in the calculations. In addition, PK data was also analyzed using a 2-compartment PK model with linear elimination from the central compartment using Phoenix 64® Win Non Lin® (Certara L.P.).
In vivo evaluation of GUCY2C T-BsAb mediated activity
In vivo efficacy studies were performed using the adoptive transfer model in NSG mice. Mice were randomized and staged at tumor size of 150–200 mm3. An initial dose of GUCY2C T-BsAb, a non-targeted CD3 bispecific control, or PBS (vehicle) was administered to animals on Day 0, and 2 × 106 cultured activated pan human T cells (containing CD8 and CD4 T cells) were inoculated the following day using methods previously described.21 Mice were dosed in 0.2 mL bolus injections weekly up to 3 doses. Tumor measurements were collected using Vernier caliper, and volumes were calculated by use of the modified ellipsoid formula ½ x length x width.2
Statistical analyses
Statistical analysis of in vitro and in vivo experiments was performed using GraphPad Prism (verson 8.02). Tumor volume data was log-transformed after an adjustment was made for zero tumor size. A separate ANOVA analysis using all groups was performed for each day. From each analysis, a one-sided p-value from a Student’s T pairwise comparison of each group with the control was reported.
Supplementary Material
Acknowledgments
The authors are grateful to the following individuals for their contributions to this work:
Arjun Bollampalli, Ying Sun and Mike Gallo for phage and hybridoma molecular biology support; Linette Rodriguez, Evan Mahan, Monika Musial-Siwek, Darren Ferguson, Xiaotian Zhong and Richard Zollner for their work supporting the expression and purification of the many thousands of antibodies and bispecifics; Matthew Lambert for consultation around phage library design and characterization; Lisa Racie, Liz DiBlasio-Smith and Chris Corcoran for their support with cell line and target protein generation; Wei Fang, Bozena Bugaj-Gaweda and Xingzhi Tan for in vitro characterization of the many T-BsAbs; Jessica Kearney for in vivo efficacy support; and Eric Bennett, Joel Bard and Joe DeBartolo for bioinformatics support.
Funding Statement
All research was funded by Pfizer Inc.
Disclosure of potential conflicts of interest
At the time the research was performed, all authors were employees of Pfizer Worldwide Research & Development. No potential conflicts of interest are disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Abbreviations
- AC-SINS
Affinity-capture self-interaction nanoparticle spectroscopy
- ADA
Anti-drug antibody
- AUC
Area under curve
- BiTE
Bispecific T cell engager
- BsAb
Bispecific antibody
- CDR
Complementarity-determining region
- CHO
Chinese hamster ovary
- cIEF
Capillary isoelectric focusing
- CL
Clearance
- CRC
Colorectal cancer
- CTL
Cytotoxic T Lymphocyte
- DELFIA
Dissociation‑enhanced lanthanide fluorescence immunoassay
- DSC
Differential scanning calorimetry
- FcRn
Neonatal Fc-receptor
- GUCY2C
Guanylate cyclase 2c
- HMMS
High molecular mass species
- IEDB
Immune epitope database
- ITA
Isothermal assembly
- mAb
Monoclonal antibody
- MS
Mass spectrometry
- NHP
Non-human primates
- NSG
Nod scid gamma
- PD
Pharmacodynamics
- PK
Pharmacokinetics
- pMHC
Peptide-major histocompatibility complex
- SPR
Surface plasmon resonance
- SSI
Site-specific integration
- STa
Heat stable enterotoxin a
- t1/2
Half-life
- T-BsAb
T cell bispecific antibody
- Tg
Transgenic
- TMDD
Target-mediated drug disposition
References
- 1.Wu Z, Cheung NV.. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther. 2018;182:161–18. PMID: 28834699182. doi: 10.1016/j.pharmthera.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69(12):4941–44. PMID: 19509221. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- 3.Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18(8):585–608. PMID: 31175342. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs PMID: 28071970. 2017;9(2):182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci. 2015;104(6):1885–98. PMID: 25821140. doi: 10.1002/jps.24430. [DOI] [PubMed] [Google Scholar]
- 6.Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, Tanaka E, Muto A, Kojima T, Kitazawa T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One. 2013;8(2):e57479. PMID: 23468998. doi: 10.1371/journal.pone.0057479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fennell BJ, McDonnell B, Tam AS, Chang L, Steven J, Broadbent ID, Gao H, Kieras E, Alley J, Luxenberg D, et al. CDR-restricted engineering of native human scFvs creates highly stable and soluble bifunctional antibodies for subcutaneous delivery. MAbs. 2013;5(6):882–95. PMID: 23995618. doi: 10.4161/mabs.26201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114(5):944–49. PMID: 28096333. doi: 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore PA, Shah K, Yang Y, Alderson R, Roberts P, Long V, Liu D, Li JC, Burke S, Ciccarone V, et al. Development of MGD007, a gpA33 x CD3-bispecific DART protein for T-cell immunotherapy of metastatic colorectal cancer. Mol Cancer Ther. 2018;17(8):1761–72. PMID: 29866746. doi: 10.1158/1535-7163.MCT-17-1086. [DOI] [PubMed] [Google Scholar]
- 10.Root AR, Cao W, Li B, LaPan P, Meade C, Sanford J, Jin M, O’Sullivan C, Cummins E, Lambert M, et al. Development of PF-06671008, a highly potent anti-P-cadherin/anti-CD3 bispecific DART molecule with extended half-life for the treatment of cancer. Antibodies (Basel). 2016;5(1):6. PMID: 31557987. doi: 10.3390/antib5010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, Godwin JE, Arellano ML, Sweet K, Emadi A, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. 2020. September 14. Online ahead of print. PMID: 32929488. doi: 10.1182/blood.2020007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, Diaz LA Jr.. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16(6):361–75. PMID: 30886395. doi: 10.1038/s41575-019-0126-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. PMID: 26742998. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 14.Birbe R, Palazzo JP, Walters R, Weinberg D, Schulz S, Waldman SA. Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum Pathol. 2005;36(2):170–79. PMID: 15754294. doi: 10.1016/j.humpath.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Danaee H, Kalebic T, Wyant T, Fassan M, Mescoli C, Gao F, Trepicchio WL, Rugge M. Consistent expression of guanylyl cyclase-C in primary and metastatic gastrointestinal cancers. PLoS One. 2017. December 19;12(12):e0189953. PMID: 29261789. doi: 10.1371/journal.pone.0189953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Currie MG, Fok KF, Kato J, Moore RJ, Hamra FK, Duffin KL, Smith CE. Guanylin: an endogenous activator of intestinal guanylate cyclase. Proc Natl Acad Sci U S A. 1992;89(3):947–51. PMID: 1346555. doi: 10.1073/pnas.89.3.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamra FK, Forte LR, Eber SL, Pidhorodeckyj NV, Krause WJ, Freeman RH, Chin DT, Tompkins JA, Fok KF, Smith CE, et al. Uroguanylin: structure and activity of a second endogenous peptide that stimulates intestinal guanylate cyclase. Proc Natl Acad Sci U S A. 1993;90(22):10464–68. PMID: 7902563. doi: 10.1073/pnas.90.22.10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulz S, Green CK, Yuen PS, Garbers DL. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell. 1990;63(5):941–48. PMID: 1701694. doi: 10.1016/0092-8674(90)90497-3. [DOI] [PubMed] [Google Scholar]
- 19.Magee MS, Kraft CL, Abraham TS, Baybutt TR, Marszalowicz GP, Li P, Waldman SA, Snook AE. GUCY2C-directed CAR-T cells oppose colorectal cancer metastases without autoimmunity. OncoImmunology. 2016;5(10):e1227897. PMID:27853651. doi: 10.1080/2162402X.2016.1227897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almhanna K, Prithviraj GK, Veiby P, Kalebic T. Antibody–drug conjugate directed against the guanylyl cyclase antigen for the treatment of gastrointestinal malignancies. Pharmacol Ther. 2017;170:8–13. PMID: 27765652. doi: 10.1016/j.pharmthera.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Mathur D, Root AR, Bugaj-Gaweda B, Bisulco S, Tan X, Fang W, Kearney JC, Lucas J, Guffroy M, Golas J, et al. A novel GUCY2C-CD3 T cell engaging bispecific construct (PF-07062119) for the treatment of gastrointestinal cancers. Clin Cancer Res. 2020;26(9):2188–202. PMID: 31996389. doi: 10.1158/1078-0432.CCR-19-3275. [DOI] [PubMed] [Google Scholar]
- 22.Chang CS, Guntas G, Katragadda M, Mathur D, Root AR, Mosyak L, LaVallie ER; Pfizer, Inc., assignee . Antibodies specific for GUCY2C and uses thereof. United States patent application 2019/417,863. 2019. May 21.
- 23.Apgar JR, Jin F, Katragadda M, Mathur D, Tchistiakova LG; Pfizer, Inc., assignee . Antibodies specific for CD3 and uses thereof. United States patent application 2019/417,589. 2019. May 21.
- 24.Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, Kufer P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother. 2010;59(8):1197–209. PMID: 20309546. doi: 10.1007/s00262-010-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore GL, Lee SH, Schubbert S, Miranda Y, Rashid R, Pong E, Phung S, Chan EW, Chen H, Endo N, et al. Tuning T cell affinity improves efficacy and safety of anti-CD38 × anti-CD3 bispecific antibodies in monkeys - a potential therapy for multiple myeloma. Blood. 2015;126231798. doi: 10.1182/blood.V126.23.1798.1798. [DOI] [Google Scholar]
- 26.Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9(7):617–21. PMID: 8844834. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- 27.Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270(1):26–35. PMID: 9231898. doi: 10.1006/jmbi.1997.1116. [DOI] [PubMed] [Google Scholar]
- 28.Kasaian MT, Raible D, Marquette K, Cook TA, Zhou S, Tan XY, Tchistiakova L. IL-13 antibodies influence IL-13 clearance in humans by modulating scavenger activity of IL-13Ralpha2. J Immunol. 2011;187(1):561–69. PMID: 21622864. doi: 10.4049/jimmunol.1100467. [DOI] [PubMed] [Google Scholar]
- 29.Lund J, Pound JD, Jones PT, Duncan AR, Bentley T, Goodall M, Levine BA, Jefferis R, Winter G. Multiple binding sites on the CH2 domain of IgG for mouse Fc gamma RII. Mol Immunol. 1992;29(1):53–59. PMID: 1530984. doi: 10.1016/0161-5890(92)90156-r. [DOI] [PubMed] [Google Scholar]
- 30.Canfield SM, Morrison SL. The binding affinity of human IgG for its high affinity Fc receptor is determined by multiple amino acids in the CH2 domain and is modulated by the hinge region. J Exp Med. 1991;173(6):1483–91. PMID: 1827828. doi: 10.1084/jem.173.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strop P, Ho WH, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, Rickert M, Appah CT, Pascua E, Radcliffe T, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol. 2012;420(3):204–19. PMID: 22543237. doi: 10.1016/j.jmb.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 32.Avery LB, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, Kavosi MS, Penn S, Cirelli D, Kurz JC, et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs. 2018;10(2):244–55. PMID: 29271699. doi: 10.1080/19420862.2017.1417718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeung YA, Krishnamoorthy V, Dettling D, Sommer C, Poulsen K, Ni I, Pham A, Chen W, Liao-Chan S, Lindquist K, et al. An optimized full-length FLT3/CD3 bispecific antibody demonstrates potent anti-leukemia activity and reversible hematological toxicity. Mol Ther. 2020;28(3):889–900. PMID: 31981494. doi: 10.1016/j.ymthe.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiller T, Schuster I, Deppe D, Siegers K, Strohner R, Herrmann T, Berenguer M, Poujol D, Stehle J, Stark Y, et al. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. MAbs. 2013;5(3):445–70. PMID: 23571156. doi: 10.4161/mabs.24218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Apgar JR, Mader M, Agostinelli R, Benard S, Bialek P, Johnson M, Gao Y, Krebs M, Owens J, Parris K, et al. Beyond CDR-grafting: structure-guided humanization of framework and CDR regions of an anti-myostatin antibody. MAbs. 2016;8(7):1302–18. PMID: 27625211. doi: 10.1080/19420862.2016.1215786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Betts A, Haddish-Berhane N, Shah DK, van der Graaf PH, Barletta F, King L, Clark T, Kamperschroer C, Root A, Hooper A, et al. A translational quantitative systems pharmacology model for CD3 bispecific molecules: application to quantify T cell-mediated tumor cell killing by P-cadherin LP DART(®). Aaps J. 2019;21(4):66. PMID: 31119428. doi: 10.1208/s12248-019-0332-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321(6069):522–25. PMID: 3713831. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- 38.Michaelson JS, Demarest SJ, Miller B, Amatucci A, Snyder WB, Wu X, Huang F, Phan S, Gao S, Doern A, et al. Anti-tumor activity of stability-engineered IgG-like bispecific antibodies targeting TRAIL-R2 and LTβR. MAbs. 2009;1(2):128–41. PMID: 20061822. doi: 10.4161/mabs.1.2.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller BR, Demarest SJ, Lugovskoy A, Huang F, Wu X, Snyder WB, Croner LJ, Wang N, Amatucci A, Michaelson JS, et al. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng Des Sel. 2010;23(7):549–57. PMID: 20457695. doi: 10.1093/protein/gzq028. [DOI] [PubMed] [Google Scholar]
- 40.Vlasak J, Bussat MC, Wang S, Wagner-Rousset E, Schaefer M, Klinguer-Hamour C, Kirchmeier M, Corvaïa N, Ionescu R, Beck A. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal Biochem. 2009. September 15;392(2):145–54. PMID: 19497295. doi: 10.1016/j.ab.2009.05.043. [DOI] [PubMed] [Google Scholar]
- 41.Schafer JR, Jesdale BM, George JA, Kouttab NM, De Groot AS. Prediction of well-conserved HIV-1 ligands using a matrix-based algorithm, EpiMatrix. Vaccine. 1998;16(19):1880–84. PMID: 9795396. doi: 10.1016/s0264-410x(98)00173-x. [DOI] [PubMed] [Google Scholar]
- 42.Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008. April 4;4(4):e1000048. PMID: 18389056. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010;11:568. PMID: 21092157. doi: 10.1186/1471-2105-11-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Walle I, Gansemans Y, Parren PW, Stas P, Lasters I. Immunogenicity screening in protein drug development. Expert Opin Biol Ther PMID: 17309332. 2007;7(3):405–18. doi: 10.1517/14712598.7.3.405. [DOI] [PubMed] [Google Scholar]
- 45.Cousens L, Terry F, Ardito M, Martin W, De Groot A. In silico high throughput pre-clinical determination of monoclonal antibody immunogenicity (P3391). J Immunol. 2013;190:135.27. [Google Scholar]
- 46.Zhang L, Inniss MC, Han S, Moffat M, Jones H, Zhang B, Cox WL, Rance JR, Young RJ. Recombinase-mediated cassette exchange (RMCE) for monoclonal antibody expression in the commercially relevant CHOK1SV cell line. Biotechnol Prog. 2015;31(6):1645–56. doi: 10.1002/btpr.2175. [DOI] [PubMed] [Google Scholar]
- 47.Hui GK, Gardener AD, Begum H, Eldrid C, Thalassinos K, Gor J, Perkins SJ. The solution structure of the human IgG2 subclass is distinct from those for human IgG1 and IgG4 providing an explanation for their discrete functions. J Biol Chem. 2019;294(28):10789–806. PMID: 31088911. doi: 10.1074/jbc.RA118.007134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Betts A, Keunecke A, van Steeg TJ, van der Graaf PH, Avery LB, Jones H, Berkhout J. Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: A comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. MAbs. 2018;10(5):751–64. PMID: 29634430. doi: 10.1080/19420862.2018.1462429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones HM, Zhang Z, Jasper P, Luo H, Avery LB, King LE, Neubert H, Barton HA, Betts AM, Webster R. A physiologically-based pharmacokinetic model for the prediction of monoclonal antibody pharmacokinetics from in vitro data. CPT Pharmacometrics Syst Pharmacol. 2019;8(10):738–47. PMID: 31464379. doi: 10.1002/psp4.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang X, Xu W, Dukleska S, Benchaar S, Mengisen S, Antochshuk V, Cheung J, Mann L, Babadjanova Z, Rowand J, et al. Developability studies before initiation of process development. MAbs. 2013;5(5):787–94. PMID: 23883920. doi: 10.4161/mabs.25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Y, Wang D, Mason B, Rossomando T, Li N, Liu D, Cheung JK, Xu W, Raghava S, Katiyar A, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11(2):239–64. PMID: 30543482. doi: 10.1080/19420862.2018.1553476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11(2):219–38. PMID: 30516432. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu L, Kohli N, Rennard R, Jiao Y, Razlog M, Zhang K, Baum J, Johnson B, Tang J, Schoeberl B, et al. Rapid optimization and prototyping for therapeutic antibody-like molecules. MAbs. 2013;5(2):237–54. PMID: 23392215. doi: 10.4161/mabs.23363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun. 2007;355(3):751–57. PMID: 17321501. doi: 10.1016/j.bbrc.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 55.Lee CC, Perchiacca JM, Tessier PM. Toward aggregation-resistant antibodies by design. Trends Biotechnol. 2013;31(11):612–20. PMID: 23932102. doi: 10.1016/j.tibtech.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 56.McConnell AD, Zhang X, Macomber JL, Chau B, Sheffer JC, Rahmanian S, Hare E, Spasojevic V, Horlick RA, King DJ, et al. A general approach to antibody thermostabilization. MAbs. 2014;6(5):1274–82. PMID: 25517312. doi: 10.4161/mabs.29680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wörn A, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305(5):989–1010. PMID: 11162109. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- 58.Ewert S, Huber T, Honegger A, Plückthun A. Biophysical properties of human antibody variable domains. J Mol Biol. 2003;325(3):531–53. PMID: 12498801. doi: 10.1016/s0022-2836(02)01237-8. [DOI] [PubMed] [Google Scholar]
- 59.Röthlisberger D, Honegger A, Plückthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol. 2005;347(4):773–89. PMID: 15769469. doi: 10.1016/j.jmb.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 60.Jung S, Plückthun A. Improving in vivo folding and stability of a single-chain Fv antibody fragment by loop grafting. Protein Eng. 1997;10(8):959–66. PMID: 9415446. doi: 10.1093/protein/10.8.959. [DOI] [PubMed] [Google Scholar]
- 61.Ewert S, Honegger A, Plückthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods. 2004;34(2):184–99. PMID: 15312672. doi: 10.1016/j.ymeth.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 62.Reiter Y, Brinkmann U, Lee B, Pastan I. Engineering antibody Fv fragments for cancer detection and therapy: disulfide-stabilized Fv fragments. Nat Biotechnol. 1996;14(10):1239–45. PMID: 9631086. doi: 10.1038/nbt1096-1239. [DOI] [PubMed] [Google Scholar]
- 63.Jung S, Honegger A, Plückthun A. Selection for improved protein stability by phage display. J Mol Biol. 1999;294(1):163–80. PMID: 10556036. doi: 10.1006/jmbi.1999.3196. [DOI] [PubMed] [Google Scholar]
- 64.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci U S A. 2009;106(29):11937–42. PMID: 19571001. doi: 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lehmann A, Wixted JH, Shapovalov MV, Roder H, Dunbrack RL Jr, Robinson MK. Stability engineering of anti-EGFR scFv antibodies by rational design of a lambda-to-kappa swap of the VL framework using a structure-guided approach. MAbs. 2015;7(6):1058–71. PMID: 26337947. doi: 10.1080/19420862.2015.1088618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Benschop RJ, Chow CK, Tian Y, Nelson J, Barmettler B, Atwell S, Clawson D, Chai Q, Jones B, Fitchett J, et al. Development of tibulizumab, a tetravalent bispecific antibody targeting BAFF and IL-17A for the treatment of autoimmune disease. MAbs. 2019;11(6):1175–90. PMID: 31181988. doi: 10.1080/19420862.2019.1624463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 2018;17(3):197–223. PMID: 29192287. doi: 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- 68.Ivleva VB, Schneck NA, Gollapudi D, Arnold F, Cooper JW, Lei QP. Investigation of sequence clipping and structural heterogeneity of an HIV broadly neutralizing antibody by a comprehensive LC-MS analysis. J Am Soc Mass Spectrom PMID: 29736600. 2018;29(7):1512–23. doi: 10.1007/s13361-018-1968-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. 2008;329(1–2):112–24. PMID: 17996249. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T, Reid F, Cao Y, Estep P, Yu Y, et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs. 2014;6(2):483–92. PMID: 24492294. doi: 10.4161/mabs.27431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krishna M, Nadler SG. Immunogenicity to biotherapeutics - the role of anti-drug immune complexes. Front Immunol. 2016;7:21. eCollection 2016 PMID: 26870037. doi: 10.3389/fimmu.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li L, Li C, Sarkar S, Zhang J, Witham S, Zhang Z, Wang L, Smith N, Petukh M, Alexov E. DelPhi: a comprehensive suite for DelPhi software and associated resources. BMC Biophys. 2012;5:9. PMID: 22583952. doi: 10.1186/2046-1682-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rocchia W, Sridharan S, Nicholls A, Alexov E, Chiabrera A, Honig B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J Comput Chem. 2002;23(1):128–37. PMID: 11913378. doi: 10.1002/jcc.1161. [DOI] [PubMed] [Google Scholar]
- 74.Rocchia W, Alexov E, Honig B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J Phys Chem B. 2001;105:6507–14. doi: 10.1021/jp010454y. [DOI] [Google Scholar]
- 75.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320(2):369–87. PMID: 12079393. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 76.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(suppl_2):W382–W388. PMID: 15980494. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sirin S, Apgar JR, Bennett EM, Keating AE. AB-Bind: antibody binding mutational database for computational affinity predictions. Protein Sci. 2016;25(2):393–409. PMID: 26473627. doi: 10.1002/pro.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Finlay WJ, Cunningham O, Lambert MA, Darmanin-Sheehan A, Liu X, Fennell BJ, Mahon CM, Cummins E, Wade JM, O’Sullivan CM, et al. Affinity maturation of a humanized rat antibody for anti-RAGE therapy: comprehensive mutagenesis reveals a high level of mutational plasticity both inside and outside the complementarity-determining regions. J Mol Biol. 2009;388(3):541–58. PMID: 19285987. doi: 10.1016/j.jmb.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 79.Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–39. PMID: 1943776. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 80.Finlay WJ, Bloom L, Cunningham O. Optimized generation of high-affinity, high-specificity single-chain Fv antibodies from multiantigen immunized chickens. Methods Mol Biol. 2011;681:383–401. PMID: 20978977. doi: 10.1007/978-1-60761-913-0_21. [DOI] [PubMed] [Google Scholar]
- 81.National Research Council . Guide for the care and use of laboratory animals. 8th ed. Washington (DC): The National Academies Press; 2011. PMID: 21595115. doi: 10.17226/12910. [DOI] [Google Scholar]
- 82.Jendeberg L, Nilsson P, Larsson A, Denker P, Uhlén M, Nilsson B, Nygren PA. Engineering of Fc(1) and Fc(3) from human immunoglobulin G to analyse subclass specificity for staphylococcal protein A. J Immunol Methods. 1997;201(1):25–34. PMID: 9032407. doi: 10.1016/s0022-1759(96)00215-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.