

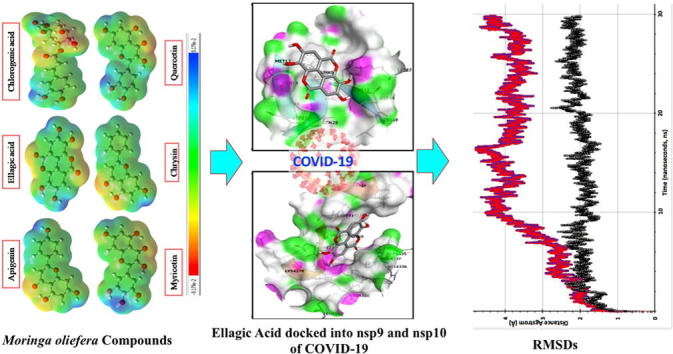
Graphical abstract
Keywords: COVID-19, Moringa oliefera, Molecular docking, Molecular dynamics, ADMET
Abstract
The interactions of two crucial proteins of COVID-19 have been investigated with potential antiviral compounds from Moringa oliefera using quantum chemical, molecular docking and dynamic methods. The results of the present investigation show that ellagic acid and apigenin possess the highest binding affinities of −7.1 and −6.5 Kcal.mol-1against nsp9 and −6.9 and −7.1 Kcal.mol−1 against nsp10, respectively. The dynamic behavior of individual proteins and their respective best docked ligand–protein complexes are also studied at 30 ns timescale. Both of these compounds also show the highest intestinal absorption and total clearance rate as compared to the other compounds under present investigation without any toxicity.
1. Introduction
In December 2019, the virus SARS-CoV-2 emerged in Wuhan city of Hubei province of China and spread speedily throughout the world [1]. The world health organization (WHO) declared it as pandemic on March 12, 2020 [2]. The worldwide number of COVID-19 cases and deaths has reached to 95,482,598 and 2,039,653 respectively as reported by January 17, 2021[3]. This novel virus belongs to the family coronaviridae and spherical in shape. It spreads primarily via saliva, droplets, or nasal discharges of an infected individual after coughing or sneezing [4]. These droplets can spread upto 1–2 m and can remain viable on surfaces for days [5].
Clinical features of COVID-19 ranges from asymptomatic state to severe respiratory distress syndrome. The common clinical symptoms include fever, sore throat, cough, fatigue, headache and breathlessness [6]. The laboratory result of patients with severe COVID-19 presents increased neutrophil count, the most abundant type of white blood cells in human body [7]. They play a major role in line of defense as the first responders against acute infection. Neutrophil chemotaxis is stimulated by chemical signals such as interleukin-6/8 (IL-6/8) and recruit to the site of infection where they assemble and release more pro-inflammatory molecules which in response stimulate further inflammatory signals, progressing to cytokine storms that leads to failure of vital organs [8]. Furthermore, a large number of neutrophils were reported to found in alveolar cavities of serious patients suffering from COVID-19, which validates their roles in cytokine storms [9]. In this way, aberrant neutrophil activity and cytokine response contributes to SARS-CoV-2 pathogenesis in COVID-19. It is reported that two non-structural proteins 9/10(nsp9/nsp10) promote the IL-6/IL-8 mediated neutrophils chemotaxis and stimulate the host inflammatory response as observed in case of COVID-19[10]. In a study, two anti-inflammatory drugs, pemirolast and eriodictyol were docked against nsp9 with the resulting binding energy of −6.5 kcal/mol and further suggest their role in inhibiting infection. It has been further reported that retonavir and remdesivir are potential drugs against nsp10, suggesting their ability to inhibit viral transcription [11]. Thus, both proteins can be proved as a potential therapeutic target in combating SARS-CoV-2 pathogenesis.
Today, computational drug discovery program has been gaining popularity and success because of its potential to discover robust molecule prior to its synthesis, whereas any traditional drug discovery approach is expensive and may take decades to accomplish [12], [13]. It is not affordable to wait till the discovery of any potential drug for COVID-19. Molecular docking is considered more useful in designing, evaluation and comparison of new drugs as it allows the examination of interaction of molecules in 3-dimensional (3D) space by considering the different forms of molecules and determines the factors that play role in vital pharmacological interaction [14]. Other approaches including in silico ADMET analysis, prediction of drug likeness and toxicity are also adopted to analyze potential drugs from several databases. These computational mediated approaches save the experimental cost and time in the process of drug discovery [15]. Moreover, frontier molecular orbitals (HOMO and LUMO) energies and molecular electrostatic potential (MEP) are widely used to determine their chemical reactivity of ligands. The current study is based on in silico analysis which was aimed to evaluate the interaction of selected components of Moringa oliefera against nsp9 and nsp10. M. oliefera is a highly valued plant because of its medicinal value. This plant has showed its efficacy against HIV, HBV, HSV, EBV, NDV, FMDV and AIDS [16]. The present study will provide several powerful insights for the researchers in quest for therapeutic intervention against COVID-19 pathogenesis.
2. Computational methodology
Autodock Vina (ADV) [17] was used for molecular docking studies to calculate the binding affinity and type of interactions between the ligands and targets (COVID-19 nsp9/10). Ligands and proteins were prepared for docking analysis by using Autodock 1.5.6 tool from MGL Tools (The Scripps Research Institute, Molecular Graphics Laboratory, 10,550 North Torrey Pines Road, CA, 92037). The substrate or protein macromolecules were treated as rigid target while ligands were kept as flexible with different rotatable bonds. The quantum chemical studies for the molecular structural analysis of ligands were performed using GAUSSIAN 09 suit of program[18]while orbitals and electrostatic potentials are visualized by GaussView5 [19]. For quantum chemical calculations of ligand structures, their chemical structures were optimized (the lowest energy conformation) by density functional theory (DFT) method with B3LYP/6-311G* level of theory. The substrate or protein macromolecules were treated as rigid target while ligands were kept as flexible with different rotatable bonds. There are various docking protocols whose details are given below:
2.1. Macromolecules/proteins preparation
Crystal structures of nsp9 (PDB ID: 6W4B) and nsp16/10 complex (PDB ID: 6WVN) were retrieved in .pdb format from the Protein Data Bank (https://www.rcsb.org/). PDB is a worldwide archive to access the 3D structure of biological macromolecules [20]. 6WVN is a heterodimer containing nsp16 and nsp10 in a complex form [21]. In this study, we used a single chain of nsp10 for docking analysis. The MGL Tools were used for protein preparation. The water molecules and co-crystallized ligands were deleted from the macromolecule and polar hydrogens were added.
2.2. Ligands
The initial 3D structures of selected ligands were retrieved in .sdf format from PubChem (https://pubchem.ncbi.nlm.nih.gov/). PubChem is a public accessible repository for chemical substances and related biological activities [22]. The optimized structures were then converted into .pdb format.
2.3. Docking protocol
The PDB files of both ligands and proteins were converted in an extended PDB format, termed PDBQT for performing molecular docking analysis using ADV. Docking was performed with the grid box sized of 40 × 40 × 40 and spacing of 1 Å. The output pdbqt files were written into a config. (Configuration) file. The conformation with the lowest binding energy was considered as the most stable conformations of the ligand with respect to the macromolecule. The results were analyzed by using free version of Biova Discovery Studio 2020 client (Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017, San Diego: Dassault Systèmes, 2016).
2.4. ADMET study and drug likeness
ADMET (Absorption, distribution, metabolism, excretion and toxicity) profile exhibits vital importance in pharmaceutical industry and required to reduce undesired effects [23]. In this study, we used online server of pkCSM to predict drug likeness and ADMET profile. The pkCSM is an open database server which provides information regarding drug pharmacokinetics [24]. The SMILES of the selected ligands were taken from PubChem and submitted to pkCSM. The server database was run by selecting toxicity ADMET module.
3. Results and discussion
3.1. Ligand selection and structural chemistry
Molecular docking was done to depict the mode of action of antiviral constituents of M. oliefera against nsp9 and nsp10. M. oleifera is known as “miracle tree” because it is highly nutritious and has the ability to improve the immune system with antiulcer, antipyretic, antiplasmodic, antioxidant, antibacterial properties [25]. It has been shown that M. oliefera contains flavonoids. Flavonoids are proved to have antiviral activity, especially against syncytial virus [26], [27]. A total of six ligands were selected based on their reported antiviral activity. Their chemical structures can be seen in Figure S1. Apigenin, quercetin, myricetin and chrysin are flavonoid constituents of M. oliefera with the reported ability to hinder viral growth. Ellagic acid and chlorogenic acid, being phenolic compounds, has been suggested as anti-HIV/HSV candidates [16], [28], [29].
3.1.1. Frontier molecular orbital (FMO) analysis of ligand structures
The pharmacological properties of ligands are also related to their electronic structures. It is also important to investigate the energy and distribution patterns of their orbitals especially the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), which are usually call frontier molecular orbitals (FMOs). To shed light on the electronic structural properties of our selected ligands, we have optimized their structures using density functional theory (DFT) method at B3LYP/6-311G* level of theory. The energy gap between HOMO and LUMO orbitals is important factor for assessing thermal stability and reactivity of a molecule [30]. According to Koopmans' theorem[31], the HOMO energy presents the ability to donate an electron and the LUMO energy is related to accept an electron. Both of these properties (HOMO and LUMO) have significant roles in thermal stability and reactivity of a compound [32]. The values of HOMO and LUMO energies and their energy gap (ΔEHL) calculated in aqueous phase can be seen in Fig. 1 . Overall, there are no drastic changes found among the ΔEHL values of selected ligands where the maximum difference of 0.29 eV was found between the ΔEHL values of Quercetin and Chrysin. The increasing order of the ΔEHL is Quercetin < Myricetin < Chlorogenic acid < Ellagic acid < Apigenin < Chrysin. It can be seen that Ellagic acid, Apigenin and Chrysin possess higher energy gaps and better thermal stabilities in aqueous phases.(See Fig. 2 )
Fig. 1.

The Frontier molecular orbitals including HOMO and LUMO for all studied ligands as calculated at B3LYP/6-311G* level of theory at iso-values of ± 0.002 a. u.
Fig. 2.

Molecular electrostatic potential (MEP) plots for all studied ligands as calculated at B3LYP/6-311G* level of theory with iso-values of ± 0.002 a. u.
3.1.2. Molecular electrostatic potential of ligand structures
The presence of partial charges on ligand and protein molecules plays a crucial role in leading the docking of ligand with protein. Quantum chemically, the molecular electrostatic potential (MEP) diagram can be helpful to get insights into the 3-dimentional structural and topological features of ligands. Molecular electrostatic potential on the total density surface of a compound indicates whether the effect of nuclei or electrons are dominant over a specific point of molecular geometry [33]. It is important to pen down here that the MEPs are used in a semi quantitative way to study the 3-dimentional structural features. For instance, blue region represents the most positive electrostatic potential, red color shows the areas with the most negative electrostatic potential and green areas with nearly zero potentials. To be more specific, the electrostatic potential decreases in the order of blue ˃ green ˃ yellow ˃ orange ˃ red color coding. In our selected ligands, the most of the negative regions (red) are around carbonyl groups owing to their electron abundance environment, which are also attractive sites for electrophilic attack. On the other hand, more positive regions (blue) are mainly towards the protonic H-atom of hydroxyl groups (OH) which will potentially act as H-bond donors in protein–ligand intermolecular interactions [34].
3.2. Binding energy and inhibition constant
The basics of designing rational drug is to utilize the knowledge of the protein–ligand binding mechanisms and structural data to explore the potential of uncovering new drug candidates. So, an extensive understanding of the molecular interaction/recognition nature is also of great value, which will provide insights about designing, developing and discovering drugs [35]. Molecular docking is an extensively used computational tool for predicting binding modes in protein–ligand interaction [36]. In present study, the molecular docking was done by adopting a grid based technique as implemented AutoDock Vina. All six ligands were docked explicitly against nps9 (Figure S2a) and nsp10 (Figure S2b) of COVID-19. Table 1 revealed that binding affinity of our ligands with COVID-19 nsp9 ranged from −6.1 to −7.1 kcal/mol. The binding affinity of antiviral ligands for nsp10 of COVID-19 was only slightly different from that of nsp9. The binding affinities for nsp10 of COVID-19ranged from –6.8 to −7.1 kcal/mol. Among all compounds, ellagic acid showed the highest binding affinity to nsp9 (Figure S3) with an inhibition constant (ki) of 5.98 µM. The ki can be used for accurate appreciation of inhibitory potential of the drug. The Ki value reflects the amount of drug required to reach the 50% inhibition [37]. Whereas, apigenin exhibited the highest affinity in case of nsp10 (Figure S4) with same ki value as in prior case. These results showed that all selected ligands exhibit good binding affinity with our target molecules. Moreover we selected Remdesivir as a standard drug [38] and Niaziminin, well known for its anticancer properties [39], docked against the both targeted proteins. The results were compared with our selected antiviral ligands (Table S2), which further suggest their affinity with the targeted proteins.
Table 1.
Molecular docking analysis of different compounds against nsp9 and nsp10 using AutoDock Vina.
| Compounds | Protein | Binding Energy (kcal/mol) | Inhibition constatnt Ki (µM) | Protein | Binding Energy (kcal/mol) | Inhibition constatnt Ki (µM) |
|---|---|---|---|---|---|---|
| Apigenin | nsp9 | −6.5 | 16.52 | nsp10 | −7.1 | 5.98 |
| Ellagic acid | −7.1 | 5.98 | −6.9 | 8.39 | ||
| Chlorogenic acid | −6.6 | 13.94 | −6.8 | 9.94 | ||
| Myricetin | −6.1 | 32.53 | −6.7 | 11.78 | ||
| Chrysin | −6.8 | 9.94 | −7.0 | 7.08 | ||
| Quercetin | −6.6 | 13.94 | −7.0 | 7.08 |
3.3. Protein and ligand interactions
To analyze the detailed interaction of best conformation, free version of Biova Discovery Studio Visualizer was used. The interaction of ellagic acid with nsp9 showed the highest affinity interaction in chain B (Fig. 3 a) and makes hydrogen bonding between oxygen of carbonyl group with the hydrogen of SER 47, ASP 48 and LYS 87 residues of nsp9. This high binding affinity can be attributed to various van der Waal forces with the respective amino acids SER 14, ASP 27 and ASN 28 (Fig. 3b). While in case of nsp10, ellagic acid makes hydrogen bond with ASP 4275 and ALA 4277. Other noticeable interactions are with CYS 4270, ALA 4271, VAL 4274, ALA 4276, PRO 4437 forming π-alkyl interactions and van der Waals forces with LEU 4267, LYS 4278, TYR 4329, ILE 4334, ASP 4335 (Fig. 3d). On the other side, apigenin binds to nsp9 surrounding by both chains A and B (Fig. 4 a), which leads to make several interactions. Its benzene ring forms π-alkyl bond with LEU 104, ALA 108, LEU 113, while VAL 8 shared both π-alkyl and π-sigma bond with apigenin (Fig. 4b). In case of nsp10, apigenin interacts with ASN 4293, CYS 4294 by H-O bond. Other interactions include van der Waals, a single π -alkyl and C-H bond (Fig. 4d). By binding to chain A of nsp9 (Fig. 5 a), chlorogenic acid shares hydrogen bond with ARG 40, VAL 42 and SER 60 (Fig. 5b). However, chlorogenic acid shows highest binding affinity with nsp10 including hydrogen bonding with Val 4274, ASP 4344 and LEU 4365. A large number of van der Waals interaction can be seen with ALA 4271, ALA 4276, ALA 4277, CYS 4327, TYR 4329, LEU 4345, THR 4364, ASN 4388 and CYS 4343, (Fig. 5d).
Fig. 3.

The Ellagic Acid as docked into the pocket of total density surface (pink) of nsp9 (a), nsp10 (c) the total density surface as represented with H-bond donor and acceptor moieties with green and pink meshes colors. 2-D plot of binding interactions of Ellagic acid with nsp9 (b), nsp10 (d).
Fig. 4.

The Apigenin as docked into the pocket of total density surface (pink) of nsp9 (a), nsp10 (c) the total density surface as represented with H-bond donor and acceptor moieties with green and pink meshes colors. 2-D plot of binding interactions of Apigenin with nsp9 (b), nsp10 (d).
Fig. 5.

The Chlorogenic Acid as docked into the pocket of total density surface (pink) of nsp9 (a), nsp10 (c) the total density surface as represented with H-bond donor and acceptor moieties with green and pink meshes colors. 2-D plot of binding interactions of Chlorogenic acid with nsp9 (b), nsp10 (d).
The docking of myricetin with the nsp9 of COVID-19 (Fig. 6 b) revealed that it shows binding affinity to chain B of nsp9 with six hydrogen bonds between SER 14, ALA 16, ASP 26, ASP 27, ARG 40, ARG 50. Moreover, its benzene rings make π-alkyl bond with PRO 58. In nsp10 (Fig. 6c), myricetin binds to core pocket region which allows it to make more interaction. Besides hydrogen bonding with CYS 4356, ASN 4358, LYS 4366, ASN 4367, TYR 4379, myricetin also makes van der Waals and π-alkyl interactions. It also makes one C-H interaction with PHE 4363 and π-alkyl bond with LEU 4284 (Fig. 6d). Results obtained by docking of chrysin with nsp9 of COVID-19 showed more π-alkyl bonds with LEU 5, LEU 104, ALA 108, LEU 113 and, with a single hydrogen bond with SER 6 (Fig. 7 b). However, chrysin shares two hydrogen bonds with ASN 4293, CYS 4294 with nsp10 (Fig. 7d). The docking of quercetin docked in nsp9 of COVID-19 shows significant binding with chain A (Fig. 8 a). The interaction between quercetin and nsp9 is characterized by hydrogen bonding with GLY 38 and ARG 40. Some of the van der Waals and π-alkyl have also been observed (Fig. 8b). Quercetin forms hydrogen bond with LEU 4284, LYS 4366, ASN 4358 and ASN 4367 of nsp10 with a single π-anion and π-alkyl bond with ASP 4359 and CYS 4356, respectively (Fig. 8d). These results indicate that docked ligands forms a stable complex with targeted proteins (nsp9/10), hence we can conclude that our selected ligands might have antiviral property.
Fig. 6.

The Myricetin as docked into the pocket of total density surface (pink) of nsp9 (a), nsp10 (c) the total density surface as represented with H-bond donor and acceptor moieties with green and pink meshes colors. 2-D plot of binding interactions of Myricetin with nsp9 (b), nsp10 (d).
Fig. 7.

The Chrysin as docked into the pocket of total density surface (pink) of nsp9 (a), nsp10 (c) the total density surface as represented with H-bond donor and acceptor moieties with green and pink meshes colors. 2-D plot of binding interactions of Chrysin with nsp9 (b), nsp10 (d).
Fig. 8.

The Quercetin as docked into the pocket of total density surface (pink) of nsp9 (a), nsp10 (c) the total density surface as represented with H-bond donor and acceptor moieties with green and pink meshes colors. 2-D plot of binding interactions of Quercetin with nsp9 (b), nsp10 (d).
3.4. MD simulations
After completing molecular docking studies by using rigid crystal structure of nsp9 and nsp10, MD simulation was done to analyze the dynamic behavior of targeted proteins and lead compounds. We performed MD simulations for 30 ns (3 × 106 fs) time scale for the best docked protein–ligand complexes comprising of ellagic acid-nsp9 and apigenin-nsp10 complexes. All molecular dynamic calculations were performed using CHARMM force-field [40] in NAMD [41] while trajectory, RMSD and other analysis were done by VMD program [42] as developed at NIH Center for Macromolecular Modeling and Bioinformatics, Theoretical and Computational Biophysics group. Further details about molecular dynamic calculations and its adopted input parameters can be seen in supporting information of the article. The values of RMSDs for the C-α atoms of apo-forms (only proteins without docked ligands) of nsp9 and nsp10 proteins and their respective complexes were evaluated to analyze thermodynamic conformational stability during 30 ns time period of MD simulations. The RMSD graphs of ellagic acid-nsp9 complex and its apo-form nsp9 were shown in Fig. 9 (a) while for the apigenin-nsp10 complex and its apo-form nsp10 were shown in Fig. 10 (a). A careful analysis of RMSD plots shows a different trend in the RMSD plots for both proteins and their respective ligand complexes. For example, the RMSD of apo-form of nsp9 indicates that after 1 ns, it is equilibrated from 1 to 30 ns and fluctuates mainly from 1.6 to 2.7 Å. Unlike its apo-form protein, the RMSD of ellagic acid-nsp9 complex indicates a different behavior where it rose upto 3.3 Å in the first 10 ns and then it is equilibrated from 10 to 30 ns and fluctuates mainly from 3.6 to 4.7 Å (see Fig. 9 (a)). The RMSD analysis of ellagic acid-nsp9 complex indicates a profound effect of ellagic acid docking, which after 10 ns shows relatively stable ligand–protein complex. Similarly, the RMSD graphs of apigenin-nsp10 complex and its apo-form nsp10 were shown in Fig. 10(a). These RMSDs are different as compared with those for nsp-9 and its complex. For example, during 6 ns time, the apigenin docking disturb the RMSD of nsp-10 and then both RMSDs have been equilibrated from 6 to 30 ns showing some fluctuations in similar range of distance which is between ~ 3.8 to ~ 5.5 Å as shown in Fig. 9 (a). Thus, we assume that apigenin might have not shown a profound impact on the RMSD of nsp10 protein. Besides this, we have also provided the structural changes during MD simulations over the 30 ns in the form of different frames of MD trajectory which are taken approximately after every ~ 3 ns. The frames were overlapped with each other to see the differences during trajectory as shown in Fig. 9(c) and 10(c) while their respective single protein structures were also shown in Fig. 9(b) and 10(b). A comparative analysis of structural changes among Fig. 9(b) and 9(c) illustrates that nsp9 show relatively more prominent changes where more fluctuations or randomness is seen towards the right side of nsp9 where ellagic acid is docked. While on the other hands, the structural changes among Fig. 10(b) and 10(c) are less obvious which might be due to less intense interactions/effects of apigenin over nsp10. Somewhat similar findings were also observed in their RMSDs which have shown more fluctuations in ellagic acid-nsp9 complex as compared with apigenin-nsp10 complex. Thus, it might be reasonable to say that the effect of ellagic acid is more obvious on nsp9 as compared to apigenin for nsp10 as studied by molecular docking and molecular dynamic studies here.
Fig. 9.

RMSDs of nsp9 (black color) and Ellagic acid-nsp9 complex (red color) (a), the apo-form of nsp9 crystal structure (b), different structural frames of nsp9-Ellagic acid complex from 0 to 30 ns MD simulations.
Fig. 10.

RMSDs of nsp10 (black color) and Apigenin-nsp10 complex (radish blue color) (a), the apo-form of nsp10 crystal structure (b), different structural frames of nsp10-Apigenin complex from 0 to 30 ns MD simulations.
3.5. Drug likeness
A candidate molecule is of no use and does not matter how good it fits with the receptor, if its absorption is poor or if it is excreted too slowly from the body. So, it is always necessary to predict the drug likeness, for which Lipinski’s rule of 5 was used. According to this rule, any ligand is considered as a drug-like if it meets certain requirements including molecular weight < 500 Dalton, number of H-bond acceptor < 10, number of H-bond donors < 5 and lipophilicity represented as log P < 5 [43]. All our selected ligands obeyed this rule with just one violation in case of chlorogenic acid and myricetin (H-bond donors greater than 5) (Table S1), which strongly suggests their suitability as a drug.
3.6. ADMET profile
In drug designing process, it is very important to predict human pharmacokinetic properties which help in identification and progression of candidate molecules that can be successful in the clinic [44].
3.6.1. Absorption
Absorbance in the small intestine is one of the vital processes for determining the drug bioavailability after oral administration [45]. It is worth mentioning that all selected constituents of M. oliefera have intestinal absorbance more than 30%, especially of apigenin and chrysin, which describes their ability to absorb easily. Moreover, all these constituents showed water solubility (log S) greater than −5, which reflect their solubility in water at 25˚C (Table 2 ) [24]. Our results also showed that none of our selected drug is P-glycoprotein I/II inhibitor which is a very essential aspect in pharmacokinetics study. Any drug that can inhibit these transporters can lead to interfere with the pharmacokinetics of other drugs [46]. So, it can be suggested that these drugs can be safe and effective adjuvant agents with any pharmaceutical drug against Sars-CoV-2 pathogenesis.
Table 2.
ADMET analysis of selected ligands using pkCSM online database server.
| ADMET | Parameters | Apigenin | Ellagic acid | Chlorogenic acid | Myricetin | Chrysin | Quercetin |
|---|---|---|---|---|---|---|---|
| Absorbance | Water solubility (log S) mol/L | −3.329 | −3.181 | −2.449 | −2.915 | −3.538 | −2.925 |
| Intestinal absorption % | 93.25 | 86.684 | 36.377 | 65.93 | 93.761 | 77.207 | |
| P-glycoprotein I/II inhibitor | No | No | No | No | No | No | |
| Distribution | log VDss (L/Kg) | 0.822 | 0.375 | 0.581 | 1.317 | 0.403 | 1.599 |
| Metabolism | CYP2D6 substrate | no | No | No | No | No | No |
| CYP3A4 substrate | no | No | No | No | No | No | |
| CYP2D6 inhibitor | No | No | No | No | No | No | |
| CYP3A4 inhibitor | No | No | No | No | No | No | |
| Excretion | Total clearance (Log ml/min/kg) | 0.566 | 0.537 | 0.307 | 0.422 | 0.405 | 0.407 |
| Renal OCT2 substrate | No | No | No | No | No | No | |
| Toxicity | AMES | No | No | No | No | No | No |
| Max. tolerable dose (log mg/kg/day) | 0.328 | 0.476 | −0.134 | 0.51 | 0.016 | 0.499 | |
| Hepatotoxicity | No | No | No | No | No | No |
3.6.2. Distribution
The steady state volume of distribution (VDss) shows the dose of drug needed to distribute uniformly as in blood and plasma. It is considered low if log VDss is less than −0.5 and high if it is more than 0.45. Higher VDss value presents greater drug distribution in plasma rather than tissue and lower VDss shows the poor ability of drug to transverse or diffuse across the cell membrane[47].Our results show that ellagic acid and chrysin are ideal candidates in respect to distribute uniformly in blood plasma (see Table 2).
3.6.3. Metabolism
Another factor that can affect the pharmacokinetics interactions is drug metabolizing enzymes. Cytochrome P450 is an important detoxification enzyme in human body, mainly in liver. Any alteration in activity of these enzymes can lead to disturbance in pharmacokinetics of drugs that are metabolized by these enzymes [48]. There are two most important isoforms of cytochrome P450: cytochrome P2D6 and cytochrome P3A4 [49]. Table 2 shows that those compounds don’t inhibit or affect these enzymes. So, it can be interpreted that all these compounds are safe as adjuvant agents with other drugs.
3.6.4. Excretion
Total clearance quantifies the removal of drug from blood or plasma. Clearance (drug elimination process) results from both kidney and liver[50]. Total clearance Log (CLtot) predicts combination of renal clearance and hepatic clearance. It is linked to bioavailability and vital to achieve steady-state concentrations by determining dosing rates [24]. A steady-state level is reached when the drug is administered at appropriate concentration and bioavailable. The higher the CLtot value, the faster the excretion process of the compound[49]. Table 2 shows the total clearance values from which their rate of excretion can be predicted. Apigenin and ellagic acid has the highest CLtot value as compared to the others. Moreover, it can be seen that all of the compounds are not substrate of renal OCT2. OCT2 (organic cation trasnsporter 2) is a protein transporter having vital role in renal drug clearance. Substrates of OCT2 can interact with OCT2 inhibitors and may lead to adverse reactions [24].
3.6.5. Toxicity
Toxicity of the compound can be predicted by AMES toxicity which predicts if any compound has mutagenic potential or not [24]. Table 2 reveals that all of our compounds do not have mutagenic effects. Another important aspect in drug development research is determining the ability of any drug in inducing liver injury. From table 2, it can be seen that all our compounds are not hepatotoxic.
4. Conclusion
Thus, a combined quantum chemical, molecular docking and dynamic study was performed for nsp9 and nsp10 of COVID-19 with antiviral constituents of M. Oliefera to assess a new potential therapeutic drug candidate. Unlike previous studies, we have firstly studied the structural chemistry of ligands in the form of their optimized geometries, HOMO-LUMO orbital energies, reactivity parameters, the 3-D orbital distribution patterns and MEPs to get insights into the structure–reactivity of ligands. All the potential ligands were docked with nsp9 and nsp10 nonstructural proteins of COVID-19. These results revealed that all selected ligands form the stable complexes with the targeted proteins and showed the highest binding affinity values of apigenin (-7.1 kcal/mol) for nsp10 and ellagic acid (-7.1 kcal/mol) for nsp9, which are in line to our previous quantum chemical results. The dynamic behavior of individual proteins and their respective best docked ligand–protein complexes are also studied at 30 ns timescale, which showed different effects of ligands on docking with these proteins as assessed by their respective RMSDs. Based on quantum chemical results, apigenin (4.17 eV) and ellagic acid (4.32 eV) showed lower HOMO-LUMO energy gaps and predicted for better chemical reactivity. Additionally, ADMET analysis by pkCSM revealed that both of these compounds also have the highest absorption and clearance rate in comparison with other ligands. The present investigation highlighted the significant potential of studied ligands for antiviral activity against SARS-COV-2. These results suggest further in vitro and in vivo studies for establishing a strong experimental evidence of activity of antiviral constituents of M. oliefera against SARS-COV-2.
Credit author statement
S. Muhammad conceived the idea, S.H. Hassan and A.G. Al-Sehemi have performed calculations, H.A. Shakir and M. Khan played important role to study docking interactions. While M. Irfan and J. Iqbal helped a lot in writing results and discussion. Besides this, S. Muhammad and S. H. Hassan also performed molecular dynamic calculations.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors from King Khalid University extend their appreciation to the Institute of Research and Consulting Studies at King Khalid University for supporting this research through grant number 2-N-20/22 and the support of Research Center for Advanced Materials Science is highly acknowledged. For computer time, this research used the resources of the Supercomputing Laboratory at King Abdullah University of Science & Technology (KAUST) in Thuwal, Saudi Arabia.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cplett.2021.138379.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Dong L., Hu S., Gao J. Drug discoveries & therapeutics. 2020;14:58. doi: 10.5582/ddt.2020.01012. [DOI] [PubMed] [Google Scholar]
- 2.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Science. 2020;368:409. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Worldometer, COVID-19 CORONAVIRUS PANDEMIC, 2021.
- 4.Pant S., Singh M., Ravichandiran V., Murty U., Srivastava H.K. J. Biomol. Struct. Dyn. 2020:1. doi: 10.1080/07391102.2020.1757510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kampf G., Todt D., Pfaender S., Steinmann E. J. Hosp. Infect. 2020;104:246. doi: 10.1016/j.jhin.2020.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y. The Lancet. 2020;395:507. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K., Zuo P., Liu Y., Zhang M., Zhao X., Xie S., Zhang H., Chen X., Liu C. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolaczkowska E., Kubes P. Nat. Rev. Immunol. 2013;13:159. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 9.C. Wang, J. Xie, L. Zhao, X. Fei, H. Zhang, Y. Tan, L. Zhou, Z. Liu, Y. Ren, L. Yuan, (2020).
- 10.Q. Liang, J. Li, M. Guo, X. Tian, C. Liu, X. Wang, X. Yang, P. Wu, Z. Xiao, Y. Qu, bioRxiv (2020).
- 11.Deshpande R.R., Tiwari A.P., Nyayanit N., Modak M. Eur. J. Pharmacol. 2020;886:173430. doi: 10.1016/j.ejphar.2020.173430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gahtori J., Pant S., Srivastava H.K. Mol. Diversity. 2019:1. doi: 10.1007/s11030-019-10015-y. [DOI] [PubMed] [Google Scholar]
- 13.Romano J.D., Tatonetti N.P. Front. Genet. 2019;10:368. doi: 10.3389/fgene.2019.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.N. Mohammadi, N. Shaghaghi, Preprint. https://doi/. org/10.26434/chemrxiv 11987475 (2020) v1.
- 15.S. Vardhan, S.K. Sahoo, arXiv preprint arXiv:2005.07955 (2020).
- 16.Biswas D., Nandy S., Mukherjee A., Pandey D., Dey A. S. Afr. J. Bot. 2020;129:272. [Google Scholar]
- 17.Trott O., Olson A.J. J. Comput. Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, Gaussian Inc, Wallingford (2009).
- 19.R. Dennington, T. Keith, J. Millam, GaussView, Version 5 (2009).
- 20.Burley S.K., Berman H.M., Kleywegt G.J., Markley J.L., Nakamura H., Velankar S. Protein Crystallography, Springer. 2017:627. doi: 10.1007/978-1-4939-7000-1_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.M. Rosas-Lemus, G. Minasov, L. Shuvalova, N.L. Inniss, O. Kiryukhina, G. Wiersum, Y. Kim, R. Jedrzejczak, N.I. Maltseva, M. Endres, bioRxiv (2020).
- 22.Bolton E.E., Wang Y., Thiessen P.A., Bryant S.H. Annual reports in computational chemistry. Elsevier. 2008:217. [Google Scholar]
- 23.Curreli F., Kwon Y.D., Belov D.S., Ramesh R.R., Kurkin A.V., Altieri A., Kwong P.D., Debnath A.K. J. Med. Chem. 2017;60:3124. doi: 10.1021/acs.jmedchem.7b00179. [DOI] [PubMed] [Google Scholar]
- 24.Pires D.E., Blundell T.L., Ascher D.B. J. Med. Chem. 2015;58:4066. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kini S.G., Wong K.H., Tan W.L., Xiao T., Tam J.P. BMC Plant Biol. 2017;17:68. doi: 10.1186/s12870-017-1014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das S., Sarmah S., Lyndem S., Singha Roy A. J. Biomol. Struct. Dyn. 2020:1. doi: 10.1080/07391102.2020.1763201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shinwari S., Ahmad M., Luo Y., Zaman W. Pak. J. Bot. 2017;49:725. [Google Scholar]
- 28.Khan M.T.H., Ather A., Thompson K.D., Gambari R. Antiviral Res. 2005;67:107. doi: 10.1016/j.antiviral.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Kurokawa M., Wadhwani A., Kai H., Hidaka M., Yoshida H., Sugita C., Watanabe W., Matsuno K., Hagiwara A. Phytother. Res. 2016;30:797. doi: 10.1002/ptr.5580. [DOI] [PubMed] [Google Scholar]
- 30.Raja M., Muhamed R.R., Muthu S., Suresh M. J. Mol. Struct. 2017;1141:284. [Google Scholar]
- 31.Koopmans T. physica. 1934;1:104. [Google Scholar]
- 32.Shafieyoon P., Mehdipour E., Mary Y.S. J. Mol. Struct. 2019;1181:244. [Google Scholar]
- 33.Kuruvilla T.K., Prasana J.C., Muthu S., George J., Mathew S.A. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018;188:382. doi: 10.1016/j.saa.2017.07.029. [DOI] [PubMed] [Google Scholar]
- 34.Collado S., Laca A., Diaz M. Chem. Eng. J. 2011;166:940. [Google Scholar]
- 35.Du X., Li Y., Xia Y.-L., Ai S.-M., Liang J., Sang P., Ji X.-L., Liu S.-Q. Int. J. Mol. Sci. 2016;17:144. doi: 10.3390/ijms17020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sousa S.F., Ribeiro A.J., Coimbra J., Neves R., Martins S., Moorthy N., Fernandes P., Ramos M. Curr. Med. Chem. 2013;20:2296. doi: 10.2174/0929867311320180002. [DOI] [PubMed] [Google Scholar]
- 37.Y. Parmentier, M.-J. Bossant, M. Bertrand, B. Walther, (2007).
- 38.N.I.o. Health, Final report confirms remdesivir benefits for COVID-19, 2020.
- 39.Charoensin S. J. Medicinal Plants Research. 2014;8:318. [Google Scholar]
- 40.Best R.B., Zhu X., Shim J., Lopes P.E., Mittal J., Feig M., MacKerell A.D., Jr J. Chem. Theory Comput. 2012;8:3257. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips J.C., Hardy D.J., Maia J.D., Stone J.E., Ribeiro J.V., Bernardi R.C., Buch R., Fiorin G., Hénin J., Jiang W. J. Chem. Phys. 2020;153:044130. doi: 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.W. Humphrey, A. Dalke, K. Schulten, VMD: Visual molecular dynamics (1996) Journal of Molecular Graphics, 14 (1). DOI 10.1016/0263-7855 (96) 00018-5. [DOI] [PubMed]
- 43.Lipinski C.A. Drug Discovery Today: Technologies. 2004;1:337. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 44.Mathialagan S., Piotrowski M.A., Tess D.A., Feng B., Litchfield J., Varma M.V. Drug Metab. Dispos. 2017;45:409. doi: 10.1124/dmd.116.074294. [DOI] [PubMed] [Google Scholar]
- 45.Radchenko E., Dyabina A., Palyulin V., Zefirov N. Russ. Chem. Bull. 2016;65:576. [Google Scholar]
- 46.Lund M., Petersen T.S., Dalhoff K.P. Drugs. 2017;77:859. doi: 10.1007/s40265-017-0729-x. [DOI] [PubMed] [Google Scholar]
- 47.Llorach-Pares L., Nonell-Canals A., Sanchez-Martinez M., Avila C. Mar. Drugs. 2017;15:366. doi: 10.3390/md15120366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tracy T.S., Chaudhry A.S., Prasad B., Thummel K.E., Schuetz E.G., Zhong X.-B., Tien Y.-C., Jeong H., Pan X., Shireman L.M. Drug Metab. Dispos. 2016;44:343. doi: 10.1124/dmd.115.067900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ekowati J., Diyah N.W., Nofianti K.A., Hamid I.S., Siswandono S. Journal of Mathematical and Fundamental Sciences. 2018;50:203. [Google Scholar]
- 50.Smith D.A., Beaumont K., Maurer T.S., Di L. J. Med. Chem. 2018;62:2245. doi: 10.1021/acs.jmedchem.8b01263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.