

Abstract
Myelodysplastic syndromes (MDS) are clonal hematological disorders arising from hematopoietic stem cells that have accumulated various genetic abnormalities. MDS are heterogeneous in nature but uniformly characterized by chronic and progressive cytopenia from ineffective hematopoiesis, dysplasia in single or multiple lineages, and transformation to acute leukemia in a subset of patients. The genomic landscape revealed by next-generation sequencing has provided a comprehensive picture of the molecular pathways involved in MDS pathogenesis. Recurrent mutational targets in MDS are the genes involved in RNA splicing, DNA methylation, histone modification, transcription, signal transduction, cohesin complex and DNA repair. Sequential acquisition of mutations in these sets of genes serves as a driver for the initiation, clonal evolution and progression of MDS. Based on these findings, novel agents targeting driver mutations of MDS are currently under development and expected to improve the clinical outcome of MDS in the coming decades.
Keywords: myelodysplastic syndromes, MDS, molecular pathogenesis, genome, epigenome, treatment
Introduction
Myelodysplastic syndromes (MDS) are clonal hematological malignancies arising from hematopoietic stem cells (HSCs) that have accumulated various genetic mutations. MDS is heterogeneous in nature but is uniformly characterized by chronic and progressive cytopenia from ineffective hematopoiesis and transformation to acute leukemia in a subset of patients. Single- or multi-lineage dysplasia of blood cells either in the peripheral blood (PB) or bone marrow (BM) and peripheral cytopenia are shared clinical features in MDS. Some patients present with increased blasts in the PB or BM, which is associated with an increased risk of transformation to acute myeloid leukemia (AML). According to a survey by the Ministry of Health and Welfare in Japan, the median age of patients with MDS is 64 years old, and prevalence rate is approximately 3 in 100,000.
The genomic landscape of MDS revealed by next-generation sequencing indicates that mutations of various genes involved in epigenetic, signaling, transcription, apoptosis, DNA repair, RNA splicing and cohesin pathways serve as drivers for MDS (1). Furthermore, the accumulation of genetic mutations has been shown to be associated with the clonal evolution and development of MDS.
This review will summarize our recent understanding regarding the molecular pathogenesis of MDS and novel approaches to their treatment.
Clinical Features and Classification of MDS
MDS are clonal disorders originating from abnormal HSCs with various genetic mutations. These abnormal HSCs have clonal advantages over wild-type (WT) HSCs and undergo clonal expansion over time in the BM. Hematopoietic cells generated from abnormal HSCs are defective in their proliferation and differentiation capacities, and many of them die from apoptosis during differentiation. This impaired differentiation process, called ineffective hematopoiesis, is a background mechanism for peripheral cytopenia in MDS patients. Half of MDS patients present with pancytopenia, and the others show only anemia or anemia with leukocytopenia or thrombocytopenia.
The World Health Organization (WHO) revised classification of MDS in 2016 did not make drastic changes to the classification in 2008 (Table 1). Notably, a mutation in SF3B1 was incorporated into the diagnosis of MDS with ring sideroblasts (MDS-RS) because of their tight association. MDS with an increased percentage of blasts, which have a high propensity for progressing to AML, were renamed MDS-EB-1, whose blast counts of either BM or PB are 5-9% or 2-4%, respectively, and MDS-EB-2, whose blast counts of either BM or PB are 10-19% or 5-19%, respectively. MDS with single- or multi-lineage dysplasia were termed MDS-SLD or MDS-MLD, respectively.
Table 1.
Classification of MDS (WHO2016).
| Name | BM and PB blasts | Ringed Sideroblasts | ||
|---|---|---|---|---|
| MDS with single lineage dysplasia (MDS-SLD) | BM<5%, PB<1%, no Auer rods | <15%* | ||
| MDS with multilineage dysplasia (MDS-MLD) | BM<5%, PB<1%, no Auer rods | <15%* | ||
| MDS with ring sideroblasts (MDS-RS) | ||||
| MDS-RS with single lineage dysplasia (MDS-RS-SLD) | BM<5%, PB<1%, no Auer rods | ≥15%** | ||
| MDS-RS with multilineage dysplasia (MDS-RS-MLD) | BM<5%, PB<1%, no Auer rods | ≥15%** | ||
| MDS with isolated del(5q) | BM<5%, PB<1%, no Auer rods | None or any | ||
| MDS with excess blasts (MDS-EB) | ||||
| MDS-EB-1 | BM 5%-9% or PB 2%-4%, no Auer rods | None or any | ||
| MDS-EB-2 | BM 10%-19% or PB 5%-19% or Auer rods | None or any | ||
| MDS, unclassifiable (MDS-U) | ||||
| with 1% blood blasts | BM<5%, PB=1%,‡no Auer rods | None or any | ||
| with single lineage dysplasia and pancytopenia | BM<5%, PB<1%, no Auer rods | None or any | ||
| based on defining cytogenetic abnormality | BM<5%, PB<1%, no Auer rods | <15% | ||
| Refractory cytopenia of childhood | BM<5%, PB<2% | None |
*<5%, if SF3B1 mutation is present. **≥5%, if SF3B1 mutation is present.
Chromosomal Abnormalities in MDS
About half of MDS patients have chromosomal abnormalities (2). Most of them are abnormalities affecting genomic copy numbers, such as deletion, monosomy or trisomy, and structural alterations of chromosomes, such as the balanced translocations frequently seen in acute leukemia, are relatively rare. Loss or deletion of chromosome 5 or 7 is particularly common in MDS. Monosomy 7 is known to be associated with a poor prognosis (3). MDS with isolated del(5q), also known as 5q- syndrome, are a unique subtype associated with a favorable outcome and characterized by macrocytic anemia with normal platelet levels (4). The frequency of 5q- syndrome is relatively high in the US and European countries, accounting for approximately 5% of MDS patients, while the frequency is only 1-2% in Japan (5). A commonly deleted chromosomal region of 5q- syndrome is 5q31-5q33, encompassing 1.5Mb (6). Previous studies have identified several pathogenic genes in this region, including RPS14, microRNA (miR)-145 and miR-146a. RPS14 is a ribosomal protein, and its haploinsufficiency is responsible for macrocytic anemia (7). miR-145 and miR-146a are involved in thrombocytosis and neutropenia through interleukin (IL)-6 regulation (8). Lenalidomide is effective for resolving anemia in 5q- syndrome (4).
Genetic Abnormalities in MDS
As stated above, approximately half of MDS patients have normal karyotype, suggesting that genetic mutations play critical roles in the pathogenesis of MDS. Indeed, recent data from next-generation sequencing have revealed a variety of recurrently mutated genes in MDS (Table 2 and Fig. 1). These include genes encoding RNA splicing factors, epigenetic regulators for DNA methylation or histone modification, transcription factors, signaling molecules, cohesin complex and DNA repair. Among these, the mutation frequencies are particularly high in RNA splicing factors and epigenetic regulators (Table 2).
Table 2.
Recurrently Mutated Gens in MDS.
| Gene | Mutation frequency (%) | Function | ||
|---|---|---|---|---|
| TET2 | 33 | DNA demethylation | ||
| SF3B1 | 33 | RNA splicing | ||
| ASXL1 | 23 | Histone modification | ||
| SRSF2 | 18 | RNA splicing | ||
| DNMT3A | 13 | DNA methylation | ||
| RUNX1 | 11 | Transcription factor | ||
| U2AF1 | 8 | RNA splicing | ||
| ZRSR2 | 8 | RNA splicing | ||
| STAG2 | 8 | Cohesin complex | ||
| TP53 | 6 | DNA repair | ||
| EZH2 | 6 | Histone modification | ||
| CBL | 5 | Signal transduction (ubiquitin ligase) | ||
| JAK2 | 5 | Signal transduction (tyrosine kinase) | ||
| BCOR | 4 | Transcriptional corepressor | ||
| IDH2 | 4 | DNA demethylation | ||
| NRAS | 4 | Signal transduction (Ras signaling) | ||
| NF1 | 3 | Signal transduction (Ras signaling) |
Figure 1.

Genetic abnormalities in MDS. Recurrently mutated genes in MDS (shown in red characters) and their roles in physiological pathways are shown. MDS: myelodysplastic syndromes
1) RNA splicing factors
RNA splicing is a complicated process governed by the unique protein complex ‘spliceosome,’ which comprises more than 50 proteins (9). Among these proteins, recurrent mutations are found in U2AF35, ZRSR2, SRSF2, SF3A1, SF3B1, PRPF40B, U2AF65 and SF1 in MDS patients (10). Interestingly, these mutations are mutually exclusive, suggesting that a single mutation is sufficient to impair the spliceosome function and drive the development of MDS, or mutations in multiple spliceosome genes are detrimental to MDS clones.
The SF3B1 mutation is highly specific to MDS-RS (11). About 70% of MDS-RS patients are positive for the SF3B1 mutation, suggesting its diagnostic significance (12). Mutations in SRSF2, U2AF1 and ZRSF2 are observed in approximately 20%, 10% and 10% of MDS patients, respectively.
The molecular mechanisms underlying how these mutations contribute to MDS pathogenesis are not clear at present. Recent studies have shown that splicing factor mutations can lead to a variety of splicing abnormalities, such as exon skipping and intron retention. By using knock-in mouse models, it was demonstrated that the SRSF2 mutation induces alternative recognition of the target sequence, resulting in disordered splicing of major epigenetic regulators, such as EZH2 (13). Mutations in splicing factors are now considered to drive MDS, at least in part, by inducing disordered epigenetic regulation through abnormal RNA splicing.
2) Regulators of DNA methylation
DNA methylation plays a critical role in transcriptional regulation. It is well known that methylation of cytosine residues in the promoter region strongly suppresses the transcription of the downstream gene. The methylation status of DNA is maintained by the fine-tuned, balanced regulation of methylation and demethylation of DNA. DNA methylation is catalyzed by DNA methyltransferase (DNMT), which comprises DNMT1, DNMT3A and DNMT3B. DNMT1 is critical for the maintenance of the genome-wide methylation pattern upon DNA replication, and DNMT3A and B are required for establishing tissue- or cell type-specific methylation patterns. TET proteins, by contrast, are essential for DNA demethylation. The TET family contains TET1, TET2 and TET3, all of which catalyze the first step of DNA demethylation by converting 5-methyl cytosine (5-mC) to 5-hydroxymethyl cytosine (5-hmC).
Dysregulated DNA methylation is critical for MDS pathogenesis, since the mutation rates of TET2 and DNMT3A are high in MDS (10-30% and 10-20%, respectively) (14-16). Notably, mutations in DNMT3A or TET2 are also frequent in AML, myeloproliferative neoplasm (MPN) and malignant lymphoma, suggesting that disordered DNA methylation is fundamental for various hematological malignancies.
TET2 mutations are considered to be loss-of-function, since nonsense mutations are clustered in the N-terminal region and missense mutations are mostly localized in C-terminal catalytic domain (15). In fact, TET2-deficient HSCs have enhanced self-renewal capacity and show clonal advantage in vivo (17,18). Furthermore, TET2-deficient mice develop hematological malignancies, such as myeloid neoplasm or malignant lymphoma, after a long latency (18).
For DNMT3A, arginine 882 (R882), located in the catalytic domain of methyltransferase, is a hotspot for mutations, and its mutation profoundly affects its catalytic activity (19). Similarly to TET2, DNMT3A plays a crucial role in the HSC function, and DNMT3A mutations confer HSCs with an enhanced stem cell capacity and clonal advantage (20,21).
Recent findings from mouse models and clonal hematopoiesis in human settings indicate that abnormal DNA methylation alone is not sufficient to cause MDS, and additional mutations are required for the development of hematological malignancies (22-26).
3) Histone modifiers
Histones are major components for chromatin that are required for maintaining its integrity and structure. Histone octamer, consisting of histone H2A, H2B, H3 and H4, serves as the core of the nucleosome, the smallest unit of chromatin structure. Histones are targets of various post-translational modifications, such as methylation, acetylation and phosphorylation. Histone modifications induce dynamic changes in the local chromatin structure, leading to the transcriptional activation or suppression of the genes in the surrounding region.
Polycomb group complex (PRC) 2 is a well-known histone modifying complex frequently targeted in MDS. PRC2 induces tri-methylation of lysine 27 of histone H3 (H3K27), which suppresses transcription of the downstream gene. The loss of function mutation of EZH2, a major component of PRC2 with histone methyltransferase activity, is reported in 5-10% of MDS patients and has been shown to be associated with a poor prognosis (27,28). Interestingly, the EZH2 gene is localized on chromosome 7q, suggesting that haploinsufficiency of EZH2 might be associated with the poor prognosis of the -7/-7q karyotype (29). ASXL1, another frequent target in MDS, is considered to be critical for the PRC function and is mutated in 20-25% of MDS patients (30).
4) Cohesin complex
Cohesin complex, consisting of four proteins (STAG2, SMC3, SMC1A and RAD21), is known to play critical roles in sister chromatid segregation during cell division (31). Recurrent mutations of cohesin complex have been reported in various myeloid malignancies, including MDS (32). Mutations in four components of cohesin complex are mutually exclusive, indicating that functional disruption of the whole complex is important for the pathogenesis of MDS. Recent studies have suggested that cohesin complex plays a role in maintaining the higher-order DNA structure, such as facilitating chromatin looping by combining DNA at distant sites, and is thereby deeply involved in transcriptional regulation. Cohesin mutations have been shown to induce abnormal myeloid differentiation and enhance the HSC function in mouse models (33,34). Therefore, it is now postulated that cohesin mutations contribute to the development of MDS through the structural alteration of chromatin and resultant transcriptional dysregulation of the genes critical for HSCs and hematopoietic differentiation (35).
5) Isocytrate dehydrogenase (IDH)
IDH is an enzyme of the tricarboxylic acid (TCA) cycle that converts isocitrate to α-ketoglutarate (α-KG). α-KG is an essential cofactor for TET and histone demethylases, such as KDM6A, and is therefore critical for epigenetic regulation, such as DNA and histone methylation. IDH has two isoforms (IDH1 and IDH2) localized in the cytoplasm and mitochondria, respectively. Heterozygous missense mutations of IDH1 and IDH2 (IDH 1/2), mostly at the critical arginine residue in the catalytic domain, have been reported in about 5-12% of MDS patients (36,37). Interestingly, this mutation confers the enzyme with novel activity to convert α-KG to the oncometabolite 2-hydroxy glutarate (2-HG) (38). 2-HG was shown to inhibit the function of α-KG-dependent enzymes, such as TET and KDM6A, by competing with α-KG (39). Therefore, IDH mutations are considered to affect the genome-wide methylation status of DNA and histones, serving as a background for MDS pathogenesis. Supporting this notion, mutations in IDH1, IDH2 and TET2 are mutually exclusive. IDH mutations in MDS are reported to be associated with mutations in DNMT3A, ASXL1 and SRSF2 (36,37).
6) Transcription factors
Transcription factors involved in hematopoietic differentiation and HSC regulation, such as RUNX1, ETV6 or GATA2, are recurrently mutated in MDS.
RUNX1 is a core binding factor (CBF) family transcription factor that plays critical roles in the emergence of HSCs during ontogeny, the function of hematopoietic stem and progenitor cells and the differentiation of megakaryocytes and lymphocytes. RUNX1 is mutated in about 10% of MDS patients, placing itself as the most frequently targeted transcription factor in MDS (40). The mutation frequency is reported to be substantially higher in radiation-associated and therapy-related MDS (41,42). Most mutations are loss-of-function, while some mutants show a dominant negative effect on the residual WT allele. RUNX1 mutations are known to be associated with a poor prognosis.
Germline mutations in transcription factors, such as RUNX1, ETV6, GATA2 or CEBPA, are known to cause familial MDS/AML syndromes (43).
7) TP53
TP53 is a well-known tumor suppressor gene mutated in a wide variety of tumors. TP53 encodes a transcription factor that induces apoptosis, cell cycle arrest and senescence in response to DNA damage or oncogenic stress (44). Mutations of TP53 are found in 5-10% of MDS patients and are particularly associated with secondary MDS and patients with del(5q) cytogenetics. TP53 mutations are well known to be associated with a poor prognosis (45).
8) Signaling molecules
Activating mutations of tyrosine kinase or serine/threonine kinase, leading to constitutive activation of the JAK-STAT or RAS-MAPK pathways, are commonly seen in various myeloid malignancies. In MDS, mutations in JAK2, CBL, NRAS and NF1 have been reported, each in about 5% of MDS patients. Active signaling evoked by these mutations simulates cell proliferation and suppresses apoptosis, contributing to the expansion of abnormal clones that have accumulated various mutations.
Clonal Evolution of MDS and AML Transformation
Understanding the clonal dynamics and the molecular basis behind the initiation, progression and transformation of MDS are crucial for developing novel therapies, better clinical care and prevention of disease progression in the future practice. It has been considered that MDS clones sequentially acquire mutations as they evolve and selected clones would expand and dominate during disease progression. However, recent studies using deep sequencing and single-cell analyses have revealed the complex and heterogeneous nature of clonal evolution in MDS.
Makishima et al. have revealed the genetic basis for the clonal evolution of MDS by sequencing DNA samples collected from 2,250 patients who had progressed from lower-risk MDS to secondary AML (46). They reported that the number of genetic mutations found in the patient BM cells increased with disease progression. Furthermore, mutations were able to be classified as type 1 or type 2, which are involved in the transformation from MDS to AML or the progression from lower-risk to higher-risk MDS, respectively (Fig. 2). Type 1 mutations include FLT3, PTPN11, WT1, IDH1, NPM1, IDH2 and NRAS; and type 2 mutations include TP53, GATA2, KRAS, RUNX1, STAG2, ASXL1, ZRSR2 and TET2. It was also shown that patients with SF3B1 mutations have a lower risk of AML transformation, whereas those with type 1 mutations have a worse outcome. These data clearly demonstrate that each mutation has a unique role in the pathogenesis and progression of MDS, and the order as well as the type of mutations critically shapes the clinical features of MDS. In addition, they also analyzed the dynamics of the clonal architecture during progression from MDS to AML. They observed two distinct patterns of clonal evolution: ‘linear evolution’ and ‘clone sweeping’. In the ‘linear evolution’ pattern, subclones that have acquired new mutations emerge recursively within the dominant population and take over the latter population. In contrast, in the ‘clone sweeping’ pattern, a new or pre-existing small subclone sweeps out the other clones and dominates during progression.
Figure 2.
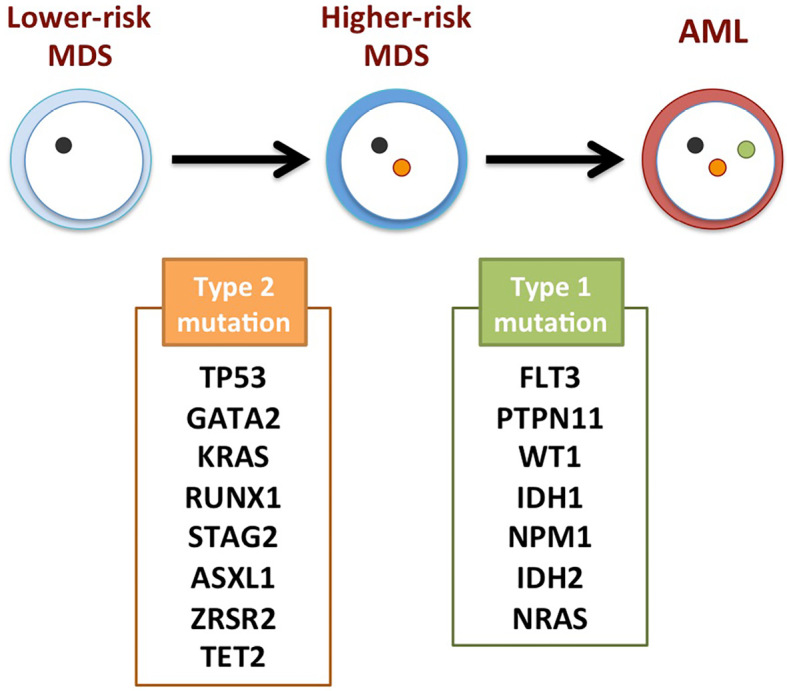
Molecular mechanism underlying the clonal evolution from lower-risk MDS to AML. Type 2 mutations are commonly seen during the evolution from lower-risk MDS to higher-risk MDS. Type 1 mutations are mainly involved in the transformation from higher-risk MDS to AML. MDS: myelodysplastic syndromes, AML: acute myeloid leukemia
Recently, a detailed picture of clonal evolution from MDS to AML was revealed by single-cell analyses of highly fractionated disease stem cells from longitudinally collected samples of the same patient (47). The study examined genetic mutations in pre-stem cell, stem cell and blast fractions in MDS and AML samples at the single-cell level and found that the MDS blasts and AML blasts were clonally distinct. In turn, it was revealed that a small subclone pre-existing in the MDS stem cell or pre-MDS stem cell fraction expanded during transformation and became dominant in AML. These findings are not compatible with the linear progression model, which predicts that MDS blasts give rise to AML blasts by acquiring additional mutations, but clearly supports the non-linear, parallel-type clonal evolution at the disease stem cell level. However, that study was conducted with only a small number of patients, and the proposed model must be confirmed in a larger-scale study.
Novel Treatments for MDS
Accumulating knowledge on the molecular pathogenesis of MDS has enabled us to develop novel agents for MDS. Drug development at present is mainly focused on epigenetics (DNA methylation and histone modification), RNA splicing and signal transduction because of the major role of these factors in MDS pathogenesis.
1) Hypomethylating agents (HMAs)
Recurrent mutations in DNA methylation pathways in MDS suggest that aberrant DNA methylation serves as a critical basis for MDS development. Indeed, genome-wide hypermethylation of DNA has been reported in MDS patients (48). Azacitidine (AZA) and Decitabine are HMAs currently approved for the treatment of MDS (only AZA is approved in Japan) and are the first choice for transplant-ineligible higher risk MDS (49-51). HMA has been shown to improve the overall survival of higher-risk MDS by delaying the transformation to AML (52). It is also effective for inducing hematopoietic recovery in less than 50% of patients with lower-risk MDS, although it does not improve the survival (53-55). The response rate of HMA in higher-risk MDS is 40-50%, and it normally takes 3-6 cycles to obtain a substantial hematological response. Factors predicting the response to HMA are reported to be mutations of TET2 or DNMT3A, DNA hypomethylation and the absence of an ASXL1 mutation.
Guadecitabine (GDAC) is a next-generation HMA with a prolonged half-life. It is a novel dinucleotide of decitabine and deoxyguanosine, which makes decitabine resistant to degradation by cytidine deaminase. It is currently under clinical trials for MDS and AML (56-58). It was shown to be effective for higher-risk MDS, CMML and low-blast-count AML cases with AZA failure in phase 2 trials (56,58).
2) Erythropoiesis stimulating agents (ESAs)
Anemia is commonly observed in MDS, and a majority of patients require frequent red blood cell (RBC) transfusions. This results in iron overload, which eventually leads to organ dysfunction due to hemosiderosis and significantly impairs the quality of life as well as the survival of the affected patients. Treatment of anemia with ESAs is a standard of care, particularly for transfusion-dependent lower-risk MDS patients. Darbepoietin, a modified recombinant human erythropoietin with a long serum half-life, is widely used for treating anemia in lower-risk MDS. In a phase 3 trial, Darbepoietin 500 μg every 3 weeks induced a significant hematological response and reduction in the transfusion frequency (59). A phase 2 trial conducted in Japan and Korea showed that the weekly administration of Darbepoietin 240 μg led to a 50% reduction in the transfusion burden in 67% of patients. Predictive factors for a response to ESA included a low serum concentration of Epo and low transfusion burden. Based on these findings, it is recommended that ESA be used in patients with a serum Epo value of <500 IU/mL (60). Response to ESA usually occurs within 3 months of treatment, and the median duration of response is 15 to 18 months (60). The combination of granulocyte-colony stimulating factor (G-CSF) with ESA was shown to enhance its efficacy in a subset of patients refractory to ESA.
3) Erythropoiesis-maturating agents (EMAs)
Luspatercept and Sotatercept are novel agents stimulating the late stage of erythropoiesis (61-64). They are fusion proteins of the human immunoglobulin G1 (IgG1) Fc domain and extracellular domain of activin receptor type IIB (Luspatercept) or IIA (Sotatercept). They act as ligand traps to neutralize TGFβ superfamily such as GDF11, which is a negative regulator for the late stage of erythropoiesis. Since Luspatercept and Sotatercept stimulate erythropoiesis through a different mechanism from Epo, which works at the early stage of erythropoiesis, they are expected to be effective for MDS patients refractory to ESA.
Luspatercept was well-tolerated and showed promising activity for increasing hemoglobin levels in lower-risk MDS patients in a phase 2 clinical trial (61). In that trial, an erythroid response and transfusion independence were observed in about 60% and 40% of the patients, respectively. Interestingly, patients with ring sideroblasts or an SF3B1 mutation showed a better response to Luspatercept than patients without them. A randomized, placebo-controlled phase 3 trial (Medalist Trial) was recently conducted by enrolling 229 transfusion-dependent, lower-risk MDS-RS patients refractory to ESA or with high serum Epo levels. The rates for an erythroid response and transfusion independence of Luspatercept vs. placebo were 53% vs. 12% and 38% vs. 13%, respectively (63). The median duration for the response to Luspatercept was 30.6 weeks.
Regarding Sotatercept, a dose-escalating phase 2 clinical trial for lower-risk MDS refractory to ESA showed favorable erythroid responses in approximately 50% of the patients both with high and low transfusion dependency (62). Similar to Luspatercept, the response rates were higher among patients with MDS-RS (58.8%) than those with other lower-risk MDS without ring sideroblasts (22.2%).
In summary, these results show that both Luspatercept and Sotatercept are promising agents for treating ESA-refractory anemia in MDS-RS patients.
4) Rigosertib
Rigosertib is a multi-kinase inhibitor that selectively induces apoptosis in tumor cells by inhibiting Ras-driven signals. It is a Ras-mimetic small molecule that binds to a variety of Ras effectors, such as Raf and phosphoinositide-3 kinase, and blocks their activation. Randomized phase 3 clinical trial of Rigosertib for HMA-refractory higher-risk MDS failed to show a survival benefit. However, a subgroup analysis suggested a potential benefit for patients with early failure of HMAs (within the first nine months) (65), and a study to confirm this finding is currently ongoing.
5) IDH inhibitors
IDH mutations are gain-of-function mutations that are capable of generating the oncometabolite 2-HG from α-KG. Since IDH mutations lead to disordered methylation of DNA and histones that are fundamental for MDS development, it is reasonable to target this mutation when developing drugs for MDS. Enasidenib (AG-221) is a covalent inhibitor of mutant IDH2 and is approved in the US for relapsed or refractory AML with IDH2 mutations. In a phase 1 study, Enasidenib was effective for a subset of MDS patients with an overall response rate of 53% (66). Based on this promising result, a phase 2 trial of Enasidenib alone or in combination with AZA is currently ongoing. The IDH1 inhibitor AG-120 (Ivosidenib) has proven its efficacy in relapsed or refractory AML (67), and other IDH1 inhibitors, such as IDH305 or FT-2102, are under clinical trials for MDS.
6) Spliceosome inhibitors
Recurrent and frequent mutations of spliceosome components suggest that alterations of RNA splicing play critical roles in MDS pathogenesis. Based on the hypothesis that inhibiting the spliceosome function may be detrimental to cells with aberrant RNA splicing, spliceosome inhibitors have been tested for their anti-tumor activity in preclinical models and clinical trials. E7107, a novel spliceosome inhibitor, showed anti-leukemic activity in mice with heterozygous SRSF2 P95H mutation (13,68). H3B-8800 is an orally available small molecule that selectively modulates SF3B complex and has been shown to selectively kill leukemia cells with an SF3B1 or SRSF2 mutation in PDX mouse models in vivo (69). H3B-8800 is currently under phase 1/2 multicenter clinical trials for patients with MDS, CMML and AML.
7) Pevonedistat
Pevonedistat is an inhibitor of the neural cell developmentally downregulated 8 (NEDD8)-activating enzyme (NAE). NAE processes NEDD8 to activate cullin-RING E3 ubiquitin ligases (70-72), which then induce proteosomal degradation of their substrates, such as p27, CDT1 and Nrf-2, through ubiquitination. Pevonedistat is thus considered to suppress the proliferation of leukemic cells by inducing the accumulation of specific substrates of cullin-RING E3 ubiquitin ligases (73). Pevonedistat in combination with AZA was shown to be safe and effective for AML in a phase 1b study (74). A randomized, controlled phase 3 study comparing AZA alone and AZA+pevonedistat for higher-risk MDS and oligoblastic AML is currently underway.
Conclusion
Next-generation sequencing has revealed the recurrent genetic mutations involved in MDS and their correlation with the clinical features. such as diagnostic classification, response to therapy and prognosis. Furthermore, the analysis of serial samples taken from the same patient has provided molecular insights into clonal evolution. Novel agents targeting the molecular pathways involved in MDS pathogenesis are currently under development and are expected to be available for improving the clinical outcomes of MDS in the coming decades.
Author's disclosure of potential Conflicts of Interest (COI).
Hideaki Nakajima: Research funding, Daiichi-Sankyo.
References
- 1.Ogawa S. Genetics of MDS. Blood 133: 1049-1059, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haase D. Cytogenetic features in myelodysplastic syndromes. Ann Hematol 87: 515-526, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haase D, Germing U, Schanz J, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood 110: 4385-4395, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Boultwood J, Pellagatti A, McKenzie AN, Wainscoat JS. Advances in the 5q- syndrome. Blood 116: 5803-5811, 2010. [DOI] [PubMed] [Google Scholar]
- 5.Bernasconi P, Klersy C, Boni M, et al. Incidence and prognostic significance of karyotype abnormalities in de novo primary myelodysplastic syndromes: a study on 331 patients from a single institution. Leukemia 19: 1424-1431, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Boultwood J, Fidler C, Strickson AJ, et al. Narrowing and genomic annotation of the commonly deleted region of the 5q- syndrome. Blood 99: 4638-4641, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Ebert BL, Pretz J, Bosco J, et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 451: 335-339, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barlow JL, Drynan LF, Hewett DR, et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat Med 16: 59-66, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701-718, 2009. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64-69, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365: 1384-1395, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malcovati L, Karimi M, Papaemmanuil E, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 126: 233-241, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim E, Ilagan JO, Liang Y, et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27: 617-630, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363: 2424-2433, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakajima H, Kunimoto H. TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci 105: 1093-1099, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 12: 599-612, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Kunimoto H, Fukuchi Y, Sakurai M, et al. Tet2 disruption leads to enhanced self-renewal and altered differentiation of fetal liver hematopoietic stem cells. Sci Rep 2: 273, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20: 11-24, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holz-Schietinger C, Matje DM, Reich NO. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem 287: 30941-30951, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayle A, Yang L, Rodriguez B, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 125: 629-638, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44: 23-31, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muto T, Sashida G, Oshima M, et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med 210: 2627-2639, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunimoto H, Meydan C, Nazir A, et al. Cooperative epigenetic remodeling by TET2 loss and NRAS mutation drives myeloid transformation and MEK inhibitor sensitivity. Cancer Cell 33: 44-59 e48, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371: 2488-2498, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet 44: 642-650, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desai P, Mencia-Trinchant N, Savenkov O, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med 24: 1015-1023, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itzykson R, Fenaux P. Epigenetics of myelodysplastic syndromes. Leukemia 28: 497-506, 2014. [DOI] [PubMed] [Google Scholar]
- 28.Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol 30: 3376-3382, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jerez A, Sugimoto Y, Makishima H, et al. Loss of heterozygosity in 7q myeloid disorders: clinical associations and genomic pathogenesis. Blood 119: 6109-6117, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thol F, Friesen I, Damm F, et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol 29: 2499-2506, 2011. [DOI] [PubMed] [Google Scholar]
- 31.Gruber S, Arumugam P, Katou Y, et al. Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell 127: 523-537, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Kon A, Shih LY, Minamino M, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 45: 1232-1237, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Mullenders J, Aranda-Orgilles B, Lhoumaud P, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med 212: 1833-1850, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazumdar C, Shen Y, Xavy S, et al. Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell 17: 675-688, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noutsou M, Li J, Ling J, et al. The cohesin complex is necessary for epidermal progenitor cell function through maintenance of self-renewal genes. Cell reports 20: 3005-3013, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DiNardo CD, Jabbour E, Ravandi F, et al. IDH1 and IDH2 mutations in myelodysplastic syndromes and role in disease progression. Leukemia 30: 980-984, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin CC, Hou HA, Chou WC, et al. IDH mutations are closely associated with mutations of DNMT3A, ASXL1 and SRSF2 in patients with myelodysplastic syndromes and are stable during disease evolution. Am J Hematol 89: 137-144, 2014. [DOI] [PubMed] [Google Scholar]
- 38.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462: 739-744, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kats LM, Reschke M, Taulli R, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell 14: 329-341, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harada Y, Harada H. Molecular pathways mediating MDS/AML with focus on AML1/RUNX1 point mutations. J Cell Physiol 220: 16-20, 2009. [DOI] [PubMed] [Google Scholar]
- 41.Harada H, Harada Y, Tanaka H, Kimura A, Inaba T. Implications of somatic mutations in the AML1 gene in radiation-associated and therapy-related myelodysplastic syndrome/acute myeloid leukemia. Blood 101: 673-680, 2003. [DOI] [PubMed] [Google Scholar]
- 42.Harada Y, Harada H. Molecular mechanisms that produce secondary MDS/AML by RUNX1/AML1 point mutations. J Cell Biochem 112: 425-432, 2011. [DOI] [PubMed] [Google Scholar]
- 43.Kennedy AL, Shimamura A. Genetic predisposition to MDS: clinical features and clonal evolution. Blood 133: 1071-1085, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2: 594-604, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Kulasekararaj AG, Mohamedali AM, Mufti GJ. Recent advances in understanding the molecular pathogenesis of myelodysplastic syndromes. Br J Haematol 162: 587-605, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Makishima H, Yoshizato T, Yoshida K, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet 49: 204-212, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J, Kao YR, Sun D, et al. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med 25: 103-110, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Figueroa ME, Skrabanek L, Li Y, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 114: 3448-3458, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 106: 1794-1803, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Silverman LR, McKenzie DR, Peterson BL, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol 24: 3895-3903, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol 28: 562-569, 2010. [DOI] [PubMed] [Google Scholar]
- 52.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 10: 223-232, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Musto P, Maurillo L, Spagnoli A, et al. Azacitidine for the treatment of lower risk myelodysplastic syndromes: a retrospective study of 74 patients enrolled in an Italian named patient program. Cancer 116: 1485-1494, 2010. [DOI] [PubMed] [Google Scholar]
- 54.Garcia-Manero G, Jabbour E, Borthakur G, et al. Randomized open-label phase II study of decitabine in patients with low- or intermediate-risk myelodysplastic syndromes. J Clin Oncol 31: 2548-2553, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jabbour E, Sasaki K, Daver N, et al. Initial results of a randomized phase II study of low dose decitabine (DAC) versus low dose azacitidine (AZA) in patients with low- or intermediate-1-risk myelodysplastic syndromes (MDS). Blood 124: 4640, 2014. [Google Scholar]
- 56.Garcia-Manero G, Roboz G, Walsh K, et al. Guadecitabine (SGI-110) in patients with intermediate or high-risk myelodysplastic syndromes: phase 2 results from a multicentre, open-label, randomised, phase 1/2 trial. Lancet Haematol 6: e317-e327, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kubasch AS, Platzbecker U. Beyond the edge of hypomethylating agents: novel combination strategies for older adults with advanced MDS and AML. Cancers (Basel) 10: 158, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sébert M, Renneville A, Bally C, et al. A phase II study of guadecitabine in higher-risk myelodysplastic syndrome and low blast count acute myeloid leukemia after azacitidine failure. Haematologica 104: 1565-1571, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Platzbecker U, Symeonidis A, Oliva EN, et al. A phase 3 randomized placebo-controlled trial of darbepoetin alfa in patients with anemia and lower-risk myelodysplastic syndromes. Leukemia 31: 1944-1950, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park S, Hamel JF, Toma A, et al. Outcome of lower-risk patients with myelodysplastic syndromes without 5q deletion after failure of erythropoiesis-stimulating agents. J Clin Oncol 35: 1591-1597, 2017. [DOI] [PubMed] [Google Scholar]
- 61.Platzbecker U, Germing U, Gotze KS, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol 18: 1338-1347, 2017. [DOI] [PubMed] [Google Scholar]
- 62.Komrokji R, Garcia-Manero G, Ades L, et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: a phase 2, dose-ranging trial. Lancet Haematol 5: e63-e72, 2018. [DOI] [PubMed] [Google Scholar]
- 63.Fenaux P, Platzbecker U, Mufti GJ, et al. The MEDALIST trial: results of a phase 3, randomized, double-blind, placebo-controlled study of luspatercept to treat anemia in patients with very low-, low-, or intermediate-risk myelodysplastic syndromes (MDS) with ring sideroblasts (RS) who require red blood cell (RBC) transfusions [abstract]. Blood 132 (Suppl): 1, 2018.29976776 [Google Scholar]
- 64.Komrokji R, Garcia-Manero G, Ades L, et al. A phase 2, dose-finding study of sotatercept (ACE-011) in patients (PTS) with lower-risk myelodysplastic syndromes (MDS) and anemia requiring transfusion. Haematologica 100 (Suppl): abstract, 2015. [Google Scholar]
- 65.Garcia-Manero G, Fenaux P, Al-Kali A, et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial. Lancet Oncol 17: 496-508, 2016. [DOI] [PubMed] [Google Scholar]
- 66.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130: 722-731, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 378: 2386-2398, 2018. [DOI] [PubMed] [Google Scholar]
- 68.Lee SC, Dvinge H, Kim E, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 22: 672-678, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seiler M, Yoshimi A, Darman R, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 24: 497-504, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brownell JE, Sintchak MD, Gavin JM, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell 37: 102-111, 2010. [DOI] [PubMed] [Google Scholar]
- 71.Milhollen MA, Traore T, Adams-Duffy J, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-κB-dependent lymphoma. Blood 116: 1515-1523, 2010. [DOI] [PubMed] [Google Scholar]
- 72.Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458: 732-736, 2009. [DOI] [PubMed] [Google Scholar]
- 73.Swords RT, Kelly KR, Smith PG, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood 115: 3796-3800, 2010. [DOI] [PubMed] [Google Scholar]
- 74.Swords RT, Coutre S, Maris MB, et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 131: 1415-1424, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]