

Abstract
Sequencing 16S rRNA gene amplicons is the gold standard to uncover the composition of prokaryotic communities. The presence of multiple copies of this gene makes the community abundance data distorted and gene copy normalization (GCN) necessary for correction. Even though GCN of 16S data provided a picture closer to the metagenome before, it should also be compared with communities of known composition due to the fact that library preparation is prone to methodological biases. Here, we process 16S rRNA gene amplicon data from eleven simple mock communities with DADA2 and estimate the impact of GCN. In all cases, the mock community composition derived from the 16S sequencing differs from those expected, and GCN fails to improve the classification for most of the analysed communities. Our approach provides empirical evidence that GCN does not improve the 16S target sequencing analyses in real scenarios. We therefore question the use of GCN for metataxonomic surveys until a more comprehensive catalogue of copy numbers becomes available.
Electronic supplementary material
The online version of this article (10.1007/s00248-020-01586-7) contains supplementary material, which is available to authorized users.
Keywords: 16S rRNA, Metataxonomic surveys, Gene
Amplicon sequencing of 16S rRNA gene is considered a gold standard to evaluate the composition of prokaryotic communities due to (i) low cost, (ii) easy availability, (iii) easy practicality of extraction and preparation kits, (iv) high taxonomic resolution as deep as the level of genera (or sometimes species) and (v) extensive databases. The concept of gold standards implies a level of perfection never attained by any biological test [1] which is why those are constantly challenged and replaced when appropriate [2]. Still, amplicon sequencing outcompetes (88,889 papers with “16S rRNA” as of June 9, 2020) other possible techniques to describe the community structure such as metagenomics (22,106), metaproteomics (1717) or metatranscriptomics (2639) with many thousand publications in recent years. The general practice as shown by the myriads of publications does not comprise the correction of the obtained raw counts by 16S rRNA gene copy numbers per bacterial genome even though it is known that bacteria can have multiple copy numbers of the 16S rRNA gene and the normalization of 16S rRNA amplicon data gave a picture closer to the metagenomes [3]. However, both amplicon and shotgun sequencing are prone to methodological biases introduced by extraction, PCR, sequencing and bioinformatics and could thus similarly diverge from the real picture [4]. Recently, it was recommended not to use GCN based on the systematic evaluation of the predictability of 16S GCNs in bacteria [5], but the validity of GCN in 16S rRNA gene amplicon sequencing has never been shown for communities with known composition. These so-called mock communities are defined mixtures of microbial cells or nucleic acids created in vitro for the simulation of the composition of a microbiome sample, or DNA mixture isolated therefrom are used as a uniform benchmark for microbiome and metagenome technology development and evaluation [6]. We believe that the comparison of amplicon data to the actual relative abundances of taxonomic groups in the community is the only way to verify the validity of GCN in 16S rRNA gene amplicon data. Admittedly, the choice of the mock community (from cells, DNA, RNA, proteins or metabolites), the sample preparation protocol, the primer pair that targets a specific region of the 16S rRNA gene and the processing pipeline can all bias the outcome and must all be considered moving forward.
Here, we processed nine bacterial mock communities from purified genomic DNA and two from cloned 16S rRNA genes in the pUC19 plasmid vector targeting the V4 region of the 16S rRNA gene provided elsewhere [7]. We used DADA2 v1.8 [8] for sequence data processing and the amplicon sequence variants (ASV) classification using the naïve Bayesian classifier method and the SILVA database (version 138) as reference [9], with or without GCN correction, based on the information available in the Ribosomal Database Project (RDP, Release 11, Update 5 from September 30, 2016) [10]. All applied methods are provided in detail as Supplementary Information.
Many pipelines exist for the processing and taxonomic annotation of 16S rRNA gene amplicon data [11, 12], but all taxonomic assignment methods are similarly limited which is why the use of a taxonomical assignment on a higher rank than species ensures both better accuracy and the detection of species without an exact match [8]. Approaches based on machine learning such as TAGME (https://github.com/gabrielrfernandes/tagme) can provide better assignments as the exact matching used by DADA2 yields several ambiguities but are as yet unpublished. The community profile derived from ASVs [13] appeared to better the expected profile than those based on a clustering method, as we reported previously in a study using three of these mock communities [14]. The strategy we used, however, harbours issues with varying gene copy numbers within the same genus that forces GCN to use averages. In addition to that, multiple copies in the same gene can diverge [15] as a result of pseudogene formation or horizontal gene transfer [16]. One of the eleven mock communities (Mock-12) showed a poor fit of the sequencing data to the mock community (shown in red in Table 1) and was therefore removed from the analysis (Fig. 1, legend is shown in Supplementary Fig. S1). Interestingly, the richness of the mock communities was overrepresented by, on average, 27.4% (n = 10, SE = 12.1%) in the sequencing data, but this was mainly caused by low-abundant genera, making up 1.4% (n = 10, SE = 0.1%) of the ASV counts (Fig. 1). Another cause of misidentification are unidentified sequences that made up 4.0% (n = 10, SE = 3.1%). However, DADA2 with RDP seemed to annotate the amplicon data more reliably as Escherichia was not mistakenly identified as Klebsiella within the family Enterobacteriaceae when operational taxonomic units (OTUs) were previously annotated using Blast [14]. Similar to the approach with blasting OTUs [14], many genera such as Bacteroides aligned better with the expected content of the mock community with normalization by GCN but other genera such as Escherichia or Nitrosomonas aligned better without GCN. Altogether, the 16S sequencing data without GCN fitted the mock community composition 7.1% (n = 10, SE = 3.6%) better than with GCN (Fig. 1). This was driven by Mock-18 where Nitrosomonas and Desulfovibrio were misidentified and by Mock-19 where the unidentified ASVs decreased the fitness GCN as the average copy number of bacteria was applied. Both mock communities derived from cloned 16S rRNA genes in the pUC19 plasmid vector while the mock communities from purified DNA showed smaller RSS to the actual community composition. As expected, unidentified ASVs will be overrepresented in the normalized data, while the bacteria with known gene copy numbers could have more, in this case, an average of 6.6 (n = 7, SE = 0.9) in Mock-19. The misrepresentation of the mock community increases with an increasing number of unidentified ASVs, which can further be improved as soon as better classification methods arise.
Table 1.
Misidentification as unidentified ASVs (NA) and ASVs assigned to other genera, the Shannon diversity and richness of bacterial genera as well as the residual sum of squares (RSS) as discrepancy of the sequenced community composition and the structure of the mock community on genus level
| Community | Misidentification | Shannon diversity | Richness | RSS to Mock | |||||
|---|---|---|---|---|---|---|---|---|---|
| NA | Other genera | Mock | Raw | GCN | Mock | Raw | Raw | GCN | |
| Mock-12 | 0.0003 | 0.0461 | 2.0736 | 0.3926 | 0.5765 | 11 | 13 | 1.3138 | 1.3274 |
| Mock-13 | 0.0005 | 0.0057 | 2.8216 | 2.6968 | 2.7120 | 18 | 32 | 0.5198 | 0.5258 |
| Mock-14 | 0.0003 | 0.0103 | 2.8216 | 2.7039 | 2.7456 | 18 | 35 | 0.5245 | 0.4991 |
| Mock-15 | 0.0001 | 0.0038 | 2.8216 | 2.6950 | 2.6591 | 18 | 30 | 0.5447 | 0.5823 |
| Mock-16 | 0.0853 | 0.0913 | 3.7543 | 3.1574 | 3.0887 | 46 | 54 | 0.8441 | 0.9053 |
| Mock-18 | 0.0019 | 0.0000 | 2.7081 | 2.6027 | 2.4329 | 15 | 15 | 0.3089 | 0.5965 |
| Mock-19 | 0.3074 | 0.0000 | 2.3581 | 2.4581 | 2.1697 | 15 | 15 | 0.8829 | 1.1353 |
| Mock-20 | 0.0000 | 0.0001 | 2.7616 | 2.5335 | 2.4519 | 17 | 17 | 0.5107 | 0.5766 |
| Mock-21 | 0.0000 | 0.0000 | 1.6901 | 1.5246 | 1.5091 | 17 | 14 | 0.4041 | 0.3534 |
| Mock-22 | 0.0004 | 0.0292 | 2.7616 | 2.7212 | 2.7024 | 17 | 20 | 0.2978 | 0.4075 |
| Mock-23 | 0.0008 | 0.0017 | 1.6901 | 1.7205 | 1.7214 | 17 | 20 | 0.1938 | 0.1566 |
The mock community (Mock) was compared with the 16S amplicon sequencing data without (raw) or with gene copy normalization (GCN). Mock-12 was removed from the analysis due to the low Shannon diversity of the sequencing data, accounting for only 20% of the real diversity (shown in red). The best fit of the sequencing data with or without GCN using RSS compared with the mock composition is shown in bold
Fig. 1.

Microbial community structure as relative abundance of microbial genera (legend shown in Supplementary Fig. S1) and the difference between the residual sum squares (RSS) between 16S rRNA sequencing data without (raw) and with gene copy number normalization (GCN) compared with the mock community structure (Mock)
The average gene copy number in bacteria in the database using 152 bacterial genera was threefold higher with 5.29 (SE = 0.21) than the previously determined average for bacteria of 1.8. Using the higher average gene copy number would result in a similar representation of Mock-19 compared with the raw sequencing data (data not shown). We therefore suggest the reconsideration of 1.8 16S gene copies in bacteria as standard for unidentified sequences. Otherwise, GCN provided a picture closer to reality in the mock communities of lowest Shannon diversity (Mock-21 and Mock-23) with Mock-14 being the only exception with high diversity and a better fit with GCN. In Mock-14, all of the 18 genera from the mock community have a known gene copy number (available in the RDP GCN database), and both unidentified ASVs (0.04%) and ASVs assigned to other genera than present in the mock community (1.02%) make up a small proportion of the total ASV counts. The scenarios where GCN provides a better picture than the raw sequencing data therefore seemed artificial, given that the α-diversity in environmental samples is much higher than in our mock communities in both terrestrial [17–20] and aquatic ecosystems [21, 22], and it appears unreasonable to assume perfect sequencing data as found for Mock-14 (Fig. 2).
Fig. 2.
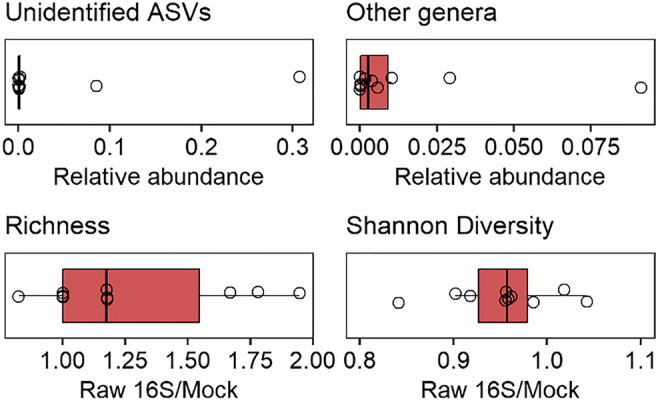
The difference between the richness and Shannon diversity of the 16S amplicon data without gene copy normalization and the mock community structure as well as the relative abundance of unidentified ASVs and ASVs assigned to other genera than present in the mock community as boxplots with median, lower and upper quartiles as well as minima and maxima
Correcting for 16S rRNA gene copy numbers in microbiome surveys still an unsolved problem [5]. The plasticity of the bacterial ribosome able to accommodate foreign 16S rRNA from diverse organisms as shown by horizontal gene transfer [23–27] also makes the use of GCN questionable. Our comparison of 16S amplicon data with the known structure of mock communities from purified DNA and plasmid vectors suggests that GCN only provided a better picture in artificial scenarios, e.g. low α-diversity or perfect sequencing data. However, different mock communities (from cells, RNA, proteins or metabolites), different primer pairs that target another region of the 16S rRNA gene and different processing pipeline may all yield different results. Importantly, predicting the as yet unknown 16S rRNA gene copy numbers [5, 28, 29] could be a viable approach to increase the fitness of GCN in amplicon sequencing. However, accounting for variance, e.g. by latent variable models, seems to be a more promising approach to understand the drivers of diversity [30] than correcting GCN sequence data. Noteworthy, we highlight the importance of quality checking publicly available mock community data to ensure high quality of future meta-analysis surveys.
Supplementary Materials and Methods
Data Generation
The community data was obtained from the mockrobiata database [7]. In total, 12 mock communities with known composition (as “taxonomy.csv” within “source” from https://github.com/caporaso-lab/mockrobiota/tree/master/data) containing both forward and reverse sequencing reads that target the 16S rRNA gene were obtained. The raw sequencing data was processed with DADA2 v1.8 [8] using the R software to yield an ASV table that provides higher resolution than the traditional OTU table and records the number of times each exact ASV was observed in each sample (more information on the R script can be found in the Supplementary Material). The reads were truncated at position 230 for the forward and position 160 for the reverse read, respectively. The recovery of reads through the pipeline was tracked for each step in each sample (Supplementary Table S1). Mock-17 failed processing in the pipeline with our parameters and was removed from the analysis. For five communities (Mock-16, Mock-18, Mock-19, Mock-22 and Mock-23), many reads were lost after chimera removal (shown in red in Supplementary Table S1). In almost all cases, this is caused by primer sequences with ambiguous nucleotides that were not removed prior to data processing with DADA2 [8]. Indeed, after removing the primers using the constant length of the forward primer 515f (n = 19) and the reverse primer 515f (n = 20) with the function trimLeft = c(19,20) within the function filterAndTrim, most of the sequences were retained after chimera removal, and the most abundant ASV length was 253. The taxonomy was assigned using the naïve Bayesian classifier method using the 16S database Silva (version 138 from May 6, 2020) and species level assignments based on exact matching between ASVs and sequenced reference strain [31, 32].
Gene Copy Number Normalization and Statistical Analysis
The absolute ASV counts were divided by known 16S rRNA gene copy numbers from bacterial genomes obtained from the Ribosomal Database Project (RDP, Release 11, Update 5 from September 30, 2016) [10]. For bacterial genera without reported gene copy number, unidentified bacteria and other bacteria than present in the mock community, the average 16S rRNA gene copy number of 1.8 (n = 45, SE = 0.13) was used. For both with and without gene GCN, the absolute ASV counts were divided by the total number of recovered reads to obtain relative ASV abundances. The pipeline with the best fitting sequencing data was determined by residual sum squares (RSS) as deviation of the predicted abundance derived from the mock community composition from the empirical values of the 16S rRNA gene amplicon data from the difference of the ith value between the mock community as yi and the 16S rRNA gene sequencing without (raw) and with normalization (GCN) both as f(xi) given by Eq. 1. The relative amount of unidentified ASVs and ASVs assigned to genera that are not present in the mock community were estimated. Taxonomic richness was calculated as the number of different genera in each sample. Alpha diversity, as Shannon diversity, was calculated on the level of bacterial genera. Mock-12 was removed from the data analysis as the raw sequencing data was dominated by Bacteroides making up 90.7% of all reads while only present at 29.6% in the mock community and tremendously changing the evenness and thereby not only impacting the Shannon diversity of the community but also showing the highest RSS of all communities (shown in red in Table 1), which deemed the comparison with the other mock communities unreliable. Visualization was carried out in R using the package ggplot2 [33].
| 1 |
Electronic Supplementary Material
{kind=link}
(PNG 918 kb)
(XLSX 9 kb)
(PDF 534 kb)
Authors’ Contributions
RS, VSP and DKM designed the experiment. RS and DM performed data analysis. The paper was written by RS, VSP and DKM who approved the final version of the manuscript.
Funding
This work was supported by the Czech Science Foundation (18-25706S and 20-02022Y) and the Brazilian Microbiome Project (http://brmicrobiome.org).
Compliance with Ethical Standards
Competing Interests
The authors declare that they have no competing interests.
References
- 1.Claassen JAHR. The gold standard: not a golden standard. BMJ. 2005;330:1121. doi: 10.1136/bmj.330.7500.1121. [DOI] [Google Scholar]
- 2.Versi E. “Gold standard” is an appropriate term [29] Br Med J. 1992;305:187. doi: 10.1136/bmj.305.6846.187-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Větrovský T, Baldrian P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One. 2013;8:e57923. doi: 10.1371/journal.pone.0057923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLaren MR, Willis AD, Callahan BJ (2019) Consistent and correctable bias in metagenomic sequencing experiments. Elife. 10.7554/elife.46923 [DOI] [PMC free article] [PubMed]
- 5.Louca S, Doebeli M, Parfrey LW. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome. 2018;6:41. doi: 10.1186/s40168-018-0420-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Highlander S (2014) Mock Community Analysis. In: Encyclopedia of Metagenomics. 10.1007/978-1-4614-6418-1_54-1
- 7.Bokulich NA, Rideout JR, Mercurio WG et al (2016) Mockrobiota: a public resource for microbiome bioinformatics benchmarking. mSystems. 10.1128/mSystems.00062-16 [DOI] [PMC free article] [PubMed]
- 8.Callahan BJ, McMurdie PJ, Rosen MJ, et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoddard SF, Smith BJ, Hein R, Roller BRK, Schmidt TM. rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015;43:D593–D598. doi: 10.1093/nar/gku1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siegwald L, Touzet H, Lemoine Y, Hot D, Audebert C, Caboche S. Assessment of common and emerging bioinformatics pipelines for targeted metagenomics. PLoS One. 2017;12:e0169563. doi: 10.1371/journal.pone.0169563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pylro VS, Roesch LFW, Morais DK, Clark IM, Hirsch PR, Tótola MR. Data analysis for 16S microbial profiling from different benchtop sequencing platforms. J Microbiol Methods. 2014;107:30–37. doi: 10.1016/j.mimet.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–2643. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starke R, Morais D (2019) Gene copy normalization of the 16S rRNA gene cannot outweigh the methodological biases of sequencing. bioRxiv. 10.1101/813477
- 15.Pylro VS, Morais DK, Kalks KHM, Roesch LFW, Hirsch PR, Tótola MR, Yotoko K. Misguided phylogenetic comparisons using DGGE excised bands may contaminate public sequence databases. J Microbiol Methods. 2016;126:18–23. doi: 10.1016/j.mimet.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 16.Kitahara K, Miyazaki K. Revisiting bacterial phylogeny: natural and experimental evidence for horizontal gene transfer of 16S rRNA. Mob Genet Elem. 2013;3:e24210. doi: 10.4161/mge.24210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bastida F, Torres IF, Andrés-Abellán M, Baldrian P, López-Mondéjar R, Větrovský T, Richnow HH, Starke R, Ondoño S, García C, López-Serrano FR, Jehmlich N. Differential sensitivity of total and active soil microbial communities to drought and forest management. Glob Chang Biol. 2017;23:4185–4203. doi: 10.1111/gcb.13790. [DOI] [PubMed] [Google Scholar]
- 18.Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng M, Zi X, Wang Q. Bacterial community diversity of oil-contaminated soils assessed by high throughput sequencing of 16s rRNA genes. Int J Environ Res Public Health. 2015;12:12002–12015. doi: 10.3390/ijerph121012002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaiser K, Wemheuer B, Korolkow V, Wemheuer F, Nacke H, Schöning I, Schrumpf M, Daniel R (2016) Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci Rep 6. 10.1038/srep33696 [DOI] [PMC free article] [PubMed]
- 21.Zhang HH, Chen SN, Huang TL, Ma WX, Xu JL, Sun X. Vertical distribution of bacterial community diversity and water quality during the reservoir thermal stratification. Int J Environ Res Public Health. 2015;12:6933–6945. doi: 10.3390/ijerph120606933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu K, Liu Y, Han BP, Xu B, Zhu L, Ju J, Jiao N, Xiong J. Bacterial community changes in a glacial-fed Tibetan lake are correlated with glacial melting. Sci Total Environ. 2019;651:2059–2067. doi: 10.1016/j.scitotenv.2018.10.104. [DOI] [PubMed] [Google Scholar]
- 23.Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol. 2004;186:2629–2635. doi: 10.1128/JB.186.9.2629-2635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardly BD, Nour SM, Van Berkum P, Selander RK. Rhizobial 16S rRNA and dnaK genes: Mosaicism and the uncertain phylogenetic placement of rhizobium galegae. Appl Environ Microbiol. 2005;71:1328–1335. doi: 10.1128/AEM.71.3.1328-1335.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller SR, Augustine S, Le Olson T, et al. Discovery of a free-living chlorophyll d-producing cyanobacterium with a hybrid proteobacterial/cyanobacterial small-subunit rRNA gene. Proc Natl Acad Sci U S A. 2005;102:850–855. doi: 10.1073/pnas.0405667102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schouls LM, Schot CS, Jacobs JA. Horizontal transfer of segments of the 16S rRNA genes between species of the Streptococcus anginosus group. J Bacteriol. 2003;185:7241–7246. doi: 10.1128/JB.185.24.7241-7246.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Zhang Z. Comparative sequence analyses reveal frequent occurrence of short segments containing an abnormally high number of non-random base variations in bacterial rRNA genes. Microbiology. 2000;146:2845–2854. doi: 10.1099/00221287-146-11-2845. [DOI] [PubMed] [Google Scholar]
- 28.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angly FE, Dennis PG, Skarshewski A, Vanwonterghem I, Hugenholtz P, Tyson GW (2014) CopyRighter: a rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome. 10.1186/2049-2618-2-11 [DOI] [PMC free article] [PubMed]
- 30.Willis AD (2019) Rarefaction, alpha diversity, and statistics. Front Microbiol [DOI] [PMC free article] [PubMed]
- 31.Edgar R (2017) Updating the 97% identity threshold for 16S ribosomal RNA OTUs. bioRxiv. 10.1101/192211 [DOI] [PubMed]
- 32.Edgar RC. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics. 2018;34:2371–2375. doi: 10.1093/bioinformatics/bty113. [DOI] [PubMed] [Google Scholar]
- 33.Wickham H (2017) ggplot2: elegant graphics for data analysis. J Stat Softw. 10.1007/978-0-387-98141-3
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PNG 918 kb)
(XLSX 9 kb)
(PDF 534 kb)