

Abstract
Background
Coronavirus disease 2019 (COVID-19) outbreaks emerged at two university-affiliated hospitals in Seoul (hospital A) and Uijeongbu City (hospital S) in the metropolitan Seoul area in March 2020. The aim of this study was to investigate epidemiological links between the outbreaks using whole genome sequencing (WGS) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Methods
Fifteen patients were enrolled in the study, including four non-outbreak (A1–A4) and three outbreak cases (A5–A7) in hospital A and eight cases (S1–S8) in hospital S. Patients' hospital stays, COVID-19 symptoms, and transfer history were reviewed. RNA samples were submitted for WGS and genome-wide single nucleotide variants and phylogenetic relationships were analyzed.
Results
The index patient (A5) in hospital A was transferred from hospital S on 26 March. Patients A6 and A7 were the family caregiver and sister, respectively, of the patient who shared a room with A5 for 4 days. Prior to transfer, A5 was at the next bed to S8 in the emergency room on 25 March. Patient S6, a professional caregiver, took care of the patient in the room next to S8's room for 5 days until 22 March and then S5 for another 3 days. WGS revealed that SARS-CoV-2 in A2, A3, and A4 belong to clades V/B.2, S/A, and G/B.1, respectively, whereas that of A5–A7 and S1-S5 are of the V/B.2.1 clade and closely clustered. In particular, SARS-CoV-2 in patients A5 and S5 showed perfect identity.
Conclusion
WGS is a useful tool to understand epidemiology of SARS-CoV-2. It is the first study to elucidate the role of patient transfer and caregivers as links of nosocomial outbreaks of COVID-19 in multiple hospitals.
Keywords: COVID-19, SARS-CoV-2, Nosocomial Outbreak, Whole Genome Sequencing, Clade, Lineage
Graphical Abstract
INTRODUCTION
Coronavirus disease 2019 (COVID-19) was first described in December 2019, in Wuhan, Hubei Province, China.1 As of December 21, 2020, there were 75,704,857cases of COVID-19 confirmed in 222 countries,2 and 45,475 cases were reported in South Korea since its introduction by a Chinese traveler on January 19, 2020.3,4 COVID-19 cases are mainly found to be community-acquired; sporadic cases of unknown origin account for 16.0%.4 A community outbreak involving 6,930 cases in the Daegu-Gyeongbuk area of South Korea emerged between mid-February and mid-March, accounting for the majority of COVID-19 cases to date nationwide.5,6 Most community cases stem from numerous outbreaks of varying sizes in Korea.7,8 COVID-19 is well known to be highly contagious in mild to asymptomatic cases, thus causing many community outbreaks and often become widespread very quickly in a community. Therefore, early detection based on rapid molecular diagnostics and thorough contact tracing are essential, basic tools to help prevent the spread of COVID-19 in a community.
Health care facilities house some of the most vulnerable and immunocompromised individuals and clearly have high morbidity and mortality related to COVID-199 and require strict infection control to prevent nosocomial outbreaks. From January to September 2020, several instances of nosocomial outbreaks of COVID-19 have been published.10,11 Korea Centers for Disease Control and Prevention (KCDC) reviewed seven major COVID-19 nosocomial outbreaks in South Korea affecting 39 to 196 patients,5 but no molecular epidemiological investigations were reported. Whole genome sequencing (WGS) is a powerful tool for epidemiological analysis of newly emerging viral disease outbreaks12,13 and currently is an established technique to classify the clades and lineages of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) to investigate the evolution and epidemiology of the virus.14,15 At the clade level, it is possible to match dominant clones to certain regions. According to information published on the GISAID Next hCoV-19 app (https://www.gisaid.org/), as of 8 October, SARS-CoV-2 clade L is the most ancestral clade from Wuhan, China, clade G is the second generation clade prevailing in the European Union, which then evolved to give rise to clade GH in the United States, and GR in Russia, Brazil, and the United Kingdom. Although initially the predominance of V clades is obvious due to a large clade V cluster outbreak in Daegu-Gyeongbuk, clades S (n = 24), V (n = 67), and G (n = 55) are found among 151 representative cases in Korea.5 WGS is also remarkably successful in tracing clonality in community outbreaks16,17 as well as nosocomial outbreaks.18 However, to date, there has been no reports of WGS analysis of epidemiologic link between nosocomial outbreaks of COVID-19 in multiple hospitals.
In late March, an outbreak of three COVID-19 cases emerged in a pediatric ward in a tertiary care hospital in Seoul,19 in which an index patient was transferred from a university-affiliated hospital in Uijeongbu City during an ongoing outbreak, comprising 60 patients.6 The two hospitals are 36 km apart in the Seoul metropolitan area. This study was to further investigate the nosocomial outbreaks of COVID-19 in both hospitals using WGS analysis of SARS-CoV-2 to elucidate epidemiologic link between them.
METHODS
Patient's enrolment
A total of 15 patients, including 3 outbreak cases (A5–A7) and 4 non-outbreak cases (A1–A4) in Asan Medical Center (hospital A) and eight cases (S1–S8) in Uijeongbu St. Mary's hospital (hospital S) were reviewed for their hospital stays, COVID-19 symptoms, and transfer history. For all patients, COVID-19 was confirmed by SARS-CoV-2 real-time reverse transcription polymerase chain reaction (rRT-PCR). We reviewed the electronic medical records for clinical symptoms and epidemiologic findings such as hospital stays and transfer history to generate an outbreak synopsis.
WGS of SARS-CoV-2
Samples for RNA extraction were obtained by nasopharyngeal swabs or sputa. RNA was extracted using eMAG (bioMérieux, Marcy-l'Etoile, France) in hospital A and AdvanSure E3 (LG Chem, Seoul, Korea) in hospital S, and stored at −70°C until further analysis. Whole metagenome sequencing was performed on library preparations prepared using TruSeq Stranded Total RNA Human/Mouse/Rat kit (Illumina, CA, USA) and sequenced using the NovaSeq 6000 (Illumina), following the scheme used by Wu et al.20 WGS results were reconstructed by aligning the sequence reads to the reference genome, SARS-CoV-2 Wuhan-Hu-1 genome (NC_045512.2).
Analysis of single nucleotide variants (SNVs): clade and lineage assignment
SNVs were assigned based on the reference-based alignment of published sequences.21 Clade assignment was performed according to the current scheme on the GISAID website14. The genome sequences were phylogenetically classified under the lineage classification scheme proposed by Rambaut et al. using Pangolin software version 1.1.14 and the lineage database version 2020-05-19.15
SARS-CoV-2 genome sequences, obtained from GISAID on June 1, 2020 (n = 34,806), were aligned pairwise to the reference genome (NC_045512.2) (Shu and McCauley, 2017). Following the sequence alignment, high-quality sequences with ≥ 99% completeness, ≤ 30 nucleotide differences relative to the reference genomes, and without frame-shift error in all open reading frames were selected (n = 24,684). The high-quality sequences were de-replicated to 15,067 unique genome sequences. Genomic positions with < 99% coverage by the non-redundant genomes were omitted from the single nucleotide polymorphism (SNP) analyses. The positions where the minor allele frequency was ≥ 1% in the non-redundant genome set were defined as SNP sites (n = 55). The 104 GISAID-obtained genomes, representing the 104 unique allelic combinations in the genome-wide SNP sites, and the 57 GISAID-obtained genomes that had the fewest genome-wide single-nucleotide differences compared to the case genomes were included in phylogenetic analysis. From the whole genome alignment of 29,903 genomic positions, we removed the positions with more than a 1% gap or ambiguous characters, resulting in the clipped alignment of 28,371 positions. The ModelFinder function of IQ-Tree version 1.6.12 was run to determine the optimal substitution model for the data set. Using the selected model, a maximum-likelihood phylogenetic tree was generated with ultrafast bootstrap (n = 1,000) for branch support estimation.
Data availability
The raw reads from the total RNA sequencing of nasopharyngeal samples (n = 15) were deposited in the NCBI SRA as BioProject number PRJNA645035. The SARS-CoV-2 genome sequences recovered in this study (n = 11) were deposited in GISAID with accession numbers EPI_ISL_485001 - EPI_ISL_485002 and EPI_ISL_485388 - EPI_ISL_485396.
Ethical statement
This study was reviewed and approved by the Institutional Review Board of Asan Medical Center (S2019-1169-0001) and the Institutional Review Board of the Catholic Medical Center (UC20SIDI0057). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
RESULTS
Outbreak synopsis
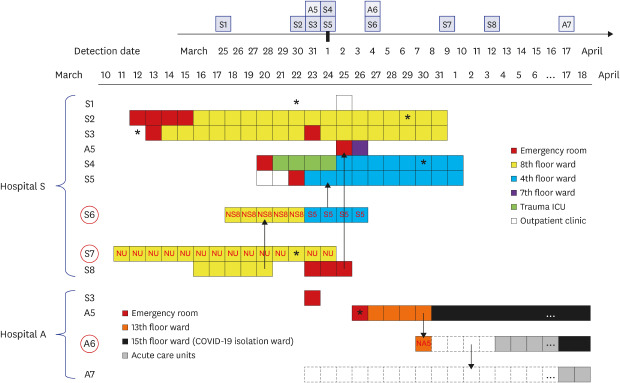
Clinical and epidemiological features of all fifteen COVID-19 patients are presented in Table 1, and ward transfer history of outbreak-related patients is detailed in Fig. 1. Patient A5 was the index patient in hospital A, who was transferred from hospital S on 26 March; A6 and A7 were the mother and elder sister, respectively, of the non-infected patient (NA5) who stayed in the bed across from A5 in a six-bed hospital room. A6 stayed overnight in the hospital room as a family caregiver of NA5 on 30 March and then went home and stayed with another daughter (A7) at home for 4 days, and then returned to provide care for NA5. A5 was negative for SARS-CoV-2 by rRT-PCR testing when she was tested in the ER upon transfer to hospital A on 26 March. Patients were sequentially diagnosed on 31 March (A5), 4 April (A6), and 17 April (A7). S1 visited the outpatient clinic in hospital S and was diagnosed with COVID-19 on 25 March. S2–S5 and S8 were inpatients in the outbreak wards located on the 8th or 4th floor in hospital S and S6 and S7 were professional caregivers in these wards. S1–S8 were diagnosed with COVID-19 between March 25 and April 12 (Table 1). A5 was originally admitted into the bed next to S8 in the emergency room (ER) in hospital S, 1 hour after S8 had departed. S3 was transferred to the ER in hospital A on 23 March but returned to the ER in hospital S on the same day. S6 was the caregiver for the non-infected patient (NS8) in a room next to S8 for 5 days during S8's stay in the 8th floor ward and then took care of S5, who was on the 4th floor.
Table 1. Clinical and epidemiological features of fifteen coronavirus disease 2019 patients.
| Case | Age/Sex | Comorbidity | Symptoms at diagnosis | Pneumonia on chest X-ray | Acquisition | Resident region |
|---|---|---|---|---|---|---|
| A1 | 73/F | Arrythmia | Fever, cough | Pneumonia, consolidation, and GGO | Community | Daegu-Gyeongbuk |
| A2 | 61/F | TB history | Fever, cough | Pneumonia, consolidation, and GGO | Community | Daegu-Gyeongbuk |
| A3 | 62/F | Dyslipidemia | Cough, headache | Pneumonia, patchy GGO | Community | Seoul |
| A4 | 16/F | VSD | Fever | Pneumonia, peribronchial infiltration | Community | United Kingdom |
| A5 | 9/F | ICH | Fevera | No active lung lesions | Nosocomial | Northern Gyeonggi |
| A6 | 40/F | None | Asymptomatic | Pneumonia, patchy increased opacities | Nosocomial | Southern Gyeonggi |
| Caregiverb | ||||||
| A7 | 2/F | None | Asymptomatic | No active lung lesions | Community | Southern Gyeonggi |
| S1 | 76/M | None | Fever, cough, sore throat, rhinorrhea | Not tested | Community | Northern Gyeonggi |
| S2 | 82/F | Tuberculosis | Fever | Pneumonia, increased opacities in LLLF | Nosocomial | Northern Gyeonggi |
| S3 | 56/M | Liver abscess | Fever | Subsegmental atelectasis | Nosocomial | Northern Gyeonggi |
| S4 | 83/M | ICH | Fever | Subsegmental atelectasis | Nosocomial | Northern Gyeonggi |
| S5 | 52/M | Spine fracture | Asymptomatic | No active lung lesions | Nosocomial | Northern Gyeonggi |
| S6 | 64/F | Hypertension | Asymptomatic | Pneumonia, consolidation, and GGO | Nosocomial | Northern Gyeonggi |
| Caregiverb | ||||||
| S7 | 59/F | None | Asymptomatic | No active lung lesions | Nosocomial | Northern Gyeonggi |
| Caregiverb | ||||||
| S8 | 66/F | DLBCL | Headache | No active lung lesions | Nosocomial | Northern Gyeonggi |
DLBCL = diffuse large B-cell lymphoma, GGO = ground-glass opacification, ICH = intracranial hemorrhage, LLLF = left lower lung field, TB = tuberculosis, VSD = ventricular septal defect.
aThe patient A5 initially had fever in the emergency room and no more fever until discharge with continuing antipyretics under impression of ICH-associated fever; bThey were caregivers without comorbidity except hypertension.
Fig. 1. Hospital stay and transfer history of 11 COVID-19 patients and caregivers associated with the outbreaks in hospitals A and S. Detection dates were denoted on the calendar in the upper corner. The wards in which patient A5, A6, and A7 from hospital A and all eight patients from hospital S (S1–S8) stayed were denoted using the color-coded squares. The red circles represents caregivers and the red letters inside squares indicated the patients cared by the caregivers. Asterisk denoted the onset of symptoms related to COVID-19 and dotted squares indicated the family home in which A6 and A7 lived. Possible transmission directions between outbreak patients were indicated by arrows.

WGS determination of clades and lineage
RNA samples from four patients were negative for SARS-CoV-2 reads, but the SARS-CoV-2 genome sequences from eleven samples were successfully constructed (Table 2).
Table 2. Quality parameters and typing results of SARS-CoV-2 genome sequences.
| Case | Sequencing run | SARS-CoV-2 coverage | Typing results | ||||
|---|---|---|---|---|---|---|---|
| Reads | Bases (Mbp) | Covered positions (bp) | Coverage breadth (%) | Mean depth | Clade | Lineagesa | |
| A1 | 6.4 × 106 | 967 | 0 | 0 | 0 | NA | NA |
| A2 | 8.8 × 106 | 1,328 | 29,869 | 99.9 | 87.3 | V | B.2 |
| A3 | 7.2 × 106 | 1,086 | 29,896 | 100 | 172 | S | A |
| A4 | 6.2 × 106 | 932 | 19,384 | 64.8 | 2.73 | G | B.1 |
| A5 | 8.1 × 106 | 1,216 | 29,715 | 99.4 | 14.5 | V | B.2.1 |
| A6 | 7.7 × 106 | 1,159 | 29,901 | 100 | 4,867 | V | B.2.1 |
| A7 | 1.4 × 107 | 2,130 | 29,862 | 99.9 | 27.8 | V | B.2.1 |
| S1 | 7.1 × 106 | 1,072 | 29,873 | 99.9 | 70 | V | B.2.1 |
| S2 | 6.0 × 106 | 908 | 27,696 | 92.6 | 27.7 | V | B.2.1 |
| S3 | 1.4 × 107 | 2,094 | 26,853 | 89.8 | 11.9 | Vb | B.2.1 |
| S4 | 6.9 × 106 | 1,047 | 28,241 | 94.4 | 23.3 | V | B.2.1 |
| S5 | 8.3 × 106 | 1,260 | 29,903 | 100 | 7,199 | V | B.2.1 |
| S6 | 7.6 × 106 | 1,141 | 0 | 0 | 0 | NA | NA |
| S7 | 6.0 × 106 | 904 | 0 | 0 | 0 | NA | NA |
| S8 | 4.6 × 107 | 7,006 | 0 | 0 | 0 | NA | NA |
NA = not available.
aLineage classification scheme proposed by Rambaut et al. (14) using Pagolin software version 1.1.14 and the lineage database version 2020-05-19; bS3 strain had the G26144T variant but not the G11083T variant.
Among the eleven SARS-CoV-2 genomes, SNVs were detected at 25 positions, 9 of which overlap with SNP positions in the global SARS-CoV-2 pandemic population (Fig. 2A). The single nucleotide variant (SNV) positions were distributed throughout the entire length of the genome including most of the SARS-CoV-2 protein-coding genes (Fig. 2A, bottom panel). Nine out of twenty-five SNVs occurred among the global SNP sites, whereas the other sixteen were presented in positions where the global frequency of variants was quite low. Three to six alleles were found to be missing due to the incompleteness of the A4, S2, S3, and S4 genomes at 25 SNV positions. Fifteen different SNV sites were observed among the eleven genomes (Supplementary Table 1). However, the eight outbreak strains in the study (A5–A7 and S1–S5) showed up to 6 SNVs. A5–A7 and S5 were the least variable with 3 SNVs. Mutation events among outbreak strains, as defined based on ancestral reconstruction, included eight genomic positions that were well separated across the genomes without a linkage to any particular protein-coding gene (Fig. 2B).
Fig. 2. SNVs among the genomes reconstructed from eleven RNA samples. (A) Allele states of the eleven genomes at the SNV sites and global SNP sites. The upper panel outlined the allele states of the 11 genomes at the 71 positions that had either SNVs from the cases in this study or from GISAID sequences. The lower panel described the minor allele frequency of the 71 SNV/SNP positions. The vertical lines connecting the allele frequency point to the bottom x-axis were colored according to the color of the genomic feature in the genomic map that was inserted in the bottom of the figure. The genomic coordinates of each of the 71 positions were mapped by line to the genomic map at the bottom. RdRp, S, M, and N stood for regions encoding the RNA-dependent RNA polymerase, the spike protein, the membrane protein, and the nucleocapsid, respectively. (B) Reconstructed mutation events among the eight epidemiologically related cases. The SNVs were phylogenetically analyzed using the maximum-likelihood tree method. Mutations on the branches were defined based on ancestral reconstruction of the allele states. Labels above the branches denoted the nucleotide changes at a given genomic coordinate; those below the branches denoted the protein-level regions where the mutated loci resided.

SNV = single nucleotide variant, SNP = single nucleotide polymorphism, nsp = nonstructural protein, N = nucleocapsid protein, M = membrane protein.
According to the popular clade classification scheme of SARS-CoV-2 genomic variants, as presented on the GISAID website, seven out of the eight outbreak genomes were unambiguously classified as the V clade, while the remaining one case (S3) showed an L-clade-associated allele (G) at position 11,083 and a V-clade-associated allele (T) at the other marker position 26,144 (Table 2). Based on the placement of the S3 GS in the whole-genome phylogenetic tree (Fig. 3) and the lineage assignment (to be described below), S3 could be robustly assigned to a V clade variant (Fig. 2B) with guanine at position 26,144 like an L clade variant. Non-outbreak cases were classified as clade S/lineage A, clade G/lineage B.1, and clade V/lineage B.2 for each of A3, A4, and A2 genomes, respectively. The A4 genome had V614G variant of spike protein but could not be further classified to the GH or GR clade due to its low coverage. According to the phylogenetic classification scheme, the genomes from outbreak-associated patients were all classified as lineage B.2.1 (n = 8).
Fig. 3. Phylogenetic analysis of the sequenced SARS-CoV-2 genomes with the global reference genomes. (A) Maximum-likelihood phylogenetic tree of the genomes recovered from the reported cases (n = 11) and the selected genomes from the GISAID database (n = 161). Epidemiological information on the patient samples, when available, were noted on the right side of the tip labels. (B) Subtree of the focal clade containing eight outbreak-related cases and the public genomes with highly similar sequences (≤ 3 nucleotide differences). To the right was the corresponding scatter plot of the collection date and geographic origin of the genomes belonging to each tip in the subtree. Each tip in the phylogenetic tree represented a cluster of identical genomes of multiple samples.

Whole genome phylogenetic analysis
A maximum-likelihood whole genome phylogeny containing the genomes representing the reported cases, the global SNP types, and those publicly available that had the highest identity to the case-derived genomes, was inspected to cross-check the epidemiological relationship for the cases. The genomes belonging to clade V and lineage B.2.1 (n = 8) appeared to be a focal clade in which the close epidemiological relationships were plausible (Fig. 3A). Genomes assigned to clade G, clade S, and clade V- lineage B.2 were distant from the focal clade on the phylogenetic tree. Furthermore, the phylogenetic tree constructed using the WGS data of all nosocomial outbreak strains from both hospitals showed that all were almost identical, which could be deduced as inter-hospital transmission mediated by a single patient (A5).
We further investigated the date of collection and the geographic origins of the genomes belonging to the focal clade containing 8 outbreak-related strains. The outbreak strains of this study revealed high similarity (< 6 nucleotide difference) with the strains sampled from March to April 2020 in Europe, United States, Jamaica, Australia, and Taiwan. Also, S2 was identical to the representative strain of the clade. Other patients belonging to S hospital outbreak had one base pair difference from patient S2, with the exception of S5, who had a difference of two base pairs. Sequence obtained from A3 was identical to that of S5, and had one base pair difference from A6 and two base pair difference from A7 (Fig. 3B).
DISCUSSION
Phylogenetic analysis revealed that the SARS-CoV-2 genomes of A5–A7 clustered with those of S1–S5 and were almost identical to S5, suggesting transmission of the virus from A5 to A6 and A7 and a link between the outbreaks in the two hospitals. These findings were consistent with the finding that patient A5 tested negative for SARS-CoV-2 by initial rRT-PCR performed in the ER in hospital A, but tested positive 5 days later in hospital A. Since S6 could explain the transmission of the same clone between S8 and S5, and A5 was identical to S5, it was most likely that A5 acquired the virus from S8 in the ER of hospital S. Therefore, the caregivers certainly played an important role in the nosocomial spread of SARS-CoV-2 between and within the two hospitals. This situation is likely explained by the fact that caregivers resided in the patient's room, that increased the density of number of humans in the room. In addition, caregivers taking care of patients professionally have more risk to facilitate the spread of SARS-CoV-2 more extensively. Thus, transmission of the virus can occur not only among hospital inpatients but also between other people in the hospital and those in the community. Patient transfer between hospitals could be an added risk that causes consecutive outbreaks between hospitals. These findings are not surprising given that numerous healthcare-associated outbreaks of COVID-19 have been reported22 and healthcare workers in hospitals and long-term care facilities are well-known as high-risk group to acquire COVID-19.23
Only a few nosocomial outbreaks of COVID-19 in pediatric patients have been reported.11,19 Children are often unnoticed cases in household outbreaks of COVID-19 since they are usually asymptomatic or have only mild to moderate symptoms.24,25 It is uncertain whether they are victims or silence spreaders, as observed in this study. Pediatric inpatients require caregivers so the risk of nosocomial transmission could be doubled. High transmissibility of SARS-CoV-2 is well known and a part of the reason is the role that asymptomatic patients play as undetected carriers of the virus.26
Among the 15 cases in this study, less than 50% displayed pneumonia or respiratory symptoms. As demonstrated by A5, asymptomatic or paucisymptomatic patients may not be suspected of having COVID-19.27,28 Due to the COVID-19 pandemic, both the university-affiliated hospitals adopted a strict policy to screen and test patients with suspected symptoms or epidemiological relevance for COVID-19 (S1 and A5 when they visited the outpatient clinic and ER, respectively). However, the outbreaks in the two hospitals clearly showed that the policies in place were not sufficient to prevent the spread of COVID-19 in patients in the acute care hospitals. Inpatients are especially vulnerable to nosocomial outbreaks due to the high-risk of close contact among patients as well as caregivers who share the same wards or even the same multiple-occupancy room with each other. The outbreak in hospital S eventually comprises 60 cases of COVID-19 from March to April 2020,6 whereas the outbreak in hospital A was limited to 3 cases. The difference between them was likely due to how quickly introductions of new COVID-19 infections were detected in hospital A. Universal testing of SARS-CoV-2 in pregnant women for childbirth has proven to be effective in controlling spread of COVID-19 infections.29 Although the prevalence is not high in the community, universal screening of all inpatients upon admission and resident caregivers may be an effective strategy to prevent outbreaks in a tertiary care hospital such as hospital A, which has a high influx of patients from other hospitals. In fact, two patients in this study had a transfer history to hospital A among the five inpatients who acquired COVID-19 in hospital S. The infection control policy for COVID-19, such as mandated use of masks and frequent use of alcohol-based hand sanitizers is currently being employed in all Korean hospitals, but it is difficult to enforce these practices among children. Thus, universal screening of inpatients and their caregivers is needed and even more so in pediatric wards in which caregivers were always needed for all inpatients.
When genotypes of the SARS-CoV-2 genomes were analyzed, it was noted that all the patients of hospital S and A5 were residents of the northern part of Gyeonggi-do in the Seoul metropolitan area, in which hospital S located. Therefore, it is possible that A5 may have acquired the virus in the community before admission to hospital S. The outbreak of COVID-19 in the community of northern Gyeonggi-do may have preceded the outbreak in hospital S since S1, the first COVID-19 patient in hospital S, was diagnosed with COVID-19 in an outpatient clinic. The SARS-CoV-2 sequences of all COVID-19 patients from hospital S including S1 belong to the V clade/B.2.1 lineage, although no epidemiological link was identified among them by contact tracing conducted by KCDC officials. The V clade/B.2.1 lineage is found globally and first identified on February 23, 2020 in Korea according to public database (https://www.gisaid.org/), and this clone is close to Daegu-Gyeongbuk clone, V calde/B.2. Although G614 variant of the spike protein, well-known for its high infectivity, is currently dominant variant,30 the outbreak strains in this study did not harbor it. Patient S1 did not have any history of hospital visits. Therefore, a probable regional spread of this lineage in the northern part of Gyeonggi-do occurred and later, a nosocomial outbreak of this strain took place in a secondary care hospital in that region. Only patients A5–A7 in hospital A belonged to the same lineage, whereas three non-outbreak strains were assigned to different clades or lineages. However, A5 more likely acquired the virus from S8 and transmitted the virus to A6. Subsequently A6 transmitted the infection to A7, considering that the S5 genome had no SNPs, and the A6 and A7 genomes had 2 SNPs and 3 SNPs, respectively, in alignment with the A5 genome. Phylogenic analysis showed that clonal evolution of each of the index strains, S2 and A3, of nosocomial outbreaks in two hospitals proceeded during virus propagation. Considering that the accumulation of SNPs is time-dependent, outbreaks in the two hospitals spanning almost one month likely originated from a single strain and evolved enough to trace the linkage between the individuals and hospitals. Genotyping using SNVs is a powerful tool for epidemiological analysis.15,31
However, there were some limitations of this study. First, virus genomes were not detected in four samples including those from S8. Therefore, the linkage between the A5 genome and the S8 and S5 genomes was not fully proven by WGS. Second, S7 and S8 were diagnosed too late after being discharged from hospital S to obtain a RNA sample with high viral load or clinical analysis of COVID-19. Therefore, genotyping and clinical data were incomplete. Last, the distribution of the uploaded genome sequences on GISAID could be unbalanced. This could lead to inadequate geographical inference of transmission sources of SARS-CoV-2.
In conclusion, WGS is a useful tool to find linkages of epidemiologically-related outbreaks. Caregivers may facilitate virus transmission within a hospital as well as between community and hospital, and patient transfer poses a risk of inter-hospital spread of COVID-19 outbreaks. Therefore, professional caregivers and patients transferred from other hospitals should be considered a high-risk group and subjected to better screening with proactive diagnostic testing to prevent nosocomial outbreaks of COVID-19, especially in tertiary care hospitals.
ACKNOWLEDGMENTS
We would like to thank Moon Jung Kim M.D. and Jaegyun Lim M.D. (Department of Laboratory Medicine, Myungji Hospital) for kindly providing RNA samples and Editage (www.editage.co.kr) for editing and reviewing this manuscript for English language.
Footnotes
Funding: This work was supported by a Bio and Medical Technology Development Program of the National Research Foundation (NRT) of Korea grant funded by the Korea government (grant number NRF-2016M3A9B6918716).
Disclosure: The authors have no potential conflicts of interest to disclose.
- Conceptualization: Kim MN, Sung H.
- Formal analysis: Park K, Lee J,1 Lee K.
- Funding acquisition: Sung H.
- Methodology: Park K, Lee K, Chalita M, Yoon SH, Chun J.
- Project administration: Kim MN, Sung H.
- Resources: Hur KH, Lee J.1
- Writing - original draft: Park K, Lee J,1 Lee K.
- Writing - review & editing: Jung J, Kim SH, Lee J,2 Sung H, Kim MN, Lee HK.
Lee J,1 Jaewoong Lee; Lee J,2 Jina Lee.
SUPPLEMENTARY MATERIAL
The fifteen sites that are variable among the case genomes and not missing in any case genome
References
- 1.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weekly operational update on COVID-19 - 21 December 2020. [Updated 2020]. [Accessed December 22, 2020]. https://www.who.int/publications/m/item/weekly-operational-update-on-covid-19---21-december-2020.
- 3.Kim JY, Choe PG, Oh Y, Oh KJ, Kim J, Park SJ, et al. The first case of 2019 novel coronavirus pneumonia imported into Korea from Wuhan, China: implication for infection prevention and control measures. J Korean Med Sci. 2020;35(5):e61. doi: 10.3346/jkms.2020.35.e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The updates on COVID-19 in Korea as of 21 December. [Updated 2020]. [Accessed December 22, 2020]. https://www.cdc.go.kr/board/board.es?mid=a30402000000&bid=0030.
- 5.The updates on COVID-19 in Korea as of 6 July. [Updated 2020]. [Accessed July 6, 2020]. http://www.cdc.kr/board/board.es?mid=a30402000000&bid=0030.
- 6.COVID-19 National Emergency Response Center; Epidemiology and Case Management Team; Korea Centers for Disease Control and Prevention. Coronavirus disease-19: the first 7,755 cases in the Republic of Korea. Osong Public Health Res Perspect. 2020;11(2):85–90. doi: 10.24171/j.phrp.2020.11.2.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The updates on COVID-19 in Korea as of 8 July. [Updated 2020]. [Accessed July 8, 2020]. https://www.cdc.go.kr/board/board.es?mid=a30402000000&bid=0030.
- 8.Park SY, Kim YM, Yi S, Lee S, Na BJ, Kim CB, et al. Coronavirus disease outbreak in call center, South Korea. Emerg Infect Dis. 2020;26(8):1666–1670. doi: 10.3201/eid2608.201274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo W, Li M, Dong Y, Zhou H, Zhang Z, Tian C, et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab Res Rev. 2020;36(7):e3319. doi: 10.1002/dmrr.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Zhou Q, He Y, Liu L, Ma X, Wei X, et al. Nosocomial outbreak of COVID-19 pneumonia in Wuhan, China. Eur Respir J. 2020;55(6):2000544. doi: 10.1183/13993003.00544-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwierzeck V, König JC, Kühn J, Mellmann A, Correa-Martínez CL, Omran H, et al. First reported nosocomial outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in a pediatric dialysis unit. Clin Infect Dis. 2020:ciaa491. doi: 10.1093/cid/ciaa491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudas G, Bedford T. The ability of single genes vs full genomes to resolve time and space in outbreak analysis. BMC Evol Biol. 2019;19(1):232. doi: 10.1186/s12862-019-1567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grubaugh ND, Faria NR, Andersen KG, Pybus OG. genomic insights into zika virus emergence and spread. Cell. 2018;172(6):1160–1162. doi: 10.1016/j.cell.2018.02.027. [DOI] [PubMed] [Google Scholar]
- 14.Clade and lineage nomenclature, July 4, 2020. [Updated 2020]. [Accessed July 7, 2020]. https://www.gisaid.org/references/statements-clarifications/clade-and-lineage-nomenclature-aids-in-genomic-epidemiology-of-active-hcov-19-viruses/
- 15.Rambaut A, Holmes EC, O'Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5(11):1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Böhmer MM, Buchholz U, Corman VM, Hoch M, Katz K, Marosevic DV, et al. Investigation of a COVID-19 outbreak in Germany resulting from a single travel-associated primary case: a case series. Lancet Infect Dis. 2020;20(8):920–928. doi: 10.1016/S1473-3099(20)30314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sekizuka T, Itokawa K, Kageyama T, Saito S, Takayama I, Asanuma H, et al. Haplotype networks of SARS-CoV-2 infections in the Diamond Princess cruise ship outbreak. Proc Natl Acad Sci U S A. 2020;117(33):20198–20201. doi: 10.1073/pnas.2006824117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meredith LW, Hamilton WL, Warne B, Houldcroft CJ, Hosmillo M, Jahun AS, et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect Dis. 2020;20(11):1263–1272. doi: 10.1016/S1473-3099(20)30562-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung J, Hong MJ, Kim EO, Lee J, Kim MN, Kim SH. Investigation of a nosocomial outbreak of coronavirus disease 2019 in a paediatric ward in South Korea: successful control by early detection and extensive contact tracing with testing. Clin Microbiol Infect. 2020;26(11):1574–1575. doi: 10.1016/j.cmi.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22(13):30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhee C, Baker M, Vaidya V, Tucker R, Resnick A, Morris CA, et al. Incidence of nosocomial COVID-19 in patients hospitalized at a large US academic medical center. JAMA Netw Open. 2020;3(9):e2020498. doi: 10.1001/jamanetworkopen.2020.20498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ran L, Chen X, Wang Y, Wu W, Zhang L, Tan X. Risk factors of healthcare workers with corona virus disease 2019: a retrospective cohort study in a designated hospital of Wuhan in China. Clin Infect Dis. 2020;71(16):2218–2221. doi: 10.1093/cid/ciaa287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong Y, Mo X, Hu Y, Qi X, Jiang F, Jiang Z, et al. Epidemiology of COVID-19 among children in China. Pediatrics. 2020;145(6):e20200702. doi: 10.1542/peds.2020-0702. [DOI] [PubMed] [Google Scholar]
- 25.Qiu H, Wu J, Hong L, Luo Y, Song Q, Chen D. Clinical and epidemiological features of 36 children with coronavirus disease 2019 (COVID-19) in Zhejiang, China: an observational cohort study. Lancet Infect Dis. 2020;20(6):689–696. doi: 10.1016/S1473-3099(20)30198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai Y, Yao L, Wei T, Tian F, Jin DY, Chen L, et al. Presumed asymptomatic carrier transmission of COVID-19. JAMA. 2020;323(14):1406–1407. doi: 10.1001/jama.2020.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamagishi T, Kamiya H, Kakimoto K, Suzuki M, Wakita T. Descriptive study of COVID-19 outbreak among passengers and crew on Diamond Princess cruise ship, Yokohama Port, Japan, 20 January to 9 February 2020. Euro Surveill. 2020;25(23):28–35. doi: 10.2807/1560-7917.ES.2020.25.23.2000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu XW, Wu XX, Jiang XG, Xu KJ, Ying LJ, Ma CL, et al. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: retrospective case series. BMJ. 2020;368:m606. doi: 10.1136/bmj.m606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sutton D, Fuchs K, D'Alton M, Goffman D. Universal screening for SARS-CoV-2 in women admitted for delivery. N Engl J Med. 2020;382(22):2163–2164. doi: 10.1056/NEJMc2009316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182(4):812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics. 2020;112(5):3588–3596. doi: 10.1016/j.ygeno.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The fifteen sites that are variable among the case genomes and not missing in any case genome
Data Availability Statement
The raw reads from the total RNA sequencing of nasopharyngeal samples (n = 15) were deposited in the NCBI SRA as BioProject number PRJNA645035. The SARS-CoV-2 genome sequences recovered in this study (n = 11) were deposited in GISAID with accession numbers EPI_ISL_485001 - EPI_ISL_485002 and EPI_ISL_485388 - EPI_ISL_485396.