

ABSTRACT
The role of the inflammation-silencing ribonuclease, MCPIP1 (monocyte chemoattractant protein-induced protein 1), in neoplasia continuous to emerge. The ribonuclease can cleave not only inflammation-related transcripts but also some microRNAs (miRNAs) and viral RNAs. The suppressive effect of the protein has been hitherto suggested in breast cancer, clear cell renal cell carcinoma, osteosarcoma, and neuroblastoma. Our previous results have demonstrated a reduced levels of several oncogenes, as well as inhibited growth of neuroblastoma cells upon MCPIP1 overexpression. Here, we investigate the mechanisms underlying the suppression of MYCN proto-oncogene, bHLH transcription factor (MYCN)-amplified neuroblastoma cells overexpressing the MCPIP1 protein. We showed that the levels of several transcripts involved in cell cycle progression decreased in BE(2)-C and KELLY cells overexpressing MCPIP1 in a ribonucleolytic activity-dependent manner. However, RNA immunoprecipitation indicated that only AURKA mRNA (encoding for Aurora A kinase) interacts with the ribonuclease. Furthermore, the application of a luciferase assay suggested MCPIP1-dependent destabilization of the transcript. Further analyses demonstrated that the entire conserved region of AURKA seems to be indispensable for the interaction with the MCPIP1 protein. Additionally, we examined the effect of the ribonuclease overexpression on the miRNA expression profile in MYCN-amplified neuroblastoma cells. However, no significant alterations were observed. Our data indicate a key role of the binding and cleavage of the AURKA transcript in an MCPIP1-dependent suppressive effect on neuroblastoma cells.
KEYWORDS: MCPIP1, Aurora A kinase, AURKA, inflammation-silencing RNases, ribonuclease, neuroblastoma
1. Introduction
MCPIP1 (monocyte chemoattractant protein-induced protein 1), alias regnase-1, was first described more than a decade ago as the negative regulator of inflammation in macrophages [1]. The best-recognized domains of the MCPIP1 protein are the PilT N‐terminus domain, exhibiting ribonuclease activity, and the Cys–Cys–Cys–His- type zinc finger domain responsible for the binding of its substrate RNA molecules [2–4]. Apart from mRNA molecules, the protein is also thought to be able to antagonize DICER by cleaving the miRNA precursors [5,6]. Studies of MCPIP1-mediated regulation of immune response revealed that the ribonuclease could bind and degrade transcripts of several pro-inflammatory cytokines [7–10]. Recently, a novel activity of the MCPIP1 protein has been observed. The ribonuclease can silence the translation of bound RNA molecules without induction of their cleavage [11]. Additionally, the protein was also found to be a part of the deubiquitinase complex that facilitates the deubiquitination of tumour necrosis factor receptor–associated factor (TRAF) proteins. Deubiquitinase-associated activity of MCPIP1 leads to inhibition of nuclear factor kappa B (NFκB) signalling in immune cells [12–14].
As tumour-promoting inflammation is one of the hallmarks of cancer, the pathways regulating immune response have been broadly studied in tumorigenesis. Moreover, selective anti-inflammatory drugs have been developed and are currently used or undergoing clinical trials [15]. However, the knowledge concerning the role of inflammation-silencing RNases in cancer is still limited. Recent publications point to the tumour-suppressive role of the MCPIP1 protein in a few cancers, such as clear cell renal cell carcinoma, breast cancer, neuroblastoma, and osteosarcoma [16, 17, 18, 19, 20, 21, 22, 23, 24, 25].
Neuroblastoma is the most common extracranial solid tumour of childhood. It arises from the sympathoadrenal lineage of the neural crest, which migrates away from the place of origin during embryogenesis. Therefore, primary tumours usually develop in the chest, head, neck, and pelvis [26]. The clinical presentation of neuroblastoma varies from spontaneous regression to rapid progression and metastasis despite multimodal therapies. This depends on the age of diagnosis, as well as genetic prognostic factors. The most important one is MYCN proto-oncogene, bHLH transcription factor (MYCN) amplification, which occurs in approximately 25% of tumours (Maris et al. 2007). High levels of MYCN transcription factor prevent cell cycle exit and differentiation of the neuroblasts, which leads to neuroblastoma tumour development. Unfortunately, due to the extensiveness of the region of MYCN interaction with its protein partners and target DNA, the inhibitors of the transcription factor were not hitherto developed. However, it is possible to diminish MYCN levels in neuroblastoma cells by the inhibition of Aurora A kinase, (encoded by the AURKA gene) stabilizing the transcription factor [27]. Recently, we have demonstrated that overexpression of MCPIP1 ribonuclease in MYCN-amplified (MNA) neuroblastoma cell lines results in decreased levels of both MYCN and Aurora A proteins [17]. The therapeutic strategies targeting Aurora A have been investigated during the last decade resulting in a phase II clinical trial of one of the kinase small molecule inhibitors, alisertib, for the treatment of high-risk neuroblastoma [28–30].
In the present study, we elaborate on the mechanisms of MCPIP1 ribonuclease action in MNA neuroblastoma cells. Based on our previous findings [17–19], we proposed several putative MCPIP1’s substrates in neuroblastoma cells. In order to verify their interaction with the ribonuclease, we demonstrated that the overexpression of MCPIP1 leads to decreased levels of a few mRNAs, encoding proteins facilitating cell cycle progression in a RNase-dependent manner. Then, we evaluated the interaction of these transcripts with the MCPIP1 protein. The analysis allowed us to suggest the binding between the ribonuclease and the AURKA transcript. Subsequently, we showed that MCPIP1 overexpression leads to the destabilization of the AURKA mRNA in BE(2)-C neuroblastoma cells. After the indication of AURKA mRNA as a novel MCPIP1’s substrate in neuroblastoma cells, we demonstrated that the whole conserved region of AURKA 3′UTR seems to be indispensable for the ribonuclease-dependent cleavage of the transcript. In addition, we investigated the effects of MCPIP1 overexpression on the miRNA expression profile in MNA neuroblastoma cells.
3. Materials and methods
3.1. Cell culture
BE(2)-C (ATCC, CRL-2268, Manassas, VA, USA) and KELLY (DSMZ, ACC 355, UK) cells were cultured as described previously [31].
3.2. Generation of genetic constructs
3′UTR of AURKA was synthesized with NheI and XhoI restriction sites added on the 3′ and 5′ ends and cloned into the pcDNA3.1 vector by GenScript (Leiden, Netherlands). Sequences of the conserved region (CR) of AURKA 3′UTR and putative binding sites (PBS) of the MCPIP1 protein with the restriction sites for NheI and SalI added on the 5′ and 3′ ends, respectively, were synthesized by Genomed (Warsaw, Poland). Sequences of the inserts are listed in Table S1. Additionally, flanking sequences were added to the PBS2 sequence in order to assure the unaltered secondary structure of the RNA on the final transcript (luciferase_PBS2). Furthermore, a sequence of mutated PBS1 with two-point mutations introduced in order to stabilize the secondary structure was included (PBS1stab). Subsequently, inserts gathered in Table S1 were cloned into the pJet1.2 vector using a CloneJET PCR Cloning Kit (K1231, Thermo Fisher Scientific, Waltham, MA, USA). Then, the pmiRGlo plasmid vector (E1330, Promega, Madison, Wisconsin, USA), pcDNA3.1_AURKA-3′UTR, pJet1.2_AURKA-CR, pJet1.2_AURKA-PBS1wt, pJet1.2_AURKA-PBS1stab, pJet1.2_AURKA-PBS2, and pJet1.2_AURKA-PBS3 were digested using NheI (R3131, LabJot, Warsaw, Poland) and SalI (R3138, LabJot, Warsaw, Poland) or NheI and XhoI (R0146, LabJot, Warsaw, Poland) restriction enzymes and separated by gel electrophoresis in 1% agarose gel for pmiRGlo, 2% agarose gel for pcDNA3.1_AURKA-3′UTR, or 5% agarose gel for pJet1.2_AURKA-CR, pJet1.2_AURKA-PBS1wt, pJet1.2_AURKA-PBS1stab, pJet1.2_AURKA-PBS2, and pJet1.2_AURKA-PBS3 and visualized by Simply Safe (E-4600, EURx, Gdańsk, Poland). Subsequently, digested pmiRGlo plasmid vector and appropriate inserts were cut from the agarose gels and isolated using a Gel-Out kit (024–50, A&A Biotechnology, Gdynia, Poland). Next, AURKA-3′UTR, AURKA-CR, AURKA-PBS1wt, AURKA-PBS1stab, AURKA-PBS2, and AURKA-PBS3 were incorporated into the pmiRGlo plasmid vector using T4 DNA ligase (M0202, LabJot, Warsaw, Poland). All genetic constructs were verified by sequencing (Genomed, Warsaw, Poland).
3.3. Cell transfection with MCPIP1-wt and MCPIP1-D141N expression vector
Vectors used to achieve transient overexpression of wild type or mutated MCPIP1 were described by Lipert and colleagues [32]. Transfection procedures for BE(2)-C and KELLY cells were depicted previously [17].
3.4. RNA isolation
Total RNA was isolated using TRI-REAGENT (TRI118, Lab Empire, Rzeszów, Poland) or a miRVana miRNA Isolation Kit (AM1560, Thermo Fisher Scientific, Waltham, MA, USA) for mRNA and miRNA analyses, respectively. The integrity of the RNA samples was verified by electrophoresis in 1% agarose gel.
3.5. mRNA reverse transcription and reverse transcription polymerase chain reaction (RT-qPCR)
Each sample of 1 μg RNA was treated with DNase I (AMPD1, Sigma-Aldrich, Darmstadt, Germany) and reverse-transcribed using M-MLV Reverse Transcriptase (28025013, Thermo Fisher Scientific, Waltham, MA, USA). RT-qPCR was carried out using a KAPA SYBR FAST qPCR Master Mix (SFUKB, Sigma-Aldrich, Darmstadt, Germany). cDNA used for RT-qPCR was diluted 50 times. Primers used for amplifications are shown in Table S2. All experiments were performed three times. For quantification of the relative mRNA level, the ‘ΔΔCq’ method was used [33].
3.6. Small RNA sequencing (RNAseq)
KELLY cells were transfected with MCPIP1-wt, MCPIP1-D141N, or empty expression vectors, as described above. On the fourth day after transfection, cells were harvested, and RNA was extracted using a miRVana miRNA Isolation Kit (AM1560, Thermo Fisher Scientific, Waltham, MA, USA). Next, deep miRNA sequencing was carried out on an Ion Torrent TM Proton machine. RNA samples were evaluated using an RNA 6000 Pico Kit, and the miRNA fraction was assessed using a Small RNA Kit on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Subsequently, libraries were generated using an Ion Total RNA-Seq Kit v2 (4479789, Thermo Fisher Scientific, Waltham, MA, USA). Total RNA (10 µg) was used as the input material. Libraries were sequenced on an Ion Proton Sequencer using an Ion PI Hi-Q Sequencing 200 chemistry and Ion PI Chips v3 (A26433, Thermo Fisher Scientific, Waltham, MA, USA).
3.7. Analysis of RNAseq data
Quality control of reads was conducted using FastQC software version 0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Raw sequencing reads were adapter-trimmed using Cutadapt version 2.08 [34] and aligned with Bowtie version 1.2.3 [35] to a miRBase (version 21) database using miRge2 [36]. MicroRNAs, in which at least ten reads were aligned in a single sample, were selected for further study. Read normalization, as well as identification of differentially expressed miRNAs, was conducted using DESeq2 version 1.24.0 [37] with Benjamini and Hochberg correction for multiple testing and 0.1 significance level. Read counts were normalized across all the samples according to the DESeq2 methodology using normalization by the median algorithm.
3.8. miRNA reverse transcription and quantitative PCR
Each sample of 20 ng RNA was reverse transcribed using a miRCURY LNATM Universal RT microRNA PCR (203907, Exiqon, Vedbaek, Copenhagen). Quantification of specific miRNAs relative expression was carried out with a miRCURY LNA microRNA PCR, ExiLENT SYBR Green master mix (203401, Exiqon, Vedbaek, Copenhagen). For the PCR reaction, cDNA was diluted 60 times. As a reference, hsa-miR-103a-3p was used. Primers for hsa-miR-324-3p (YP00204303), hsa-miR-328-3p (YP00204364), hsa-miR-99b-5p (YP00205983), hsa-miR-1260a (YP00205892), hsa-miR-361-5p (YP00206054), hsa-miR-484 (YP00205636), and the reference hsa-miR-103a-3p (YP00204063) were provided by Exiqon. All experiments were carried out three times. For the relative quantification of miRNA levels, the ‘ΔΔCq’ method was used [33].
3.9. Protein extraction and western blot analyses
Protein extraction and western blot analyses of MCPIP1 and the reference protein (GAPDH) were performed as described previously [19].
3.10. RNA immunoprecipitation (RIP)
BE(2)-C and KELLY cells were transfected with pCMV2-Flag and pCMV2-MCPIP1-D141N-Flag expression vectors depicted by Lipert and colleagues [32] using the transfection procedure described earlier. On the fourth day, cells were collected and washed twice using a cold phosphate buffer solution. Cells were lysed in a lysis buffer [TRIS 50 mM, NaCl 150 mM, 0.5% Nonidet P-40 (NON505, Lab Empire, Rzeszów, Poland)], supplemented with an RNase inhibitor (037, A&A Biotechnology, Gdynia, Poland) and a protease inhibitor cocktail (P8340, Sigma-Aldrich, Darmstadt, Germany), incubated 20 min on ice, and centrifuged at 16000 x g for 20 min. Subsequently, supernatants were transferred to fresh tubes, and the protein concentration was assessed using the bicinchoninic acid assay method [38]. Next, beads from a Dynabead Protein A Immunoprecipitation Kit (10006D, Thermo Fisher Scientific, Waltham, MA, USA) were incubated with gentle rotation for 30 min at room temperature (20–25 °C) with 10 μl anti-FLAG rat monoclonal antibody (SAB4200071, Sigma-Aldrich, Darmstadt, Germany) diluted in a phosphate buffer solution with 0.01% TWEEN-20 (TWN508, Lab Empire, Rzeszów, Poland). Beads conjugated with antibodies were incubated for 2 h with gentle rotation at 4°C with 1 mg of protein isolated from cells transfected with an empty vector (negative control) or the MCPIP1-D141N-Flag expression vector. After binding Flag-tag or MCPIP1-D141N-Flag protein, beads were washed with the lysis buffer and treated with DNase I (AMPD1, Sigma-Aldrich, Darmstadt, Germany). Subsequently, beads were washed four times with the lysis buffer. Following the RIP procedure, immunoprecipitated RNA was extracted from the beads with TRI-REAGENT.
3.11. Actinomycin D treatment
BE(2)-C cells were transfected with the empty control vector or the MCPIP1-wt expression vector following the procedure described in the subchapter 3.3. On the fourth day after transfection the medium was changed and replaced with complete medium containing actinomycin D (ACT001, Lab Empire, Rzeszow, Poland) at the concentration of 5 µg/ml or the even volume of DMSO (D2650, Sigma, Darmstadt, Germany). Cells were harvested at 4 time points (0 hours, 1 hour, 2 hours, 3 hours), and RNA was extracted from the cells. Subsequently, RNA samples were reverse transcribed and the relative levels of AURKA mRNA at different time points versus 0 hours’ time point were assessed by RT-qPCR.
3.12. Luciferase assays
BE(2)-C and KELLY cells were transfected in 6-well plates following the transfection procedure depicted earlier. For each well, 2 μg of the pmirGlo dual-luciferase reporter vector (pmirGlo_AURKA-3′UTR, pmirGlo_AURKA-CR, pmirGlo_AURKA-PBS1wt, pmirGlo_AURKA-PBS1stab, pmirGlo_AURKA-PBS2, pmirGlo_AURKA-PBS3, or the empty pmirGlo) and 2 μg of MCPIP1-wt, MCPIP1-D141N, or the empty expression vector were used. 48 hours after transfection cells were lysed. Next, firefly (reporter gene) and renilla (internal control) luciferase activities were assessed using a Dual-Luciferase Reporter Assay System (Promega, Madison, Wisconsin, USA). Relative luciferase activities were calculated compared to the corresponding values for the empty reporter vector.
3.13. Bioinformatics analyses of sequences interacting with MCPIP1 protein
Conservation of the RNA sequences interacting with the MCPIP1 protein in mammalian species was assessed with ClustalW (https://www.genome.jp/tools-bin/clustalw). Accessibility, defined as probability of a lack of interaction with other nucleotides in the analysed sequence (being unpaired), of trinucleotides in the MCPIP1 binding sites together with 40 nucleotide flanking regions were determined using RNAplfold 2.4.14 [39]. Sequence motifs enriched in the sequences interacting with the MCPIP1 protein were identified with the MEME 5.1.0 algorithm [http://meme-suite.org/tools/meme; 40].
3.14. Statistical analyses
Statistical analyses were performed using Statistica 13 (StatSoft, Kraków, Polska). Exact tests performed for each set of experiments are indicated in the figure legends.
4. Results
4.1. Six transcripts associated with cell cycle progression are possible MCPIP1’s ribonuclease substrates in neuroblastoma cells
Elucidation of a ribonuclease action in cells requires the determination of its substrate RNAs. For better control of the ribonucleolytic activity of MCPIP1, we applied a point mutated form of the MCPIP1 protein (MCPIP1-D141N) instead of the deletion mutant, devoid of the ribonuclease domain, used by us so far [17–19,31]. Substitution of aspartic acid residue 141 with asparagine in the catalytic centre of the MCPIP1 protein results in the abolishment of ribonucleolytic activity, while the ability to bind specific RNAs is retained [9]. Transfection of neuroblastoma cells with the expression vector for wild-type (MCPIP1-wt), and the mutated (MCPIP1-D141N) form of MCPIP1 was shown to be highly effective as upregulation of the protein was observed at 10-fold for BE(2)-C cells (Figure 1 A and C) and 8-fold for KELLY cells (Fig. 1 B and D).
Figure 1.

Overexpression levels of the MCPIP1 protein in BE(2)-C and KELLY neuroblastoma cells. Relative MCPIP1 protein levels were measured by western blot 4 days after transfection with either MCPIP1-wt or MCPIP1-D141N expression vectors in BE(2)-C (A) and KELLY (B) cells. Representative images of western blot signals for MCPIP1 and the reference protein (GAPDH) for BE(2)-C (C) and KELLY (D) cells. The experiments were performed three times. Graphs represent mean ± SEM. The one-way analysis of variance was implemented with Tukey’s post hoc test. C, cells transfected with an empty vector; MCPIP1-wt/Wt, cells transfected with the wild type MCPIP1 expression vector; MCPIP1-D141N/D141N, cells transfected with the mutated MCPIP1 expression vector; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. *P < 0.05, **P < 0.01, ***P < 0.001
The previously used experimental model contained the control for both, the MCPIP1 overexpression (cells transfected with an empty expression vector) and the protein’s ribonucleolytic activity (cells overexpressing the mutated form of MCPIP1). Using this model we have shown a broad range of alteration in transcriptome and proteome of neuroblastoma cells overexpressing MCPIP1 protein via application of microarrays, RT-qPCR and western blot [17–19,31]. The decreased transcript expression in the cells overexpressing the wild-type form of MCPIP1, and simultaneously, unaltered levels in the cells with overexpression of the mutated form of the protein, points to the indispensability of the ribonucleolytic activity of MCPIP1 for the observed downregulation of a mRNA. Moreover, the decrease of a specific transcript levels in a manner dependent on the MCPIP1’s ribonucleolytic activity constitutes a premise of the possible interaction between the mRNA and the ribonuclease. According to the described requisites we were able to select twelve putative MCPIP1’s substrates based on our hitherto published data concerning the expression in the cells overexpressing wild type and mutated form of the ribonuclease. In depth, we have chosen Minichromosome maintenance 10 replication initiation factor (MCM10) [18]; Cell division control protein 2 (CDC2), AURKA, Mammalian target of rapamycin (MTOR), Rapamycin-insenstivie companion of mTOR (RICTOR), Regulatory associated protein of mTOR (RPTOR), MYCN [17]; and CCND1, CCND3, CCNE1, CCNE2, Retinoblastoma (RB) [19]. The expression of the genes from this group was decreased in cells overexpressing MCPIP1-wt, but unaltered in cells overexpressing the deletion mutant of the protein in at least one of the studied neuroblastoma cell lines [17–19]. To estimate the probability of interaction between MCPIP1 and the candidate transcripts, we analysed the 3′UTRs to find evolutionarily conserved stem-loop structures with pyrimidine-purine-pyrimidine sequences in the loop, as such structures were described as recognized by the MCPIP1 protein [9]. We obtained candidate structures for nine of the transcripts (Table S3).
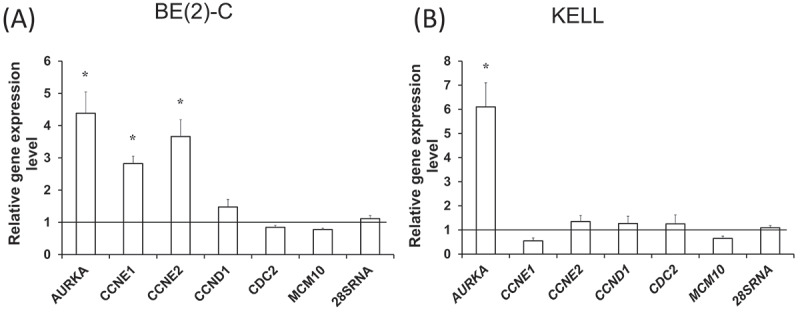
As MCPIP1 overexpression was found to suppress breast cancer progression via destabilization of RELB, BCL2A1, BCL2L1, BCL3, and BIRC3 [21], we decided to include these genes in the assessment of relative expression in our experimental model in addition to the previously mentioned twelve candidate substrates of the ribonuclease. Additionally, we evaluated the expression of C-REL mRNA, which was identified as MCPIP1’s substrate by Mino and colleagues [9]. Decreased mRNA level in cells overexpressing MCPIP1-wt, together with unchanged expression in cells overexpressing MCPIP1-D141N (control for the ribonucleolytic activity of the protein), points to a possible degradation of the transcript by the MCPIP1 protein. Described downregulation of the mRNA levels, dependent on the ribonucleolytic activity of MCPIP1, was found for AURKA, CCND1, CCNE1, CCNE2, CDC2, MCM10, and MYCN in BE(2)-C cells (Fig. 2 A). The results were consistent with the data obtained for KELLY cells, apart from the MYCN transcript, which was also decreased in the KELLY cells overexpressing MCPIP1-D141N (Fig. 2 B). Of note, all the transcripts, which were decreased in the manner dependent on the ribonucleolytic activity of MCPIP1, and therefore possibly degraded by the ribonuclease, encode proteins facilitating cell cycle progression [19]. The remaining transcripts, ie. RB, MTOR, RICTOR, RPTOR, RELB, C-REL, BCL2L1, and BIRC3, were either unchanged or slightly increased in our experimental model (Fig. 2). Interestingly, potent upregulation of BCL2A1 transcript levels was observed in cells overexpressing MCPIP1 (Fig. 2 C and D).
Figure 2.

Analysis of putative MCPIP1 target mRNAs expression in neuroblastoma cells overexpressing MCPIP1 protein. Relative expression levels of selected genes encoding proteins involved in cell cycle regulation in BE(2)-C (A) and KELLY (B) cells overexpressing MCPIP1 assessed by RT-qPCR. Relative expression levels of genes encoding proteins involved in apoptosis, mTOR, and NFκB pathways in BE(2)-C (C) and KELLY (D) cells overexpressing MCPIP1 protein. RPS13 (40S ribosomal protein S13) was used as a reference. All experiments were performed three times. The results were calculated compared to the values for the control cells, set as 1 (black baseline). Graphs represent mean ± SEM. The one-way analysis of variance was implemented with Tukey’s post hoc test. MCPIP1-wt, cells transfected with the wild type MCPIP1 expression vector; MCPIP1-D141N, cells transfected with the mutated MCPIP1 expression vector. *P < 0.05, **P < 0.01, ***P < 0.001
Apart from its RNase activity, the MCPIP1 protein is also able to facilitate the deubiquitination of several proteins involved in the NFκB pathway [14]. To assess whether the deubiquitinase-associated activity of MCPIP1 contributes to its suppressive effect on neuroblastoma cells, we measured the levels and phosphorylation status of several proteins involved in the NFκB pathway. However, no changes were found between control cells and cells overexpressing the MCPIP1 protein (Fig. S1), ruling out the involvement of the NFκB pathway in the observed effect. Therefore, the MCPIP1 effect on neuroblastoma cells seems to be mostly dependent on the RNase activity of the protein.
4.2. MCPIP1 protein binds AURKA transcript in neuroblastoma cells
For further investigation of possible MCPIP1-dependent degradation of AURKA, CCND1, CCNE1, CCNE2, CDC2, and MCM10 mRNAs, decay rates of the transcripts in cells overexpressing MCPIP1-wt or MCPIP1-D141N were assessed after treatment with actinomycin D. Unexpectedly, the observed toxic effect of the antibiotic on the KELLY cells was severe. Even the cells treated with a minute concentration of the antibiotic exhibited some motions of decreased viability, such as reduced adherence. Since the six investigated transcripts encode proteins involved in the cell cycle progression, the cytostatic effect of the antibiotic might mask the impact of MCPIP1 protein on the mRNAs stability. Therefore, the results of these experiments were not conclusive (Fig. S2; Table S4).
An alternative method to assess the degradation of a transcript by a ribonuclease is the evaluation of binding between RNA and ribonuclease. To evaluate the physical interaction of MCPIP1 and the transcripts possibly degraded by the ribonuclease (ie. AURKA, CDC2, MCM10, CCNE1, CCNE2, CCND1), cells were transfected with the Flag-tagged MCPIP1-D141N (MCPIP1-D141-Flag) expression vector. Transfection efficacy was assessed by western blot (Fig S4. A and B). After 48 h, RNA immunoprecipitation (RIP) was performed. Compared to the negative control samples, significant enrichment of the AURKA transcript was found in the RNA immunoprecipitated with MCPIP1-D141N-Flag protein (Fig. 3), indicating that AURKA mRNA might be the preferred substrate of MCPIP1. Additionally, for BE(2)-C cells, CCNE1 and CCNE2 transcripts were also enriched in the RNA immunoprecipitated with MCPIP1-D141N-Flag (Fig. 3 A). However, as the enrichment of CCNE1 and CCNE2 mRNAs occurs only in the RNA immunoprecipitated with MCPIP1 in the BE(2)-C cells, we decided to focus on further evaluation of only the interaction between the protein and AURKA transcript. Other transcripts levels were similar to those in the negative control group (Fig. 3).
Figure 3.
Assessment of MCPIP1 protein binding of several transcripts in neuroblastoma cells. Evaluation of relative levels of selected transcripts in the RNA immunoprecipitated with MCPIP1-D141N-Flag protein or the Flag tag (negative control) in BE(2)-C (C) and KELLY (D) cells. RPS13 (40S ribosomal protein S13) was used as a reference. All experiments were performed three times. Graphs represent mean ± SEM and were calculated compared to the negative control values, set as 1 (black baseline). For the determination of statistical significance, a T-test was performed. *P < 0.05
4.3. MCPIP1 overexpression in neuroblastoma cells results in destabilization of AURKA 3′UTR
Following the establishment of the interaction between the MCPIP1 protein and AURKA transcript, we investigated possible MCPIP1-dependent destabilization of AURKA 3′UTR. To this end we have performed AURKA mRNA stability assay using BE(2)-C neuroblastoma cells treated with actinomycin D. A faster degradation of AURKA transcript correlating with MCPIP1 overexpression was observed (Fig. 4 A). Treatment of the cells with DMSO did not affect AURKA levels in both control cells and cells overexpressing MCPIP1 protein (Fig. 4 B). To further investigate MCPIP1-dependent destabilization of AURKA mRNA a luciferase reporter system was applied, in which 3′UTR of AURKA was attached downstream of the luciferase coding sequence. BE(2)-C and KELLY cells overexpressing MCPIP1-wt exhibited significantly decreased luciferase activity (Fig. 4 C and D). Moreover, in the MCPIP1-D141N overexpressing cells of both cell lines, the luciferase activity was similar to the control cells (Fig. 4 C and D). The levels of achieved MCPIP1 overexpression were measured by western blot (Fig. S4 C and D). MCPIP1-dependent decrease of luciferase activity suggests, that although the mutated form of MCPIP1, lacking the ribonucleolytic activity was able to bind AURKA mRNA (Fig. 3), the active catalytic centre of the protein is indispensable for the destabilization of the transcript (Fig. 4 C and D). The recognition of sequences in the 3ʹUTR of specific transcripts by MCPIP1 followed by the endonucleolytic cleavage catalysed by the ribonuclease is well documented in the literature [7–10,21,32,41,42]. Therefore, presented results strongly indicate, that AURKA is a preferred substrate of MCPIP1 protein.
Figure 4.
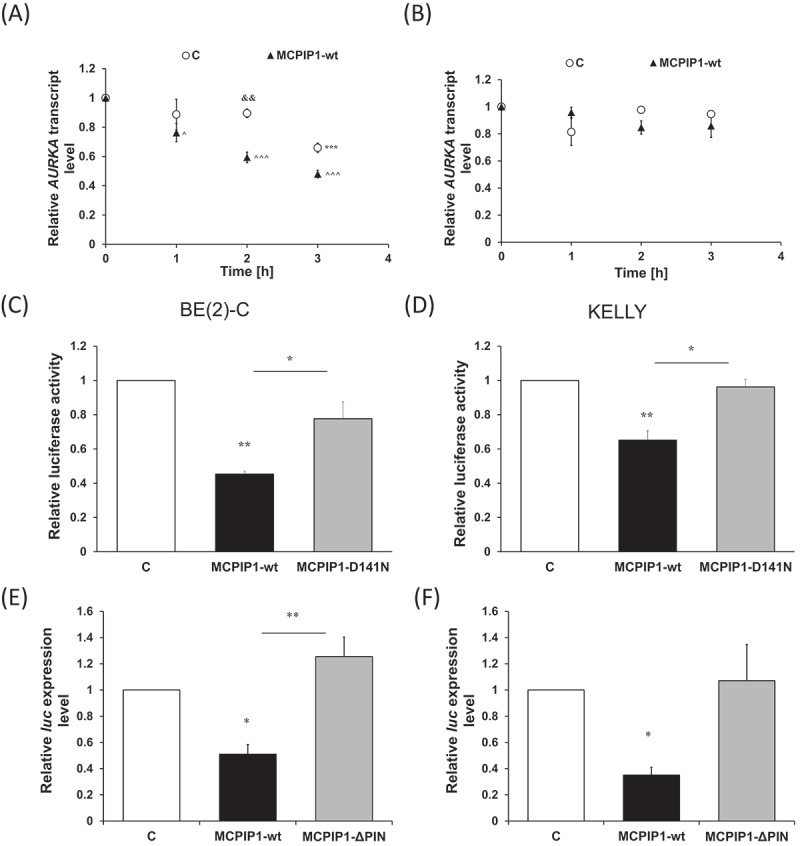
Determination of AURKA 3′UTR stability in neuroblastoma cells overexpressing MCPIP1 protein. Relative AURKA transcript levels following the treatment with (A) actinomycin D and (B) DMSO. For the determination of statistical significance two-way analysis of variance with Tukey’s post-hoc test was performed. Relative luciferase activity of BE(2)-C (C) and KELLY (D) cells co-transfected with MCPIP1-wt, MCPIP1-D141N or control expression vectors and pmiRGlo-AURKA_3′UTR reporter vector calculated compared to the relative activity of corresponding cells transfected with an empty pmiRGlo vector. Relative luciferase transcript levels in BE(2)-C (E) and KELLY (F) cells transfected with MCPIP1-wt, MCPIP1-D141N or control expression vectors and pmiRGlo-AURKA_3′UTR reporter vector assessed by RT-qPCR. Renilla luciferase was used as a reference. All experiments were performed three times. Graphs represent mean ± SEM. The one-way analysis of variance was implemented with Tukey’s post hoc test. C, cells co-transfected with the empty expression vector and pmiRGlo-AURKA_3′UTR reporter vector; MCPIP1-wt, cells transfected with wild type MCPIP1 expression vector and pmiRGlo-AURKA_3′UTR reporter vector; MCPIP1-D141N, cells transfected with mutated MCPIP1 expression vector and pmiRGlo-AURKA_3′UTR reporter vector.*P < 0.05, **P < 0.01; ^ P < 0.05, ^^^ P < 0.001; && P < 0.01
Degradation of AURKA 3′UTR was further analysed by assessing levels of luciferase mRNA in parallel with luciferase activity. The levels of the luciferase transcript were diminished in BE(2)-C and KELLY cells overexpressing MCPIP1-wt, whereas overexpression of MCPIP1-D141N does not alter the luciferase expression (Fig. 4 E and F). Thus, the decrease of luciferase activity depended on the amount of the transcript. Consequently, to investigate the cleavage of the AURKA transcript by the MCPIP1 protein, further analyses to determine which specific regions of AURKA 3′UTR interact with MCPIP1 protein were performed.
4.4. Previously described sequences cleaved by MCPIP1 protein share common sequence and structure motifs
As MCPIP1 protein was first described more than a decade ago, several detailed sequences showing interaction with the RNase were reported in human and murine settings by different researchers (Table 1). All sequences were highly conserved in mammalian species (data not shown). Detailed bioinformatics analysis of MCPIP1-interacting elements, together with the 40 nucleotide flanking regions, revealed that the pyrimidine-purine-pyrimidine sequence does not necessarily form a loop with a thermodynamically stable hairpin structure (Fig. S3). Nevertheless, the assessment of the probability of being unpaired (i.e., accessibility) of pyrimidine-purine-pyrimidine sequences recognized by MCPIP1 protein revealed that these sequences are significantly more accessible than the neighbouring six nucleotides downstream and upstream (Fig. 5 A). All sequences found in literature share a common, significantly enriched motif-containing the pyrimidine-purine-pyrimidine sequence (U[G/A]U) (Fig. 5 B). This motif was also found in the conserved region of AURKA 3′UTR.
Table 1.
Previously determined sequences from human and mouse transcriptome interacting with MCPIP1 protein.
| Gene | Sequence interacting with MCPIP1 | Reference | |
|---|---|---|---|
| Human | BCL2L1 | UUUAUGUGUGAGGAGCUGCUGG | [21] |
| BIRC3 | GUGUGCAUAUAUGUUGAAUGAC | [21] | |
| IER3 | AGCGACUGUCGAGAUCGCCUAGU | [41] | |
| IL6 | UUAUGUUGUUCUCUAUGGAGAAC | [41] | |
| NFKBIZ | AGUUGUUUCUAUGAAACAAACAUAUUUAGUUCACUAUUAUAUAG | [9] | |
| TM2D3 | UUUUUAAUGUACAGCAUCUGUACUU | [9] | |
| Mouse | cRel | AAAACGUGUAAUGGCUAUGCCAUU | [10] |
| Il2 | GAUAAAUAUGGAUCUUUAAAGAUU | [7] | |
| Il6 | GACUUAUGUUGUUCUCUACGAAGA | [7] | |
| Mcpip1 | UGAUCACCCUGUUGAUACACAUUG | [42] | |
| Ptgs2 | CCGUUUCUCGUGGUCACUUUACUA | [9] | |
| Stat3 | UCAGUGCAGUGGCUUGUGUUCUGG | [8] | |
| Tnf | GACAGACAUGUUUUCUGUGAAAAC | [9] |
Figure 5.

Characterization of known MCPIP1 target sequences. (A) The correlation between the distance from nucleotides bound by MCPIP1 and their accessibility calculated using the RNAplfold algorithm. Nucleotides bound by MCPIP1 were set as 0. (B) Representation of consensus RNA motif recognized by MCPIP1 protein as position weight matrix logos generated using the MEME algorithm. [E-value = 1.2e-003]. Letter size corresponds to relative nucleotide entropy, represented as bits
4.5. Complete conserved region of AURKA 3′UTR seems to be required for MCPIP1-dependent cleavage of the transcript
Although AURKA 3′UTR consists of 768 bases, only a 95-base region is highly conserved across mammalian species (Fig. 6 A). Thus, we hypothesized that a binding site recognized by the MCPIP1 protein is localized within this conserved region (CR). Analysis of base accessibility and the sequence of the AURKA 3′UTR CR allowed for the proposal of three putative binding sites (PBS) of MCPIP1 ribonuclease (Fig. 6 A and B). Among the chosen sites, PBS1 and PBS2 were characterized by base accessibility pattern common within the hitherto described MCPIP1 binding sites (Fig. 6 A). However, the sequential motif shared between all known MCPIP1 target sites was found only in PBS2 and PBS3 sequences (Fig. 6 B).
Figure 6.
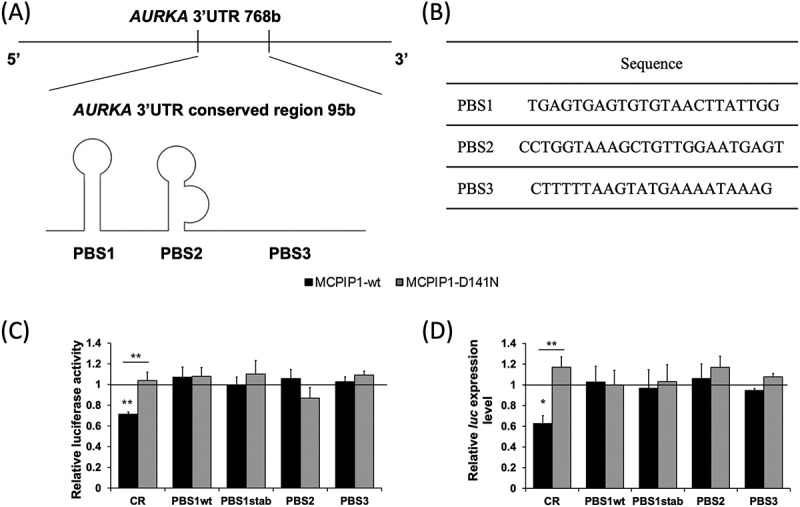
Assessment of stability of the conserved region (CR) of AURKA 3′UTR and putative binding sites (PBS) of MCPIP1 protein in BE(2)-C cells overexpressing MCPIP1. (A) Visualization of AURKA 3′UTR CR position and secondary structure in the 3′UTR. (B) Sequences of MCPIP1 putative binding sites. (C) Relative luciferase activity of BE(2)-C cells co-transfected with MCPIP1-wt, MCPIP1-D141N or control expression vectors and pmiRGlo_AURKA-CR, pmiRGlo_AURKA-PBS1wt, pmiRGlo_AURKA-PBS1stab, pmiRGlo_AURKA-PBS2, or pmiRGlo_AURKA-PBS3 reporter vectors calculated compared to the relative activity of corresponding cells transfected with the empty pmiRGlo vector. (D) Relative luciferase transcript levels in BE(2)-C cells transfected with MCPIP1-wt, MCPIP1-D141N or control expression vectors and pmiRGlo_AURKA-CR, pmiRGlo_AURKA-PBS1wt, pmiRGlo_AURKA-PBS1stab, pmiRGlo_AURKA-PBS2, or pmiRGlo_AURKA-PBS3 reporter vectors assessed by RT-qPCR. Renilla luciferase was used as a reference. All experiments were performed three times. Graphs represent mean ± SEM. Black baseline illustrates values for the control cells (transfected with the empty expression vector), set as 1. The one-way analysis of variance was implemented with Tukey’s post hoc test. MCPIP1-wt, cells transfected with wild type MCPIP1 expression; MCPIP1-D141N, cells transfected with mutated MCPIP1 expression vector; CR, cells transfected with pmiRGlo_AURKA-CR reporter vector; PBS1wt, cells transfected with pmiRGlo_AURKA-PBS1wt reporter vector; PBS1stab, cells transfected with pmiRGlo_AURKA-PBS1stab reporter vector; PBS2, cells transfected with pmiRGlo_AURKA-PBS2 reporter vector; PBS3, cells transfected with pmiRGlo_AURKA-PBS3 reporter vector. **P < 0.01
In order to determine if any of the proposed sites could be functional, we applied a luciferase reporter system in which the CR of AURKA or the putative binding sites were cloned downstream of the luciferase coding sequence. Since MCPIP1 protein requires a spacing sequence of at least 20 nucleotides from the stop codon in order to recognize the specific binding site [9], we have chosen the restriction site located 28 nucleotides downstream from the luciferase coding sequence. Furthermore, as placement of the PBS1 sequence in the pmiRGlo reporter vector resulted in a decrease of the secondary structure stability, we introduced a point mutant of the sequence (PBS1stab). The mutation was brought into the stem sequence of PBS1 and increased the stability of the stem loop structure in the plasmid-derived mRNA. After the BE(2)-C cells co-transfection, achieved overexpression of MCPIP1 wild type and mutated form was evaluated by western blot (Fig. S4 E-J). The series of luciferase assay experiments revealed that only BE(2)-C cells co-transfected with the MCPIP1-wt expression vector, and the pmiRGlo_AURKA-CR reporter vector exhibited significantly decreased luciferase activity (Fig. 6 C). Moreover, this phenomenon was dependent on MCPIP1 ribonucleolytic activity, as we did not observe the inhibition of luciferase activity in cells co-transfected with the MCPIP1-D141N expression vector and the pmiRGlo_AURKA-CR reporter vector (Fig. 6 C). Simultaneously, the luciferase activity of the cells co-transfected with MCPIP1 expression vectors and reporter vectors containing putative binding sites was similar to the control cells (Fig. 6 C). Therefore, MCPIP1 overexpression does not seem to affect the stability of any of the putative MCPIP1 binding sites within the AURKA 3′UTR conserved region. Luciferase transcript stability was further assessed by RT-qPCR (Fig. 6 D). The experiments revealed a decrease in the luciferase transcript level in cells co-transfected with the MCPIP1-wt expression vector and the pmiRGlo_AURKA-CR reporter vector (Fig. 6 D). The luciferase expression in the remaining groups of studied cells was similar to control (Fig. 6 D). Thus, the RT-qPCR experiments further confirmed the results obtained by the luciferase assays. All the described data indicate, that the entire AURKA conserved region is required for MCPIP1-dependent decay of the transcript. The exact mechanism of the binding between MCPIP1 ribonuclease and AURKA mRNA, however, remains to be elucidated.
4.6. Overexpression of MCPIP1 protein does not significantly alter miRNA expression profile in neuroblastoma cells
Some reports point to the possibility of the cleavage of specific pre-miRNAs by the MCPIP1 protein [6,43]. Moreover, deregulation of the miRNA profile was found to affect the biology and prognosis of neuroblastoma tumours [44]. Thus, we hypothesized that some aspects of the suppressive effect of MCPIP1 protein upregulation on neuroblastoma cells might be attributed to MCPIP1-dependent miRNA dysregulation.
To investigate this hypothesis, we applied small RNA sequencing (RNAseq) of RNA extracted from KELLY cells transfected with the MCPIP1-wt and MCPIP1-D141N expression vectors or the empty expression vector as a control. Interestingly, no significant changes in the miRNA expression profile were observed between MCPIP1-wt or MCPIP1-D141N against the control cells (data not shown). Nevertheless, 14 miRNAs were found to be differentially expressed in the cells overexpressing MCPIP1-D141N as compared to the cells overexpressing MCPIP1-wt (Table 2). Twelve of the miRNAs decreased in cells overexpressing point MCPIP1-D141N (Table 2). Simultaneously, only two of the altered miRNAs exhibited higher levels in cells with MCPIP1-D141N overexpression (Table 2).
Table 2.
List of 14 miRNAs differentially expressed in MCPIP1-D141N compared to MCPIP1-Wt KELLY cells based on small RNA sequencing analysis
| miRNA | Fold change | Standard Error | P-value | P-value adjusted |
|---|---|---|---|---|
| hsa-miR-1180-3p | 0.4034 | 0.0929 | 1.18E-05 | 0.0027 |
| hsa-miR-1260a/1260b | 0.3955 | 0.1235 | 0.00064 | 0.021 |
| hsa-miR-1306-5p | 0.3269 | 0.1067 | 7.64E-05 | 0.0036 |
| hsa-miR-3180-5p | 0.3534 | 0.1092 | 0.00011 | 0.0046 |
| hsa-miR-324-3p | 0.4187 | 0.0971 | 3.00E-05 | 0.002 |
| hsa-miR-328-3p | 0.0862 | 0.0987 | 0.0013 | 0.037 |
| hsa-miR-4286 | 0.3671 | 0.1545 | 0.0043 | 0.096 |
| hsa-miR-4454 | 0.3722 | 0.1024 | 4.82E-05 | 0.0027 |
| hsa-miR-4485-3p | 0.3522 | 0.1015 | 3.78E-05 | 0.0027 |
| hsa-miR-484 | 0.5387 | 0.1233 | 0.0027 | 0.071 |
| hsa-miR-766-3p | 0.3749 | 0.1129 | 0.00019 | 0.0069 |
| hsa-miR-99b-5p | 0.4812 | 0.0916 | 2.72E-05 | 0.0027 |
| hsa-miR-129-1-3p/129-2-3p | 1.3454 | 0.1492 | 0.0047 | 0.098 |
| hsa-miR-361-5p | 1.9501 | 0.4947 | 0.0031 | 0.075 |
In order to select miRNAs to be validated via RT-qPCR, we performed a thorough literature review. We chose six miRNAs based on their potential to regulate intracellular pathways important in neuroblastoma pathogenesis or their described role in cancer development and diagnosis. Two of the selected miRNAs, hsa-miR-328-3p and hsa-miR-99b-5p, were found to affect Akt/mTOR signalling in cancer cells [45,46]. Another two of the miRNAs, hsa-miR-324-3p and hsa-361-5p, contribute to the activation and inhibition of the Wnt pathway, respectively [47,48]. Another miRNA, hsa-miR-484, is involved in apoptosis suppression [49]. Moreover, we decided to include in our quantitative analysis hsa-miR-1260a because of its prognostic value in a few cancer types [50–52].
Surprisingly, RT-qPCR experiments did not confirm any of the changes detected by RNAseq (Fig. 7 A). The only alteration in miRNA expression was the increase of the hsa-miR-484 level in KELLY cells overexpressing MCPIP1-D141N (Fig. 7 A). However, since the statistically significant changes detected with RNAseq concerned only the comparison between MCPIP1-D141N and MCPIP1-wt groups, we reanalysed the obtained data to calculate the fold change in MCPIP1-D141N cells vs MCPIP1-wt cells. A comparative analysis of the PCR results between MCPIP1-D141N and MCPIP1-wt uncovered decreased hsa-miR-1260a levels in KELLY cells overexpressing mutated form of the protein (Fig. 7 A). The observed downregulation of hsa-miR-1260a was in accordance with the RNAseq results (Table 2).
Figure 7.

Determination of selected miRNA expression in neuroblastoma cells overexpressing MCPIP1 protein. Relative expression levels of selected miRNAs in KELLY (A) and BE(2)-C (B) cells overexpressing MCPIP1 normalized to control cells. The values for the control cells are represented as the black baseline, set as 1 (black baseline). Relative expression levels of selected miRNAs in MCPIP1-D141N KELLY (C) and BE(2)-C (D) cells normalized to MCPIP1-wt cells. Black baseline illustrates the values for MCPIP1-wt cells (set as 1). Relative expression levels were assessed by RT-qPCR. Hsa-miR-103a-3p was used as a reference. All experiments were performed three times. Graphs represent mean ± SEM. The one-way analysis of variance was implemented with Tukey’s post hoc test. MCPIP1-wt, cells transfected with the wild type MCPIP1 expression vector; MCPIP1-D141N, cells transfected with the mutated MCPIP1 expression vector. *P < 0.05
As MCPIP1 action in cells is often dependent on the cell type, we decided to investigate the expression of the selected miRNAs in BE(2)-C cells with MCPIP1 overexpression. Here, we observed altered expression of three of the miRNAs (Fig. 7 B). Hsa-miR-99b-5p and hsa-miR-1260a were upregulated in BE(2)-C cells overexpressing MCPIP1-D141N, while hsa-miR-361-5p was increased in cells overexpressing MCPIP1-wt (Fig. 7 B). Analysis of miRNA levels in BE(2)-C MCPIP1-D141N cells compared to MCPIP1-wt cells revealed an increase of hsa-miR-324-3p in cells overexpressing the point mutant of the ribonuclease.
As the observed changes in miRNA expression were minute, miRNA deregulation does not seem to play an important role in MCPIP1-dependent neuroblastoma cell growth suppression. However, since the results obtained by different experimental methods exhibited significant discrepancies, further studies are necessary to elucidate the relationship between the miRNA expression profile and MCPIP1 protein activity in neuroblastoma.
5. Discussion
The suppressive effect of MCPIP1 on cancer cells continues to emerge. Among MCPIP1 substrates that are involved in immune response, apart from cytokines and chemokines, were transcripts of genes exhibiting oncogenic potential such as ID1 and C-REL [9]. Analysis of transcripts bound to the MCPIP1 protein in the human breast cancer cell line MDA-MB-231 led to the discovery of five anti-apoptotic mRNAs as the ribonuclease’s substrates [21]. Suppression of breast cancer tumorigenesis by MCPIP1 is also dependent on the facilitation of the deubiquitination of RGS2 protein [53] . The effect of MCPIP1 overexpression in breast cancer cells was further studied in vivo and showed the potential to elicit complete tumour regression [21]. The protein is also involved in the regulation of growth, metastasis and vascularization of clear cell renal cell carcinoma. Overexpression of MCPIP1 in the human Caki-1 cell line results in apoptosis activation, a decrease of signalling via the Akt/mTOR pathway, cell cycle arrest, and inhibition of angiogenesis and metastasis [16; 20, 22]. Moreover, the protein contributes to MG-132 proteasome inhibitor toxicity by inhibiting the genotoxic activation of NFκB signalling in a deubiquitinase-dependent manner in several human cancer cell lines [13,24]. Recently, Ren and colleagues reported that low levels of MCPIP1 in osteosarcoma tumours correlate with poor prognosis [23].
Our previous data on the effects of MCPIP1 protein overexpression on neuroblastoma cells further confirm the suggestion of the tumour-suppressive role of the protein [17–19,24]. Overexpression of the ribonuclease inhibits the proliferation of MNA neuroblastoma cells. Analysis of the transcriptome of neuroblastoma cells exhibiting MCPIP1 overexpression revealed significant downregulation of many known oncogenes [17–19]. One of the oncogenes, MYCN, was the key driver of neuroblastoma tumorigenesis. Similarly to the findings of another group in the clear cell renal cell carcinoma Caki-1 cell line (Ligeza i in., 2017), we observed decreased Akt/mTOR activity upon MCPIP1 overexpression in BE(2)-C and KELLY MNA-neuroblastoma cells [17]. Our studies on MNA neuroblastoma cells overexpressing MCPIP1 protein demonstrated decreased expression of several genes involved in cell cycle regulation, such as CDC2, AURKA, CCNA2, CCNB1, CCND1, CCNE1, CCNE2, and MCM10 [17–19]. Moreover, we found the downregulation of phosphorylation levels of CDC2 and Aurora A kinases [17]. A thorough investigation of the biology of MCPIP1 overexpressing MNA neuroblastoma cells revealed cell cycle arrest at the G1/S checkpoint [19].
All data described above points to the crucial role of the ribonucleolytic activity of MCPIP1 protein in the suppression of MNA neuroblastoma cells [17–19,24]. Considering that the mechanisms of any ribonuclease action in the cells can only be delineated by the establishment of its substrates, we selected 18 proposed MCPIP1’s substrates based on our previous studies [17–19,24] and a literature review [21]. Only six of the proposed substrates exhibited decreased levels in MNA neuroblastoma in a ribonucleolytic activity-dependent manner (Fig. 2). Interestingly, all transcripts encode proteins involved in the regulation of cell cycle progression. Aurora A, a protein product of the AURKA gene, is essential for correct chromosome segregation during cell division [54]. Three decreased transcripts, CCND1, CCNE1, and CCNE2, encode cyclins D1, E1, and E2, whereas the protein product of CDC2 is CDK1 (cyclin-dependent kinase 1). Cell cycle progression is controlled by the activity of specific holoenzymes, consisting of cyclins and their partner CDKs. CDK1 is a M-phase cyclin-dependent kinase, while cyclins from D and E families act in the G1 phase of the cell cycle [19]. Another of the six transcripts, MCM10, encodes a protein crucial for the DNA replication in the S phase [18]. Therefore, the ribonucleolytic activity of the protein seems to be indispensable for the inhibition of the cell cycle in MNA neuroblastoma cell lines overexpressing MCPIP1.
Although six transcripts were decreased in MNA neuroblastoma in the ribonucleolytic activity-dependent manner, RIP experiment suggested the interaction only between the MCPIP1 protein and AURKA mRNA in both studied cell lines (Fig. 3). Furthermore, assessment of stability of the transcript with application of actinomycin D treatment and the luciferase assay indicated the destabilization of AURKA 3′UTR upon MCPIP1 overexpression in the MNA-neuroblastoma cells (Fig. 4). Hence, the obtained data constitute a strong premise of the recognition of the ribonuclease binding site in the 3′UTR of AURKA transcript followed by the binding and cleavage of the mRNA.
Negative regulation of AURKA expression by MCPIP1 protein might have a key role in the observed cell growth arrest of MNA neuroblastoma cells manifesting ribonuclease overexpression. High levels and activity of the protein product of the AURKA gene, Aurora A kinase, are some of the crucial factors driving the tumorigenesis of MNA neuroblastoma. The decrease of the AURKA gene and Aurora A protein expression has been observed after the treatment of neuroblastoma cells with chemotherapy, as well as with anti-GD2 ganglioside antibodies [55,56]. Moreover, the small-molecule inhibitor of Aurora A kinase, alisertib, which can inhibit the Aurora A-dependent stabilization of MYCN, is undergoing clinical trials as a treatment for high-risk neuroblastoma [29,57]. Amplification of AURKA loci was observed in 19% of neuroblastoma tumours and was closely correlated with MYCN amplification [58]. Aurora A kinase stabilizes the MYCN protein, which results in inhibition of the differentiation of neuroblasts and the promotion of uncontrolled cell division leading to the rapid progression of neuroblastoma. Additionally, MYCN transcription factor was found to enhance AURKA expression, therefore creating a positive loop, which adds to the tumorigenic potential of MNA neuroblastoma cells [59]. The interaction between MYCN and Aurora A proteins is physical; hence, the catalytic activity of the kinase is dispensable for the stabilization of the transcription factor [27]. Therefore, the ribonucleolytic activity of the MCPIP1 protein might suppress the oncogenic effect of Aurora A kinase. Additionally, the kinase contribution to the inhibition of NFκB pathway activity has been described [56]. As MCPIP1 synthesis is induced by the NFκB transcription factor [60], there is a possibility of a negative feedback loop between the MCPIP1 protein and Aurora A kinase. However, in our experimental model, no alterations of NFκB activation was observed (Fig. S1). It would be interesting to study the relationship between MCPIP1 and Aurora A kinase in a cellular setting exhibiting high activity of the NFκB pathway.
The interaction of MCPIP1 ribonuclease with its substrate RNAs has been studied by a number of research groups [3,7–10,21,41,42]. Mino and colleagues described a consensus sequence and detailed RNA structure recognized by MCPIP1. Their analyses revealed that sequences forming structures with 3–7 stem nucleotides and pyrimidine-purine-pyrimidine three-nucleotide loops were significantly enriched among RNA molecules bound to the MCPIP1 protein in HeLa cells [9]. Furthermore, the unwinding of the stem-loop structures by UPF1 helicase is required for the RNA cleavage catalysed by MCPIP1 protein [61]. However, our visualization of the structures of RNA sequences hitherto reported as interacting with MCPIP1 protein suggested that the characteristic stem-loop structure might not be necessary for recognition by the ribonuclease (Fig. S3). Our bioinformatics analyses of the sequences established as MCPIP1 binding sites revealed a common sequential motif, which exhibited similar features to the consensus sequence of MCPIP1’s substrates reported by Mino and colleagues (Fig. 5 B; [9]. All of the described motifs contained a relatively stable U[G/A]U sequence (Fig. 5 B) [9]. Nevertheless, the analysis of the structure of the hitherto reported MCPIP1’s binding sites did not reveal a stem-loop structure, but rather a significant difference in the accessibility of unpaired nucleotides in the U[G/A]U sequence, as compared to inaccessible nucleotides upstream and downstream of the sequence (Fig. 6 A). The discrepancy of the secondary structure of the sequences interacting with the MCPIP1 protein might be attributed to the inclusion of the 40 nucleotide flanking sites from the investigated transcripts in our analysis. However, it is difficult to predict the structures of the described binding sites due to many protein-RNA interactions occurring in the cytoplasm of the cells. Nevertheless, we believe that taking into consideration the 40 nucleotide flanking regions allows for a more general analysis of the secondary structure of the MCPIP1 binding sites.
Human AURKA 3′UTR length varies from 768 nucleotides for most of the transcript variants to 785 nucleotides in case of the transcript variant 3 (according to NM_198433.3, NM_003600.4, NM_198434.2, NM_198435.3, NM_198436.3, NM_198437.3, NM_001323303.2, NM_001323304.2, and NM_001323305.2). However, only a 95-nucleotide region of the 3′UTR is highly conserved across mammalian species (Fig. 6 A), while the rest of the 3′UTR exhibits very low conservation. The interaction of MCPIP1 with its substrates is generally well conserved between human and mouse [7,41]. Hence, it is unlikely that the cis-acting element responsible for the interaction between the ribonuclease and the transcript is located outside of the conserved region. Furthermore, we have found two sequences matching the common sequential motif found by us for the RNA sequences interacting with MCPIP1 in the conserved region of AURKA 3′UTR (Fig. 6 A and B). Additionally, analysis of the secondary structure of the region revealed the presence of U[G/A]U within a firm stem-loop (Fig. 6 A and B). However, a series of luciferase assays signalled a lack of interaction between MCPIP1 and the putative binding sites within the conserved region of the AURKA 3ʹUTR. Nevertheless, the ribonuclease overexpression led to the destabilization of luciferase mRNA conjugated with the conserved region of AURKA 3′UTR. The result points to the binding of the conserved region by the MCPIP1 protein and subsequent degradation of the mRNA. The exact sequence indispensable for the observed interaction remains to be established.
MicroRNA deregulation contributes to all the aspects of cancer development. Several miRNA molecules were described as either tumour suppressors or oncogenes in neuroblastoma [44]. Moreover, MCPIP1 was found to degrade the substrate pre-miRNA forms of miRNAs from the hsa-miR-200 family in pancreatic cancer cells [5]. Additionally, cleavage of pre-hsa-miR-155 by the ribonuclease has been reported [6,62]. Furthermore, our previous studies revealed the possible regulation of hsa-miR-3613-3p levels by MCPIP1 in MNA neuroblastoma cells [18,31]. However, the application of small RNA sequencing did not result in the detection of significant changes in the miRNA expression profile upon the ribonuclease overexpression in BE(2)-C and KELLY neuroblastoma cells (Table 2; Fig. 7). Hence, the miRNA deregulation does not seem to play an important role in the suppressive effect of MCPIP1 overexpression in MNA neuroblastoma cells. However, elucidation of the role of MCPIP1 in regulating miRNA profile of cancer cells requires further studies.
Our results constitute further indication of the importance of ribonucleolytic activity in the suppressive effect of MCPIP1 on MNA neuroblastoma cells. MCPIP1 overexpression did not significantly affect the miRNA expression profile in our experimental model, which suggests the crucial role of the mRNA cleavage by the ribonuclease. Accordingly, we have shown a significant body of data pointing to binding and degradation of AURKA mRNA by MCPIP1. As AURKA exhibits oncogenic potential, the observed activity of the protein might be crucial in the MCPIP1-dependent cell cycle arrest of MNA neuroblastoma cell lines. Additionally, we specified that the MCPIP1 binding site is located within the 95-nucleotide conserved region of the 3′UTR of the AURKA transcript. However, to determine the exact sequence in AURKA 3′UTR interacting with MCPIP1 protein further studies are necessary.
Supplementary Material
Acknowledgments
The study was supported by the grant no 2017/25/N/NZ1/01254 from the Polish National Science Center awarded to IN.
Funding Statement
This work was supported by the Polish National Science Center [2017/25/N/NZ1/01254].
Disclosure statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the here.
References
- [1].Liang J, Wang J, Azfer A, et al. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J Biol Chem. 2008;283(10):6337–6346. [DOI] [PubMed] [Google Scholar]
- [2].Fu M, Blackshear PJ.. RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat Rev Immunol. 2017;17(2):130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wilamowski M, Gorecki A, Dziedzicka-Wasylewska M, et al. Substrate specificity of human MCPIP1 endoribonuclease. Sci Rep. 2018;8(1):7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Xu J, Peng W, Sun Y, et al. Structural study of MCPIP1 N-terminal conserved domain reveals a PIN-like RNase. Nucleic Acids Res. 2012;40(14):6957–6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Boudouresque F, Siret C, Dobric A, et al. Ribonuclease MCPiP1 contributes to the loss of micro-RNA-200 family members in pancreatic cancer cells. Oncotarget. 2018;9(89):35941–35961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Suzuki HI, Arase M, Matsuyama H, et al. MCPIP1 ribonuclease antagonizes dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol Cell. 2011;44(3):424–436. [DOI] [PubMed] [Google Scholar]
- [7].Li M, Cao W, Liu H, et al. MCPIP1 down-regulates IL-2 expression through an ARE-independent pathway. PLoS One. 2012;7(11):e49841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Masuda K, Ripley B, Nyati KK, et al. Arid5a regulates naive CD4+ T cell fate through selective stabilization of Stat3 mRNA. J Exp Med. 2016;213(4):605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mino T, Murakawa Y, Fukao A, et al. Regnase-1 and roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell. 2015;161(5):1058–1073. [DOI] [PubMed] [Google Scholar]
- [10].Uehata T, Iwasaki H, Vandenbon A, et al. Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell. 2013;153(5):1036–1049. [DOI] [PubMed] [Google Scholar]
- [11].Behrens G, Winzen R, Rehage N, et al. A translational silencing function of MCPIP1/Regnase-1 specified by the target site context. Nucleic Acids Res. 2018;46(8):4256–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li Z, Jia Y, Han S, et al. Klf4 alleviates lipopolysaccharide-induced inflammation by inducing expression of MCP-1 induced protein 1 to deubiquitinate TRAF6. Cell Physiol Biochem. 2018;47(6):2278–2290. [DOI] [PubMed] [Google Scholar]
- [13].Niu J, Shi Y, Xue J, et al. USP10 inhibits genotoxic NF-κB activation by MCPIP1-facilitated deubiquitination of NEMO. Embo J. 2013;32(24):3206–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang W, Huang X, Xin HB, et al. TRAF family member-associated NF-κB activator (TANK) inhibits genotoxic nuclear factor κB activation by facilitating deubiquitinase USP10-dependent deubiquitination of TRAF6 ligase. J Biol Chem. 2015;290(21):13372–13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hanahan D, Weinberg RA.. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- [16].Ligeza J, Marona P, Gach N, Lipert B, Miekus K, Wilk W, Jaszczynski J, Stelmach A, Loboda A, Dulak J, Branicki W, Rys J, Jura J. MCPIP1 contributes to clear cell renal cell carcinomas development. Angiogenesis. 2017. August;20(3):325-340. doi: 10.1007/s10456-017-9540-2. Epub 2017 Feb 14. PMID: 28197812; PMCID: PMC5511613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boratyn E, Nowak I, Durbas M, et al. MCPIP1 exogenous overexpression inhibits pathways regulating MYCN oncoprotein stability in neuroblastoma. J Cell Biochem. 2017;118(7):1741–1755. [DOI] [PubMed] [Google Scholar]
- [18].Boratyn E, Nowak I, Horwacik I, et al. Monocyte chemoattractant protein-induced protein 1 overexpression modulates transcriptome, including MicroRNA, in human neuroblastoma cells. J Cell Biochem. 2016;117(3):694–707. [DOI] [PubMed] [Google Scholar]
- [19].Boratyn E, Nowak I, Karnas E, et al. MCPIP1 overexpression in human neuroblastoma cell lines causes cell-cycle arrest by G1/S checkpoint block. J Cell Biochem. 2020;121(5–6):3406–3425. [DOI] [PubMed] [Google Scholar]
- [20].Lichawska-Cieslar A, Pietrzycka R, Ligeza J, et al. RNA sequencing reveals widespread transcriptome changes in a renal carcinoma cell line. Oncotarget. 2018;9(9):8597–8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lu W, Ning H, Gu L, et al. MCPIP1 selectively destabilizes transcripts associated with an antiapoptotic gene expression program in breast cancer cells that can elicit complete tumor regression. Cancer Res. 2016;76(6):1429–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Marona P, Górka J, Mazurek Z, et al. MCPIP1 downregulation in clear cell renal cell carcinoma promotes vascularization and metastatic progression. Cancer Res. 2017;77(18):4905–4920. [DOI] [PubMed] [Google Scholar]
- [23].Ren Z, He M, Shen T, et al. MiR-421 promotes the development of osteosarcoma by regulating MCPIP1 expression. Cancer Biol Ther. 2020;21(3):231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Skalniak A, Boratyn E, Tyrkalska SD, et al. Expression of the monocyte chemotactic protein-1-induced protein 1 decreases human neuroblastoma cell survival. Oncol Rep. 2014;31(5):2385–2392. [DOI] [PubMed] [Google Scholar]
- [25].Zhuang J, Wu Y, Chen L, et al. Single-cell mobility analysis of metastatic breast cancer cells. Adv Sci (Weinh). 2018;5(12):1801158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Louis CU, Shohet JM. Neuroblastoma: molecular pathogenesis and therapy. Annu Rev Med. 2015;66(1):49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Brockmann M, Poon E, Berry T, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell. 2013;24(1):75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Michaelis M, Selt F, Rothweiler F, et al. Aurora kinases as targets in drug-resistant neuroblastoma cells. PLoS One. 2014;9(9):e108758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mossé YP, Fox E, Teachey DT, et al. A phase II study of alisertib in children with recurrent/refractory solid tumors or leukemia: children’s oncology group phase I and pilot consortium (ADVL0921). Clin Cancer Res. 2019;25(11):3229–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shang X, Burlingame SM, Okcu MF, et al. Aurora A is a negative prognostic factor and a new therapeutic target in human neuroblastoma. Mol Cancer Ther. 2009;8(8):2461–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nowak I, Boratyn E, Durbas M, et al. Exogenous expression of miRNA-3613-3p causes APAF1 downregulation and affects several proteins involved in apoptosis in BE(2)-C human neuroblastoma cells. Int J Oncol. 2018;53(4):1787–1799. [DOI] [PubMed] [Google Scholar]
- [32].Lipert B, Wilamowski M, Gorecki A, et al. MCPIP1, alias Regnase-1 binds and cleaves mRNA of C/EBPβ. PLoS One. 2017;12(3):e0174381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- [34].Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12(10):671–682. [DOI] [PubMed] [Google Scholar]
- [35].Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lu Y, Baras AS, Halushka MK. miRge 2.0 for comprehensive analysis of microRNA sequencing data. BMC Bioinformatics. 2018;19(1):275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Smith P, Krohn RI, Hermanson GT. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150(1):76–85. [DOI] [PubMed] [Google Scholar]
- [39].Lorenz R, Bernhart SH, Hoener Zu Siederdissen C, et al. (2011), ViennaRNA package 2.0, Algorithms for Molecular Biology: 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bailey T, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, 28–36, AAAI Press, Menlo Park, California. [PubMed] [Google Scholar]
- [41].Kochan J, Wawro M, Kasza A. IF-combined smRNA FISH reveals interaction of MCPIP1 protein with IER3 mRNA. Biol Open. 2016;5(7):889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Uehata T, Akira S. mRNA degradation by the endoribonuclease Regnase-1/ZC3H12a/MCPIP-1. Biochim Biophys Acta. 2013;1829(6–7):708–713. [DOI] [PubMed] [Google Scholar]
- [43].Losko M, Lichawska-Cieslar A, Kulecka M, et al. Ectopic overexpression of MCPIP1 impairs adipogenesis by modulating microRNAs. Biochim Biophys Acta Mol Cell Res. 2018;1865(1):186–195. [DOI] [PubMed] [Google Scholar]
- [44].Zhi F, Wang R, Wang Q, et al. MicroRNAs in neuroblastoma: small-sized players with a large impact. Neurochem Res. 2013;39(4):613–623. [DOI] [PubMed] [Google Scholar]
- [45].Li W, Chang J, Wang S, et al. miRNA-99b-5p suppresses liver metastasis of colorectal cancer by down-regulating mTOR. Oncotarget. 2015;6(27):24448–24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang C, Wang S, Ma F, et al. miRNA‑328 overexpression confers cisplatin resistance in non‑small cell lung cancer via targeting of PTEN. Mol Med Rep. 2018;18(5):4563–4570. [DOI] [PubMed] [Google Scholar]
- [47].Sun GL, Li Z, Wang WZ, et al. miR-324-3p promotes gastric cancer development by activating Smad4-mediated Wnt/beta-catenin signaling pathway. J Gastroenterol. 2018;53(6):725–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tian L, Zhao Z, Xie L, et al. MiR-361-5p inhibits the mobility of gastric cancer cells through suppressing epithelial-mesenchymal transition via the Wnt/β-catenin pathway. Gene. 2018;675:102–109. [DOI] [PubMed] [Google Scholar]
- [49].Li T, Ding ZL, Zheng YL, et al. MiR-484 promotes non-small-cell lung cancer (NSCLC) progression through inhibiting Apaf-1 associated with the suppression of apoptosis. Biomed Pharmacother. 2017;96:153–164. [DOI] [PubMed] [Google Scholar]
- [50].Latchana N, DiVincenzo MJ, Regan K, et al. Alterations in patient plasma microRNA expression profiles following resection of metastatic melanoma. J Surg Oncol. 2018;118(3):501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Latchana N, Regan K, Howard JH, et al. Global microRNA profiling for diagnostic appraisal of melanocytic spitz tumors. J Surg Res. 2016;205(2):350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Said R, Garcia-Mayea Y, Trabelsi N, et al. Expression patterns and bioinformatic analysis of miR-1260a and miR-1274a in prostate cancer tunisian patients. Mol Biol Rep. 2018;45(6):2345–2358. [DOI] [PubMed] [Google Scholar]
- [53].Lyu JH, Park D-W, Huang B, Kang SH, Lee SJ, Lee C, Bae Y-S, Lee J-G, Baek S-H. RGS2 suppresses breast cancer cell growth via a MCPIP1-dependent pathway. J Cell Biochem. 2015. February;116(2):260–267. doi: 10.1002/jcb.24964. PMID: 25187114. [DOI] [PubMed] [Google Scholar]
- [54].Damodaran AP, Vaufrey L, Gavard O, et al. Aurora A kinase is a priority pharmaceutical target for the treatment of cancers. Trends Pharmacol Sci. 2017;38(8):687–700. [DOI] [PubMed] [Google Scholar]
- [55].Horwacik I, Durbas M, Boratyn E, et al. Targeting GD2 ganglioside and aurora A kinase as a dual strategy leading to cell death in cultures of human neuroblastoma cells. Cancer Lett. 2013;341(2):248–264. [DOI] [PubMed] [Google Scholar]
- [56].Yan M, Wang C, He B, et al. Aurora-A Kinase: A Potent Oncogene and Target for Cancer Therapy. Med Res Rev. 2016;36(6):1036–1079. [DOI] [PubMed] [Google Scholar]
- [57].DuBois SG, Mosse YP, Fox E, et al. Phase II trial of alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma. Clin Cancer Res. 2018;24(24):6142–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Inandiklioğlu N, Yilmaz D, Erdoğan S, et al. Chromosome imbalances and alterations of AURKA and MYCN genes in children with neuroblastoma. Asian Pac J Cancer Prev. 2012;13(11):5391–5397. [PubMed] [Google Scholar]
- [59].Otto T, Horn S, Brockmann M, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15(1):67–78. [DOI] [PubMed] [Google Scholar]
- [60].Skalniak L, Mizgalska D, Zarebski A, et al. Regulatory feedback loop between NF-kappaB and MCP-1-induced protein 1 RNase. Febs J. 2009;276(20):5892–5905. [DOI] [PubMed] [Google Scholar]
- [61].Mino T, Iwai N, Endo M, et al. Translation-dependent unwinding of stem-loops by UPF1 licenses Regnase-1 to degrade inflammatory mRNAs. Nucleic Acids Res. 2019;47(16):8838–8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Li Z, Han S, Jia Y, et al. MCPIP1 regulates RORα expression to protect against liver injury induced by lipopolysaccharide via modulation of miR-155. J Cell Physiol. 2019. DOI: 10.1002/jcp.28327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.