

ABSTRACT
Hypoxia causes a series of responses supporting cells to survive in harsh environments. Substantial post-transcriptional and translational regulation during hypoxia has been observed. However, detailed regulatory mechanism in response to hypoxia is still far from complete. RNA m6A modification has been proven to govern the life cycle of RNAs. Here, we reported that total m6A level of mRNAs was decreased during hypoxia, which might be mediated by the induction of m6A eraser, ALKBH5. Meanwhile, expression levels of most YTH family members of m6A readers were systematically down-regulated. Transcriptome-wide analysis of m6A revealed a drastic reprogramming of m6A epitranscriptome during cellular hypoxia. Integration of m6A epitranscriptome with either RNA-seq based transcriptome analysis or mass spectrometry (LC-MS/MS) based proteome analysis of cells upon hypoxic stress revealed that reprogramming of m6A epitranscriptome reshaped the transcriptome and proteome, thereby supporting efficient generation of energy for adaption to hypoxia. Moreover, ATP production was blocked when silencing an m6A eraser, ALKBH5, under hypoxic condition, demonstrating that m6A pathway is an important regulator during hypoxic response. Collectively, our studies indicate that crosstalk between m6A and HIF1 pathway is essential for cellular response to hypoxia, providing insights into the underlying molecular mechanisms during hypoxia.
KEYWORDS: Hypoxia, m6A epitranscriptome, trancriptome, proteome, ATP production
Introduction
Hypoxia occurs in a range of either physiological or pathological processes such as embryogenesis and development of solid tumours [1].This is one of the major factors promoting tumour progression, metastasis, generation of cancer stem cells, and resistance to radiotherapy and chemotherapy [1]. Hypoxia-inducible factor-1 (HIF1), consisting of HIF1A as an O2-responsive subunit and HIF1B as a constitutively expressed subunit, is a core transcription factor that is activated upon hypoxia [2]. Under normoxic condition, HIF1A is hydroxylated by proline hydroxylase domain proteins (PHDs), subsequently ubiquitinated by von Hippel-Lindau (VHL) protein, and finally degraded by the proteasome degradation pathway [3]. Under hypoxic condition, PHDs activity is inhibited. HIF1A accumulates and enters into the nucleus to form the active HIF1 complex with HIF1B [3]. The HIF1 complex binds to the hypoxic response elements (HREs) in the genome [4] to promote reprogramming of the transcriptome and proteome of cells systematically. Together, this facilitates metabolic rewiring that shifts production of cellular energy from high mitochondrial efficiency of ATP production to lowly efficient glycolysis making cells adaptive to hypoxia [5]. Meanwhile, due to insufficient ATP generation by glycolysis, cells reduce energy consumption [e.g., suppression of transcription [6] and translation [7]] to preserve energy for obligatory functions necessary for cell survival [6], which in turn add complexity to reprogramming of transcriptome and proteome. Detailed mechanisms underlying transcriptome and proteome reprogramming during hypoxic process are still unclear.
Epitranscriptome consists of diverse covalent RNA modifications that shape cellular transcriptome and proteome via regulation of RNA metabolism including processing, decay and translation of RNA [8]. Among these modifications, m6A is the most prevalent internal mRNA modification, occurring at a consensus motif (DRACH), enriching in the 3ʹ UTRs near the stop codon [9]. m6A modification is decorated by a multicomponent methyltransferase complex containing METTL3, METTL14 and WTAP and demethylated by the demethylase FTO or ALKBH5 [10], which makes methylation process dynamic and reversible. Notably, FTO also demethylates m6Am in mRNA and snRNA as well as m1A in tRNA [11,12]. The effects of m6A modification depend on various reader proteins [13], like YTH (YT521-B homology) family members, which participate in the process of RNA splicing, location, stability and protein translation efficiency. Among them, YTHDF2 expedites the half-life of mRNA; YTHDF1 mediates mRNA translation promotion; YTHDF3 together with YTHDF1 and YTHDF2 facilitates their processing in context. It should be noted that the functions of YTHDFs are still controversial [14,15]. YTHDC1 is required for mRNA splicing and nuclear export, and YTHDC2 improves translation efficiency while also decreases its targets [13]. Regulation of m6A is important in haematopoietic system [16,17], cell fate determination [18] and neuronal functions [19,20]. YTHDF2-mediated mRNA decay of notch1a and rhoca is required for development of haematopoietic stem/progenitor cells as shown in zebrafish [17]. Accurate m6A level is crucial for behaviour and electrophysiological properties of mouse cortex in response to acute stress [19]. Moreover, m6A also plays a vital role in cellular response to external stimuli such as viral infection [21,22], DNA damage [23] and heat shock response [24,25]. For example, m6A modifications on transcripts rapidly recruit DNA polymerase to ultraviolet (UV) induced damage sites to facilitate DNA repair and cell survival [23]. m6A pathway may be important for hypoxic regulation by HIFs. Previous studies reported that hypoxic induction of ALKBH5 was dependent on HIFs and contributed to the breast cancer stem cell phenotype [26]. In the hypoxia/reoxygenation-treated cardiomyocytes, METTL3 is responsible for inhibiting autophagic flux and promoting apoptosis [27]. However, detailed regulatory mechanisms of cellular response to hypoxia by m6A pathway are still unclear.
In this study, to elucidate the role of m6A in the context of cellular hypoxic stress, we performed m6A-seq, RNA-seq and data dependent acquisition (DDA)-based LC-MS/MS of cells upon hypoxic stress. Through integrated analysis of high-throughput epitranscriptome, transcriptome and proteome data, a dramatic transition of RNA m6A epitranscriptome was noted during hypoxic response, contributing to reshaping the transcriptome and proteome to support efficient cellular responses to hypoxia. Our results indicate that the m6A pathway is crucial for cellular adaption to hypoxia.
Materials and methods
Cell culture and culture conditions
HeLa, SMMC7721, Huh7, HepG2 and Hep3B cell lines were obtained from the Shanghai Cell Bank of Chinese Academy of Sciences. All cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1 × penicillin-streptomycin (Beyotime, #C0222) and maintained in a 5% CO2 and 95% air incubator [20% (vol/vol) O2] at 37°C. For hypoxia exposure, cells were placed in a modular incubator chamber (MART) filled with a hypoxic gas mixture containing 1% O2, 5% CO2 and 94% N2 for indicated time points.
RNA isolation
Total RNA from indicated samples was extracted with TRIzol reagent (Invitrogen Life Technologies, #15596018) according to the manufacturer’s instruction. All samples were treated with DNase I to avoid genomic DNA contaminations. The purified RNA pellet was stored at −80°C for later use.
RNA m6A dot blots
Polyadenylated (poly(A)+) RNA was isolated from total RNA using Oligotex mRNA Kits (QIAGEN, #70022). RNA was denatured at 70°C for 2 min and immediately transferred on ice. Samples were spotted onto the Hybond-N+ membrane (Amersham) and cross-linked by UV 254 nm. The membrane was then blocked with 5% non-fat milk in 1× PBST for 1 h at room temperature and incubated with a specific anti-m6A antibody (Abcam, ab151230) for overnight at 4°C, followed by incubation with the HRP-conjugated anti-rabbit secondary antibodies (Transgen Biotech, HS101-01) for 1 h at room temperature and the membrane was developed with enhanced chemiluminescent (ECL) substrate (Thermo Fisher Scientific, #34096).
Immunoblot assay
HeLa cells treated for indicated time points were washed twice with Phosphate Buffer solution (PBS), and then lysed in RIPA buffer (150 mM NaCl, 50 mM tris-HCl, pH 8.0, 5 mM EDTA, 0.5% NP-40). After sonication, lysates were subjected to electrophoresis on a NuPage 4–12% Bis-Tris gel and transferred onto a PVDF membrane. The membrane was blocked for 1 h in 5% non-fat milk in 1× PBST and incubated overnight at 4°C with primary antibodies. Antibodies used include: anti-HIF1A (Proteintech, 20960-1-AP), anti-HIF2A (Novus Biologicals, NB100-122), anti-METTL3 (Proteintech, 15073-I-AP), anti-METTL14 (Atlas antibodies, HPA038002), anti-WTAP (Proteintech, 10200-I-AP), anti-ALKBH5 (Abcam, ab69325), anti-FTO (Phosphosolution, 597-FTO), anti-ACTB (Transgen Biotech, HC201-02), anti-YTHDF1 (Proteintech, 17479-I-AP), anti-YTHDF2 (Abcam, ab176846), anti-YTHDF3 (Santa Cruz, sc-377119), anti-YTHDC1 (Cell Signalling Technology, 87459S), anti-YTHDC2 (Abcam, ab176846), anti-SLC2A1 (Proteintech, 21829-1-AP), anti-MTCH2 (Proteintech, 16888-1-AP), HRP-conjugated anti-rabbit (Transgen Biotech, HS101-01) and anti-mouse (Transgen Biotech, HS201-01) secondary antibodies.
m6A-seq and RNA-seq assay
For m6A immunoprecipitation, procedure was modified from previously reported methods [28]. In brief, poly(A)+ RNA was isolated from total RNA using Oligotex mRNA Kits (QIAGEN, #70022) and subsequently fragmented into about 150 nt fragments using RNA fragmentation buffer (20 mM Tris-HCl, pH 7.4, 20 mM ZnCl2) at 94°C for 40 s. Reaction was stopped with 0.05 M EDTA. For m6A-IP, 2 μg fragmented RNA was incubated with 3 μg anti-m6A antibody (Abcam) in immunoprecipitation buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% Igepal CA-630) supplemented with RNase inhibitor (Promega) for 2 h at 4°C. Above mixture was incubated with 20 μl protein A/G beads (Thermo Fisher Scientific, #88803) for an additional 2 h at 4°C on a rotating wheel. After washing five times with immunoprecipitation buffer, bound RNA was extracted by proteinase K digestion, phenol-chloroform extraction followed by standard ethanol precipitation. Libraries were constructed by Truseq Stranded mRNA Sample Prep Kit (Illumina) according to the manufacturer’s instructions and quantified by BioAnalyzer High Sensitivity DNA chip (Agilent), and then deeply sequenced on the Illumina HiSeq X10 to generate 150-bp paired-end reads. For RNA-seq library, mRNA enrichment, cDNA synthesis, adaptor addition, circularization, PCR amplification and library examination were performed on the BGISEQ 500 at Beijing Genome Institute (BGI; Shenzhen, China).
Processing of m6A-seq and RNA-seq
Adaptor sequences for all raw reads were removed using cutadapt software (version 3.5.1). Sequences shorter than 20 nt in length or reads of which more than 10% presented a quality score less than 25 were filtered. The remaining sequences were aligned to human genome hg19 with TopHat 2.0 program as described previously [29] and the longest isoform was used if multiple isoforms existed. The uniquely mapping reads were used for the subsequent analysis. For m6A-seq, the m6A modification peaks were identified by exomePeak with FDR (false discovery rate) < 0.0001 [30], and the corresponding RNA-seq profiles were used for normalization [31]. m6A peaks that satisfied 1) peak read counts > 10 and 2) enrichment score > 1.5 as described previously [32] were considered for subsequent analysis. DiffBind was used to search the common and unique peaks having m6A modification among more than two samples. CoverageBed of BedTools with ‘-F 0.50’ parameters was used to calculate the read count of each peak. Subsequently, the ‘IP FPKM’, ‘input FPKM’ and ‘Enrichment score’ of peaks were calculated as previously reported [20]. Alternatively, differential m6A peaks identified by exomePeak between the corresponding treated and control samples were considered to be significant with peak read counts in any sample more than 10 and P value < 0.01. Motifs enriched with m6A peaks were identified by HOMER (version 3.5.1) [33] and lengths were restricted to 4–6 nucleotides. For RNA-seq, uniquely mapping reads were counted as FPKM of each gene to represent RNA expression level using Cufflink [29].
Characterization of m6A peak distribution patterns
The m6A peaks were annotated with GTF file. m6A tagged transcripts were split into protein-coding genes and noncoding RNAs according to the GTF file. To characterize the distribution patterns of m6A peaks, the 5ʹ UTR, CDS and 3ʹ UTR regions of each protein-coding gene or the entire transcripts of noncoding RNAs were split into equal length with 100 bins as previously reported [34,35]. Percentage of m6A peaks in each bin indicated occupancy of m6A peaks along the overall transcripts [36].
MazF-qPCR and analysis
100 ng of poly(A)+ RNA was denatured at 70°C for 2 min and immediately transferred on ice. RNA was then digested with MazF enzyme (Takara, 2415A) at 37°C for 30 min following the manufacturer’s instruction and stopped by placing on ice [37]. Digested RNA was purified with MyOne SILANE Dynabeads (Invitrogen, 37002D). For quantifying methylation in two conditions, designation of primer pairs and calculation of the relative ratio of m6A abundance were performed as described in ref [37].
MeRIP-QPCR and analysis
Total RNA was extracted with TRIzol reagent (Invitrogen Life Technologies) according to the manufacturer’s instructions and fragmented into length of 300 ~ 500 nt with RNA fragmentation buffer. A 100 μg aliquot of fragmented RNA was incubated with either 3 μg m6A specific antibody (Abcam, ab151230) or normal IgG (negative control). RNA was eluted according to the above protocol. Reverse transcription was carried out with an equal ratio of RNA from input and IP product by using PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, R047A). Quantitative real-time PCR was performed using FS universal SYBR Green (Roche, #4887352001-1). Percentage of a target gene in IP sample was calculated relative to in input sample as previously reported [20,38]. Sequences used are listed in the Supplementary Table S1.
siRNA Knockdown and plasmid transfection
The siRNA sequences used were as follows:
Non-specific small interfering RNA (siRNA):
sense strand: 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ
antisense strand: 5ʹ- ACGUGACACGUUCGGAGAATT −3ʹ
siRNA targeting ALKBH5#1:
sense strand: 5ʹ-GCUGCAAGUUCCAGUUCAATT-3ʹ
antisense strand: 5ʹ-UUGAACUGGAACUUGCAGCTT −3ʹ
siRNA targeting ALKBH5#2:
sense strand: 5ʹ-GCUUCAGCUCUGAGAACUATT-3ʹ
antisense strand: 5ʹ-UAGUUCUCAGAGCUGAAGCTT-3ʹ
siRNA targeting ALKBH5 and control scrambled siRNA were purchased from GenPharma.lnc. Human CDS of YTHDF1, YTHDF2 and YTHDF3 were cloned into pcDNA4.0/TO-SBP-Flag-S protein-tagged (SFB) vector backbone. Human CDS of HIF1A was cloned into pSIN vector backbone. Transfection was achieved using Lipofectamine 2000 reagent (Thermo Fisher Scientific, #11668-019) for siRNA, and ViaFect transfection reagent (Promega, #E4982) for plasmids following the manufacturer’s protocols.
LC-MS/MS and Protein quantification
Proteome data were quantified by label-free quantitation. Briefly, 100 μg of protein from normoxia and oxygen deprivation (24 h) were solubilized in RIPA buffer and sonicated for 10 min. Protein lysis was digested with trypsin, desalted with Oasis HLB (Waters), dissolved in 0.1% formic acid, followed by quantified using a peptide quantification kit (Thermo Fisher Scientific, #23275). 1 μg eluted peptides per sample were prepared for the LC-MS/MS analysis. Raw MS proteomics data obtained from Orbitrap were analysed by MaxQuant software. Andromeda search engine was used to search against the UniProt human database for MS/MS spectra. Relative protein abundance was determined as previously reported [39].
Detection of cellular ATP levels
ATP levels of cells were measured using a firefly luciferase-based ATP-enhanced assay kit (Beyotime, China) according to the manufacturer’s instructions. Briefly, after the indicated treatment, cells were lysed and centrifuged at 13,000 g for 5 min. Supernatant (20 μl) was mixed with 100 ul of ATP working solution in a white 96-well plate. Luminescence (RLU) was measured by a GloMax microplate reader. Protein concentration of each treatment group was determined using BCA protein assay. Total ATP levels were considered as nmol/mg protein. These experiments were repeated twice.
Gene ontology and KEGG pathway analysis
Gene Ontology (GO) analysis was accomplished using either ConsensusPathDB website (http://cpdb.molgen.mpg.de) [40] or cytoscape software [41]. Top 8 enriched GO terms of biological processes were depicted in figures with R software (version 3.4.0).
Statistical analysis
All statistical analyses were performed with GraphPad Prism (version 7.0) or R software (version 3.4.0). Two-tailed Student’s t-test was used for both LC-MS/MS analysis and real-time PCR. p value <0.05 was considered statistically significant.
Results
m6A epitranscriptome was suppressed upon hypoxic stress
To investigate whether m6A epitranscriptome was involved in regulation of cellular hypoxic response, HeLa and SMMC7721 cells were cultured in a low-oxygen sealed container for 24 h (1% O2), causing dramatically elevated HIF1A (Fig. 1C). Intriguingly, total m6A level of poly(A)+ RNAs was decreased after hypoxia (Fig. 1A, Supplementary Figure S1). Cellular m6A levels are determined by m6A modification enzymes. Expression levels of core subunits of m6A writers and easers of mRNA were examined in cells after hypoxic treatment. Immunoblot assays revealed that protein level of only one m6A eraser, ALKBH5, was obviously increased after cells exposed to hypoxia (24 h) in both HeLa and SMMC7721 cells. In contrast, expression levels of either other main writers or eraser including METTL3, METTL14, WTAP and FTO, were not affected (Fig. 1B,C). Hence, down-regulation of total RNA m6A level may result from up-regulation of ALKBH5. Intriguingly, in coordination with down-regulation of total RNA m6A level, the protein levels of m6A readers including YTHDF1, YTHDF2, YTHDF3 and YTHDC2 were extensively down-regulated, while YTHDC1 was up-regulated, in HeLa, SMMC-7721, as well as Huh7, HepG2 and Hep3B upon hypoxic stress (Fig. 1C), implying that the hypoxic stress silenced the m6A pathway in cells through down-regulation of both m6A level and m6A readers. Analysis of HIF1A binding site in the promoter region of all m6A related genes using ChIP-seq datasets for HeLa and T47D (generated in previous studies [42,43],), only ALKBH5 was a hypoxic responsive gene with HIF1A binding site in its promoter (Fig. 1D). Consistently, ectopic expression of HIF1A in Hela cells just up-regulated the expression of ALKBH5 but not other YTH proteins (Fig. 1E). QPCR assays also showed that only the RNA level of ALKBH5 was significantly increased upon hypoxic stress (Fig. 1F). Analysis of 10 RNA-Seq datasets generated from 8 different cell lines upon hypoxic stress in GEO database showed that the mRNA expression level ALKBH5 is consistently up-regulated in all cell lines upon hypoxia, indicating that this phenomenon is general [44–49] (Fig. 1G). We further silenced ALKBH5 in HeLa cells under hypoxic condition. m6A dot blot assay showed that the total m6A level of mRNA was partly rescued (Fig. 1H), demonstrating that ALKBH5 mediated the decreased total m6A levels of mRNA in cells upon hypoxia. (Fig. 1H). These results suggested that suppression of m6A pathway is synergistic with HIF1-mediated hypoxic response during hypoxia.
Figure 1.
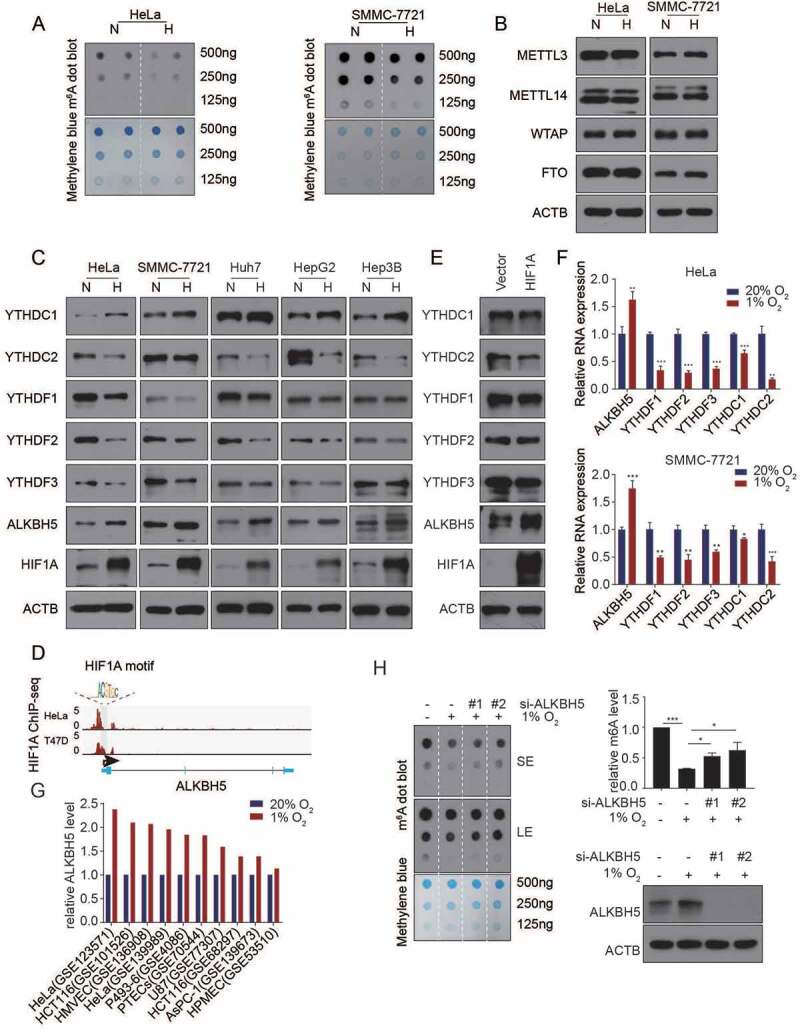
m6A regulatory system is restrained under hypoxic condition. (A) HeLa and SMMC7721 cells were exposed to either 20% or 1% O2 for 24 h. Poly(A)+ RNA was extracted and m6A levels were determined by dot blot. MB, methylene blue staining (as loading control). (B) HeLa and SMMC7721 cells were exposed to either 20% or 1% O2 for 24 h; whole cell lysates were prepared; and immunoblot assays were performed to analyse levels of protein expression of METTL3, METTL14, WTAP and FTO. (C) HeLa, SMMC7721, Huh7, HepG2 and Hep3B cells were exposed to either 20% or 1% O2 for 24 h; whole cell lysates were prepared; and immunoblot assays were performed to analyse levels of protein expression of HIF1A, ALKBH5, YTHDF1, YTHDF2, YTHDF3, YTHDC1 and YTHDC2. (D) HIF1A ChIP-seq peak signals of ALKBH5. (E) The effects of HIF1A overexpression on the level of ALKBH5 and YTHs readers. (F) HeLa (upper panel) and SMMC7721 (lower panel) after their exposure to either 20% or 1% O2 for 24 h, RT-qPCR assays were performed to determine mRNA levels of m6A regulatory system relative to RPLP0. Results were normalized to normoxia (mean ± SEM; n = 3; * p < 0.05, ** p < 0.01 and *** p < 0.001). (G) Column chart displaying ALKBH5 level upon hypoxic stress within 10 GEO hypoxia-related transcriptome datasets. The y-coordinates represent fold changes of ALKBH5 level relative to normoxic condition per dataset. (H) Left panel, cells with knockdown of ALKBH5 were exposed to either 20% or 1% O2 for 24 h. m6A dot blot was performed to determine total m6A levels of poly(A)+ RNA (SE, short exposure, LE, long exposure); upper right panel, statistical analysis of m6A dot blot with grey values by ImageJ (*** p < 0.001; * p < 0.05, one-way ANOVA test); lower right panel, ALKBH5 protein levels were detected by immunoblot assays
Transcriptome-wide sequencing of hypoxia-related m6A epitranscriptome
To further explore the detailed role that RNA m6A modification may play in the regulation of hypoxic process, HeLa cells were grown under either normoxic or hypoxic conditions (oxygen deprivation for 6 h, 12 h and 24 h) (Supplementary Figure S2A), and deep sequencing of the transcriptome and m6A methylome using poly(A)+ RNAs isolated from HeLa cells. Differentially expressed genes identified by RNA-seq were enriched in HIF1A signalling pathway, as well as glycolysis (Supplementary Figure S2B, S2C), demonstrating successful induction of hypoxia in HeLa cells. Total amount and quality of m6A/RNA-seq datasets are shown in Supplementary Table S2. In total, 26,057, 39,502, 39,728 and 41,653 m6A peaks from 10,843, 9,309, 10,200 and 10,717 transcripts, respectively, were identified at the four time points using exomePeak [30] (Supplementary Table S3). De novo motif search by HOMER [33] showed that m6A sites of all samples were highly enriched in DRACH consensus motif (Fig. 2A). To confirm the m6A peaks, genes were randomly selected for meRIP-qPCR assays. Enrichment scores of the candidate genes were significantly higher in m6A antibody than in control IgG samples (Fig. 2B).
Figure 2.

Transcriptome-wide m6A landscape upon hypoxic stress was determined using m6A-seq. (A) Top consensus motif identified by HOMER with m6A peaks under either normoxic (20% O2) or hypoxic (1% O2) conditions for 6 h, 12 h and 24 h. (B) meRIP-qPCR assays validated methylation levels of the representative genes. (EEF1A1-PC as positive control and EEF1A1-NC as negative control). (C) Box plot showing the methylation level of RNAs under hypoxic (1% O2) and normoxic (20% O2) conditions. ****p value < 2.2e-16 (Wilcoxon test). (D) Venn diagrams showing number and relationship of m6A peaks in response to oxygen deprivation (1% O2) at 0 h, 6 h, 12 h and 24 h time points. (E) Number of genes contained only specific m6A peaks in m6A-seq in response to hypoxia (1% O2)
Consistent with m6A dot blot, m6A methylation levels (evaluated by the enrichment scores) after exposure to hypoxia for 24 h were significantly decreased compared to cells under normoxia (Fig. 2C). Furthermore, to make these peaks comparable among the four time points, diffBind was used to identify the common peaks (appeared in all time points) and specific peaks (only appeared at one time point) at the indicated time point (Fig. 2D). Among them, a total of 7,993 common peaks continuously appeared overall during hypoxic conditions, whereas, 579, 1,025, 1,062 and 1,452 specific peaks only appeared in normoxia and hypoxia for 6 h, 12 h, 24 h, respectively (Fig. 2D). Meanwhile, 145, 0, 0, 436 genes contained only specific m6A peaks were identified (Fig. 2E). It is interesting that cells upon hypoxia have higher ALKBH5 level but have more specific m6A peaks than cells upon normoxia (Fig. 2D). Sicong Zhang et al. have found similar result that silencing of ALKBH5 in Glioblastoma Stem-like Cells (GSCs) reduced the number of m6A peaks [50]. The possible explanation for the contradiction might be that the new m6A sites under hypoxic condition did not contribute much to the total m6A level. Congruently, we observed the normalized level of hypoxia-specific peaks is significantly lower than that of normoxia-specific peaks (Supplementary Figure S3A).
Reprogramming of m6A epitranscriptome during cellular hypoxic response
Among the 7,993 common peaks during oxygen deprivation, m6A modification levels or a specific peak from one gene showed dramatic changes (Fig. 3A), which suggested that specific methylations in transcripts were indeed an actively regulated mechanism during hypoxia. Consistent with previous findings [17,35], m6A modifications were not randomly distributed along mRNAs but mainly enriched in 3ʹ UTR region near the stop codon; along noncoding RNAs, they were nearly uniformly distributed (Fig. 3B, Supplementary Figure S3B). m6A modification at different loci along transcripts might have distinct functions [25,51]. Intriguingly, the distribution of m6A along mRNAs increased slightly in coding region (CDS), while decreased in 3ʹ UTR region during hypoxic response (Fig. 3B). Similarly, m6A modifications along noncoding RNAs were increased at the 5ʹ end but decreased at the 3ʹ end region (Supplementary Figure S3B). These results indicated that cells underwent reprogramming of m6A epitranscriptome by altering both the m6A level at specific sites and their global distribution patterns in response to hypoxic stress. To investigate the effects of m6A at different positions on RNA expression, total transcripts were classified into four categories: 5ʹ UTR, CDS and 3ʹ UTR-tagged and non-tagged with m6A. The m6A-tagged transcripts, especially those which were 5ʹ UTR tagged, tended to be more stable at the RNA level than non-m6A-tagged transcripts upon hypoxic stress (Fig. 3C-E).
Figure 3.

Reprogramming of m6A epitranscriptome upon hypoxic stress. (A) Heatmap representing enrichment scores for all methylated RNAs upon hypoxic stress (1% O2) for different durations. (B) Metagene profiles of enrichment of all m6A peaks across mRNA transcriptome. (C-E) Cumulative frequency of mRNA log2 FC for transcripts containing m6A located at 5ʹ UTR, CDS, 3ʹ UTR regions or non-methylated transcripts upon hypoxic stress. (F) Venn diagrams showing the number of either common m6A-hypo or m6A-hyper transcripts (|log2 (FC)| > 1) at all time points in response to oxygen deprivation (1% O2). (G) Gene ontology (GO) enrichment analysis of either common m6A-hypo or m6A-hyper genes in response to hypoxia (1% O2)
Various mRNA modifications including m6A determine the protein output by influencing either metabolism of mRNA or translation machinery [52]. Upon hypoxic stress, 165 genes showed reduced m6A modifications (called m6A-hypo genes, fold change (FC) < 0.5) and 601 genes increased m6A modifications (called m6A-hyper genes, fold change (FC) > 2) (Fig. 3F). GO analysis showed that m6A-hypo genes were enriched in biological processes sensitive to oxygen concentration including system development and cellular response to stress (e.g., oxygen levels). In contrast, m6A-hyper genes were closely related to cellular metabolic processes (Fig. 3G). These analyses suggested that m6A might play two-side role in response to hypoxia.
Reprogramming of m6A epitranscriptome is crucial for response to hypoxia as related to transcriptome and proteome
To investigate the effects of altered m6A modification on RNA expression, we focused on hypoxic treatment for 24 h, which resulted in 717 genes showing m6A-hypo modifications (m6A-hypo genes) and 1,762 genes showing m6A-hyper modifications (m6A-hyper genes). Among m6A-hypo genes, 48 had reduced mRNA levels (called m6A-hypo-down genes) and 62 had increased mRNA levels (called m6A-hypo-up genes). Among m6A-hyper genes, 54 had reduced mRNA levels (called m6A-hyper-down genes) and 68 had increased mRNA levels (called m6A-hyper-up). The others had no change in RNA levels (Fig. 4A). Remarkably, 11 genes among the m6A-hypo-up genes were closely associated with HIF1A transcription factor network (e.g. VEGFA, SLC2A1, SERPINE1, NDRG1, their m6A modification and mRNA expression level were confirmed using MazF-qPCR [37], meRIP-qPCR and qPCR, respectively) (Fig. 4B, 4C, Supplementary Figure S3C), but not observed in the other categories of genes. Since m6A modification decreases stability of RNA as previously reported [53], we speculated that up-regulation of these hypo-up genes under hypoxic condition were dependent on demethylation caused by up-regulation of ALKBH5. To verify this hypothesis, ALKBH5 was silenced with two independent siRNAs under normoxic and hypoxic conditions (Fig. 4D). QPCR assays revealed that hypoxic induction of VEGFA, SLC2A1, SERPINE1, NDRG1, was partly abrogated after silencing ALKBH5 (Fig. 4E-H) under hypoxic conditions, but this phenomenon was not observed under normoxia. Moreover, the decreased m6A levels of these transcripts under hypoxic condition were partly rescued after silencing ALKBH5 (Fig. 4I). These results demonstrated that reprogramming of m6A epitranscriptome was involved in cellular hypoxic response.
Figure 4.

m6A epitranscriptome reshapes the transcriptome upon hypoxic stress. (A) Distribution of genes with a significant change in both the m6A and RNA levels under hypoxia (1% O2, 24 h) compared with normoxia (20% O2). (B) Bars represent the relative methylation levels at the selected m6A sites under hypoxic condition (1% O2) relative to normoxic condition (20% O2) measured via MazF-qPCR. The level of a targeted sequence (labelled ‘T’) is measured against a negative control sequence that does not contain any ACA motif (labelled ‘C’) in a MazF digested sample and normalized against a non-digested sample. (C) Integrative Genomics Viewer (IGV) plots showing methylation levels of representative genes upon oxygen deprivation (1% O2, 24 h) (light blue indicates input data, yellow orange indicates IP data). (D) Knockdown of ALKBH5 with two independent siRNAs, cells then exposed to either 20% or 1% O2 for 24 h. Efficiency of knockdown was validated using immunoblot assay. (E-H) Knockdown of ALKBH5 (two independent siRNAs) in cells exposed to either 20% or 1% O2 for 24 h. RT-qPCR was performed to determine levels of candidate genes relative to RPLP0. (I) Knockdown of ALKBH5 with two independent siRNAs, cells then exposed to either 20% or 1% O2 for 24 h. Bars represent the relative methylation levels at the candidate m6A sites (SERPINE1.chr7: 100,781,216, SLC2A1.chr1: 43,392,309, VEGFA. chr6: 43,753,243, NDRG1.chr8: 134,250,675) relative to siNC under normoxic condition (20% O2) measured via MazF-qPCR (NDRG1. chr8: 134,250,837 as the negative control)
A study suggested that m6A regulates gene expression not only at the post-transcriptional level but also at the translational level [25]. Interestingly, we found that among m6A-altered RNAs, majority of genes (2,248/2,479 = 90.6%) showed no change in RNA levels (Fig. 4A). Our studies using label-free quantitation-based proteomics analysis of HeLa cells upon hypoxia identified 479 genes differentially expressed at the protein levels (Fig. 5A, 5B, Supplementary Figure S4A), of which 124 proteins (called protein-only genes) did not change in RNA levels but changed in their m6A modification levels (Fig. 5C). Among them, 94 genes (including 51 up-regulated and 43 down-regulated) increased their m6A levels, while 30 genes reduced their m6A levels (Fig. 5C, Supplementary Figure S4B). We validated the protein levels of two randomly selected genes, SLC2A1 and MTCH2, using immunoblot assays. Protein changes of SLC2A1 and MTCH2 upon hypoxic stress were congruent with the proteome data (Fig. 5D). Level of MTCH2 (one of the protein-only genes) mRNA was not changed (Fig. 5E), whereas the m6A level was up-regulated upon hypoxic stress (Supplementary Figure S4C). To investigate whether protein expression of the protein-only genes was regulated by m6A modification under stress condition, ALKBH5 was silenced under both normoxic and hypoxic conditions, and changes of MTCH2 at both the mRNA and protein levels were examined. mRNA level of MTCH2 did not change after ALKBH5 knockdown, whereas MTCH2 protein levels were down-regulated under hypoxic condition (Fig. 5F, 5G). Since m6A-mediated effects are dependent on various readers and most of these YTH family readers were down-regulated upon hypoxic stress (described above), we hypothesized that reduction of MTCH2 under hypoxic condition was associated with down-regulation of the readers. When these readers were overexpressed, the reduction of MTCH2 induced by hypoxia was rescued compared with overexpression of empty vector under hypoxic condition (Supplementary Figure S4D). Taken together, these results indicated that both the level of m6A and the readers that mediate m6A effects were involved in regulating the hypoxic proteome, which is important for the regulation of hypoxic response.
Figure 5.

m6A epitranscriptome reshapes the proteome upon hypoxic stress. (A) Correlation between duplicates of differential proteome samples. (B) Volcano plots displaying differentially expressed proteins upon hypoxic stress (1% O2, 24 h) (blue indicates down-regulated proteins; red indicates up-regulated proteins). (C) Venn diagrams showing overlap between genes with altered m6A but unchanged RNA levels and those genes with 1.5-fold protein change in expression upon hypoxia (1% O2, 24 h). Tables show data of overlapping genes and their locations of differential m6A sites. (D) HeLa cells were exposed to either 20% or 1% O2 for 24 h; whole cell lysates were prepared, and immunoblot assays were performed to analyse SLC2A1 and MTCH2 protein expression. (E) HeLa cells were exposed to either 20% or 1% O2 for 24 h. RT-qPCR was used to determined mRNA level of MTCH2 relative to RPLP0. (F) HeLa cells either with or without knockdown of ALKBH5 were exposed to either 20% or 1% O2 for 24 h, and immunoblot assays were performed to analyse MTCH2 protein expression. (G) HeLa cells either with or without knockdown of ALKBH5 were exposed to either 20% or 1% O2 for 24 h; RT-qPCR was performed to analyse MTCH2 RNA expression
Reprogramming of m6A epitranscriptome is required for efficient energy metabolism during cellular hypoxic response
GO analysis was conducted to reveal potential biological functions of genes with alteration of m6A modification upon hypoxic stress. Genes with both m6A and RNA alterations were enriched in metabolism, including response to oxygen levels, some metabolic processes, and regulation of steroid biosynthesis (Fig. 6A). Subsequently, we performed GO analysis of m6A-hypo-up, m6A-hypo-down, m6A-hyper-up andm6A-hyper-down genes, respectively. Interestingly, only m6A-hypo-up genes were enriched in pyruvate metabolism including glycolysis and NADH regeneration (Fig. 6B, Supplementary Figure S5A-C). Next, the functions of 124 protein-only genes identified by proteome upon hypoxic stress were studied by GO analysis. These genes were involved in regulation of ATP metabolic process including citrate cycle (TCA cycle) and gluconeogenesis (Fig. 6C). In addition, genes showing both m6A-hyper and down-regulated at the protein level (called m6A-hyper∩protein-down) under hypoxic condition were enriched in respiratory electron transport chain process (Supplementary Figure S5D). Genes showing m6A-hyper and up-regulation of protein level (called m6A-hyper∩protein-up) were enriched in mRNA splicing process (Supplementary Figure S5E). Therefore, reprogramming of the m6A epitranscriptome might facilitate the energy metabolic process like ATP synthesis. Of interest, total ATP levels were impaired under both normoxic and hypoxic conditions when ALKBH5 was silenced (compared with control) (Supplementary Figure S5F), demonstrating that m6A pathway is critical for energy metabolism during cellular response to hypoxia.
Figure 6.

m6A pathway regulates cellular energy metabolism. (A) GO analysis of genes with both m6A and RNA alteration upon hypoxic stress. (B) GO analysis of the m6A-hypo-up genes upon hypoxic stress. (C) GO analysis of the protein-only genes
Discussion
Cellular response to hypoxia is essential for cell survival. Low oxygen promotes extensive reprogramming of transcriptome and proteome which alters metabolism of cells to produce and utilize energy economically [54]. However, detailed regulatory mechanisms mediating shape of specific transcriptome and proteome utilized for hypoxic response are far from being understood. In this study, we found that hypoxia systematically reprogramed m6A epitranscriptome of cells, characterized by reduction of total m6A level in poly(A)+ RNA, extensive down-regulation of m6A readers, and systematically changing m6A levels of many transcripts. Massive reprogramming of m6A remodels the transcriptome and proteome to facilitate cellular accommodation to limitation of energy caused by cellular hypoxia. Knockdown of one of the key m6A erasers (ALKBH5) significantly reduced cellular ATP levels under hypoxic condition.
Transcriptional activation by HIFs is the main pathway for hypoxic adaptation [1]. Consistent with a prior report [26], we observed up-regulation of ALKBH5 upon hypoxic stress. Moreover, the total m6A level in poly(A)+ RNAs was down-regulated during this process. We further found that protein levels of most YTH family members were also down-regulated. m6A modification has been reported to facilitate mRNA decay [53]. Down-regulation of total m6A levels of mRNA and protein levels of m6A readers under hypoxic condition might promote mRNA stability, reducing the need to produce new mRNAs. Additionally, we found that m6A-tagged transcripts (especially in the 5ʹ UTR) had a lower ratio of RNA change than the non-m6A-tagged transcripts (Fig. 3C-E). Meanwhile, the majority of transcripts with m6A alteration showed no change in RNA level. Similarly, among the 124 protein-only genes, the differential m6A modification region of 71 genes (57.3%) was located in 5ʹ UTR region (Fig. 5C). This finding also is consistent with the above finding that m6A (especially located in 5ʹ UTR) stabilize their transcripts upon hypoxic stress. Alteration of m6A further contributes to regulating gene expression at the protein level. Taken together, we speculate that to reduce unnecessary energy demand to sustain fundamental cellular activities, cells may add the m6A epitranscriptome regulational layer to increase utilization of existing RNAs to decrease energy consumption.
Therefore, we hypothesized a model of m6A involved in the regulation of cellular hypoxic response (Fig. 7). Upon hypoxic stress, HIF1A is stabilized, enters the nucleus, heterodimers with HIF1B to bind to the HRE elements in the promoters and activates target genes including ALKBH5. Considering that the repertoire of m6A regulator is far from complete, new m6A regulators are continually to be identified [55], we speculate that additional regulators are also involved in regulation of hypoxia. Reprogramming of m6A epitranscriptome further reshapes transcriptome and proteome to promote glycolysis and gluconeogenesis, and inhibit mitochondria oxidative respiratory chain, facilitating cells to response efficiently to hypoxia.
Figure 7.
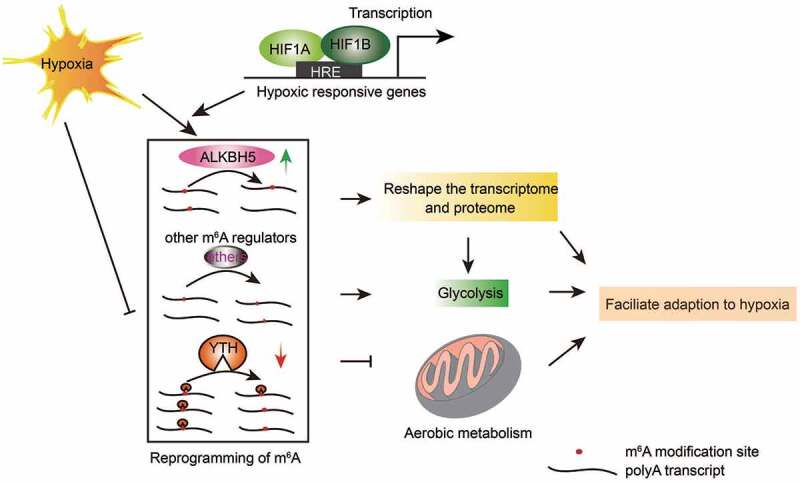
Schematic model of regulation of hypoxic response by m6A pathway
Both m6A modification and RNA alternative splicing events occur co-transcriptionally [56,57]. Cells have been reported to use alternative splicing intensively to survive under hypoxic condition [58]. Several studies revealed that m6A modulates pre-mRNA splicing through the m6A reader YTHDC1 [56,59]. In this study, YTHDC1 was induced upon hypoxic stress (Fig. 1C). Moreover, genes which increased both their m6A levels and protein levels (called m6A-hyper∩protein-up) were enriched in alternative mRNA splicing process (Supplementary Figure S5E). These data implied that m6A might be involved in regulation of activity of alternative splicing in response to hypoxic stress.
Collectively, we provide a global view of m6A epitranscriptome upon hypoxic stress, which reshapes the transcriptome and proteome. Although modulation at the post-transcriptional or translational levels could provide a more sensitive layer of gene regulation, transcriptional activity dictates the initial level of protein abundance. Hypoxia has been reported to reprogram the chromatin by inducing changes in histone methylation to determine transcriptional activity, a process independent of HIFs [60,61]. Meanwhile, m6A deposition was reported to occur co-transcriptionally guided by H3K36me3 (histone H3 trimethylation at lysine 36) [57]. Further studies of hypoxic stress are warranted to investigate whether m6A modification is involved in histone methylation-regulated transcripts, which may enhance our understandings of the molecular mechanism of hypoxia.
Supplementary Material
Funding Statement
This work was supported by the Natural Science Foundation of China [81872140, 81420108026, 81572484, 81621004 to D.Y., 81872155, 81672621 to J.Y.L.]; Guangzhou Bureau of Science and Information Technology [201704030036 to D.Y., 202002020070 to Y.P.]; Guangdong Science and Technology Department [2019B020226003 to D.Y.]; Tip-top Scientific and Technical Innovative Youth Talents of Guangdong special support program [No. 2016TQ03R686 to J.Y.L.]; National Research Foundation Singapore under its Singapore Translational Research (STaR) Investigator Award to H.P.K. [NMRC/STaR/0021/2014]; Guangdong Basic and Applied Basic Research Foundation for Distinguished Young Scholars [2020B1515020027 to Y.P.].
Data availability
Data accession: all the raw data have been deposited in the Gene Expression Omnibus, accessible number GSE141941.
Supplemental material
Supplemental data for this article can be accessed here.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- [1].Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Semenza GL. The hypoxic tumor microenvironment: a driving force for breast cancer progression. Biochim Biophys Acta. 2016;1863:382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yang F, Zhang H, Mei Y, et al. Reciprocal regulation of HIF-1alpha and lincRNA-p21 modulates the Warburg effect. Mol Cell. 2014;53:88–100. [DOI] [PubMed] [Google Scholar]
- [4].Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. 2014;49:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. 2020;21:268–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cavadas MAS, Cheong A, Taylor CT. The regulation of transcriptional repression in hypoxia. Exp Cell Res. 2017;356:173–181. [DOI] [PubMed] [Google Scholar]
- [7].Chee NT, Lohse I, Brothers SP. mRNA-to-protein translation in hypoxia. Mol Cancer. 2019;18:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Roundtree IA, Evans ME, Pan T, et al. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3ʹ UTRs and near stop codons. Cell. 2012;149:1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tong J, Flavell RA, Li HB. RNA m(6)A modification and its function in diseases. Front Med. 2018;12:481–489. [DOI] [PubMed] [Google Scholar]
- [11].Wei J, Liu F, Lu Z, et al. Differential m(6)A, m(6)Am, and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71(973–985):e975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mauer J, Luo X, Blanjoie A, et al. Reversible methylation of m(6)Am in the 5ʹ cap controls mRNA stability. Nature. 2017;541:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Berlivet S, Scutenaire J, Deragon JM, et al. Readers of the m(6)A epitranscriptomic code. Biochim Biophys Acta Gene Regul Mech. 2019;1862:329–342. [DOI] [PubMed] [Google Scholar]
- [14].Zaccara S, Jaffrey SR. A unified model for the function of YTHDF proteins in regulating m(6)A-modified mRNA. Cell. 2020;181(1582–1595):e1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang Z, Luo K, Zou Z, et al. Genetic analyses support the contribution of mRNA N(6)-methyladenosine (m(6)A) modification to human disease heritability. Nat Genet. 2020. DOI: 10.1038/s41588-020-0644-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee H, Bao S, Qian Y, et al. Stage-specific requirement for Mettl3-dependent m(6)A mRNA methylation during haematopoietic stem cell differentiation. Nat Cell Biol. 2019;21:700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang C, Chen Y, Sun B, et al. m(6)A modulates haematopoietic stem and progenitor cell specification. Nature. 2017;549:273–276. [DOI] [PubMed] [Google Scholar]
- [18].Wu R, Li A, Sun B, et al. A novel m(6)A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019;29:23–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Engel M, Eggert C, Kaplick PM, et al. The role of m(6)A/m-RNA methylation in stress response regulation. Neuron. 2018;99(389–403):e389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ma C, Chang M, Lv H, et al. RNA m(6)A methylation participates in regulation of postnatal development of the mouse cerebellum. Genome Biol. 2018;19:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gokhale NS, Horner SM, Evans MJ. RNA modifications go viral. PLoS Pathog. 2017;13:e1006188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yang J, Wang H, Zhang W. Regulation of virus replication and T cell homeostasis by N(6)-methyladenosine. Virol Sin. 2019;34:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xiang Y, Laurent B, Hsu CH, et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543:573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhou J, Wan J, Gao X, et al. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Meyer KD, Patil DP, Zhou J, et al. 5ʹ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang C, Samanta D, Lu H, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Song H, Feng X, Zhang H, et al. METTL3 and ALKBH5 oppositely regulate m6A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy. 2019;15(1–19):1419–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, et al. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc. 2013;8:176–189. [DOI] [PubMed] [Google Scholar]
- [29].Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Meng J, Cui X, Rao MK, et al. Exome-based analysis for RNA epigenome sequencing data. Bioinformatics. 2013;29:1565–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhou J, Wan J, Shu XE, et al. N(6)-methyladenosine guides mRNA alternative translation during integrated stress response. Mol Cell. 2018;69(636–647):e637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].McIntyre ABR, Gokhale NS, Cerchietti L, et al. Limits in the detection of m6A changes using MeRIP/m6A-seq. Sci Rep. 2020;10:6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. [DOI] [PubMed] [Google Scholar]
- [35].Xiao S, Cao S, Huang Q, et al. The RNA N(6)-methyladenosine modification landscape of human fetal tissues. Nat Cell Biol. 2019;21:651–661. [DOI] [PubMed] [Google Scholar]
- [36].Chang M, Lv H, Zhang W, et al. Region-specific RNA m6A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol. 2017;7(7):170166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Garcia-Campos MA, Edelheit S, Toth U, et al. Deciphering the “m(6)A Code” via antibody-independent quantitative profiling. Cell. 2019;178(731–747):e716. [DOI] [PubMed] [Google Scholar]
- [38].Zeng Y, Wang S, Gao S, et al. Refined RIP-seq protocol for epitranscriptome analysis with low input materials. PLoS Biol. 2018;16:e2006092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shah AD, Goode RJA, Huang C, et al. LFQ-Analyst: an easy-to-use interactive web-platform to analyze and visualize label-free proteomics data preprocessed with MaxQuant. J Proteome Res. 2020;19:204–211. [DOI] [PubMed] [Google Scholar]
- [40].Kamburov A, Wierling C, Lehrach H, et al. ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37:D623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].de Bruin A, PW AC, Kirchmaier BC, et al. Genome-wide analysis reveals NRP1 as a direct HIF1alpha-E2F7 target in the regulation of motorneuron guidance in vivo. Nucleic Acids Res. 2016;44:3549–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang J, Wang C, Chen X, et al. EglN2 associates with the NRF1-PGC1alpha complex and controls mitochondrial function in breast cancer. Embo J. 2015;34:2953–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu M, Wang Y, Yang C, et al. Inhibiting both proline biosynthesis and lipogenesis synergistically suppresses tumor growth. J Exp Med. 2020;217(3):e20191226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Galbraith MD, Andrysik Z, Pandey A, et al. CDK8 kinase activity promotes glycolysis. Cell Rep. 2017;21:1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lin J, Zhang X, Xue C, et al. The long noncoding RNA landscape in hypoxic and inflammatory renal epithelial injury. Am J Physiol Renal Physiol. 2015;309:F901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Perez-Perri JI, Dengler VL, Audetat KA, et al. The TIP60 complex is a conserved coactivator of HIF1A. Cell Rep. 2016;16:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. [DOI] [PubMed] [Google Scholar]
- [49].Gao Y, Zhang E, Liu B, et al. Integrated analysis identified core signal pathways and hypoxic characteristics of human glioblastoma. J Cell Mol Med. 2019;23:6228–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang S, Zhao BS, Zhou A, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(591–606):e596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tang C, Klukovich R, Peng H, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3ʹ-UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A. 2018;115:E325–E333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Peer E, Moshitch-Moshkovitz S, Rechavi G, et al. The epitranscriptome in translation regulation. Cold Spring Harb Perspect Biol. 2019;11(8):a032623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Du H, Zhao Y, He J, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schito L, Rey S. Cell-autonomous metabolic reprogramming in hypoxia. Trends Cell Biol. 2018;28:128–142. [DOI] [PubMed] [Google Scholar]
- [55].Chen J, Fang X, Zhong P, et al. N6-methyladenosine modifications: interactions with novel RNA-binding proteins and roles in signal transduction. RNA Biol. 2019;16:991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhou KI, Shi H, Lyu R, et al. Regulation of co-transcriptional Pre-mRNA splicing by m(6)A through the low-complexity protein hnRNPG. Mol Cell. 2019;76(70–81):e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Huang H, Weng H, Zhou K, et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature. 2019;567:414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hirschfeld M, Zur Hausen A, Bettendorf H, et al. Alternative splicing of Cyr61 is regulated by hypoxia and significantly changed in breast cancer. Cancer Res. 2009;69:2082–2090. [DOI] [PubMed] [Google Scholar]
- [59].Haussmann IU, Bodi Z, Sanchez-Moran E, et al. m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–304. [DOI] [PubMed] [Google Scholar]
- [60].Batie M, Frost J, Frost M, et al. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science. 2019;363:1222–1226. [DOI] [PubMed] [Google Scholar]
- [61].Chakraborty AA, Laukka T, Myllykoski M, et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science. 2019;363:1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data accession: all the raw data have been deposited in the Gene Expression Omnibus, accessible number GSE141941.