

Abstract
Objective
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent for coronavirus disease 2019 (COVID-19), is responsible for the recent global pandemic. As there are no effective drugs or vaccines available for SARS-CoV-2, we investigated the potential of flavonoids against SARS-CoV-2 main protease 6YNQ.
Methods
In silico molecular simulation study against SARS-CoV-2 main protease 6YNQ.
Results
Among the 21 selected flavonoids, rutin demonstrated the highest binding energy (− 8.7 kcal/mol) and displayed perfect binding with the catalytic sites.
Conclusions
Our study demonstrates the inhibitory potential of flavonoids against SARS-CoV-2 main protease 6YNQ. These computational simulation studies support the hypothesis that flavonoids might be helpful for the treatment of COVID-19.
Key words: COVID-19, SARS-CoV-2, Protease 6YNQ, In silico, Molecular simulation, Virtual drug screening, Flavonoids
1. Introduction
The unprecedented coronavirus disease 2019 (COVID-19) outbreak has had a critical impact on countries across the globe and on people from every walk of life. As of the beginning of October 2020, the world has recorded 1 111 998 deaths due to COVID-19 and more than 39 944 882 confirmed cases [ 1 ]. The causative agent of COVID-19, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), belongs to the β-coronavirus group. Antiviral drugs can target diverse phases of viral infection. In the case of SARS-CoV-2, both structural and nonstructural proteins have been identified as potential drug targets. The main proteases (Mpro or 3CLpro) of corona viruses tend to be highly conserved and are critical for viral replication. These proteins are responsible for the maturation of both nonstructural and structural viral proteins, making them a very attractive target for novel anti-coronavirus drugs. Thus, any inhibitor against these proteases (Mpro or 3CLpro) that can block the replication of SARS-CoV-2 would be effective for the development of therapeutic agents or antiviral drugs against SARS-CoV-2 [ 2 ].
The main protease of SARS-CoV-2, 6YNQ, is a homodimer bound to 2-methyl-1-tetralone, which has been expressed in Escherichia coli and has a known crystalline structure, comprising 306 amino acids [ 3 ]. One important finding is that there have been no mutations in this protein to date. This viral protease is a multifunctional protein involved in the transcription and replication of various viral RNAs and is responsible for the cleavage of the functional replicase polyproteins at 11 different sites. Additionally, 6YNQ [ 3 ] was shown to share 100% identity and similarity with 6LU7 [ 4 ] using blast-2-seq, Smith-Waterman and Needleman-Wunsch sequence alignments. In addition, Protein Data Bank (PDB) structures of 6YNQ demonstrate a better resolution (1.8 Å), which ensures better protein structure evaluations. Furthermore, R and R free, who assess the similarity between the calculated values and the observed structural factor amplitudes, were lower for these structures than 6LU7. Although the major protease, spike protein, RdRp, and the papain-like protease have been evaluated as antiviral targets, 6YNQ has not yet been evaluated for anti-COVID 19 activity. Co-crystallized ligand P6N (PDB ID 6YNQ) has been shown to interact with the binding site of Mpro, indicating its association with 6YNQ demonstrating the main interaction site for drug targeting [ 5 ]. Similarly, 6YNQ has been recently listed as a possible target for evaluating the efficacy of certain agents against COVID-19 [ 6 ]. Thus, 6YNQ with its superior protein structure predictions is far more likely to yield reliable docking results, which is essential in the drug discovery process.
Recently, the United States Food and Drug Administration approved the use of Gilead’s Remdesivir, marketed under the brand name Veklury, for treatment of COVID-19 for hospitalized patients. This is the first and only drug approved for adults and pediatric patients (age 12 years and older, weighing at least 40 kg) in the treatment of COVID-19 patients requiring hospitalization 7 , 8. Hydroxychloroquine was under early investigation for use in COVID-19 treatment; however, initial clinical trials were withdrawn amid safety concerns raised by the WHO [ 9 ]. In order to identify any potential interactions with the SARS-CoV-2 protease enzymes, many existing drugs such as Lopinavir, Oseltamivir, Ritonavir and Favipiravir using computational analysis have been evaluated 10 , 11.
Interestingly, many bioactive compounds derived from plants that are known to exert some antiviral effects, have attracted researchers’ attention, with a large panel of these compounds under evaluation for SARS-CoV-2 12 , 13. Among them, flavonoids have been revealed as the most promising antiviral agents [ 14 ]. Flavonoids are a large class of food additives that have a positive impact on health and which have been extensively evaluated against a wide range of DNA and RNA viruses. For example, the flavones apigenin, has been reported to exert an antiviral effect against picornavirus (RNA virus), while the flavonol quercetin-3-β-galactoside was found to competitively inhibit SARS-CoV 3CLpro in an in vitro assay [ 15 ]. Additionally, an in silico docking simulation established that Biflavone adheres to the SARS-CoV 3CLpro binding pocket [ 16 ]. The role of flavonoids and their interactions with diverse cellular targets and pathways involved in the viral life cycle have been widely demonstrated, and when these features are considered in conjunction with the structural diversity and degree of hydroxylation in flavonoids, it is obvious that these compounds could be used against SARS-CoV-2.
In this study, we assessed the docking of 21 flavonoids and evaluated their potential as inhibitory compounds against the SARS-CoV-2 main protease 6YNQ using the AutoDock Vina software. Furthermore, we confirmed our initial findings and evaluated the structural flexibility of the docked poses for the best flavonoidsusing CABS-flex 2.0 software [ 17 ].
2. Materials and Methods
2.1. Platform for molecular docking
The computational docking assessment of 21 flavonoid ligands for the SARS-CoV-2 main protease 6YNQ was performed using the AutoDock Vina software, and comparative docking was performed using Swiss dock (http://www.swissdock.ch/), an online server that uses EADock DSS software 18 , 19.
2.2. Preparation of proteins and grids
In silico analysis of 21 flavonoids was performed using a 1.80 Å crystal structure of 6YNQ from the SARS-CoV-2 main protease in complex with an inhibitor 2-methyl-1-tetralone (PDB ID: 6YNQ, with a resolution < 2 Å, R-value free < 0.25, R-value work < 0.25), which was retrieved from the PDB (https://www.rcsb.org). The 6YNQ protein containsan a chain with (2∼{S})-2-methyl-3,4-dihydro-2∼{H}-naphthallen-1-one, which was used in the macromolecule preparation [ 4 ]. The protein preparation parameters of AutoDock were then used to prepare the whole structure by deleting water molecules, adding hydrogen, and assigning partial charges using Kollman and Gasteiger, and the binding sites were identified after deleting the ligand.
2.3. Ligand preparations
For this investigation, the ligand (2∼{S})-2-methyl-3, 4-dihydro-2∼{H}-naphthalen-1-one and the structures of each of the flavonoids under investigation were retrieved from PubChem (https://pubchem.ncbi.nlm.nih.gov/) and saved in SDF. Since the PDB, Partial Charge (Q) and Atom Type (T) (PDBQT) formats can all be used as input in the AutoDock Vina software, Open Babel (version 3.0.0) 21 was used to convert these SDF files to PDB [ 20 ]. A total of 24 compounds were selected to target the main protease of SARS-CoV-2, including two approved anti-RdRp compounds (Remdesivir) [ 21 ] and a recently identified compound used to treat a mild to moderate cases of coronavirus (Favipiravir) [ 22 ].
2.4. Determining compound active sites
The active sites were defined as the coordinates of the ligand in the original target protein grids, and these active binding sites in the target protein were identified using the computed atlas for surface topography of proteins (CASTp) [ 23 ] and Biovia Discovery Studio 4.5 [ 24 ]. The amino acids in the active site were used to evaluate the Grid box and docking evaluation results. PyMol software (version 1.7.4) [ 25 ] and the Protein-Ligand Interaction Profiler (PLIP) web server [ 26 ] were used to profile the interactions between the ligand-protein complexes showing the lowest binding score and RMSD < 2.0 Å.
2.5. Protein ligand docking and visualization
AutoDock Vina was used in all the docking experiments, using the optimized model as the docking target. Computational docking is executed to generate a population of promising orientations and conformations of the ligand within the binding site. The grid center for docking was set at X = 3.789, Y = – 1.789, and Z = 18.846, with the grid box set to 40 Å × 40 Å × 40 Å. Flavonoids were individually evaluated in the molecular docking, and prior to their first interactions, the classical MM2 force field was applied to optimize the structures of these small molecules, ensuring that their active sites were rigid. After validation of the docking protocol, virtual screening was accomplished using rigid molecular docking into the active site of the partner proteins. Throughout the virtual screening, both the macromolecule and the ligands were kept rigid. Finally, the binding energy limits were removed from the software, and the investigation of the 2D hydrogen-bond interactions could be completed using the Biovia Discovery Studio 4.5 program. This analysis produces a graphical output describing the hydrophobic bonds, hydrogen bonds, and their bond lengths in each docking pose.
2.6. Physicochemical properties
Lipinski’s rule was used to assess the physicochemical properties of all the selected flavonoids and predict their drug-like properties, and the Swiss ADME (http://www.swissadme.ch/) was used to compute the SMILES structures of each compound [ 27 ].
2.7. Molecular dynamics simulations
Molecular dynamics simulations were performed using the CABS Flex 2.0 server and were based on the coarse-grained simulations of protein motion [ 28 ] over 50 cycles and 50 trajectory frames of 10 ns each with some additional distance restraints including a global weight of 1.0 applied. These were built with Poisson-Boltzmann/Generalized Born (PB/GB) molecular mechanics, and the solvent probe radius was set to 1.4 Å, the minimum atomic radius was 1 Å, the salt radius was 2 Å, the ionic strength was 0.15, and the temperature of the simulation was 1.4. These restraints allowed us to analyze the conformational stability of the receptor-ligand complex system.
3. Results
3.1. Molecular docking
The results of the molecular docking study using AutoDock Vina revealed the binding energies of the selected compounds, inhibitors and reference molecules (Table 1 ). Additionally, these results were compared with SwissDock. The algorithm consisted of several steps, including the generation of as many binding modes in the local docking as possible, estimating CHARMM energies on the grid, the evaluation of the most favored binding modes using FACTS, and then clustering evaluation. Cluster “0” has the best full fitness (FF) score. We submitted both our protein and phytochemicals one by one. After each docking run, the output clusters were identified and the individual conformer from each cluster with the most favorable binding mode and most negative FF score were chosen for further evaluation as these had the best fit.
Table 1.
Ligands with their binding energy and Lipinski rule parameters from the PDB for 6YNQ from SARS-CoV-2
| Sr. No. | Ligand | Docking score (kcal/mol) | MW (g/mol) | Rotatable bonds | H-bond acceptors | H-bond donors | TPSA | LOGP | Follow lipinski | Violations |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Inhibitor | – 5.6 | 160.21 | 0 | 1 | 0 | 17.07 | 2.11 | YES | 0 |
| 2 | Favipiravir | – 5.5 | 157.10 | 1 | 4 | 2 | 88.84 | 0.39 | YES | 0 |
| 3 | Remdesivir | – 8.6 | 602.58 | 14 | 12 | 4 | 213.36 | 3.40 | NO | 2 |
| 4 | Rutin | – 8.7 | 610.52 | 6 | 16 | 10 | 269.43 | 0.46 | NO | 3 |
| 5 | Baicalein | – 7.9 | 270.24 | 1 | 5 | 3 | 90.90 | 2.43 | YES | 0 |
| 6 | Fisetin | – 7.3 | 286.24 | 1 | 6 | 4 | 111.13 | 1.50 | YES | 0 |
| 7 | Astragalin | – 8.5 | 448.38 | 4 | 11 | 7 | 190.28 | 1.29 | NO | 2 |
| 8 | Chrysin | – 7.1 | 254.24 | 1 | 4 | 2 | 70.67 | 2.27 | YES | 0 |
| 9 | Epigallocatechin | – 6.8 | 306.27 | 1 | 7 | 6 | 130.61 | 0.98 | YES | 1 |
| 10 | Icaritin | – 6.3 | 368.38 | 4 | 6 | 3 | 100.13 | 3.26 | YES | 0 |
| 11 | Isoquercetin | – 8.1 | 464.38 | 4 | 12 | 8 | 210.51 | 0.94 | NO | 2 |
| 12 | Isorhamnetin | – 7.4 | 316.26 | 2 | 7 | 4 | 120.36 | 2.35 | YES | 0 |
| 13 | Isovitexin | – 7.5 | 432.38 | 3 | 10 | 7 | 181.05 | 1.97 | YES | 1 |
| 14 | Kaempferol | – 6.6 | 286.24 | 1 | 6 | 4 | 111.13 | 1.70 | YES | 0 |
| 15 | Laricitrin | – 7.8 | 332.26 | 2 | 8 | 5 | 140.59 | 2.24 | YES | 0 |
| 16 | Laricitrin 3 glucoside | – 8.4 | 494.4 | 5 | 13 | 8 | 219.74 | 1.89 | NO | 2 |
| 17 | Luteolin | – 6.9 | 286.24 | 1 | 6 | 4 | 111.13 | 1.86 | YES | 0 |
| 18 | Myricetin | – 7.1 | 318.24 | 1 | 8 | 6 | 151.59 | 1.08 | YES | 1 |
| 19 | Quercetin | – 7.2 | 302.24 | 1 | 7 | 5 | 131.36 | 1.63 | YES | 0 |
| 20 | Quercetin 7 galactoside | – 7.8 | 464.38 | 4 | 12 | 8 | 210.51 | 1.84 | NO | 2 |
| 21 | Quercitrin | – 8.3 | 448.38 | 3 | 11 | 7 | 190.28 | 1.60 | NO | 2 |
| 22 | Syringetin | – 7.3 | 346.29 | 3 | 8 | 4 | 129.59 | 1.77 | YES | 0 |
| 23 | Trifolin | – 8.5 | 448.38 | 4 | 11 | 7 | 190.28 | 1.44 | NO | 2 |
| 24 | Vitexin | – 8.0 | 432.38 | 3 | 10 | 7 | 181.05 | 1.63 | YES | 1 |
The inhibitor 2-methyl-1-tetralone binds to the active site of 6YNQ with the lowest binding energy (– 5.6 kcal/mol), where CYS A: 145 and HIS A: 163 represent the catalytic residues (Figure 1 A). Favipiravir (binding energy was – 5.5 kcal/mol) docked with a confirmation that forms conventional hydrogen bonds with HIS A: 163, GLU A: 166, MET A: 165 and ASN A: 142 along with pi donor hydrogen bonds with CYS A: 145 and LEU A: 141 (Figure 1B), suggesting its interaction with the catalytic residues. In contrast, Remdesivir had the lowest binding energy of – 8.6 kcal/mol, more than the inhibitor and Favipiravir, but failed to make hydrogen bonds with the catalytic residues and rather formed bonds with GLU A: 166, HIS A: 41, and THR A: 25 along with pialkyl interactions with PRO A: 168 (Figure 1C).
Fig. 1.

Docking Interactions of reference compound and inhibitors with SARS-CoV-2 main protease 6YNQ. A, 2 methyl 1 tetralone. B, favipiravir. C, remdesivir.
Among the 21 flavonoids, rutin was shown to bind with the lowest binding energy, – 8.7 kcal/mol, which was close to the Remdesivir binding energy, forming conventional hydrogen bonds with the catalytic active site residues CYS A: 145, HIS A: 163, SER A: 46, THR A: 45, HIS A: 41, GLU A: 166, and GLN A: 189 and additional Van der Waals interactions with ASN A: 142. In addition, it produced a pialkyl interaction with MET A: 49, re-enforcing its binding (Figure 2 ). However, the other flavonoid baicalein demonstrated a binding energy of – 7.9 kcal/mol and formed hydrogen bonds with the active site HIS A: 163 and GLU A: 166, a pi alkyl interaction with the other catalytic residue CYS A: 145, various Van der Waals interactions with LEU A: 141, and a pi bond with MET A: 49 and HIS A: 41 (Figure 3 ). Fisetin presented with a binding energy of – 7.3 kcal/mol and was shown to interact with various amino acid residues producing hydrogen bonds with active site HIS A: 163, LEU A: 141, GLU A: 166, ARG A: 188, GLN A: 192, THR A: 190, PRO A: 168 and GLN A: 189, and pi alkyl interactions with the other catalytic residues CYS A: 145 and PRO A: 168 (Figure 4 ). The other flavonoids presented with binding energy values ranging from – 8.4 kcal/mol to – 5.6 kcal/mol, although there were no hydrogen bonds with the catalytic site.
Fig. 2.
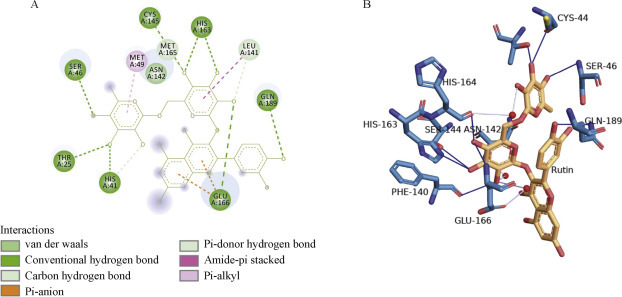
Interactions after docking SARS-CoV-2 main protease 6YNQ with rutin, A, 2D image. B, 3D image.
Fig. 3.
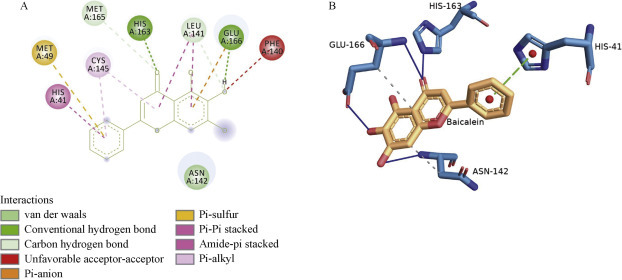
Interactions established after docking SARS-CoV-2 main protease 6YNQ with baicalein, A, 2D image. B, 3D image.
Fig. 4.
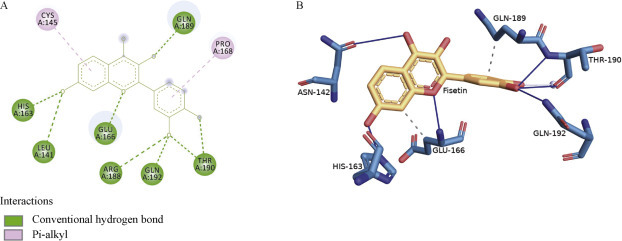
Interactions established after docking SARS-CoV-2 main protease 6YNQ with fisetin, A, 2D image. B, 3D image.
3.2. Physicochemical characterization
Furthermore, the physicochemical properties of the compounds were studied to predict the pharmacokinetics of the drugs, using Lipinski’s rule. Lipinski’s rules describe orally active drug compounds as having a molecular weight (MW) of < 500 Da, an octanol-water partition coefficient (Log P) of < 5, a polar surface area (PSA) of < 150 Å, number of hydrogen bond donors (HBDs) < 5, number of hydrogen bond acceptors (HBAs) < 10, and number of rotatable bonds (RBs) < 10 [ 29 ]. The Lipinski values for each of the selected compounds are listed in Table 1.
3.3. Molecular dynamics
The structural flexibility of the best three phytoconstituents in complex with 6YNQ was evaluated using CABS-flex 2.0. To validate the docking results, the structural PDB file was provided to the server with default parameters to obtain the maximum simulation output [ 30 ]. The root mean square fluctuation (RMSF) values (Figure 5 ) explain the fluctuation of each amino acid residue in the best docked ligand in order to validate the conformational stability of the protein-ligand docked complexes (Figure 6 ).
Fig. 5.
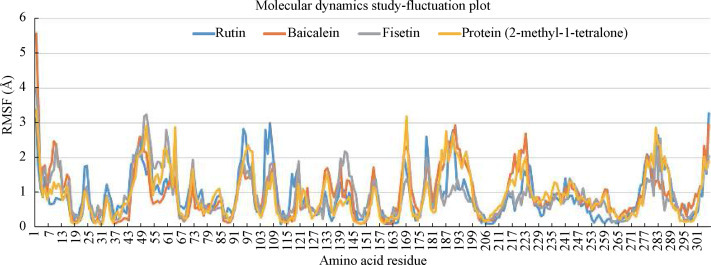
Root mean square fluctuations in protein structures in response to specific substrates
Fig. 6.
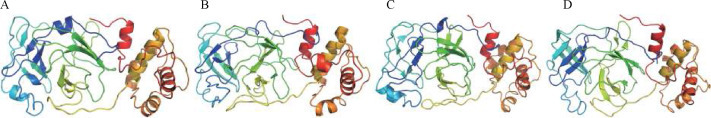
Stable protein structures generated following molecular dynamics simulations. A, 2-methyl-1-tetralone. B, rutin. C, baicalein. D, fisetin.
4. Discussion
Molecular docking studies of flavonoids with SARS-CoV-2 main protease 6YNQ exhibited promising results based on their binding energies, as determined by AutoDock Vina. In this study, some known antiviral and other flavonoids were selected for targeting SARS-CoV-2 main protease 6YNQ, and molecular docking studies were carried out to assess their potential antiviral effect. To evaluate the binding between the flavonoids and the targets, we selected 21 flavonoids against 6YNQ, along with their known inhibitor 2-methyl-1-tetralone, and reference compounds Remdesivir and Favipiravir. Our results suggest that most of the ligands present with nearly the same score in either docking method, with a corresponding correlation coefficient of 0.752 7 between docking scores obtained using AutoDock vina and Delta G by SwissDock, supporting the accuracy of the AutoDock vina predictions. Based on these results, three flavonoids, rutin, baicalein and fisetin, should be considered potential inhibitors of SARS-CoV-2 main protease 6YNQ acting via Mpro 6YNQ inhibition.
Rutin demonstrated strong inhibition of 6YNQ, forming conventional hydrogen bonds with the catalytic active site residue CysA 145 and having the lowest binding energy (− 8.7 kcal/mol) of any of the compounds. Taken together, this suggests that it exhibits the strongest and most stable binding. This result is in agreement with previously published data that suggest that rutin (docking score: − 9.16 kcal/mol) is the most potent inhibitor for 6LU7 [ 31 ]. Other researchers have also reported that rutin is an effective inhibitor of various targets of the SARS-CoV-2 proteases [ 32 ]. These studies have confirmed that CysA 145 is a critical residue within the binding pocket of these proteases falling within a 6 Å radius around the catalytic center of these proteins 33 , 34, and support the application of rutin as a competitive SARS-CoV 3CLpro inhibitor that interacts via hydrogen bonding with the catalytically active residue CysA 145.
One of the most studied flavonoids, baicalein, also forms hydrogen bonds with these proteins, targeting the other catalytic residue, i.e., histidine. Numerous studies have reported that baicalein and its analogs are strong inhibitors of SARS-CoV-2 3CLpro and helicase, suggesting that baicalein is a potential candidate for combating coronavirus disease 35 , 36. In addition, a traditional Chinese medicine formulation containing baicalein was evaluated in a neutralization study using a fRhK4 cell line infected with 10 strains of SARS-CoV-2 from 10 different patients and shown to effectively neutralize these viruses, supporting the potential clinical application of this product [ 37 ]. The flavonol fisetin also produced both hydrogen and pi alkyl bonds with the catalytic center of 6YNQ, although its binding energy was somewhat lower than rutin and baicalein. Other studies have reported binding of fisetin with 6LU7 [ 38 ]. Given these results, we propose that a combination of rutin, baicalein and fisetin may produce a synergistic inhibition of both catalytically active residues in 6YNQ, improving its overall inhibition.
Lipinski’s rule is a major deciding factor when evaluating the potential of drug candidates and is often used to determine whether a compound with particular pharmacological or biological actions possesses the necessary physical and chemical properties for administration in humans. Evaluation of the molecular properties of the compounds based on the computed partition coefficient (Log P) demonstrated that these compounds have relatively good lipophilicity, as the Log P values were less than 5 39 , 40. These results also demonstrated that both baicalein and fisetin strictly followed Lipinski’s rule with zero violations, indicating that both compounds are likely to possess active drug characteristics
Low RMSF values imply limited motion within a system, while high values in the molecular dynamics simulations reflect more flexibility [ 41 ]. The results of the molecular dynamics simulations show that there are appropriate secondary structure residues with the α-helix and β-sheet of the protein that present with minimal fluctuation when evaluated using efficient constraints in the all-atom molecular dynamics algorithm, a classical simulation approach for proteins. Rutin, baicalein and fisetin were shown to maintain their molecular interactions with the target protein under all of these conditions, confirming their likely interaction.
Interestingly, these three flavonoids are nutraceuticals and act as vital nutritional component of various fruits and vegetables. Thus, we anticipate that this nutraceutical has the potential to enhance immunity and inhibit COVID-19 infections in the population [ 42 ]. Furthermore, combination therapy of synthetic drugs with flavonoids often results in superior outcomes for antiviral treatments 43 , 44. Most flavonoid evaluations for COVID-19 have focused on 3CL as the main viral protease [ 45 ]; however, our study demonstrates the potential for flavonoid treatments to affect other targets including 6YNQ of SARS-CoV-2.
Conclusions
Human health and safety is intrinsically linked with the need to find and test novel interventions for COVID-19 (SARS-CoV-2), making any study related to these endeavors critical to global concerns. Here, we have used computational docking studies of various flavonoids against the SARS-CoV-2 main protease 6YNQ to help identify novel therapeutic effectors. We evaluated a library of 21 flavonoids and revealed that rutin, baicalein and fisetin bind the target efficiently and may have value as potential inhibitors. Thus, we conclude that these phytochemicals can be used as potential antiviral candidates and suggest that further in vitro or in vivo experiments may provide better insight into the optimal flavonoid structure for preventing and treating COVID-19.
Competing Interests
The authors declare no conflict of interest.
Acknowledgements
The authors are thankful to the Principal, Gurunanak College of Pharmacy, Principal, Priyadarshini J. L. College of Pharmacy and management of the Sikh Education Society for extending facilities.
References
- 1.WHO. WHO COVID-19 dashboard. 2020:1–8.
- 2.VLACHAKIS D., PAPAKONSTANTINOU E., MITSIS T. Molecular mechanisms of the novel coronavirus SARS-CoV-2 and potential anti-COVID19 pharmacological targets since the outbreak of the pandemic. Food and Chemical Toxicology. 2020;146:111805. doi: 10.1016/j.fct.2020.111805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.RCSB PDB - 6YNQ: structure of SARS-CoV-2 main protease bound to 2-Methyl-1-tetralone. Available from: rcsb.org/structure/6YNQ.
- 4.JIN Z., DU X., XU Y. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 5.NAVEJA J.J., MADARIAGA-MAZÓN A., FLORES-MURRIETA F. Union is strength: antiviral and anti-inflammatory drugs for COVID-19. Drug Discovery Today. 2020 doi: 10.1016/j.drudis.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.CHELLAPANDI P., SARANYA S. Genomics insights of SARS-CoV-2 (COVID-19) into target-based drug discovery. Medicinal Chemistry Research. 2020;29(10):1777–1791. doi: 10.1007/s00044-020-02610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.FDA NEWS RELEASE. FDA approves first treatment for COVID-19. US FDA, 2020.10.22.
- 8.BBC NEWS. Covid: US gives full approval for antiviral remdesivir drug. BBC NEWS, 2020.10.23.
- 9.Coronavinrus disease (COVID-19): hydroxychloroquin. WHO, 2020.06.19. Available from: who.int/news-room/q-a-detail/coronavirus-disease-covid-19-hydroxychloroquine.
- 10.XUE H., LI J., XIE H. Review of drug repositioning approaches and resources. International Journal of Biological Sciences. 2018;14(10):1232–1244. doi: 10.7150/ijbs.24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MURALIDHARAN N., SAKTHIVEL R., VELMURUGAN D. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics. 2020;38(4):1143–1157. doi: 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- 12.SAWIKOWSKA A. Meta-analysis of flavonoids with antiviral potential against coronavirus. Biometrical Letters. 2020;57(1):13–22. [Google Scholar]
- 13.JO S., KIM S., SHIN D.H. Inhibition of SARS-CoV 3CL protease by flavonoids. Journal of Enzyme Inhibition and Medicinal Chemistry. 2020;35(1):145–151. doi: 10.1080/14756366.2019.1690480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.SOLNIER J., FLADERER J.P. Flavonoids: a complementary approach to conventional therapy of COVID-19? Phytochemistry Reviews. 2020 doi: 10.1007/s11101-020-09720-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.CHEN L., LI J., LUO C. Binding interaction of quercetin-3-β-galactoside and its synthetic derivatives with SARS-CoV 3CLpro: structure-activity relationship studies reveal salient pharmacophore features. Bioorganic & Medicinal Chemistry. 2006;14(24):8295–8306. doi: 10.1016/j.bmc.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.RYU Y.B., JEONG H.J., KIM J.H. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CLpro inhibition. Bioorganic & Medicinal Chemistry. 2010;18(22):7940–7947. doi: 10.1016/j.bmc.2010.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.KURIATA A., GIERUT A.M., OLENIECKI T. CABS-flex 2.0: a web server for fast simulations of flexibility of protein structures. Nucleic Acids Research. 2018;46(W1):W338–W343. doi: 10.1093/nar/gky356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MORRIS G.M., HUEY R., LINDSTROM W. AutoDock 4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.GROSDIDIER A., ZOETE V., MICHIELIN O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Research. 2011;39(2):W270–W277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’BOYLE N.M., BANCK M., JAMES C.A. Open Babel: an open chemical toolbox. Journal of Cheminformatics. 2011;3(1):33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.GORDON C.J., TCHESNOKOV E.P., FENG J.Y. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. Journal of Biological Chemistry. 2020;295(15):4773–4779. doi: 10.1074/jbc.AC120.013056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.UDWADIA Z.F., SINGH P., BARKATE H. Efficacy and safety of favipiravir, an oral RNA-dependent RNA polymerase inhibitor, in mild-to-moderate COVID-19: a randomized, comparative, open-label, multicenter, phase 3 clinical trial. International Journal of Infectious Diseases. 2020;100(11):292–297. doi: 10.1016/j.ijid.2020.11.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.TIAN W., CHEN C., LEI X. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Research. 2018;46(W1):W363–W367. doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.BIOVIA DS. Discovery Studio Modeling Environment. San Diego: Dassault Systèmes, 2017.
- 25.DELANO W.L. Pymol: an open-source molecular graphics tool. CCP4 Newsletter on Protein Crystallography. 2002;40(1):82–92. [Google Scholar]
- 26.SALENTIN S., SCHREIBER S., HAUPT V.J. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Research. 2015;43(W1):W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DAINA A., MICHIELIN O., ZOETE V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports. 2017;7(1):42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.KURCINSKI M., OLENIECKI T., CIEMNY M.P. CABS-flex standalone: a simulation environment for fast modeling of protein flexibility. Bioinformatics. 2019;35(4):694–695. doi: 10.1093/bioinformatics/bty685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.BENET L.Z., HOSEY C.M., URSU O. BDDCS, the rule of 5 and drugability. Advanced Drug Delivery Reviews. 2016;101:89–98. doi: 10.1016/j.addr.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.JAMROZ M., OROZCO M., KOLINSKI A. Consistent view of protein fluctuations from all-atom molecular dynamics and coarse-grained dynamics with knowledge-based force-field. Journal of Chemical Theory Computation. 2013;9(1):119–125. doi: 10.1021/ct300854w. [DOI] [PubMed] [Google Scholar]
- 31.XU Z., YANG L., ZHANG X. Discovery of potential flavonoid inhibitors against COVID-19 3CL proteinase based on virtual screening strategy. Frontiers in Molecular Biosciences. 2020;7(September):1–8. doi: 10.3389/fmolb.2020.556481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ADEM S., EYUPOGLU V., SARFRAZ I. Identification of potent COVID-19 main protease (Mpro) inhibitors from natural polyphenols: an in silico strategy unveils a hope against CORONA. Preprint. 2020 doi: 10.20944/preprints202003.0333.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.JACOBS J., GRUM-TOKARS V., ZHOU Y. Discovery, synthesis, and structure-based optimization of a series of N-(tert-Butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. Journal of Medicinal Chemistry. 2013;56(2):534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.PARK J.Y., KO J.A., KIM D.W. Ezones Isolated from Angelica keiskei inhibit cysteine proteases of SARS-CoV. Journal of Enzyme Inhibition and Medicinal Chemistry. 2016;31(1):23–30. doi: 10.3109/14756366.2014.1003215. [DOI] [PubMed] [Google Scholar]
- 35.KEUM Y.S., LEE J., YU M.S. Inhibition of SARS coronavirus helicase by baicalein. Bulletin of the Korean Chemical Society. 2013;34(11):3187–3188. [Google Scholar]
- 36.LIU H., YE F., SUN Q. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. bioRxiv. 2020 doi: 10.1101/2020.04.10.035824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.CHEN F., CHAN K.H., JIANG Y. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. Journal of Clinical Virology. 2004;31(1):69–75. doi: 10.1016/j.jcv.2004.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oladele J.O., OYELEKE O.M., OLADELE O.T. Kolaviron ( Kola avanone ), apigenin , setin as potential coronavirus inhibitors: in silico investigation. Preprint. 2020 doi: 10.21203/rs.3.rs-51350/v1. [DOI] [Google Scholar]
- 39.LIPINSKI C.A., LOMBARDO F., DOMINY B.W. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 1997;23(1-3):3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 40.HUGHES J.D., BLAGG J., PRICE D.A. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorganic & Medicinal Chemistry Letters. 2008;18(17):4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- 41.SAHU A., PRADHAN D., RAZA K. In silico library design, screening and MD simulation of COX-2 inhibitors for anticancer activity. Proceedings of the 12th International Conference. 2020;70:21–32. [Google Scholar]
- 42.LIN JK, WENG MS. Flavonoids as nutraceuticals. The Science of Flavonoids. Springer, New York, 2006: 213-238.
- 43.MUCSI I., GYULAI Z., BÉLÁDI I. Combined effects of flavonoids and acyclovir against herpesviruses in cell cultures. Acta Microbiologica Hungarica. 1992;39(2):137–147. [PubMed] [Google Scholar]
- 44.CHANG J., SCHUL W., BUTTERS T.D. Combination of α-glucosidase inhibitor and ribavirin for the treatment of dengue virus infectionin vitro andin vivo. Antiviral Research. 2011;89(1):26–34. doi: 10.1016/j.antiviral.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jo S., KIM H., KIM S. Characteristics of flavonoids as potent MERS-CoV 3C-like protease inhibitors. Chemical Biology & Drug Design. 2019;94(6):2023–2030. doi: 10.1111/cbdd.13604. [DOI] [PMC free article] [PubMed] [Google Scholar]