

Abstract
Coronavirus disease 2019 (COVID-19) has caused more than 840,000 deaths as of 31 August 2020 in the whole world. The COVID-19 main protease (Mpro) has been validated as an attractive target for drug design. In this work, the binding mechanisms of five protease inhibitors (e.g., danoprevir, darunavir, ASC09, lopinavir and ritonavir) to COVID-19 Mpro were investigated. Based on the docking score, five protease inhibitors structures were selected for further evaluation. It is found that most of the selected drug molecules bind stably to the COVID-19 Mpro from the molecular dynamics simulation. Moreover, the MM/PBSA free energy calculations suggest that lopinavir with positive charge might be most active against COVID-19 Mpro.
Keywords: COVID-19, SARS-CoV-2, Protease Inhibitor, molecular modeling evaluation
1. Introduction
The 2019 novel coronavirus (COVID-19) has spread rapidly worldwide [1], [2], [3], [4], [5] . Up to now (31 August 2020), COVID-19 has caused more than 840,000 deaths in the whole world, and confirmed infection cases have been reported more than 25,000,000 (https://covid19.who.int/). Unfortunately, the number of infection cases is still growing rapidly, but there are no approved effective drugs. Therefore, there is an urgent need for developing effective anti-COVID-19 drugs.
The COVID-19 is by far the most serious, but not the first coronavirus outbreak that endangers human health. As far as is known, including severe acute respiratory syndrome (SARS-CoV) and Middle East respiratory syndrome-coronavirus (MERS-CoV) both have a huge impact on humans [6], [7], [8], [9], [10]. Currently, it was found that the main protease (Mpro, also called 3CLpro) plays a pivotal role in CoV (e.g., SARS-CoV and MERS-CoV) gene expression and replication [11]. Mpro has been validated as an attractive target for anti-SARS-CoV and anti-MERS-CoV drug design, and a variety of inhibitors drugs have been developed [12], [13], [14]. More importantly, previous genome sequences analyses have revealed that COVID-19 share a high level of sequence similarity with the corresponding SARS-CoV and MERS-CoV [15]. Therefore, Mpro is suggested to be an attractive target for anti-COVID-19 drug design, especially considering the re-use of existing MERS and SARS Mpro inhibitors for COVID-19.
Recently, approved inhibitors including danoprevir [16], darunavir [17], ASC09 [ChiCTR2000029603], lopinavir/ritonavir [ChiCTR2000029539] have been reported to treat COVID-19 patients. At present, as an anti-HIV protease inhibitor, lopinavir/ritonavir has been widely used despite lack of sufficient efficacy data [18]. And the combination of lopinavir/ritonavir is a recommended antiviral regimen in the latest version of the Diagnosis and Treatment of Pneumonia Caused by COVID‐19 issued by the National Health Commission of the People's Republic of China. However, it is debatable whether HIV protease inhibitors could effectively inhibit the COVID-19 Mpro [19]. Moreover, inhibitors may have different tautomerization and protonation states, and the effects of different tautomeric and ionization states of inhibitors on their binding to protease are still unknown.
Molecular docking and molecular dynamics (MD) simulation have been confirmed as a powerful tool to understand the binding models of inhibitors to protease [20], [21], [22], [23], [24]. They have been successfully used to study the interaction between Mpro and promising inhibitors. For example, Khan et al. found that several FDA approved anti-virus drugs can inhibit the function of Mpro of COVID-19 [20]. Elfiky revealed that inhibitors such as ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir can bind tightly the RNA dependent RNA polymerase of COVID-19 [21]. However, the interaction details between Mpro and some drugs that have participated in clinical trials are still scarce. In this study, five protease inhibitors (e.g., danoprevir, darunavir, ASC09, lopinavir and ritonavir) that have participated in clinical trials were selected for molecular docking analysis. As shown in Table 1 , the drug structures with different tautomeric and ionization states were pre-processed in Schrodinger software package using LigPrep [25]. Furthermore, based on docking scores, five filtered drug structures were subsequently imported into a detailed study through MD simulation and binding free energy calculation.
Table 1.
Five Mpro binding drugs for docking analysis.
| Drug name | System | Drug structure | Charged states | Docking score (kJ/mol) |
|---|---|---|---|---|
| Danoprevir | DAN1 | 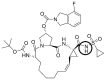 |
0 | −17.16 |
| DAN2 | 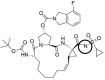 |
−1 | −6.29 | |
| Darunavir | DAR | 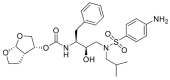 |
0 | −17.99 |
| ASC09 | ASC1 | 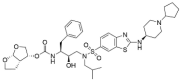 |
0 | −18.83 |
| ASC2 | 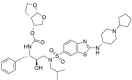 |
0 | −10.46 | |
| Ritonavir | RIT | 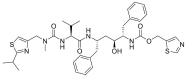 |
0 | −21.76 |
| Lopinavir | LOP1 | 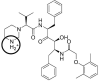 |
+1 | −29.13 |
| LOP2 | 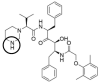 |
0 | −19.51 |
2. Computational methods and simulation details
2.1. Molecular docking
The main protease (Mpro) with structure 6LU7 from the PDB [26] was prepared by Protein Preparation Wizard tool in Schrodinger software package. As shown in Table 1, five drugs (e.g., danoprevir, darunavir, ASC09, lopinavir and ritonavir) were processed by Schrodinger’s LigPrep module, and seven structures were chosen, which mainly includes prediction of protonation states and generation of three-dimensional conformation. The seven systems were named as DAN1/DAN2 (danoprevir with neutral/negative charge), DAR (darunavir), ASC1/ASC2 (ASC09 with two conformers), RIT (ritonavir) and LOP1/LOP2 (lopinavir with positive/neutral charge), respectively. The grid area for docking were determined according to original substrate coordinates in 6LU7 crystal structure. All other settings remained default, and the Glide docking was carried out with the standard precision (standard precision, SP) mode. Each molecule was retained at most 100 docking conformations.
2.2. Molecular dynamics simulation
All MD simulations were performed with GROMACS 5.0.7 package [27], and the time-step was set to be 2 fs. CHARMM force field were used to parameterize the drugs and protein, respectively [28]. Each system was immersed in a periodic boundary conditions (PBC) box of TIP3P water, and the box size was 9.0 × 9.0 × 9.0 nm3. A rational number of counter ions (Na+ or Cl-) were added to solution to neutralize the systems. In each system, the cut-off distance of van der Waals (vdW) interaction was set to be 1.2 nm. The cross interaction and electrostatic interactions between different atoms were calculated through Lorentz-Berthelot rule [29] and particle mesh Ewald (PME) method [30], respectively. The velocity rescaling with a stochastic term [31] was used to keep system temperature at 300 K. The Berendsen method [32] was used to couple the pressure at 1 bar. In each system, a 50,000 step energy minimization was firstly performed, followed by a 200 ps pre-equilibration under an NVT ensemble. After energy minimization and pre-equilibration of the system, each simulation was conducted for 50 ns in NPT ensemble, where the last 10 ns were used for binding free energy calculation with using MM/PBSA method [33]. PyMOL software [34] was used for molecular visualization.
3. Results and discussion
In the present study, a structure-based docking screening was performed with seven systems (DAN1, DAN2, DAR, ASC1, ASC2, RIT, LOP1 and LOP2) against main protease (Mpro) of COVID-19. Recently, the crystal structure of COVID-19 Mpro has been uncovered by Liu et al. with PDB ID 6LU7 [26]. Based on this crystal structure Khan et al showed that original substrate binding site of Mpro consists of conserved catalytic dyad [20]. Inspired by Khan et al.’s work, herein, the docking grid area was placed on the original substrate coordinates to cover all the active site residues. As shown in Table 1, seven systems (DAN1, DAN2, DAR, ASC1, ASC2, RIT, LOP1 and LOP2) were docked more than 100 times, and the docking scores for these systems are: −17.16, −6.29, −17.99, −18.83, −10.46, −21.76, −29.13 and −19.51 kJ/mol, respectively. According to the docking score, one system with strong binding strength was selected for each drug, and they are DAN1, DAR, ASC2, RIT and LOP1, respectively. Fig. 1 shows the probability density of docking scores for the five selected systems. One could find that the peak position of system LOP1 (lopinavir with positive charge) is around −29.13 kJ/mol, and the value is much lower than other compounds. The docking results indicated that lopinavir with positive charge showed highest binding affinity to compare with other compounds.
Fig. 1.

Probability density of docking score for five selected molecular structures.
To estimate the stability of these compounds, the selected hits were subsequently imported into a detailed 50 ns MD simulation study. The Root Mean Square Deviation (RMSD) of drug molecules as a function of simulation time were displayed in Fig. 2 . It could be used to estimate the binding stability between drug molecules and protein. Fig. 2 showed that the RMSD value of all the systems were significantly stable with small deviation except for ASC1 system. It can be found that the overall RMSD value of ASC1 system (solid blue line) is ca. 3.0 Å, and it is highest to compare with other systems. It indicates that the binding stability of ASC1 system is weaker than other systems. Besides, the overall RMSD value of the other four systems is lower than 3 Å, and similar results have been shown by Khan et al. [20]. Specially, Khan et al. showed that the RMSD value of the system with Darunavir (DAR) is 2.59 Å, which is in according with the results from our study. Overall, the results from RMSD value indicate that most selected drugs can bind to the protein stably. The differences in stability between binding drugs and protein could also be reflected from the snapshots. As displayed in Fig. 3 , the formed hydrogen bonds between drugs and protein were shown in these systems. Fig. 3(a) showed that amino acid residues (e.g., PHE140, GLY143, CYS145, HIS164 and GLU166) play a key role in the original substrate binding, and it can form hydrogen bonding with the substrate. Besides, among these amino acid residues, GLU166 and GLN189 could form hydrogen bonding with most of selected drugs. A similar finding was previously reported by Xu et al. [35], who revealed that residue GLU166 and GLN189 maintained the binding between drug nelfinavir and COVID-19 Mpro. The results here indicate that the selected hits would stably bind to COVID-19 Mpro in a similar way to that of the original substrate against COVID-19 Mpro. The stability of these systems was also characterized by monitoring the Root Mean Square Fluctuation (RMSF) of protein residues. As shown in Fig. 4 , these systems have a quite similar RMSF fluctuation trend, and one could also found that most binding residues (e.g., ASN142, GLY143, GLU166, GLN189, etc., as shown in Fig. 3) were quite stable during the simulation. These observations further demonstrate that binding of selected hits stabilizes the COVID-19 Mpro.
Fig. 2.

RMSD of binding drugs calculated versus simulation time.
Fig. 3.

The binding model of original substrate (green) in 6LU7 and several drugs (yellow) against COVID-19 Mpro (white cartoon). (a) original substrate; (b) DAN1; (c) DAR; (d) ASC1; (e) RIT; (f) LOP1. Hydrogen bonding formed between ligands and associated residues (white) in the COVID-19 Mpro pocket were shown in black dash line.
Fig. 4.

RMSF of COVID-19 Mpro residues in five systems.
To provide insight into the binding mechanisms of the selected hits, the binding free energies (ΔG) were calculated by MM/PBSA method according to the equation (1),
| (1) |
where ΔEVDW and ΔEele refer to van der Waals and electrostatic energy contributions, respectively. ΔGgb and ΔGnp represents polar and non-polar desolvation free energies, respectively. TΔS refer to conformational entropy contribution at temperature T. As shown in Table 2 , the binding free energies (ΔG) of DAN1, DAR, ASC1, RIT and LOP1 systems are −97.10 ± 16.10, −53.22 ± 14.23, −35.16 ± 10.68, −71.78 ± 12.78 and −267.76 ± 40.53 kJ/mol, respectively. It can be found that LOP1 (lopinavir with positive charge) is the most active one, consistent with the docking results. Especially, the ΔEele of LOP1 system make a significant contribution to ΔG. While for DAN1, DAR, ASC1 and RIT systems, the contributions of ΔEVDW were higher than ΔEele. The results suggest that the electrostatic interaction play an important role in the binding of lopinavir. It mainly because of the strong electrostatic interaction between positively charged amine group of lopinavir and negatively charged residue of Mpro (e.g., GLU166, as shown in Fig. 3(f)). The binding free energy between all protein residues and selected drugs were calculated to provide more binding details. As shown in Fig. 5 (a), one could find that the residue GLU166 in LOP1 system make the most contribution to the binding free energy, and the value is much higher than other systems. Other negatively charged residues (e.g., GLU47, ASP48, ASP187) also make a great contribution to the binding free energy. Fig. 5(b) shows that these negatively charged residues are close to positively charged lopinavir, and lopinavir is able to form hydrogen bonds with GLU166 and GLN189. The results suggest that lopinavir with positive charge might be active against COVID-19 Mpro. Lopinavir/ritonavir may be a potential treatment option for COVID‐19. Two studies showed the positive effects of lopinavir/ritonavir therapy [36], [37]. However, the efficacy of lopinavir/ ritonavir antiviral treatment warrants further verification in future studies.
Table 2.
Components of the Binding Free Energy (kJ/mol) Calculated by MM/PBSA Approach.
| Energy term | DAN1 | DAR | ASC1 | RIT | LOP1 |
|---|---|---|---|---|---|
| Evdw | −153.27 ± 12.36 | −149.24 ± 7.96 | −62.79 ± 10.53 | −151.96 ± 8.59 | −101.78 ± 14.50 |
| Eele | −45.52 ± 16.52 | −50.21 ± 15.00 | −28.07 ± 9.55 | −50.86 ± 14.20 | −304.15 ± 79.19 |
| Ggb | 118.67 ± 9.98 | 164.87 ± 4.79 | 75.73 ± 11.32 | 151.72 ± 15.34 | 153.82 ± 64.82 |
| Gnp | −17.97 ± 1.01 | −18.64 ± 0.78 | −20.03 ± 1.18 | −20.67 ± 1.04 | −15.64 ± 1.43 |
| ΔG | −97.10 ± 16.10 | −53.22 ± 14.23 | −35.16 ± 10.68 | −71.78 ± 12.78 | −267.76 ± 40.53 |
Fig. 5.

(a) The bind free energy between all protein residues and selected five drugs. (b) Interaction details between negatively charged residues and positively charged lopinavir (LOP1 system). The data in red is the interaction distance (Å).
4. Conclusions
In this work, the binding mechanisms of five protease inhibitors (e.g., danoprevir, darunavir, ASC09, lopinavir and ritonavir) to COVID-19 Mpro were investigated by molecular docking, molecular dynamics simulation and free energy calculation. Based on the docking score, five drugs structures were selected for further evaluation. RMSD, RMSF, and typical snapshots of these systems suggest that most of the selected drugs can bind stably to the COVID-19 Mpro in a similar way to that of the original substrate against COVID-19 Mpro. Further MM/PBSA free energy calculations suggest that lopinavir with positive charge might be active against COVID-19 Mpro.
CRediT authorship contribution statement
Jian Liu: Conceptualization, Formal analysis, Investigation, Writing - review & editing, Project administration. You Zhai: Data curation, Writing - original draft, Validation. Lijun Liang: Methodology, Software, Validation, Writing - original draft, Resources. Danyan Zhu: Investigation. Qingwei Zhao: Project administration. Yunqing Qiu: Supervision, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This study was funded by the New Drug Creation Project of The 13th Five-Year National Science and Technology Major Special Project (2020ZX09201-003) and The Key R&D Plan Emergency Project of Zhejiang Science and Technology Department (2020C03123-8).
References
- 1.Dong L., Hu S., Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19) Drug Discov Ther. 2019;14(2020):58–60. doi: 10.5582/ddt.2020.01012. [DOI] [PubMed] [Google Scholar]
- 2.Hui D.S., Azhar E.I., Madani T.A., Ntoumi F., Kock R., Dar O., Ippolito G., Mchugh T.D., Memish Z.A., Drosten C. The continuing COVID-19 epidemic threat of novel coronaviruses to global health-The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2019;91(2020):264–266. doi: 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phelan L., Katz R., Gostin L.O. The novel coronavirus originating in Wuhan, China: challenges for global health governance. JAMA. 2020;323:709–710. doi: 10.1001/jama.2020.1097. [DOI] [PubMed] [Google Scholar]
- 4.Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., Hlh A.S.C. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollander J.E., Carr B.G. Virtually Perfect? Telemedicine for Covid-19. N. Engl. J. Med. 2020;382(18):1679–1681. doi: 10.1056/NEJMp2003539. [DOI] [PubMed] [Google Scholar]
- 6.Liu J., Zheng X., Tong Q., Li W., Wang B., Sutter K., Trilling M., Lu M., Dittmer U., Yang D. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-CoV. J. Med. Virol. 2020;92:491–494. doi: 10.1002/jmv.25709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26(4):450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar V., Shin J.S., Shie J., Ku K.B., Kim C., Go Y.Y., Huang K., Kim M., Liang P. Identification and evaluation of potent Middle East respiratory syndrome coronavirus (MERS-CoV) 3CLPro inhibitors. Antivir. Res. 2017;141:101–106. doi: 10.1016/j.antiviral.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding Y., He L.I., Zhang Q., Huang Z., Che X., Hou J., Wang H., Shen H., Qiu L., Li Z. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J. Pathol. 2004;203:622–630. doi: 10.1002/path.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao X., Chakraborti S., Dimitrov A.S., Gramatikoff K., Dimitrov D.S. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan Y., Cao D., Zhang Y., Ma J., Qi J., Wang Q., Lu G., Wu Y., Yan J., Shi Y. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017;8:1–9. doi: 10.1038/ncomms15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coleman M., Sisk J.M., Mingo R.M., Nelson E.A., White J.M., Frieman M.B. Abelson kinase inhibitors are potent inhibitors of severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus fusion. J. Virol. Methods. 2016;90:8924–8933. doi: 10.1128/JVI.01429-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheahan T.P., Sims A.C., Leist S.R., Schäfer A., Won J., Brown A.J., Montgomery S.A., Hogg A., Babusis D., Clarke M.O. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020;11:1–14. doi: 10.1038/s41467-019-13940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao T.T., Qian J.D., Zhu W.Y., Wang Y., Wang G.Q. A systematic review of lopinavir therapy for SARS coronavirus and MERS coronavirus—A possible reference for coronavirus disease-19 treatment option. J. Med. Virol. 2020;92:556–563. doi: 10.1002/jmv.25729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu F., Zhao S., Yu B., Chen Y., Wang W., Song Z., Hu Y., Tao Z., Tian J., Pei Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.H. Chen, Z. Zhang, L. Wang, Z. Huang, F. Gong, X. Li, Y. Chen, First clinical study using HCV protease inhibitor danoprevir to treat naive and experienced COVID-19 patients, medRxiv (2020), https://doi.org/10.1101/2020.03.22.20034041. [DOI] [PMC free article] [PubMed]
- 17.De Meyer S., Bojkova D., Cinatl J., Van Damme E., Meng C.B., Van Loock M., Woodfall B., Ciesek S. Lack of antiviral activity of darunavir against SARS-CoV-2. Int. J. Infect. Dis. 2020 doi: 10.1016/j.ijid.2020.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Z., Fu J., Shu Q., Chen Y., Hua C., Li F., Lin R., Tang L., Wang T., Wang W. Diagnosis and treatment recommendations for pediatric respiratory infection caused by the 2019 novel coronavirus. World J. Pediatr. 2019;2020:1–7. doi: 10.1007/s12519-020-00345-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.G. Li, E. De Clercq, Therapeutic options for the 2019 novel coronavirus (2019-nCoV), Nat. Rev. Drug Discov. 19 (2020) 149-150, https://doi.org/10.1038/d41573-020-00016-0. [DOI] [PubMed]
- 20.Khan S.A., Zia K., Ashraf S., Uddin R., Ul-Haq Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1751298. [DOI] [PubMed] [Google Scholar]
- 21.A. Elfiky, Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study, Life Sci. (2020) 117592, https://doi.org/10.1016/j.lfs.2020.117592. [DOI] [PMC free article] [PubMed]
- 22.Islam R., Parves M.R., Paul A.S., Uddin N., Rahman M.S., Mamun A.A., Hossain M.N., Ali M.A., Halim M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1761883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D. Gentile, V. Patamia, A. Scala, M. T. Sciortino, A. Piperno, A. Rescifina, Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: A virtual screening and molecular modeling study, Mar. Drugs 18 (2020) 225, https://doi.org/10.3390/md18040225. [DOI] [PMC free article] [PubMed]
- 24.Fathima A.J., Murugaboopathi G., Selvam P. Pharmacophore Mapping of ligand based virtual screening, molecular docking and molecular dynamic simulation studies for finding potent NS2B/NS3 protease inhibitors as potential anti-dengue drug compounds. Curr. Bioinform. 2018;13:606–616. doi: 10.2174/1574893613666180118105659. [DOI] [Google Scholar]
- 25.LigPrep, Version 2.5 Schrödinger, LLC., (2014), https://www.schrodinger.com.
- 26.Liu X., Zhang B., Jin Z., Yang H., Rao Z. The crystal structure of COVID-19 main protease in complex with an inhibitor N3. Protein Data Bank. 2020 doi: 10.2210/pdb6LU7/pdb. [DOI] [Google Scholar]
- 27.Van Der Spoel E., Lindahl B., Hess G., Groenhof A.E., Mark H.J. Berendsen, GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 28.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.J. O. Hirschfelder, C. F. Curtiss, R. B. Bird, M. G. Mayer, Molecular theory of gases and liquids, Wiley New York, 1964, https://doi.org/10.1063/1.306194.
- 30.Darden T., York D., Pedersen L. Particle mesh Ewald: An N∙log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 31.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:14101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 32.Berendsen H.J., Postma J.V., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 33.The PyMOL molecular graphics system, Version 2.0 Schrödinger, LLC., (2015), https://pymol.org/.
- 34.Srinivasan J., Cheatham T.E., Cieplak P., Kollman P.A., Case D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate−DNA helices. J. Am. Chem. Soc. 1998;120:9401–9409. doi: 10.1021/ja981844+. [DOI] [Google Scholar]
- 35.Z. Xu, C. Peng, Y. Shi, Z. Zhu, K. Mu, X. Wang, W. Zhu, Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation, bioRxiv (2020) 2020-2021, https://doi.org/10.1101/2020.01.27.921627.
- 36.Liu F., Xua A., Zhang Y., Xuan W., Yan T., Pan K., Yu W., Zhang J. Patients of COVID-19 may benefit from sustained Lopinavir-combined regimen and the increase of Eosinophil may predict the outcome of COVID-19 progression. International Journal of Infectious Diseases. 2020;95:183–191. doi: 10.1016/j.ijid.2020.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z., Chen X., Lu Y., Chen F., Zhang W. Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. Biosci Trends. 2020;14(1):E1. doi: 10.5582/bst.2020.E1. [DOI] [PubMed] [Google Scholar]