

Abstract
The December 2019 outbreak of a novel respiratory virus, SARS-CoV-2, has become an ongoing global pandemic due in part to the challenge of identifying symptomatic, asymptomatic, and pre-symptomatic carriers of the virus. CRISPR diagnostics can augment gold-standard PCR-based testing if they can be made rapid, portable, and accurate. Here, we report the development of an amplification-free CRISPR-Cas13a assay for direct detection of SARS-CoV-2 from nasal swab RNA that can be read with a mobile phone microscope. The assay achieved ∼100 copies/μL sensitivity in under 30 min of measurement time and accurately detected pre-extracted RNA from a set of positive clinical samples in under 5 min. We combined crRNAs targeting SARS-CoV-2 RNA to improve sensitivity and specificity and directly quantified viral load using enzyme kinetics. Integrated with a reader device based on a mobile phone, this assay has the potential to enable rapid, low-cost, point-of-care screening for SARS-CoV-2.
Keywords: CRISPR-Cas13, CRISPR Dx, SARS-CoV-2, COVID-19, point-of-care diagnostics, mobile phone microscopy
Graphical Abstract
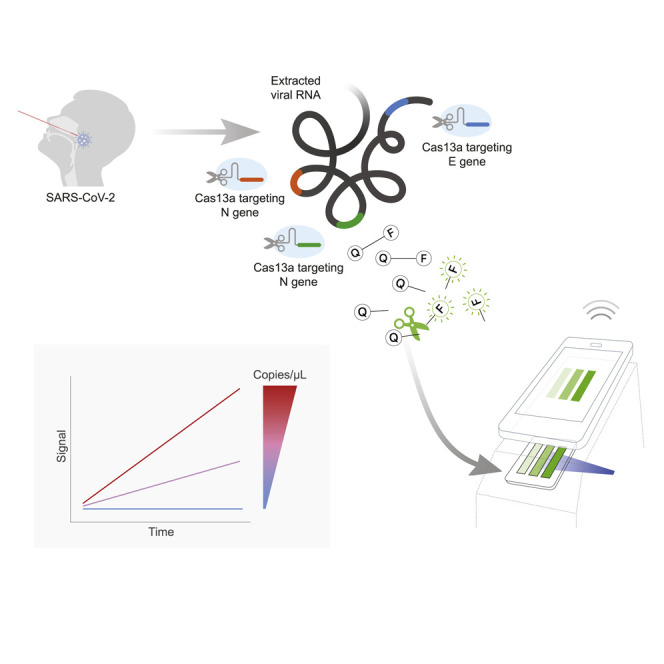
Fozouni et al. devise a way to use CRISPR-Cas13a to detect and quantify SARS-CoV-2 RNA from patient samples without the need for a pre-amplification step. They then show how the assay’s signal can be efficiently detected with a portable, mobile phone-based device.
Introduction
In late 2019, a novel infectious respiratory RNA virus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), emerged in the human population, likely from a zoonotic source (Wang, et al., 2020a; Zhu et al., 2020). In most people, SARS-CoV-2 infection causes mild or no symptoms. Critically, however, asymptomatic or lowly symptomatic carriers spread the virus, leading to delayed isolation of carriers and worldwide spread (Bai et al., 2020; Lavezzo et al., 2020). In particular, this silent transmission has led to the infection of individuals who are at increased risk of severe illness due to age or pre-existing conditions such as obesity, diabetes, cancer, immunosuppression, or cardiac, pulmonary, and kidney disease (Williamson et al., 2020).
The current gold-standard diagnostic for SARS-CoV-2 infection, quantitative reverse transcription polymerase chain reaction (RT-qPCR), is well established and widely used for screening. Based on primers directed against the nucleocapsid (N), envelope (E), and open reading frame 1ab (ORF1ab) genes, RT-qPCR has an analytical limit of detection (LOD) of 1,000 viral RNA copies/mL (1 copy/μL) (Vogels et al., 2020). However, recent modeling of viral dynamics suggests that frequent testing with a fast turnaround time is required to break the current pandemic (Larremore et al., 2020). Notably, the model ranked sensitivity of the test as a lower priority and estimated that an LOD of 100,000 copies/mL (100 copies/mL) would be sufficient for screening (Larremore et al., 2020). Although there is not yet broad consensus on the exact target LOD that is necessary, frequent testing and rapid turnaround times will allow less sensitive tests to help reduce viral transmission. In clinical studies, when viral load drops below a million copies/mL (1,000 copies/μL), few infectious particles are detected and consequently the risk of transmission is low (La Scola et al., 2020; Quicke et al., 2020; Wölfel et al., 2020).
The need for SARS-CoV-2 tests that are rapid, widespread, and able to identify infectious individuals has motivated efforts to explore new strategies for viral RNA detection based on CRISPR technology. Cas12 and Cas13 proteins are RNA-guided components of bacterial adaptive immune systems that directly target single- and double-stranded DNA or single-stranded (ss)RNA substrates, respectively (Abudayyeh et al., 2016; Chen et al., 2018; East-Seletsky et al., 2016; Zetsche et al., 2015). Cas13 is complexed with a CRISPR RNA (crRNA) containing a programmable spacer sequence to form a nuclease-inactive ribonucleoprotein complex (RNP). When the RNP binds to complementary target RNA, it activates the HEPN (higher eukaryotes and prokaryotes nucleotide-binding domain) motifs of Cas13 that then indiscriminately cleave any surrounding ssRNAs. Target RNA binding and subsequent Cas13 cleavage activity can be detected with a fluorophore-quencher pair linked by an ssRNA, which fluoresces after cleavage by active Cas13 (East-Seletsky et al., 2016). To date, four type VI CRISPR-Cas13 subtypes have been identified: Cas13a (previously known as C2c2) (Abudayyeh et al., 2016; East-Seletsky et al., 2016; Shmakov et al., 2015), Cas13b (Smargon et al., 2017), Cas13c (Shmakov et al., 2017), and Cas13d (Konermann et al., 2018; Yan et al., 2018).
What initially evolved as a successful strategy in bacteria to induce cellular dormancy to reduce phage transmission (Meeske et al., 2019) is now being harnessed for viral diagnostics (Chen et al., 2018; East-Seletsky et al., 2016; Gootenberg et al., 2018; 2017; Myhrvold et al., 2018). To achieve high sensitivity, current CRISPR diagnostics (CRISPR Dx) rely on pre-amplification of target RNA for subsequent detection by a Cas protein. In the case of RNA-sensing Cas13 proteins, this entails the conversion of RNA to DNA by reverse transcription, DNA-based amplification (i.e., isothermal amplification, loop-mediated isothermal amplification [LAMP]), and transcription back to RNA for detection by Cas13a or Cas13b, an approach named “SHERLOCK” (Gootenberg et al., 2018; 2017). This was recently adapted for SARS-CoV-2 detection (Joung et al., 2020b) and further developed as “SHINE” for testing unextracted samples (Arizti-Sanz et al., 2020). The conversion of amplified DNA back into RNA can be avoided by using the DNA-sensing Cas12 for detection, a method called “DETECTR” (Chen et al., 2018), which has recently been adapted for SARS-CoV-2 detection (Broughton et al., 2020). Both SHERLOCK and DETECTR take approximately an hour to complete and can be read with paper-based lateral flow strips appropriate for point-of-care use, although current FDA-approved protocols are still laboratory based.
Here, we report the development and demonstration of a rapid CRISPR-Cas13a-based assay for direct detection of SARS-CoV-2 RNA. This assay, unlike previous CRISPR diagnostics, does not require pre-amplification of the viral genome for detection. By directly detecting the viral RNA without additional manipulations, the test yields quantitative RNA measurements rather than simply a positive or negative result. To demonstrate the simplicity and portability of this assay, we measure fluorescence with a mobile phone camera in a compact device that includes low-cost laser illumination and collection optics. The high sensitivity of mobile phone cameras, together with their connectivity, GPS, and data-processing capabilities, have made them attractive tools for point-of-care disease diagnosis in low-resource regions (Breslauer et al., 2009; D’Ambrosio et al., 2015; Kamgno et al., 2017; Wood et al., 2019). By combining multiple crRNAs to increase Cas13a activation and analyzing the change in fluorescence over time rather than solely endpoint fluorescence, we are able to achieve detection of ∼100 copies/μL of pre-isolated SARS-CoV-2 RNA within 30 min of measurement time on the device. We also correctly identified all SARS-CoV-2-positive patient RNA samples tested (Ct values 14.37 to 22.13) within 5 min of measurement time on the device. This approach has the potential to enable a fast, accurate, portable, and low-cost option for point-of care SARS-CoV-2 screening.
Results
Quantitative direct detection of viral SARS-CoV-2 RNA with Cas13a
When the SARS-CoV-2 sequence became public in January 2020, we set out to develop a Cas13-based direct detection assay for viral RNA that would avoid the need for amplification and enable point-of-care testing. To do this, we needed to optimize Cas13 activation through careful crRNA selection and develop a sensitive and portable fluorescence detection system for our assay (Figure 1 A). Initially, we designed 12 crRNAs (Table S1) along the N gene of SARS-CoV-2, corresponding to the three Centers for Disease Control and Prevention (CDC) N qPCR primer sets and a N qPCR primer set developed in Wuhan, China (Zhu et al., 2020). Every Cas13-crRNA RNP should detect a single 20-nucleotide region in the N gene (Figure 1B).
Figure 1.

Quantitative direct detection of viral SARS-CoV-2 RNA with Cas13a
(A) Schematic of a Cas13a (beige)-crRNA (red) RNP complex binding target RNA (black), resulting in activation of the HEPN nuclease (denoted by scissors) domain. Upon target recognition and RNP activation, Cas13a indiscriminately cleaves a quenched-fluorophore RNA reporter, allowing for fluorescence detection as a proxy for Cas13a activation and target RNA.
(B) Schematic of the SARS-CoV-2 N gene, and the corresponding location of each crRNA spacer region.
(C) Cas13a RNPs made individually with each crRNA were tested against 2.89 × 105 copies/μL (480 fM) of SARS-CoV-2 IVT N gene RNA in a total 20 μL reaction volume. Background fluorescence by the individual RNP in the absence of target RNA is shown as “RNP.” Raw fluorescence values over 2 h is shown. Data are represented as mean ± standard deviation (SD) of three technical replicates. See also Figure S1A.
(D) Limit of detection of crRNA 2 and crRNA 4 was determined by testing 100 nM of each RNP individually against 105, 104, and 103 copies/μL of N gene IVT RNA. “RNP 2” and “RNP 4” represent no target RNA RNP alone controls. Background correction of fluorescence was performed by subtraction of reporter alone fluorescence values. Data are represented as mean ± standard error of the difference between means of three technical replicates. See also Figures S1B–S1D.
(E) Slope of the curve over 2 h from Figure 1D was calculated by performing simple linear regression to data merged from replicates and is shown as slope ± 95% confidence interval. Slopes were compared to the RNP alone control through an analysis of covariance (ANCOVA): ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ns = not significantly higher than RNP control.
We first tested each crRNA individually in a direct detection assay on a plate reader. We selected the Cas13a homolog from Leptotrichia buccalis (Lbu) as it demonstrated the highest sensitivity and robust trans-cleavage activity relative to other characterized Cas13a homologs (East-Seletsky et al., 2017; 2016). The assay used purified LbuCas13a (East-Seletsky et al., 2017; 2016) and a quenched fluorescent RNA reporter (East-Seletsky et al., 2017; 2016), together with in vitro transcribed (IVT) target RNA corresponding to the viral N gene (nucleotide positions 28274–29531). At a target RNA concentration of 480 fM (2.89 × 105 copies/mL), we identified 10 crRNAs with reactivity above the RNP alone control containing the same RNP and probe but no target RNA (Figure 1C). The use of RNase-free buffers minimized background fluorescence, and the plate reader gain and filter bandwidth settings were optimized to capture low-level reporter cleavage. Similar trends in specific crRNA performance were observed when genomic SARS-CoV-2 RNA was used as target RNA, with lower overall activities corresponding to a lower concentration of RNA (Figure S1 A). For initial studies, we selected two crRNAs (crRNAs 2 and 4) that generated the greatest Cas13a activation as determined by the fluorescent reporter while maintaining low levels of target-independent fluorescence (indicated by the RNP alone curve).
Figure S1.

Individual crRNAs quantitatively detect SARS-CoV-2 RNA, related to Figure 1
(A) Cas13a RNPs made individually with each N gene crRNA (final RNP complex concentration of 100 nM) were tested against extracted SARS-CoV-2 viral RNA. Background fluorescence by the individual RNP in the absence of target RNA is shown as “RNP Only.” Raw fluorescence values over 2 h is shown. Data are represented as mean ± SD of three technical replicates.
(B and C) Cas13a reaction rate is linearly proportional to the target RNA concentration.
(B) The reaction rate of Cas13a was measured by adding a range of concentrations of IVT N gene RNA to reactions that contain 100 nM of Cas13a RNP with crRNA 2 and 400 nM of polyU reporter (error bars indicate the standard deviation of triplicate measurements). The reaction rate is determined by fitting a linear curve to the data (black curves).
(C) The reaction rate of Cas13a for a range of IVT N gene RNA concentrations as measured in Figure S1B but with crRNA 4 used in place of crRNA 2.
(D) Cas13a reaction rate – either with crRNA 2 or crRNA 4 – scales linearly with the target IVT RNA concentration (R2 = 0.990 for crRNA 2 and 0.996 for crRNA 4). Assuming that Cas13a enzymatic activity can be described by the Michaelis-Menten kinetics model, and that the amount of IVT RNA sets the upper limit of active Cas13a RNP, we predict the ratio of active Cas13a to IVT RNA in for Kcat = 600/s and KM = 1 μM or 3 μM, which is the range of KM previously found for Cas13b (Slaymaker et al., 2019).
We next carried out serial dilutions of the target RNA to independently determine the limit of detection for each crRNA. LbuCas13a exhibits detectable reporter cleavage with as little as 10 fM (∼6,000 copies/μL) of target RNA (East-Seletsky et al., 2017). SHERLOCK had previously reported a limit of detection of ∼50 fM without pre-amplification (Gootenberg et al., 2017). Consistent with this, we found that RNPs made with either crRNA 2 or crRNA 4 did not appear to generate signals above the RNP controls for an IVT target RNA concentration of 1,000 copies/μL (Figure 1D). The signal generated by direct detection with Cas13a appeared proportional to the concentration of target RNA in the assay. Given that the signal generated depends solely on the RNase activity of Cas13a, the linear rate of the reaction should approximate Michaelis-Menten enzyme kinetics. To determine if our assay was indeed quantitative, we compared the slopes determined by linear regression for different target RNA concentrations, using 1 to 3 μM KM and 600/s Kcat for the modeling (Slaymaker et al., 2019). The results confirmed that crRNA 2 and 4 each facilitated detection of at least 10,000 copies/μL of IVT N gene RNA (Figure 1E). Since the measured slopes are proportional to the concentration of activated Cas13a, we could confirm that the rate of Cas13a activity scaled with concentration of target RNA (Figures S1B–S1D). This ability to estimate target RNA concentration from the measured slope allows for direct quantification of viral load in unknown samples.
Combining crRNAs improves sensitivity of Cas13a
We next evaluated whether combining crRNAs to form two different populations of RNPs in the same reaction could enhance overall Cas13a activation and, therefore, the sensitivity of the assay. In theory, a single target RNA could activate multiple Cas13a RNPs if each RNP is directed to different regions of the same viral target RNA, effectively doubling the active enzyme concentration (Figure 2 A). Targeting multiple sites is especially beneficial in cases where target RNA is the limiting reagent, i.e., in the absence of target pre-amplification. To test this, we combined crRNAs 2 and 4 in the same reaction, keeping the total concentration of Cas13a RNPs constant but divided equally between RNPs made with each crRNA. We found that combining crRNA 2 and 4 markedly increased the slope of the detection reaction and the sensitivity of the reaction when measured with a fixed IVT target RNA concentration (480 fM) (Figure 2B). The slope increased from 213 AU/min (SE ± 1.6) (crRNA 2) and 159 AU/min (SE ± 1.7) (crRNA 4), individually, to 383 AU/min (SE ± 3.0) in combination, without increasing the slope of the RNP control reactions. This nearly doubling of the average slope compared to the individual crRNA reactions demonstrates the advantage of crRNA combinations.
Figure 2.

Combining crRNAs improves sensitivity of Cas13a
(A) Schematic of two different RNPs binding to different locations of the same SARS-CoV-2 RNA, leading to cleavage of the RNA reporter and increased fluorescence.
(B) RNPs made with crRNA 2 and crRNA 4 individually and in combination (50 nM total RNP concentration per reaction) were tested against 2.89 × 105 copies/μL (480 fM) of SARS-CoV-2 IVT N gene RNA and compared to fluorescence from no target RNA RNP alone controls (“RNP 2,” “RNP 4,” and “RNP 2+4”). Background correction of fluorescence was performed by subtraction of reporter-alone fluorescence values. Data are represented as mean ± standard error of the difference between means of three technical replicates.
(C) Limit of detection of crRNA 2 and crRNA 4 in combination was determined by combining 50 nM of RNP 2 and 50 nM of RNP 4 (100 nM total) against 1,000, 100, and 1 copy/μL of SARS-CoV-2 IVT RNA (n = 3, technical replicates). Slope of the curve over 2 h was calculated by performing simple linear regression of data merged from replicates and is shown as slope ± 95% confidence interval. Slopes were compared to the no target RNA RNP alone control using ANCOVA: ∗∗∗∗p < 0.0001, ∗∗p = 0.0076, ns = not significant.
(D) Limit of detection of crRNA 2 and crRNA 4 in combination was determined by combining 50 nM of RNP 2 and 50 nM of RNP 4 (100 nM total) against 1.35 × 103, 5.4 × 102, 2.7 × 102, and 1.8 × 102 copies/μL of genomic SARS-CoV-2 viral RNA as quantified by qPCR (n = 3, technical replicates). Slope of the curve over 2 h was calculated by performing simple linear regression of data merged from replicates and is shown as slope ± 95% confidence interval. Slopes were compared to the no target RNA RNP alone control using ANCOVA: ∗∗∗∗p < 0.0001, ∗∗∗p = 0.0002, ∗∗p = 0.0023, ns = not significant.
To determine how crRNA combinations affect the limit of detection, we tested crRNA 2+4 with a dilution series of IVT N gene RNA. The RNP combination improved the limit of detection, compared to the no target RNP control (RNP 2+4), to at least 100 copies/μL of IVT target RNA (Figure 2C). We performed the same assay with genomic SARS-CoV-2 RNA isolated from the supernatant of SARS-CoV-2-infected Vero CCL-81 cells and found that the assay could detect at least 270 viral copies/μL (slope 12.4 ± SE 0.3) (Figure 2D). Viral copy numbers were determined by standard RT-qPCR. The difference between the N gene IVT and full viral genome limits of detection could be explained by different quantification techniques of the target RNA or by considerable secondary structure in the viral RNA (Manfredonia et al., 2020; Sanders et al., 2020) that could reduce the accessibility of the target RNA for the RNP (Abudayyeh et al., 2017; Abudayyeh et al., 2016).
A major advantage of CRISPR diagnostics is that they can be highly specific. To confirm the specificity of our crRNAs, we tested them against a set of other respiratory viruses, including alphacoronavirus HCoV-NL63 and betacoronaviruses HCoV-OC43 and Middle East respiratory syndrome coronavirus (MERS-CoV), which are among seven coronaviruses that infect human hosts and cause respiratory diseases (Fung and Liu, 2019). We extracted RNA from the supernatant of Huh 7.5.1-ACE2 or Vero E6 cells infected with HCoV-NL63 or HCoV-OC43, respectively, and produced IVT N gene RNA from MERS-CoV. In our Cas13a direct detection assay with crRNA 2 and 4, individually and in combination, we detected no signal above RNP background for any of viral RNAs tested (Figure 3 A). Similarly, no signal was detected with H1N1 influenza A or influenza B viral RNA or with RNA extracted from primary human airway organoids (Figure 3B).
Figure 3.

Cas13a directly detects SARS-CoV-2 RNA in patient samples
(A) crRNA 2 and crRNA 4 were tested individually (100 nM total RNP concentration) and in combination (100 nM total RNP concentration: 50 nM each of RNP 2 and RNP 4) against RNA isolated from HCoV-NL63 viral supernatant (left) and HCoV-OC43 viral supernatant (center) or the IVT N gene RNA from MERS-CoV (right). No target RNA RNP alone controls are denoted as “RNP 2,” “RNP 4,” and “RNP 2+4.” Background correction of fluorescence was performed by subtraction of reporter alone fluorescence values. Data are represented as mean ± standard error of the difference between means of three technical replicates.
(B) crRNA 2, crRNA 4, and crRNA 21 were tested individually (100 nM total RNP concentration) and in combination (100 nM total RNP concentration: 33 nM each of RNP 2, RNP 4, and RNP 21) against RNA isolated from human airway organoids (left), H1N1 influenza A (center), and influenza B (right). No target RNA RNP alone controls are denoted as “RNP 2,” “RNP 4,” “RNP 21,” and “RNP 2+4+21.” Background correction of fluorescence was performed by subtraction of reporter alone fluorescence values. Data are represented as mean ± standard error of the difference between means of three technical replicates. See also Figures S2A–S2D.
(C) RNA from five nasopharyngeal swabs confirmed negative for SARS-CoV-2 by RT-qPCR was tested against RNP 2+4+21 (100 nM total RNP concentration). The no target RNA RNP control is denoted as “RNP 2+4+21.” Raw fluorescence values over 2 h is shown. Data are represented as mean ± SD of three technical replicates. See also Figure S2E.
(D) Dilutions of genomic SARS-CoV-2 RNA independently quantified by BEI using ddPCR was tested against RNP 2+4+21 to determine the limit of detection (n = 20, technical replicates). Slope of the raw fluorescence curve over 2 h was calculated by performing simple linear regression of data merged from replicates and is shown as slope ± 95% confidence interval (left). Slopes were compared to the no target RNA RNP alone control using ANCOVA: ∗∗∗∗p < 0.0001. An individual reaction containing the diluted SARS-Cov-2 RNA was compared with the reaction without the target RNA and the number of true positives was counted at the 95% confidence level (right).
(E) Pre-extracted RNA from five nasopharyngeal swabs confirmed positive for SARS-CoV-2 by RT-qPCR was tested against RNP 2+4+21 (100 nM total RNP concentration) (n = 3, technical replicates). A confirmed negative swab was tested against RNP 2+4+21 for comparison. We added 0.3 μL of RNA from Patient Swabs 1–4, 0.26 mL of RNA from Patient Swab 5, and 0.3 μL of RNA from a confirmed negative swab to each 20 μL Cas13a reaction (in triplicate). Slope of the raw fluorescence curve over 2 h was calculated by performing simple linear regression of data merged from replicates and is shown as slope ± 95% confidence interval. Slopes were compared to the negative swab RNP background control using ANCOVA: ∗∗∗∗p < 0.0001. See also Figure S2F.
(F) The Ct value (average Ct count using CDC N1/N2 primers in RT-qPCR), the copies/mL of the original sample determined by qPCR, and the copies/μL in the Cas13a reaction are described for the RNA samples from each positive swab used in Figure 3E.
Cas13a directly detects SARS-CoV-2 RNA in patient samples
We then examined if our assay could be used with patient samples, where the swab and patient matrix (e.g., mucous from a nasal swab) could contribute additional background signal and reduce sensitivity. To increase assay performance prior to testing patient samples, we examined an additional set of crRNAs (crRNAs 19–22) (Table S1) targeting the viral E gene, based on published qPCR primer and Cas12 guide sets (Broughton et al., 2020; Corman et al., 2020) (Figure S2 A). When tested against genomic SARS-CoV-2 RNA, the crRNA that performed best, both individually (Figure S2B) and in combination (Figure S2C) with crRNA 2 and 4, was crRNA 21. Adding crRNA 21 to the combination also allowed us to have at least one crRNA in the assay at all times with perfect complementarity to 4,115 out of 4,118 sequenced genomes of SARS-CoV-2 (Figure S2D). When tested on RNA from five nasal swab samples confirmed negative for SARS-CoV-2, the triple combination (RNP 2+4+21) did not exhibit signal above the RNP control reaction (Figure 3C; Figure S2E).
Figure S2.

Combining crRNAs improves SARS-CoV-2 detection, related to Figure 3
(A) Schematic of the SARS-CoV-2 envelope (E) gene, and the corresponding location of each crRNA spacer region.
(B) Cas13a RNPs made individually with each E gene crRNA (final RNP complex concentration of 100 nM) were tested against genomic SARS-CoV-2 RNA. Background fluorescence by the RNP alone in the absence of target RNA is shown as “RNP Only.” Raw fluorescence values over 2 h are shown. Data are represented as mean ± SD of three technical replicates.
(C) RNPs made with crRNA 2, crRNA 4, and crRNA 21 individually and in combination (100 nM total RNP concentration for each reaction) were tested against 1.5 × 104 copies/mL of extracted SARS-CoV-2 RNA, and compared to fluorescence from no target RNA RNP alone controls (“RNP 2,” “RNP 4,” “RNP 21,” and “RNP 2+4+21”). Raw fluorescence values over 2 h is shown. Data are represented as mean ± SD of three technical replicates.
(D) 4118 complete SARS-CoV-2 genome sequences deposited in NCBI RefSeq under taxonomy ID (2697049) were downloaded on 06/05/2020. Each crRNA was compared against the downloaded genomes for genomes with zero mismatches to each individual crRNA. The Venn diagram shows how many complete genomes have 100% homology to crRNAs 2, 4, and 21, as well as the overlap between crRNAs.
(E) Slope of the curves in Figure 3C over two h was calculated by performing simple linear regression of data from each replicate (n = 3) individually. The mean of the replicate slopes is shown as slope ± 95% confidence interval. Slopes were compared to the no target RNA RNP alone control using repeated-measures one-way analysis of variance (ANOVA) and Dunnett’s multiple comparisons test: ns = not significant.
(F) The same swabs as in Figure 3E were tested against a non-targeting crRNA (RNP NT) (final RNP complex concentration of 100 nM). Slope of the raw fluorescence curve over 2 h was calculated by performing simple linear regression of data merged from replicates (n = 3) and is shown as slope ± 95% confidence interval. Slopes were compared to the no target RNA RNP alone control using ANCOVA: ns = not significant.
To determine if adding crRNA 21 would improve the limit of detection of our assay, we tested a combination reaction with crRNAs 2+4+21 on precisely titered SARS-CoV-2 genomic RNA obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources). In serial dilution experiments using 20 replicate reactions, the triple combination detected as few as 31 copies/μL (Figure 3D, left), based on the viral copy number independently determined by BEI with digital droplet (dd) PCR. By comparing the slope of an individual reaction with that of the RNP control, we determined that, for all dilutions, 20/20 individual tests (100%) would be correctly identified as “positive” with the 95% confidence level (Figure 3D, right).
Finally, we obtained five de-identified RNA samples extracted from nasal swabs taken from SARS-CoV-2-positive individuals. Clinical RT-qPCR measurements resulted in Ct values of 14.37–22.13 for the patient samples, correlating to copy numbers 2.08 × 107–1.27 × 105 copies/μL. Using the direct detection assay on a plate reader, we correctly identified all five positive samples (ranging from 3.2 × 105–1.65 × 103 copies/μL in the Cas13a reaction), which showed slopes significantly above that of the negative swab control (Figure 3E). The positive sample slopes correlated significantly with their input copy number (Pearson r coefficient = 0.9966, two-tailed p value = 0.0002), reinforcing the quantitative nature of the assay. Including the negative swab allows for us to account for potential matrix effects in clinical samples (McNerney et al., 2019). We also tested all samples with a non-targeting control crRNA and did not detect any significant signal (Figure S2F).
Harnessing the mobile phone camera as a portable plate reader
To demonstrate that SARS-CoV-2 screening with Cas13a would be possible outside of laboratory settings, we designed a mobile phone-based fluorescence microscope and reaction chamber to quantify the fluorescent signal generated by the Cas13a direct detection assay (Figure 4 A; Figures S3 A–S3C). The goal was to show that mass-produced consumer electronics, rather than specialized laboratory equipment, are sufficient to capture the small fluorescent signals generated by Cas13a direct detection. Interestingly, we found that our device was approximately an order of magnitude more sensitive than the plate reader used in the development of this assay due to reduced measurement noise and the ability to collect more time points, which decreased the uncertainty in slope estimations and therefore enabled us to distinguish smaller slopes relative to the control (Figure S3D).
Figure 4.

Harnessing the mobile phone camera as a portable plate reader
(A) Schematic of mobile phone-based microscope for fluorescence detection showing illumination and image collection components (left). Picture of assembled device used for data collection and sample image taken by the mobile phone camera after running a Cas13a assay (right). See also Figures S3A–S3C.
(B) Results from the Cas13a assay run on the mobile device with two different dilutions of genomic SARS-CoV-2 viral RNA isolated from infected Vero CCL-81 cells (500 and 200 copies/μL) and RNP alone, using three combined crRNAs (crRNA 2, crRNA 4, and crRNA 21). y axis is the fluorescent signal of each sample normalized by the first time point. The error bars indicate the root-mean-square error (RMSE) of simple linear regression to individual curves. See also Figures S3D and S3E.
(C) Slope of the curve over 30 min of measurement on the device from Figure 4B was calculated by simple linear regression and is shown as slope ± 95% confidence interval.
(D) Detection accuracy of the Cas13a assay is characterized in the mobile device using genomic SARS-CoV-2 viral RNA. For each target dilutions, the slope at three different times—10, 20, and 30 min of measurement time on the device—were compared to the slope of the no target RNA RNP alone controls, and the detection accuracy was determined at the 95% confidence level. The number of replicates for each concentration is 8 (500 copies/μL), 7 (200 copies/μL), 9 (100 copies/μL), and 11 (50 copies/μL). See also Figures S4A–S4C.
(E) Results from a Cas13a assay run on mobile device with two different nasal swab samples, confirmed positive for SARS-CoV-2 using RT-qPCR, and the RNP alone control, all using the crRNA combination of crRNA 2, crRNA 4, and crRNA 21. The error bars indicate the RMSE of simple linear regression to individual curves. See also Figure S3F.
(F) Slope of the curve over 30 min from Figure 4E was calculated by simple linear regression and is shown as slope ± 95% confidence interval.
(G) Detection accuracy of Cas13a assay for 5 nasal swab samples, confirmed positive by RT-qPCR. Detection accuracy was evaluated at four different time points: 5, 10, 20, and 30 min of measurement time on the device.
Figure S3.

Comparison of the Cas13a reaction measured in the plate reader and the mobile phone device, related to Figure 4
(A–C) Schematic of the reaction chamber and the sample region-of-interest (ROI)
(A) Reaction chamber dimensions are described here.
(B) Photo of a reaction chamber loaded with an artificial green dye (Scale bar = 5 mm).
(C) Raw image of patient sample and the sample ROIs (black rectangle) (Scale bar = 5 mm).
(D) The measurement error of the plate reader (left) versus the mobile device (right) for typical conditions used for Cas13a reaction (37°C, measurement interval: 30 s).
(E) The triple crRNA combination with 500 copies/μL of genomic SARS CoV-2 viral RNA was measured in the plate reader (left) and in the mobile phone device (right).
(F) The triple crRNA combination with Positive Swab 4 was measured in the plate reader (left) and in the mobile phone device (right).
We tested the performance of the mobile phone-based device for detecting SARS-CoV-2 RNA using the triple crRNA Cas13a assay and a dilution series with genomic viral RNA isolated from supernatants of infected Vero CCL-81 cells (Figures 4B–4D). Fluorescence generated in each reaction chamber was collected simultaneously and quantified over time, with measurements every 30 s. As with the plate reader, the data showed a steady increase in fluorescence for genomic SARS-CoV-2 virus concentrations of 200–500 copies/μL, compared to RNP controls (Figure 4B; Figure S3E). As before, the slopes of each curve can be calculated, along with the 95% confidence interval indicated by the error bars (Figure 4C). To determine the limit of detection of the direct detection assay on the device, several replicates of dilutions of virus corresponding to 500, 200, 100, and 50 copies/μL were measured, as determined by RT-qPCR. Slopes were calculated based on data for the first 10, 20, and 30 min of the assay, and each slope was then compared to the RNP control slope calculated over the same time (see STAR Methods). For each dilution and assay time, the ability of the assay to detect the target RNA relative to the RNP control was quantified as percent accuracy, with eight positive tests out of eight replicates for 500 copies/μL for all assay times corresponding to 100% accuracy (Figure 4D). The results over all dilutions indicate 100% accuracy for 200 copies/μL over 30 min of measurement, with accuracy dropping to 50% at 50 copies/μL.
Next, we analyzed the same RNA from patient samples as in Figure 3E to compare detection on the plate reader to that on the mobile phone-based device. We imaged each reaction for 60 min, along with the RNP control (Figure 4E), and the slope for a patient with Ct = 17.65 (Positive Swab 3, 3.71 × 104 copies/μL) is significantly greater than the slope for a patient with Ct = 20.37 (Positive Swab 4, 6.21 × 103 copies/μL) (Figure 4F; Figure S3F), as expected. To assess the detection accuracy, we performed a simple linear regression using data from the first 5, 10, 15, and 20 min of the assay and compared the slope of each sample to the RNP control. We determined all five samples to be positive within the first 5 min of measurement, indicating that the device can provide a very fast turnaround time of results for patients with clinically relevant viral loads (Figure 4G). This result highlights the inherent tradeoff between sensitivity and time in the Cas13a direct detection assay. High viral loads can be detected very rapidly because their high signals can be quickly determined to be above the control, and low viral loads can be detected at longer times once their signal can be distinguished above the control (Figure S4 ).
Figure S4.

Limit of detection of the mobile phone device, related to Figure 4
(A) We simulated the signal of mobile phone device for triple crRNA combination with the target viral RNA at 50 copies/μL (red dots) or without the target (RNP alone) (black dots) (see Methods). The red and black lines indicate the linear fit of the simulated signal.
(B) For each simulated measurement, we estimated the slope and the 95% confidence interval of the slope and tested whether the slope of the positive sample was significantly larger than the slope of the RNP alone (red line). Similarly, we tested whether the endpoint signal of the positive sample was significantly larger than the endpoint signal of the RNP alone (blue line). By repeating this procedure 1,000 times for different assay times, we estimated the difference in detection accuracy of the two methods.
(C) The simulation using the slope analysis in panel (B) was repeated for varying amounts of target viral RNA, and the time where detection accuracy reached 95% was determined.
Discussion
Here, we show that direct detection of SARS-CoV-2 RNA with CRISPR-Cas13a and a mobile phone offers a promising option for rapid, point-of-care testing. A key advance in this work is demonstrating that combinations of crRNAs can increase sensitivity by activating more Cas13a per target RNA. We show that combinations of two or three crRNAs can be used to detect viral target RNA in the attomolar range, detecting as few as ∼30 copies/μL. The use of multiple crRNAs that target different parts of the genome can also safeguard against a potential loss of detection due to naturally occurring viral mutations.
A second key advance is the ability to directly translate the fluorescent signal into viral loads. Other CRISPR Dx assays, such as CRISPR-COVID, achieve high sensitivity via isothermal amplification but provide only qualitative information rather than viral copy numbers. Fluorescent signal from 7,500 copies/μL to 7.5 copies/μL are remarkably similar, despite three orders of magnitude difference in copy number (Hou et al., 2020). By avoiding amplification and employing direct detection, we show that the reaction rate directly correlates with viral copy number and may be used for quantification. When coupled with frequent testing, quantitative data are potentially beneficial: the course of a patient’s infection can be monitored and can determine if the infection is increasing or waning. In symptomatic cases, viral loads follow the course of the infection (Wölfel et al., 2020). Notably, samples with viral loads below 106 copies/mL or 1,000 copies/μL did not yield infectious viral isolates in one study in Germany (Wölfel et al., 2020). SARS-CoV-2 transmission from asymptomatic carriers has also been documented (Bai et al., 2020), and they have viral RNA loads similar to those of symptomatic patients (Chamie et al., 2020; Lee et al., 2020). Monitoring viral loads quantitatively would allow estimation of infection stage and help predict infectivity, recovery, and return from quarantine in real time.
A third key advance in our work is the demonstration that a compact microscope based on a mobile phone and low-cost optics can accurately read the Cas13a direct detection assay, enabling ∼100 copies/μL sensitivity in 30 min of measurement and accurate diagnosis of a set of pre-extracted RNA from patient samples in 5 min of measurement time on the device. This suggests that a portable diagnostic device based on consumer electronics, rather than specialized laboratory technology, can be built to work with the Cas13a assay. Mobile phones were an attractive choice for evaluating the requirements of reading the Cas13a assay because of their high-quality sensors, intuitive user interface, and powerful computational capabilities, as well as communication capabilities. For similar reasons, previous diagnostic efforts have utilized mobile phones to detect fluorescent signals from LAMP (Chen et al., 2017; Ganguli et al., 2017; Kong et al., 2017; Priye et al., 2017; Sun et al., 2020), PCR (Angus et al., 2015; Gou et al., 2018; Jiang et al., 2014), next-generation DNA sequencing (Kühnemund et al., 2017), and recombinase polymerase amplification (Chan et al., 2018). Combined with efficient contact tracing and HIPAA-compliant upload into cloud-based systems, a mobile SARS-CoV-2 diagnostic could play an important role in the current and future pandemics.
The Cas13a direct detection assay reported here could fulfill the need for a test that provides rapid results and can be administered frequently (Larremore et al., 2020). Other tests in this category include Abbott Lab’s ID NOW and Roche’s cobas Liat System, both portable PCR-based tests, and several antigen tests, such as Quidel’s Sofia 2 SARS Antigen FIA and Abbot Lab’s BinaxNOW Antigen Test. In the case of influenza, antigen tests span a wide range of sensitivities (e.g., 51%–67.5%) (Babin et al., 2011; Chartrand et al., 2012; Chu et al., 2012). Due to the low-to-moderate sensitivity of these tests, the CDC still recommends re-testing samples that are negative with a more sensitive test, such as RT-qPCR (Green and StGeorge, 2018). Notably, none of the current rapid testing options provide precise quantitative results, which could help assess viral dynamics and evaluate an individual’s level of infection and disease progression.
While we demonstrate rapid detection with reasonable sensitivity using crRNAs based on existing PCR primers, we anticipate further improvement by systematically searching for the best crRNA combinations across the entire viral RNA genome. As more information becomes available about viral variants (Osório and Correia-Neves, 2020; Vanaerschot et al., 2020), crRNA design can be adapted to avoid false negatives. However, while combining crRNAs improves sensitivity, it also offers more opportunities for unintentional off-target detection, and lower viral loads could be registered when one crRNA in the combination does not precisely match the viral sequence in the sample. Further improvements are also anticipated in the reporter, the choice of Cas13 proteins, and in device and camera sensitivity. These advancements can improve the rate of the reaction, allowing for improvements in detection accuracy and limit of detection in shorter periods of time.
A recent national survey of over 19,000 respondents showed that the average wait time for nasal swab-based qPCR test results was 4.1 days, with 31% of tests taking more than 4 days and 10% of tests taking 10 days or more (Lazer et al., 2020). The national backlog in processing these laboratory-based tests clearly illustrates the need for rapid, point-of-care tests that can reliably detect SARS-CoV-2 RNA. As the long-term immunity induced by natural infection or vaccination may decay in as little as 2–4 months (Ibarrondo et al., 2020; Long et al., 2020), the need for rapid and frequent testing for SARS-CoV-2 will likely remain. In the future, direct detection by Cas13a as outlined here could be quickly modified to target the next respiratory pathogen that emerges, hopefully in time to help curb global spread.
Limitations of study
This study demonstrates a proof-of-concept for sensitive and rapid SARS-CoV-2 RNA detection by a mobile phone-based device. Additional work will be necessary to fully translate this work to a widely available point-of-care device. First, while the current study used a lab-based RNA extraction step, an extraction-free protocol for sample preparation (Arizti-Sanz et al., 2020; Joung et al., 2020a; Myhrvold et al., 2018) will be necessary to minimize the number of steps in the assay and the overall sample-to-answer turnaround time. Second, while we chose to integrate a mobile phone into our detection device as a way of rapidly demonstrating what mass-produced electronics could achieve, other configurations of an image-based diagnostic, including use of an embedded sensor that wirelessly connects to a mobile phone, could be built at scale with at least comparable performance. Finally, while we demonstrate a limit of detection of ∼100 copies/μL in 30 min of measurement on the device, we expect improvements to the sensitivity and turnaround time with optimization of crRNA combinations, Cas13 protein engineering, and additional device advancements. Furthermore, the sensitivity of the assay can be adjusted by changing the measurement time. This ability to tune sensitivity could make Cas13a direct detection useful for screening applications as well as more sensitive diagnostic applications.
STAR★methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| SARS-CoV-2, Isolate USA-WA1/2020 | BEI Resources | Cat#NR-52281 |
| Human Coronavirus, NL63 | BEI Resources | Cat#NR-470 |
| Betacoronavirus 1 (Human Coronavirus OC43) | ATCC | Cat#VR-1558 |
| H1N1 Influenza Virus A | Virapur | Strain: California/04/2009 |
| Influenza Virus B | Virapur | Strain: Brisbane/60/2008 |
| Biological Samples | ||
| Human Nasopharyngeal Swab RNA (Negative and Positive for SARS-CoV-2) | Chan Zuckerberg Biohub | https://www.czbiohub.org |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RNA STAT-60 | AMSBIO | Cat#CS-110 |
| Q5 High-Fidelity DNA Polymerase | NEB | Cat#M0491 |
| AMV Reverse Transcriptase | Promega | Cat#M5101 |
| Oligo(dt)18 | Thermo Scientific | Cat#S0131 |
| Random hexamers | Thermo Scientific | Cat#S0142 |
| Murine RNase Inhibitor | NEB | Cat#M0314 |
| HEPES solution | Millipore Sigma | Cat#H3537 |
| Potassium chloride solution | Millipore Sigma | Cat#60142 |
| Magnesium chloride solution | Millipore Sigma | Cat#63069 |
| Glycerol | Invitrogen | Cat#15514011 |
| Tween 20 | Millipore Sigma | Cat#P9416 |
| Critical Commercial Assays | ||
| RNeasy Plus Mini Kit | QIAGEN | Cat#741434 |
| Direct-Zol RNA MiniPrep Kit | Zymo Research | Cat#R2052 |
| HiScribe T7 Quick High Yield RNA Synthesis Kit | NEB | Cat#E2050S |
| PrimeTime Gene Expression Master Mix | IDT | Cat#1055771 |
| Experimental Models: Cell Lines | ||
| Huh7.5.1-ACE2 | Wang et al., 2020b | Senior Author: Andreas Puschnik |
| Vero CCL-81 | ATCC | Cat#CCL-81 |
| Vero E6 | ATCC | Cat#CRL-1586 |
| Oligonucleotides | ||
| ssRNA oligos | Synthego | See Table S1 |
| DNA oligos | Various | See Table S1 |
| RNase Alert Substrate | IDT | Cat#11-04-03-03 |
| Recombinant DNA | ||
| p2CT-His-MBP-Lbu_C2c2_WT | East-Seletsky et al., 2016 | Addgene #83482 |
| MERS-CoV Control Plasmid | IDT | Cat#10006624 |
| 2019-nCoV_N_Positive Control Plasmid | IDT | Cat#10006625 |
| Software and Algorithms | ||
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| MATLAB R2020a | MathWorks | https://www.mathworks.com/products/matlab.html?s_tid=hp_products_matlab |
| R (Version 3.6.0) | R | https://www.r-project.org |
| VennDiagram Package (Version 1.6.20) | R | https://cran.r-project.org/web/packages/VennDiagram/index.html |
| Custom MATLAB code | This paper | https://doi.org/10.17632/r3vwyr2w5x.1 |
| Custom Android application | This paper | N/A |
| Other | ||
| 384 Well Plates | Corning | Cat#3820 |
| Absolute qPCR Plate Seals | Thermo Fisher | Cat#AB1170 |
| Infinite 200 Pro M Plex | Tecan | Cat#INF-MPLEX |
| TRH127-020-A-ML - Ø1/2” Achromatic Triplet lens | Thorlabs | Part #: TRH127-020-A-ML |
| Emission filter | Chroma | Cat#AT535/40 m |
| Unmounted Reflective Ø25 mm ND Filter, Optical Density: 4.0 | Thorlabs | Part #: ND40B |
| Adafruit Feather M0 Bluefruit LE | Adafruit | Product ID: 2995 |
| Powerboost 1000 Charger – Rechargeable 5V Lipo USB boost | Adafruit | Product ID: 2465 |
| Lithium Ion Battery Pack – 3.7V 6600mAh | Adafruit | Product ID: 353 |
| Sharp 488nm GH04850B2G 55mW laser diode in 12mm copper module with driver and DTR-G-8 glass lens | DTR’s Laser Shop | https://sites.google.com/site/dtrslasershop/home/diodes/sharp-gh04850b2g-55mw |
| Google Pixel 4 XL | https://store.google.com/us/product/pixel_4 | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Melanie Ott (melanie.ott@gladstone.ucsf.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The custom MATLAB code for image processing and data analysis has been deposited to Mendeley Data: https://doi.org/10.17632/r3vwyr2w5x.1.
Experimental model and subject details
Mammalian cell lines and culture conditions
Human hepatocellular carcinoma cells (Huh7.5.1, gift from Frank Chisari) and monkey kidney epithelial cells (Vero, ATCC CCL-81, and Vero E6, ATCC CRL-1586) were cultured in DMEM (Corning) supplemented with 10% fetal bovine serum (FBS, Sigma), penicillin/streptomycin (Corning), and L-glutamine (Corning) at 37°C and 5% CO2. Cell lines were tested negative for mycoplasma contamination.
Generation of Huh7.5.1-ACE2 cell line
Huh7.5.1-ACE2 cells were generated as described previously in (Wang, et al., 2020b). Briefly, hACE2 (Addgene, #1786, gift from Hyeryun Choe) was amplified and cloned into EcoRV-cut plenti-CMV-Hygro-DEST (Addgene, #17454, gift from Eric Campeau & Paul Kaufman) using NEBuilder HiFi DNA Assembly Master Mix (NEB). Lentivirus was produced in HEK293FT by co-transfection of plenti-hACE2-Hygro together with pCMV-dR8.2 dvpr (Addgene, #8455, gift from Bob Weinberg), pCMV-VSV-G (Addgene, #8454, gift from Bob Weinberg) and pAdVAntage (Promega) using FugeneHD (Promega). Supernatant was collected 48 h post-transfection, filtered and added to Huh7.5.1 cells. Transduced cells were subsequently selected using Hygromycin for 7 days.
Viral strains
SARS-CoV-2 virus culture
Isolate USA-WA1/2020 of SARS-CoV-2 was used for genomic SARS-CoV-2 RNA. All live virus experiments were performed in a Biosafety Level 3 laboratory. SARS-CoV-2 stocks were propagated in Vero CCL-81 cells. Viral supernatant was collected by centrifugation and stored at −80°C.
HCoV-NL63 virus culture
Isolate Amsterdam I of HCoV-NL63 (NR-470, BEI Resources) was propagated in Huh7.5.1-ACE2 cells. Supernatant was harvested 5 days post infection, filtered and stored at −80°C.
HCoV-OC43 virus culture
HCoV-OC43 (VR-1558, ATCC) was propagated in Vero E6 cells. Supernatant was harvested 6 days post infection, filtered and stored at −80°C.
Influenza virus
H1N1 Influenza virus A (California/04/2009) and Influenza virus B (Brisbane/60/2008) in chicken allantoic fluid was purchased from Virapur and used directly for RNA extraction (see below).
Human airway organoid culture
Human airway organoids are generated using upper bronchia/trachea cells from lung resection tissue. They are cultured using the protocol previously described in (Sachs et al., 2019). Airway organoids are seeded in drops of diluted basement membrane matrix (Cultrex, diluted 3:4 with basal media) in flat-bottom, low-attachment plates. Airway Organoid (AO) media is added after the drops have solidified. Organoids are cultured at 37°C, 5% CO2, and passaged every two weeks. During passaging, the airway organoids drops are collected with cold basal media, washed and dissociated manually and chemically with TryPLE 10X (GIBCO) and Trypsin-EDTA (Corning), then re-seeded into new plates. Basal media and AO media compositions are taken from (Zhou et al., 2018).
Patient samples
De-identified RNA samples from nasopharyngeal swabs from patients testing positive and negative for SARS-CoV-2 were obtained from the Chan Zuckerberg Biohub. The institutional review board at University of California, San Francisco, approved this study under IRB #17-24056. Positive samples were quantified previously using CDC N1 and N2 SARS-CoV-2 primers.
Method details
Cas13a protein expression and purification
Expression vectors deposited with Addgene (Plasmid #83482) were used for expression of LbuCas13a. The codon-optimized Cas13a genomic sequences are N-terminally tagged with a His6-MBP-TEV cleavage site sequence, with expression driven by a T7 promoter. Purification of was based off of a previously published protocol with some modifications (East-Seletsky et al., 2017; East-Seletsky et al., 2016). Briefly, expression vectors were transformed into Rosetta2 DE3 or BL21 E. coli cells grown in Terrific broth at 37°C, induced at mid-log phase (OD600 ∼0.6) with 0.5 mM IPTG, and then transferred to 16°C for overnight expression. Cell pellets were resuspended in lysis buffer (50 mM Tris-Cl pH 7.0, 500 mM NaCl, 5% glycerol, 1 mM TCEP, 0.5 mM PMSF, and EDTA-free protease inhibitor (Roche)), lysed by sonication, and clarified by centrifugation at 35,000xg. Soluble His6-MBP-TEV-Cas13a was isolated over metal ion affinity chromatography, and in order to cleave off the His6- MBP tag, the protein-containing eluate was incubated with TEV protease at 4°C overnight while dialyzing into ion exchange buffer (50 mM Tris-Cl pH 7.0, 250 mM KCl, 5% glycerol, 1 mM TCEP). Cleaved protein was loaded onto a HiTrap SP column (GE Healthcare) and eluted over a linear KCl (0.25-1.0M) gradient. Cas13a-containing fractions were pooled, concentrated, and further purified via size-exclusion chromatography on a S200 column (GE Healthcare) in gel filtration buffer (20 mM HEPES-K pH 7.0, 200 mM KCl, 10% glycerol, 1 mM TCEP) and were subsequently flash frozen for storage at −80°C.
In vitro RNA transcription
SARS-CoV-2 N gene was transcribed off a single-stranded DNA oligonucleotide template (IDT). HCoV-MERS N gene was transcribed off of a MERS-CoV Control plasmid (IDT, Cat# 10006624) by first adding a T7 promoter via PCR using Q5 High-Fidelity DNA Polymerase (NEB) (see Table S1 for primers). A single PCR product was confirmed via gel electrophoresis. In vitro transcription was performed using HiScribe T7 Quick High Yield RNA Synthesis Kit (NEB) following manufacturer’s recommendations. Template DNA was removed by addition of DNase I (NEB), and IVT RNA was subsequently purified using RNA STAT-60 (AMSBIO) and the Direct-Zol RNA MiniPrep Kit (Zymo Research). RNA concentration was quantified by Nanodrop and copy number was calculated using full transcript length and concentration.
RNA extraction
RNA was extracted from SARS-CoV-2, HCoV-NL63, HCoV-OC43, Influenza A, and Influenza B viral supernatant via RNA STAT-60 (AMSBIO) and the Direct-Zol RNA MiniPrep Kit (Zymo Research). RNA was extracted from human airway organoid cells using the RNeasy Mini Kit (QIAGEN).
Quantitative polymerase chain reaction
RNA from SARS-CoV-2 viral supernatant was quantified via qPCR. Briefly, RNA was reverse transcribed to cDNA via AMV Reverse Transcriptase (Promega) using oligo(dt)18 and random hexamers (Thermo Scientific). cDNA was added to the qPCR reaction using PrimeTime Gene Expression Master Mix (IDT). N and E gene standards were used to generate a standard curve for copy number quantification. N gene standard was generated by PCR from the 2019-nCoV_N_Positive Control Plasmid (IDT, Cat# 10006625). E gene standard was generated by PCR using extracted genomic SARS-CoV-2 RNA as template. A single product was confirmed by gel electrophoresis and DNA was quantified by Nanodrop. cDNA was analyzed using the 7500 Fast Real-Time PCR system (Applied Biosystems). See Table S1 for primers.
crRNA design parameters
20 nucleotide crRNA spacer sequences targeting the N gene were chosen using previously published qPCR sequences from the Centers for Disease Control and Prevention (CDC) and based on (Zhu et al., 2020). For spacer sequences targeting the E gene, we based our spacers on a previously published qPCR primer set as well as a published Cas12 guide (Broughton et al., 2020; Corman et al., 2020). We confirmed the specificity of each crRNA to SARS-CoV-2 using NCBI BLAST, setting a threshold of 16/20 sequence identity to human transcripts to reduce the odds of off-target detection from human tissue. We used the following 30 nucleotide crRNA stem sequence: 5'-GACCACCCCAAAAAUGAAGGGGACUAAAAC-3'. See Table S1 for full sequences.
4118 complete SARS-CoV-2 genome sequences deposited in NCBI RefSeq under the taxonomy ID (2697049) were downloaded on 06/05/2020. We searched each guide against the downloaded SC2 genomes with no mismatches (using grep command) and the Venn diagram (Figure S2D) was generated using R package VennDiagram (version 1.6.20).
Fluorescent Cas13a nuclease assays
LbuCas13a-crRNA RNP complexes were individually preassembled by incubating 1.33 mM of LbuCas13a with 1.33 mM of crRNA for 15 min at room temperature. In Figures 1C and 2B, 677 nM of crRNA was used. These complexes were then diluted to 100 nM LbuCas13a and 100 nM (or 50 nM for Figures 1C and 2B) crRNA in cleavage buffer (20 mM HEPES-Na pH 6.8, 50 mM KCl, 5 mM MgCl2, and 5% glycerol) in the presence of 400 nM of reporter RNA (5'-FAM-rUrUrUrUrU-IowaBlack FQ-3'), 1 U/mL Murine RNase Inhibitor (NEB, Cat# M0314), and varying amounts of target RNA. In Figures 1C and 2B, the final RNP complex concentration was 50 nM. In all other figures, the final RNP complex concentration was 100 nM. In Figures 1C and 2B, 167 nM of RNase Alert substrate (IDT) was used as the reporter RNA, and in Figure 2D, 400 nM of RNase Alert substrate was used. In Figures 3D and 3E, the complexes were also diluted in 0.1% Tween-20 (Sigma) and the cleavage buffer was pH 7.1. These reactions were loaded into a 384-well plate (Corning, Cat# 3820) incubated in a fluorescence plate reader (TECAN, Infinite 200 Pro M Plex) for up to 120 min at 37°C with fluorescence measurements taken every 5 min (or every 2.5 min in Figure 3E) (λex:485 nM; λem:535 nM; Gain: 130). Background-corrected fluorescence values were obtained by subtracting fluorescence values obtained from reactions carried out containing only reporter and buffer. For assays containing more than one crRNA simultaneously, the LbuCas13a-crRNA RNP complexes were separately assembled by incubating for 15 min at room temperature, then combined in the reaction at half (in 2 RNP combinations) or one-third (in 3 RNP combinations) the volume to keep the total or combined concentration of RNP constant. Representative graphs of experiments are shown. Most experiments were replicated at least twice, with the exception of Figure 3E (only Patient Swabs 1-3 and 5 were repeated twice) due to limited sample material. For Patient Swab 5, a slightly smaller quantity of swab material was used compared to other swabs (0.26 mL rather than 0.3 mL) to achieve a lower viral concentration in the Cas13a reaction. This allowed us to demonstrate a lower copy number that the Cas13a reaction could detect in patient samples.
Mobile phone fluorescent microscope
We built a mobile phone fluorescent microscope using a 488 nm diode laser (GH04850B2G, Sharp Microelectronics), a green fluorescence interference filter (Chroma Technology AT 535/40), and a Pixel 4 XL phone camera (12.2 mega-pixel, pixel size 1.4mm, aperture f/1.7, Google). The laser beam was expanded using a glass collimation lens (10° divergence half-angle), directed toward the sample plane using two ND4 filters used as mirrors (ND40B, Thorlabs), and reduced by an elliptical aperture to fill the circular image field-of-view with a uniform field intensity. The sample was illuminated with an oblique epi configuration and the illumination power was 18 mW at the sample plane; illuminated area at the sample plane was 15 × 15 mm2. The imaging optics consist of an f = 20mm compact triplet lens (TRH127-020-A, Thorlabs) followed by the interference filter for selection of the fluorescence reporter emission wavelength and the Pixel 4 XL camera lens. Total magnification from object to image plane is ∼1/4.5 and the numerical aperture is 0.06. All optical and illumination components were enclosed in a custom-made dark box, into which a sample chip is loaded for imaging. Automated time-lapse imaging was implemented by a custom Android application and a Bluetooth receiver (Bluefruit Feather M0, Adafruit), which triggered the laser at the time of image acquisition. The Cas13a reaction was performed by placing the device in a 37°C incubator for temperature control and the reaction curve was obtained by analyzing the image time series offline using a custom MATLAB (Mathworks) code. When measuring the Cas13 reporter background, the signal-to-noise ratio of our system that is limited by the noise of diode laser is ∼1000 (Figure S3D).
Sample chip fabrication
Sample chips containing three fluid channels were made by casting polydimethylsiloxane (PDMS, Ellsworth adhesives) on an acrylic mold. The acrylic mold was assembled by adhering three laser cut acrylic lanes on a flat acrylic base. The width, height, and length of each acrylic lane were 1.6 mm, 2 mm, and 12.5 mm, respectively, resulting in a fluid channel volume of 40 μL (Figures S3A–S3C). Inlet and outlet ports were created on both ends of the channels after curing and demolding the PDMS using a biopsy punch. The PDMS chips were subsequently adhered to a siliconized cover glass (Hampton research) to close the fluid channels. To avoid generation of bubbles in the chip during the measurement, both the Cas13a reaction mix and the sample chip were degassed in a house vacuum for 15 min before loading the samples and starting the measurement.
Mobile phone image acquisition and analysis
During typical device operation, a ∼1 s exposure RGB image was acquired every 30 s for a period of 1 h and the images were analyzed offline using custom MATLAB code. First, the RGB image of 2016 × 2512 pixels was demosaiced to a greyscale image. Second, the saturated pixels or pixels exhibiting two very different green submosaic values were excluded. Third, a rectangular image region-of-interest (ROI) of 400 × 90 pixels was manually drawn within an area of each fluid channel and the reporter signal in each ROI was determined by averaging the pixel values (Figure S3C). The ROI values are accumulated in time and analyzed for slope determination (see Quantification and Statistical Analysis).
Comparison of data with enzyme kinetics
We analyzed the Cas13a reaction with a single crRNA (Figures S1B–S1D) using the Michaelis-Menten enzyme kinetics model with the quasi-steady-state approximation to estimate , the ratio of active Cas13 RNP to target RNA, from the measured reaction rate per target RNA, (below). We first converted the plate reader signal to the molar concentration of cleaved reporter and determined the Cas13a reaction rate for varying concentrations of target RNA . We then determined the reaction rate per target RNA, , by fitting a linear curve to the data. We used the total reporter concentration [S] = 400 nM, Kcat = 600/s, KM from 1 μM to 3 μM (Slaymaker et al., 2019) to calculate from .
We estimated that, for both crRNA 2 and crRNA 4, the amount of active Cas13a RNP can be as small as 31 – 37% of total target RNA (for KM = 1 μM), or as large as 75 – 89% (for KM = 3 μM).
Simulation of mobile device data
We used MATLAB to simulate the mobile device data. We simulated the data by including the random noise with standard deviation of 0.002 to conservatively emulate the measured noise ∼0.0007 in our system (Figure S3D). We assumed that for 50 copies/μL of target viral RNA the signal increases at a rate of 0.0002 fold/min at 37°C, based on Figure 4C showing the difference of ∼0.002 fold/min between reaction rates at 500 copies/μL and RNP alone, and that reaction rate is constant over time as we validated with experimental data (Figures S1B–S1D). Although we observe slightly positive slopes in RNP alone control reactions, we simulated the control reaction as a flat line with a standard deviation of 0.002 after subtracting its slope from the positive reaction. In each simulation, we tested whether the positive sample curve was significantly different from the RNP alone curve based on the comparison of either the endpoint signal or the signal slope. In both cases, we considered the signal positive if it did not overlap with the control within the 95% confidence interval. For each measurement time, we repeated the simulation 1,000 times and determined the detection accuracy by counting the positive tests.
Quantification and statistical analysis
The number of experiments and replicates are indicated in the individual Figure Legends. The slope of the Cas13a reaction is calculated by simple linear regression of raw data from time 0 for the set duration. For all the plate reader samples and the patient samples measured in a mobile device, the measurement began immediately after sample loading. For the genomic SARS-COV-2 RNA samples measured in a mobile device, the sample was equilibrated for 10 min in the device before measurement began. The slope error bar indicates the 95% confidence intervals (CI) of linear regression performed either to individual data or data merged from the group of replicates. To access significance between groups of positive Cas13a reaction versus the RNP only control, we employed a two-way ANCOVA to the slope during linear regression. When the slope of positive reaction is smaller than that of the RNP only control, it is considered non-significant. To evaluate the accuracy of our detection of positive samples, we tested whether the slope of an individual positive sample exceeded that of an RNP only control by greater than the 95% CI. The detection accuracy is quantified as the ratio of samples tested positive out of the total number of tests. The CI can be adjusted to enhance either the sensitivity or specificity of the test. For example, tightening to 99% CI will incur more potential false negatives, or loosening to 90% will risk more false positives. Data in Figures 1, 2, and 3 were processed and visualized using GraphPad Prism 8. Data in Figure 4 were processed using MATLAB.
Acknowledgments
We thank all members of the Ott, Fletcher, and Doudna laboratories for sharing reagents, expertise, and feedback in the preparation of this manuscript. We thank Stephen Floor and Nadia Roan for helpful discussions. We also thank Michael Frumkin, Michael Brenner, Ellen Klein, Alex Schiffhauer, Daniel Harbuck, Francois Bleibel, Paul Rohde, Jon Barron, and Sam Hasinoff for helpful discussions. We thank Lauren Weiser and Veronica Fonseca for administrative support, Gary Howard for editorial support, and John Carroll for graphical support. We are grateful to Kevin Mullane, Stephen B. Freedman, and Robert Wicks at the Gladstone Institutes, and JF Van Kerckhove, Savi Baveja, and Daniel LeClerc at Bain & Company for their guidance and advice. We thank Dr. Paula Ladd, Dr. Cristina Tato, Dr. Larry Brilliant, and Dr. Harvey Fineberg for helpful discussions and support. We thank the Gladstone Assay Development and Drug Discovery Core, particularly Michael Jobling, for assistance with high-throughput assays. We gratefully acknowledge support from the NIH: NIH/NIAID grant 5R61AI140465-03 to J.A.D., D.A.F., and M.O. and NIH/NIDA grant 1R61DA048444-01 to M.O. This project is supported by the NIH Rapid Acceleration of Diagnostics (RADx) program and has been funded in whole or in part with Federal funds from National Heart, Lung and Blood Institute, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, and Department of Health and Human Services under grant no. 3U54HL143541-02S1. P.F. was supported by the UCSF Medical Scientist Training Program (NIH/NIGMS T32GM007618) and the NIH/NIAID (F30AI143401). M.D.d.L.D. was supported by the UC MEXUS-CONACYT Doctoral Fellowship. G.J.K. is supported by an NHMRC Investigator Grant (EL1, APP1175568) and previously an American Australian Association Fellowship. J.A.D. is an HHMI Investigator. We are grateful for philanthropic support from Fast Grants, the James B. Pendleton Charitable Trust, the Roddenberry Foundation, and multiple individual donors. This work was made possible by a generous gift from an anonymous private donor in support of the ANCeR diagnostics consortium. Purified LbuCas13a was a kind gift from Shanghai ChemPartner. We thank Synthego for support with synthetic crRNAs. The following reagents were deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: Genomic RNA from SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52285; SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281; and Human Coronavirus, NL63, NR-470.
Author contributions
P.F., S.S., M.D.d.L.D, D.A.F., and M.O. conceived and designed the study. P.F., S.S., M.D.d.L.D., C.N.G., S.I.S., D.B., C.-L.T., P.-Y.C., and J.S. performed experiments and data analysis. C.Z. and K.S.P. performed bioinformatics analysis. S.S., M.D.d.L.D., M.V.D., N.A.S., A.B, M.A., and A.R.H. developed the mobile phone-based device and reaction chambers. G.R.K. and J.M.O supervised and provided feedback. A.M.-F. provided assistance with high-throughput assays. B.J., K.W., and A.S. assisted with BSL-3 work. A.S.P provided HCoV-NL63 and HCoV-OC43 viral supernatants. C.L., E.D.C., M.P., A.K., and J.L.D. provided patient samples and reagents. G.J.K. and J.A.D. provided reagents and critical expertise and feedback. D.A.F. and M.O. supervised the study design and data collection. J.A.D., D.A.F., and M.O. secured funding. P.F., S.S., M.D.d.L.D., D.A.F., and M.O. wrote the manuscript with input from G.J.K. and J.A.D.
Declaration of interests
P.F., S.S., G.J.K., J.A.D., D.A.F., and M.O. have filed patent applications related to this work. The Regents of the University of California have patents issued and pending for CRISPR technologies on which J.A.D. is an inventor. J.A.D. is a cofounder of Caribou Biosciences, Editas Medicine, Scribe Therapeutics, Intellia Therapeutics, and Mammoth Biosciences. J.A.D. is a scientific advisory board member of Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Mammoth Biosciences, Synthego, Algen Biotechnologies, Felix Biosciences, and Inari. J.A.D. is a director at Johnson & Johnson and has research projects sponsored by Biogen, Pfizer, AppleTree Partners, and Roche.
Published: December 4, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.12.001.
Supplemental information
References
- Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B., Shmakov S., Makarova K.S., Semenova E., Minakhin L., et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh O.O., Gootenberg J.S., Essletzbichler P., Han S., Joung J., Belanto J.J., Verdine V., Cox D.B.T., Kellner M.J., Regev A., et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus S.V., Cho S., Harshman D.K., Song J.Y., Yoon J.Y. A portable, shock-proof, surface-heated droplet PCR system for Escherichia coli detection. Biosens. Bioelectron. 2015;74:360–368. doi: 10.1016/j.bios.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arizti-Sanz J., Freije C.A., Stanton A.C., Boehm C.K., Petros B.A., Siddiqui S., Shaw B.M., Adams G., Kosoko-Thoroddsen T.F., Kemball M.E., et al. Integrated sample inactivation, amplification, and Cas13-based detection of SARS-CoV-2. bioRxiv. 2020 doi: 10.1038/s41467-020-19097-x. 2020.05.28.119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babin S.M., Hsieh Y.H., Rothman R.E., Gaydos C.A. A meta-analysis of point-of-care laboratory tests in the diagnosis of novel 2009 swine-lineage pandemic influenza A (H1N1) Diagn. Microbiol. Infect. Dis. 2011;69:410–418. doi: 10.1016/j.diagmicrobio.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Yao L., Wei T., Tian F., Jin D.Y., Chen L., Wang M. Presumed Asymptomatic Carrier Transmission of COVID-19. JAMA. 2020;323:1406–1407. doi: 10.1001/jama.2020.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslauer D.N., Maamari R.N., Switz N.A., Lam W.A., Fletcher D.A. Mobile phone based clinical microscopy for global health applications. PLoS ONE. 2009;4:e6320. doi: 10.1371/journal.pone.0006320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton J.P., Deng X., Yu G., Fasching C.L., Servellita V., Singh J., Miao X., Streithorst J.A., Granados A., Sotomayor-Gonzalez A., et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020;38:870–874. doi: 10.1038/s41587-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamie G., Marquez C., Crawford E., Peng J., Petersen M., Schwab D., Schwab J., Martinez J., Es D.J., Black D., et al. CLIAhub Consortium SARS-CoV-2 Community Transmission disproportionately affects Latinx population during Shelter-in-Place in San Francisco. Clin. Infect. Dis. 2020:ciaa1234. doi: 10.1093/cid/ciaa1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K., Wong P.Y., Parikh C., Wong S. Moving toward rapid and low-cost point-of-care molecular diagnostics with a repurposed 3D printer and RPA. Anal. Biochem. 2018;545:4–12. doi: 10.1016/j.ab.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartrand C., Leeflang M.M., Minion J., Brewer T., Pai M. Accuracy of rapid influenza diagnostic tests: a meta-analysis. Ann. Intern. Med. 2012;156:500–511. doi: 10.7326/0003-4819-156-7-201204030-00403. [DOI] [PubMed] [Google Scholar]
- Chen W., Yu H., Sun F., Ornob A., Brisbin R., Ganguli A., Vemuri V., Strzebonski P., Cui G., Allen K.J., et al. Mobile Platform for Multiplexed Detection and Differentiation of Disease-Specific Nucleic Acid Sequences, Using Microfluidic Loop-Mediated Isothermal Amplification and Smartphone Detection. Anal. Chem. 2017;89:11219–11226. doi: 10.1021/acs.analchem.7b02478. [DOI] [PubMed] [Google Scholar]
- Chen J.S., Ma E., Harrington L.B., Da Costa M., Tian X., Palefsky J.M., Doudna J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360:436–439. doi: 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H., Lofgren E.T., Halloran M.E., Kuan P.F., Hudgens M., Cole S.R. Performance of rapid influenza H1N1 diagnostic tests: a meta-analysis. Influenza Other Respir. Viruses. 2012;6:80–86. doi: 10.1111/j.1750-2659.2011.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman V.M., Landt O., Kaiser M., Molenkamp R., Meijer A., Chu D.K., Bleicker T., Brünink S., Schneider J., Schmidt M.L., et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25 doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio M.V., Bakalar M., Bennuru S., Reber C., Skandarajah A., Nilsson L., Switz N., Kamgno J., Pion S., Boussinesq M., et al. Point-of-care quantification of blood-borne filarial parasites with a mobile phone microscope. Sci. Transl. Med. 2015;7:286re4. doi: 10.1126/scitranslmed.aaa3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A., O’Connell M.R., Knight S.C., Burstein D., Cate J.H., Tjian R., Doudna J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538:270–273. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A., O’Connell M.R., Burstein D., Knott G.J., Doudna J.A. RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Mol Cell. 2017;66:373–383.e3. doi: 10.1016/j.molcel.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung T.S., Liu D.X. Human Coronavirus: Host-Pathogen Interaction. Annual Review of Microbiology. 2019;73:529–557. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- Ganguli A., Ornob A., Yu H., Damhorst G.L., Chen W., Sun F., Bhuiya A., Cunningham B.T., Bashir R. Hands-free smartphone-based diagnostics for simultaneous detection of Zika, Chikungunya, and Dengue at point-of-care. Biomed. Microdevices. 2017;19:73. doi: 10.1007/s10544-017-0209-9. [DOI] [PubMed] [Google Scholar]
- Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A., et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–442. doi: 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360:439–444. doi: 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou T., Hu J., Wu W., Ding X., Zhou S., Fang W., Mu Y. Smartphone-based mobile digital PCR device for DNA quantitative analysis with high accuracy. Biosens. Bioelectron. 2018;120:144–152. doi: 10.1016/j.bios.2018.08.030. [DOI] [PubMed] [Google Scholar]
- Green D.A., StGeorge K. Rapid Antigen Tests for Influenza: Rationale and Significance of the FDA Reclassification. J. Clin. Microbiol. 2018;56:e00711-18. doi: 10.1128/JCM.00711-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou T., Zeng W., Yang M., Chen W., Ren L., Ai J., Wu J., Liao Y., Gou X., Li Y., et al. Development and evaluation of a rapid CRISPR-based diagnostic for COVID-19. PLoS Pathog. 2020;16:e1008705. doi: 10.1371/journal.ppat.1008705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarrondo F.J., Fulcher J.A., Goodman-Meza D., Elliott J., Hofmann C., Hausner M.A., Ferbas K.G., Tobin N.H., Aldrovandi G.M., Yang O.O. Rapid Decay of Anti-SARS-CoV-2 Antibodies in Persons with Mild Covid-19. N. Engl. J. Med. 2020;383:1085–1087. doi: 10.1056/NEJMc2025179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L., Mancuso M., Lu Z., Akar G., Cesarman E., Erickson D. Solar thermal polymerase chain reaction for smartphone-assisted molecular diagnostics. Sci. Rep. 2014;4:4137. doi: 10.1038/srep04137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J., Ladha A., Saito M., Kim N.G., Woolley A.E., Segel M., Barretto R.P.J., Ranu A., Macrae R.K., Faure G., et al. Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. N. Engl. J. Med. 2020;383:1492–1494. doi: 10.1056/NEJMc2026172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J., Ladha A., Saito M., Segel M., Bruneau R., Huang M.W., Kim N.G., Yu X., Li J., Walker B.D., et al. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. medRxiv. 2020 [Google Scholar]
- Kamgno J., Pion S.D., Chesnais C.B., Bakalar M.H., D’Ambrosio M.V., Mackenzie C.D., Nana-Djeunga H.C., Gounoue-Kamkumo R., Njitchouang G.R., Nwane P., et al. A Test-and-Not-Treat Strategy for Onchocerciasis in Loa loa-Endemic Areas. N. Engl. J. Med. 2017;377:2044–2052. doi: 10.1056/NEJMoa1705026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S., Lotfy P., Brideau N.J., Oki J., Shokhirev M.N., Hsu P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell. 2018;173:665–676.e14. doi: 10.1016/j.cell.2018.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J.E., Wei Q., Tseng D., Zhang J., Pan E., Lewinski M., Garner O.B., Ozcan A., Di Carlo D. Highly Stable and Sensitive Nucleic Acid Amplification and Cell-Phone-Based Readout. ACS Nano. 2017;11:2934–2943. doi: 10.1021/acsnano.6b08274. [DOI] [PubMed] [Google Scholar]
- Kühnemund M., Wei Q., Darai E., Wang Y., Hernández-Neuta I., Yang Z., Tseng D., Ahlford A., Mathot L., Sjöblom T., et al. Targeted DNA sequencing and in situ mutation analysis using mobile phone microscopy. Nat. Commun. 2017;8:13913. doi: 10.1038/ncomms13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Scola B., Le Bideau M., Andreani J., Hoang V.T., Grimaldier C., Colson P., Gautret P., Raoult D. Viral RNA load as determined by cell culture as a management tool for discharge of SARS-CoV-2 patients from infectious disease wards. Eur. J. Clin. Microbiol. Infect. Dis. 2020;39:1059–1061. doi: 10.1007/s10096-020-03913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larremore D.B., Wilder B., Lester E., Shehata S., Burke J.M., Hay J.A., Tambe M., Mina M.J., Parker R. Test sensitivity is secondary to frequency and turnaround time for COVID-19 surveillance. medRxiv. 2020 doi: 10.1101/2020.06.22.20136309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzo E., Franchin E., Ciavarella C., Cuomo-Dannenburg G., Barzon L., Del Vecchio C., Rossi L., Manganelli R., Loregian A., Navarin N., et al. Imperial College COVID-19 Response Team Suppression of a SARS-CoV-2 outbreak in the Italian municipality of Vo’. Nature. 2020;584:425–429. doi: 10.1038/s41586-020-2488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazer D., Santillana M., Perlis R.H., Ognyanova K., Baum M.A., Quintana A., Druckman J., Della Volpe J., Chwe H., Simonson M. THE STATE OF THE NATION: A 50-STATE COVID-19 SURVEY REPORT #8: FAILING THE TEST. 50-STATE COVID-19 SURVEY. 2020 doi: 10.31219/osf.io/gj9x8. [DOI] [Google Scholar]
- Lee S., Kim T., Lee E., Lee C., Kim H., Rhee H., Park S.Y., Son H.J., Yu S., Park J.W., et al. Clinical Course and Molecular Viral Shedding Among Asymptomatic and Symptomatic Patients With SARS-CoV-2 Infection in a Community Treatment Center in the Republic of Korea. JAMA Intern. Med. 2020 doi: 10.1001/jamainternmed.2020.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q.X., Tang X.J., Shi Q.L., Li Q., Deng H.J., Yuan J., Hu J.L., Xu W., Zhang Y., Lv F.J., et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020;26:1200–1204. doi: 10.1038/s41591-020-0965-6. [DOI] [PubMed] [Google Scholar]
- Manfredonia I., Nithin C., Ponce-Salvatierra A., Ghosh P., Wirecki T.K., Marinus T., Ogando N.S., Snider E.J., Hemert M.J.v., Bujnicki J.M., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 2020 doi: 10.1093/nar/gkaa1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNerney M.P., Zhang Y., Steppe P., Silverman A.D., Jewett M.C., Styczynski M.P. Point-of-care biomarker quantification enabled by sample-specific calibration. Sci Adv. 2019;5:eaax4473. doi: 10.1126/sciadv.aax4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske A.J., Nakandakari-Higa S., Marraffini L.A. Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature. 2019;570:241–245. doi: 10.1038/s41586-019-1257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhrvold C., Freije C.A., Gootenberg J.S., Abudayyeh O.O., Metsky H.C., Durbin A.F., Kellner M.J., Tan A.L., Paul L.M., Parham L.A., et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science. 2018;360:444–448. doi: 10.1126/science.aas8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osório N.S., Correia-Neves M. Implication of SARS-CoV-2 evolution in the sensitivity of RT-qPCR diagnostic assays. Lancet Infect. Dis. 2020 doi: 10.1016/S1473-3099(20)30435-7. S1473-3099(20)30435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priye A., Bird S.W., Light Y.K., Ball C.S., Negrete O.A., Meagher R.J. A smartphone-based diagnostic platform for rapid detection of Zika, chikungunya, and dengue viruses. Sci. Rep. 2017;7:44778. doi: 10.1038/srep44778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quicke K., Gallichote E., Sexton N., Young M., Janich A., Gahm G., Carlton E.J., Ehrhart N., Ebel G.D. Longitudinal Surveillance for SARS-CoV-2 RNA Among Asymptomatic Staff in Five Colorado Skilled Nursing Facilities: Epidemiologic, Virologic and Sequence Analysis. medRxiv. 2020 doi: 10.1101/2020.06.08.20125989. [DOI] [Google Scholar]
- Sachs N., Papaspyropoulos A., Zomer-van Ommen D.D., Heo I., Böttinger L., Klay D., Weeber F., Huelsz-Prince G., Iakobachvili N., Amatngalim G.D., et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019;38:e100300. doi: 10.15252/embj.2018100300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders W., Fritch E.J., Madden E.A., Graham R.L., Vincent H.A., Heise M.T., Baric R.S., Moorman N.J. Comparative analysis of coronavirus genomic RNA structure reveals conservation in SARS-like coronaviruses. bioRxiv. 2020 doi: 10.1101/2020.06.15.153197. 2020.06.15.153197. [DOI] [Google Scholar]
- Shmakov S., Abudayyeh O.O., Makarova K.S., Wolf Y.I., Gootenberg J.S., Semenova E., Minakhin L., Joung J., Konermann S., Severinov K., et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S., Smargon A., Scott D., Cox D., Pyzocha N., Yan W., Abudayyeh O.O., Gootenberg J.S., Makarova K.S., Wolf Y.I., et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017;15:169–182. doi: 10.1038/nrmicro.2016.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker I.M., Mesa P., Kellner M.J., Kannan S., Brignole E., Koob J., Feliciano P.R., Stella S., Abudayyeh O.O., Gootenberg J.S., et al. High-Resolution Structure of Cas13b and Biochemical Characterization of RNA Targeting and Cleavage. Cell Rep. 2019;26:3741–3751.e5. doi: 10.1016/j.celrep.2019.02.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargon A.A., Cox D.B.T., Pyzocha N.K., Zheng K., Slaymaker I.M., Gootenberg J.S., Abudayyeh O.A., Essletzbichler P., Shmakov S., Makarova K.S., et al. Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol Cell. 2017;65:618–630.e7. doi: 10.1016/j.molcel.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F., Ganguli A., Nguyen J., Brisbin R., Shanmugam K., Hirschberg D.L., Wheeler M.B., Bashir R., Nash D.M., Cunningham B.T. Smartphone-based multiplex 30-minute nucleic acid test of live virus from nasal swab extract. Lab Chip. 2020;20:1621–1627. doi: 10.1039/d0lc00304b. [DOI] [PubMed] [Google Scholar]
- Vanaerschot M., Mann S.A., Webber J.T., Kamm J., Bell S.M., Bell J., Hong S.N., Nguyen M.P., Chan L.Y., Bhatt K.D., et al. Identification of a polymorphism in the N gene of SARS-CoV-2 that adversely impacts detection by a widely-used RT-PCR assay. bioRxiv. 2020 doi: 10.1101/2020.08.25.265074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogels C.B.F., Brito A.F., Wyllie A.L., Fauver J.R., Ott I.M., Kalinich C.C., Petrone M.E., Casanovas-Massana A., Catherine Muenker M., Moore A.J., et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 2020;5:1299–1305. doi: 10.1038/s41564-020-0761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Simoneau C.R., Kulsuptrakul J., Bouhaddou M., Travisano K., Hayashi J.M., Carlson-Stevermer J., Zengel J.R., Richards C.M., Fozouni P., et al. Functional genomic screens identify human host factors for SARS-CoV-2 and common cold coronaviruses. Cell. 2020 doi: 10.1016/j.cell.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson E.J., Walker A.J., Bhaskaran K., Bacon S., Bates C., Morton C.E., Curtis H.J., Mehrkar A., Evans D., Inglesby P., et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature. 2020;584:430–436. doi: 10.1038/s41586-020-2521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölfel R., Corman V.M., Guggemos W., Seilmaier M., Zange S., Müller M.A., Niemeyer D., Jones T.C., Vollmar P., Rothe C., et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–469. doi: 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- Wood C.S., Thomas M.R., Budd J., Mashamba-Thompson T.P., Herbst K., Pillay D., Peeling R.W., Johnson A.M., McKendry R.A., Stevens M.M. Taking connected mobile-health diagnostics of infectious diseases to the field. Nature. 2019;566:467–474. doi: 10.1038/s41586-019-0956-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W.X., Chong S., Zhang H., Makarova K.S., Koonin E.V., Cheng D.R., Scott D.A. Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol Cell. 2018;70:327–339.e5. doi: 10.1016/j.molcel.2018.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A., et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Li C., Sachs N., Chiu M.C., Wong B.H., Chu H., Poon V.K., Wang D., Zhao X., Wen L., et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc. Natl. Acad. Sci. USA. 2018;115:6822–6827. doi: 10.1073/pnas.1806308115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., et al. China Novel Coronavirus Investigating and Research Team A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The custom MATLAB code for image processing and data analysis has been deposited to Mendeley Data: https://doi.org/10.17632/r3vwyr2w5x.1.