

Abstract
Aims
Heart failure with preserved ejection fraction (HFpEF) is associated with reduced exercise capacity elicited by skeletal muscle (SM) alterations. Up to now, no clear medical treatment advice for HFpEF is available. Identification of the ideal animal model mimicking the human condition is a critical step in developing and testing treatment strategies. Several HFpEF animals have been described, but the most suitable in terms of comparability with SM alterations in HFpEF patients is unclear. The aim of the present study was to investigate molecular changes in SM of three different animal models and to compare them with alterations of muscle biopsies obtained from human HFpEF patients.
Methods and results
Skeletal muscle tissue was obtained from HFpEF and control patients and from three different animal models including the respective controls—ZSF1 rat, Dahl salt‐sensitive rat, and transverse aortic constriction surgery/deoxycorticosterone mouse. The development of HFpEF was verified by echocardiography. Protein expression and enzyme activity of selected markers were assessed in SM tissue homogenates. Protein expression between SM tissue obtained from HFpEF patients and the ZSF1 rats revealed similarities for protein markers involved in muscle atrophy (MuRF1 expression, protein ubiquitinylation, and LC3) and mitochondrial metabolism (succinate dehydrogenase and malate dehydrogenase activity, porin expression). The other two animal models exhibited far less similarities to the human samples.
Conclusions
None of the three tested animal models mimics the condition in HFpEF patients completely, but among the animal models tested, the ZSF1 rat (ZSF1‐lean vs. ZSF1‐obese) shows the highest overlap to the human condition. Therefore, when studying therapeutic interventions to treat HFpEF and especially alterations in the SM, we suggest that the ZSF1 rat is a suitable model.
Keywords: Skeletal muscle, Heart failure with preserved ejection fraction, Animal models
Introduction
About 50% of heart failure patients are afflicted with heart failure with preserved ejection fraction (HFpEF) exhibiting exercise intolerance [due to alterations in the skeletal muscle (SM)], 1 congestion, oedema, and increased fibrosis, a figure that is projected to increase due to the changing risk factor landscape, in particular the ageing population. Comparison of the overall mortality between patients with HFpEF and patients with heart failure with reduced ejection fraction (HFrEF) is discussed controversial, some reporting similar rates of hospitalization, 2 , 3 whereas others report higher rates in HFrEF. 4 , 5 , 6 Patients with HFpEF have more comorbid conditions such as hypertension, diabetes mellitus, and obesity (reviewed in Gevaert et al. 7 ). Unfortunately, classic treatment regimens that have proven effective in patients with HFrEF have failed to improve survival in HFpEF. 8 , 9 , 10 Up to now, no clear medical treatment advice for HFpEF is available.
The urgent clinical symptom of heart failure patients (HFrEF and HFpEF) is exercise intolerance leading to reduced quality of life. 11 , 12 , 13 Research during the last years confirmed that reduced exercise capacity is at least partially related to alterations in the peripheral SM, including molecular changes like the switch of the fibre type, muscle atrophy, and mitochondrial energy production (reviewed in Adams et al. 14 ). Recent studies revealed that exercise training could improve those alterations in the peripheral SM of both, HFrEF, 15 , 16 , 17 , 18 and HFpEF. 19
The limited number of authentic HFpEF animal models poses a major limitation for the investigation of its pathophysiology and potential therapies for HFpEF. Available rodent models of HFpEF, including the ageing accelerate mouse, 20 the Dahl salt‐sensitive (DSS) rat, 19 , 21 the Zucker fatty/spontaneously hypertensive heart failure F1 hybrid (ZSF‐1) rat, 22 , 23 the db/db mouse, 24 and the transverse aortic constriction (TAC) surgery/deoxycorticosterone acetate (TAC/DOCA) mouse 25 have been described. However, which one is most suitable in terms of comparability with SM alterations in HFpEF patients remains unclear.
Therefore, aim of the present study was to investigate molecular changes in SM of 3 different animal models and to compare them with alterations occurring in muscle biopsies obtained from human HFpEF patients. This study helps identifying the most suitable animal model mimicking the condition in HFpEF patients with respect to SM alterations.
Methods and Materials
Patients and animal models
Heart failure with preserved ejection fraction patients and healthy controls
HFpEF patients, analysed in the present study (n = 15), were randomly selected from the subgroup of HFpEF patients included into the OptimEx trial 26 of which SM biopsy specimens of the quadriceps muscle were available at the begin of the study (n = 42). The healthy control group (n = 9) included patients, who were randomly selected from the healthy control group randomized for the LEICA trial. 27 Biopsies of the quadriceps muscle were available at the beginning of the study. Patients recruited for the OptimEx trail had to be stable and under optimal medication for the last 6 weeks (detailed patient inclusion criteria see clinicaltrainls.gov—NCT02078947).
Transverse aortic constriction surgery/deoxycorticosterone acetate mouse model
Male C57BL/6J mice at 12 weeks of age underwent TAC surgery with concomitant DOCA pellet implantation (n = 7). Sham operated mice with placebo pellet implantation served as control mice (n = 6). Minimally invasive TAC was performed as described previously with modification. 25 In brief, mice were anaesthetized using a single injection of ketamine and xylazine (120 and 12 mg/kg, intraperitoneal), and a 5‐mm horizontal incision was made at the first left intercostal space. The thymus was temporarily retracted to visualize the aortic arch, and a 7–0 silk suture was passed under the aorta between the right innominate and left carotid arteries. The suture was ligated around a blunted 27‐gauge needle, and the needle was quickly removed. Sham animals underwent the same procedure without ligation around the aorta. The chest wall and skin were closed. An additional incision was made in the right flank of the animal, and a subcutaneous pocket was created by blunt dissection. A DOCA (DOCA 50 mg per pellet, 21 d release) or placebo pellet (Innovative Research of America) was implanted. The skin was closed, and the mice were allowed to recover in a ThermoCare warmer. Sham animals underwent the same procedure without ligation around the aorta or implantation of a DOCA pellet. Four weeks after TAC/DOCA or sham operation, the animals were sacrificed and the extensor digitorum longus (EDL) muscles were removed and snap frozen in liquid nitrogen.
Dahl salt‐sensitive rat model
Female DSS rats (n = 23) were randomized at the age of 7 week into the following groups: (i) control: fed with a chow diet containing 0.3% NaCl (n = 12); (ii) HFpEF: fed with a chow diet containing 8% NaCl (n = 11). Rats were exposed to identical conditions in a 12 h light–dark cycle, with food and water provided ad libitum. After 20 weeks, echocardiography and invasive haemodynamic measurements were performed to elucidate the degree of diastolic dysfunction. Rats were subsequently sacrificed, and SM tissue (EDL muscle) was removed and snap frozen in liquid nitrogen for molecular analysis. All procedures and experiments were approved by the Norwegian Council for Animal Research, which was in accordance with Use of Laboratory Animals by the European Commission Directive 86/609/EEC.
ZSF1 rat model
Female ZSF1 lean (control, n = 14) and ZSF1 obese (n = 14) rats were purchased from Charles River (Charles River Laboratories, USA) and included into the study. Rats were exposed to identical conditions in a 12 h light–dark cycle, with food and water provided ad libitum. At the age of 32 weeks the development of HFpEF was confirmed by echocardiography and invasive haemodynamic measurements. Rats were subsequently sacrificed, and SM tissue (EDL muscle) was removed and snap frozen in liquid nitrogen for molecular analysis. All animal procedures were approved by the local animal research council, TU Dresden, and the Landesbehörde Sachsen (TVV 42/2018).
A schematic summary of all animal and patient groups included into the present study is shown in Figure 1 .
Figure 1.

An overview of the study design. Skeletal muscle biopsies (Quadriceps muscle) obtained from heart failure with preserved ejection fraction (HFpEF) patients and healthy controls were compared with skeletal muscle tissue [extensor digitorum longus (EDL) muscle] obtained from three different HFpEF animal models: ZSF1 rat model (control: ZSF1‐lean rats; HFpEF: ZSF1‐obese rats, 31 weeks); Dahl salt‐sensitive (DSS) rats [control: low salt (0.3%) in drinking water, HFpEF: high salt (8%) in drinking water for 21 weeks]; TAC/DOCA mouse model (control: sham operated mouse, HFpEF: TAC operations with DOCA releasing pellet for 4 weeks). TAC, transverse aortic constriction; DOCA, deoxycorticosterone acetate.
Protein expression
For western blot analysis, SM tissue was homogenized in Relax buffer (90 mmol/L HEPES, 126 mmol/L potassium chloride, 36 mmol/L sodium chloride, 1 mmol/L magnesium chloride, 50 mmol/L EGTA, 8 mmol/L ATP, 10 mmol/L creatine phosphate, pH 7.4) containing a protease inhibitor mix (Inhibitor mix M, Serva, Heidelberg, Germany) and sonicated. Protein concentration was determined (BCA assay, Pierce, Bonn, Germany) and aliquots (5–20 μg) were separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis. Proteins were transferred to a polyvinylidene fluoride membrane and incubated overnight at 4°C using the following primary antibodies: Mitochondrial porin (1:1000; Abcam, Cambridge, United Kingdom), MuRF‐1 (1:1000; Abcam), Mafbx (1:1000; Abcam), PGC1‐α (1:200; Santa Cruz, Heidelberg, Germany), Telethonin (1:1000; Abcam), gp91 (1:1000; Abcam), ubiquitin linkage‐specific K48 (1:1000; Abcam), superoxide dismutase (SOD) 1 (1:200; Santa Cruz, Heidelberg, Germany), SOD 2 (1:200; Santa Cruz, Heidelberg, Germany), LC‐3B (1:1000; Sigma‐Aldrich, St. Louis, Missouri), Myosin heavy chain (MHC) (1:1000; Sigma‐Aldrich, St. Louis, Missouri), TNFα (1:500, Thermo Fisher Scientific, Waltham USA). Membranes were subsequently incubated with a horseradish peroxidase‐conjugated secondary antibody and specific bands were visualized by enzymatic chemiluminescence (Super Signal West Pico, Thermo Fisher Scientific Inc., Bonn, Germany). Bands were densitometrically quantified using a Bio1D software package (Version 15.08b. Vilber Lourmat, France). Measurements were normalized to GAPDH (1/30000; HyTest Ltd, Turku, Finland). All data are presented as fold change relative to the respective control group.
Enzyme activity measurements
For enzyme activity measurements, the SM tissue was homogenized in Relax buffer and aliquots were used for enzyme activity measurements. Enzyme activities for lactate dehydrogenase (LDH), malate dehydrogenase (MDH), creatine kinase (CK), succinate dehydrogenase (SDH) were measured spectrophotometrically as previously described in detail. 28 , 29 , 30 , 31 Enzyme activity data are presented as the fold change relative to the respective control group.
Statistical analysis
Data are presented as mean ± SEM. Differences between the HFpEF and the respective control group were analysed using Student t‐test. Analyses were performed by GraphPad Instat. P‐values <0.05 were considered statistically significant.
Results
Patient and animal characteristics
Characteristics of all subjects included into the present comparison document that patients and animals exhibit clear signs of HFpEF compared with their respective controls (Table 1 ).
Table 1.
Patient and animal characteristics
| Human samples | Control; n = 9 | HFpEF patient; n = 15 |
|---|---|---|
| Age (years) | 64.0 ± 2.7 | 69.7 ± 1.6 |
| BMI (kg/m2) | 27.8 ± 1.0 | 33.1 ± 1.4** |
| Systolic BP (mmHg) | 132 ± 3 | 138 ± 3 |
| NT‐pro BNP (pg/mL) | 71 ± 2 | 915 ± 198*** |
| Diabetes mellitus (%) | 11.1 | 33.3 |
| LVEF (%) | 64 ± 2 | 64 ± 1 |
| E/é ratio | 9.6 ± 0.9 | 20.1 ± 1.3*** |
| ZSF1 rat | Control (ZSF1‐lean); n = 14 | HFpEF (ZSF1‐obese); n = 14 |
|---|---|---|
| Body weight (g) | 265 ± 4 | 559 ± 9*** |
| Heart weight (mg/mm TL) | 23.54 ± 0.31 | 35.51 ± 0.54*** |
| Mean arterial BP (mmHg) | 104.8 ± 5.6 | 135.6 ± 3.0*** |
| LVEF (%) | 79.2 ± 1.1 | 74.3 ± 1.2** |
| E/é ratio | 21.63 ± 0.41 | 27.87 ± 0.65 *** |
| LVEDP (mmHg) | 15.26 ± 0.86 | 21.92 ± 1.45 *** |
| Dahl salt‐sensitive (DSS) rat | Control (low salt); n = 12 | HFpEF (high salt); n = 11 |
|---|---|---|
| Body weight (g) | 292 ± 5 | 283 ± 5 |
| Heart weight (mg) | 940 ± 10 | 1,250 ± 20** |
| Systolic BP (mmHg) | 149 ± 5 | 213 ± 5** |
| LVEF, % | 82.7 ± 1.1 | 69.2 ± 1.8* |
| E/é ratio | 10.6 ± 1.5 | 19.4 ± 1.1 * |
| LVEDP, mmHg | 5.6 ± 0.9 | 13.0 ± 2.6 * |
| TAC/DOCA mouse | Control n = 6 | HFpEF, n = 7 |
|---|---|---|
| Body weight (g) | 29.7 ± 0.5 | 28.9 ± 0.8 |
| Heart weight (mg/mm TL) | 7.15 ± 0.12 | 9.76 ± 0.21*** |
| Systolic BP (mmHg) | 87.9 ± 1.4 | 127.4 ± 2.5*** |
| LVEF (%) | 53.4 ± 1.0 | 50.9 ± 0.9 |
| E/é ratio | 34.81 ± 1.45 | 42.70 ± 1.70** |
| LVEDP (mmHg) | 3.32 ± 0.48 | 6.23 ± 0.63** |
BMI, body mass index; BNP, brain natriuretic peptide; BP, blood pressure; HFpEF, heart failure with preserved ejection fraction; LVEDP, left ventricular end‐diastolic pressure; LVEF, left ventricular ejection fraction; TAC/DOCA, transverse aortic constriction/deoxycorticosterone acetate.
P < 0.05,
P < 0.01.
P < 0.001 vs. control.
In the human cohort, HFpEF patients presented diastolic dysfunction (E/é ratio 20.4 ± 1.8 vs. 8.2 ± 0.8, P < 0.001) and raised levels of circulating NT‐pro brain natriuretic peptide (1,673 ± 671 vs. 73 ± 1, P < 0.05) compared with the healthy control group. No differences were observed in age, body mass index, systolic blood pressure, presence of diabetes mellitus and, most importantly, left ventricular ejection fraction (LVEF 64 ± 1 vs. 63 ± 1).
Regarding the HFpEF cohorts of the 3 different animal models, all presented with a significantly elevated heart weight, a higher mean arterial pressure, an increased E/é ratio and an elevated left ventricular end‐diastolic pressure (LVEDP) when compared with the controls. It has to be mentioned, that HFpEF‐groups of all animal models presented with a reduced LVEF; however, still in the preserved range of >50%. With respect to exercise intolerance a reduced VO2max was evident in the ZSF1‐obese (ZSF1‐lean: 49.1 ± 0.8 vs. ZSF1‐obese: 38.1 ± 1.2; P < 0.001) 23 and a reduced exercise capacity was detected in the DSS rats feed wit high salt. 32 Also the TAC/DOCA mice exhibited exercise intolerance since maximal running distance was significantly impaired as reported earlier. 25
Anabolic proteins
Expression levels of proteins involved in protein degradation, either via the ubiquitin proteasome system (MuRF1 or MafBx) or the autophagy pathway (LC3‐I/LC3‐II ratio) were analysed (Figure 2 ). Similar patterns were observed between the human SM and the SM obtained from ZSF1 rats (Figure 2 A‐D). Regarding the protein expression of MuRF1 (Figure 2A ), a significant elevation was seen in the HFpEF samples obtained either from humans (con: 1.00 ± 0.19 vs. HFpEF: 1.76 ± 0.22; P < 0.05 x‐fold vs. con), ZSF1 (con: 1.00 ± 0.06 vs. HFpEF: 1.22 ± 0.07 x‐fold vs. con; P < 0.05) and DSS rats (con: 1.00 ± 0.05 vs. HFpEF: 1.15 ± 0.03 x‐fold vs. con; P < 0.05) compared with the control samples, respectively. No increase in MuRF1 protein expression was evident in the TAC/DOCA HFpEF model.
Figure 2.

Protein expression of MuRF1 (A), MafBx (B), protein ubiquitinylation (C) and LC3 (D) was quantified by western blot analysis in skeletal muscle tissue obtained from humans [heart failure with preserved ejection fraction (HFpEF) and controls] and three different HFpEF animal models [ZSF1, DSS, transverse aortic constriction/deoxycorticosterone acetate (TAC/DOCA)]. Values are shown as mean ± SEM expressed as x‐fold vs. control. Statistical comparison was made between control and HFpEF of the respective model. Representative examples of Western blots are depicted on top of the figure (C, control; H, HFpEF).
Regarding the protein expression of MafBx (Figure 2B ), only the human SM showed enhanced expression levels (con: 1.00 ± 0.09 vs. HFpEF: 1.45 ± 0.15 x‐fold vs. con; P < 0.05). None of the animal models of HFpEF presented an altered MafBx protein expression.
The main task of MuRF1 and MafBx is to attach ubiquitin molecules to proteins, thereby marking them for degradation by the UPS system. Therefore, we analysed the expression of ubiquitinylated SM proteins. As shown in Figure 2C , a significant increase of ubiquitinylated proteins was seen in the HFpEF group of humans (con: 1.00 ± 0.05 vs. HFpEF: 1.15 ± 0.05 x‐fold vs. con; P < 0.05) and ZSF1 rats (con: 1.00 ± 0.09 vs. HFpEF: 1.44 ± 0.11 x‐fold vs. con; P < 0.05). A trend was observed in the TAC/DOCA HFpEF model (P = 0.06), whereas no change was evident in the DSS HFpEF model.
Besides the UPS system, autophagy is another important protein degradation pathway, with LC3‐I and LC3‐II being markers of increased autophagy. Analysing the ratio of LC3‐I/LC3‐II (Figure 2D ) a significant downregulation by 33% was observed in the human samples (con: 1.00 ± 0.15 vs. HFpEF: 0.67 ± 0.04 x‐fold vs. con; P < 0.05). A tendency of a lower LC3‐I/LC3‐II ratio was also seen in the ZSF1 model (con: 1.00 ± 0.05 vs. HFpEF: 0.85 ± 0.02 x‐fold vs. con; P = 0.06). No difference was detected in the DSS and TAC/DOCA model of HFpEF.
Structural proteins
Quantifying the protein expression of structural proteins like myosin heavy chain (MHC, Figure 3A ), actin (Figure 3B ) and telethonin (Figure 3C ) a significant upregulation was only detected for actin (con: 1.00 ± 0.03 vs. HFpEF: 1.17 ± 0.06 x‐fold vs. con; P < 0.05) and telethonin (con: 1.00 ± 0.05 vs. HFpEF: 1.34 ± 0.11 x‐fold vs. con; P < 0.05) in the HFpEF group of the DSS rat. In the human samples as well as in SM samples obtained from the ZSF1 and TAC/DOCA animals no changes were observed (Figure 3 A‐C).
Figure 3.
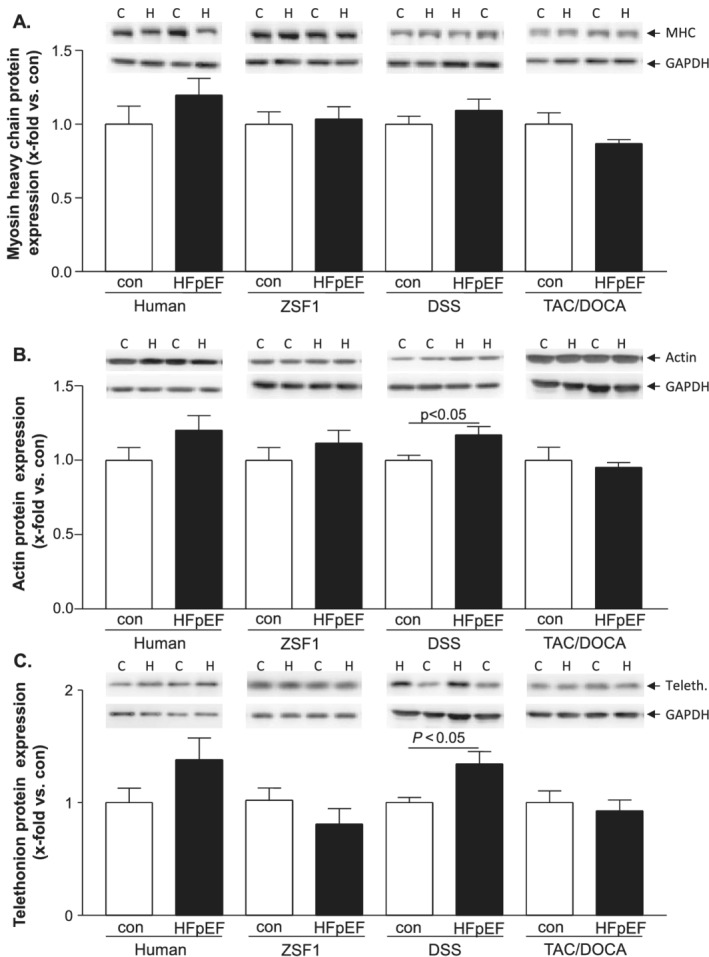
Protein expression of myosin heavy chain (A), actin (B), and telethonin (C) was quantified by western blot analysis in skeletal muscle tissue obtained from humans [heart failure with preserved ejection fraction (HFpEF) and controls] and three different HFpEF animal models [ZSF1, DSS, and transverse aortic constriction/deoxycorticosterone acetate (TAC/DOCA)]. Values are shown as mean ± SEM expressed as x‐fold vs. control. Statistical comparison was made between control and HFpEF of the respective model. Representative examples of Western blots are depicted on top of the figure (C, control; H, HFpEF).
Mitochondrial proteins and metabolic enzymes
The assessment of mitochondria and enzymes involved in energy production and transfer in the SM revealed similarities between alterations in humans and ZSF1 animals (Figure 4 ). The expression of porin, a protein of the outer mitochondrial membrane and marker to estimate mitochondrial content, was significantly elevated in SM samples of human HFpEF and ZSF1 rats (48% and 50%, respectively), whereas no changes were observed between control and HFpEF in DSS and TAC/DOCA animals (Figure 4A ). In the human HFpEF samples, the increased mitochondria content might be triggered by enhanced PGC1α expression while no changed PGC1α expression was detected in the SM across all 3 animal models (Figure 4B ). Enzyme activities of SDH and MDH revealed a significant decrease in SM samples obtained from HFpEF patients (Figure 4C, D ). This decrease was also observed in HFpEF samples of ZSF1 rats (Figure 4C, D ). With respect to creatine kinase (Figure 4E ) and LDH (Figure 4F ) a significant increase was only detected in human SM samples of HFpEF patients. None of the animal models showed alterations between control and HFpEF (Figure 4E, F ).
Figure 4.

Protein expression of porin (A) and PGC1a (B) was quantified by western blot analysis in skeletal muscle tissue obtained from humans [heart failure with preserved ejection fraction (HFpEF) and controls] and three different HFpEF animal models [ZSF1, Dahl salt‐sensitive (DSS), transverse aortic constriction/deoxycorticosterone acetate (TAC/DOCA)]. In addition, specific enzyme activity of succinate dehydrogenase (SDH) (C), malate dehydrogenase (MDH) (D), creatine kinase (CK) (E) and lactate dehydrogenase (LDH) (F) was measured. Values are shown as mean ± SEM expressed as x‐fold vs. control. Statistical comparison was made between control and HFpEF of the respective model. Representative examples of Western blots are depicted on top of the figure (C, control; H, HFpEF).
Proteins involved in reactive oxygen species production and inflammation
The presence of reactive oxygen species (ROS) represents an imbalance of ROS production and clearance by scavenger proteins. An important protein involved in ROS generation is the NADPH oxidase, whereas SOD catalyzes the dismutation of superoxide anion free radical (O2 •−) into molecular oxygen and hydrogen peroxide (H2O2). Assessment of subunit gp91 of the NADPH‐oxidase (Figure 5A ), SOD1 (Figure 5B ), and SOD2 (Figure 5C ) revealed no differences between HFpEF and control in SM either from human samples or all investigated animal models. Because it is well documented in the current literature that the development of HFrEF is associated with increased inflammation in the peripheral SM, 33 we analysed protein expression of TNFα. An increase by 84% was detected in the human HFpEF SM (con: 1.00 ± 0.01 vs. HFpEF: 1.84 ± 0.31; P < 0.05) (Figure 5D ) while no changes were observed in all three animal models (Figure 5D ).
Figure 5.

Protein expression of NADPH oxidase subunit gp91phox (A) SOD1 (B), SOD2 (C) and TNFα (D) was quantified by western blot analysis in skeletal muscle tissue obtained from humans [heart failure with preserved ejection fraction (HFpEF) and controls] and three different HFpEF animal models [ZSF1, succinate dehydrogenase (DSS), transverse aortic constriction surgery/deoxycorticosterone acetate (TAC/DOCA)]. Values are shown as mean ± SEM expressed as x‐fold vs. control. Statistical comparison was made between control and HFpEF of the respective model. Representative examples of Western blots are depicted on top of the figure (C, control; H, HFpEF).
Discussion
For the investigation of novel treatment options for HFpEF, it is of upmost interest to identify suitable animal model mimicking human pathophysiology in the peripheral SM as tool for translational research. Therefore, the present study compared molecular alterations in the peripheral SM of three different animal models and HFpEF patients. Our results suggest that among all three animal models, the ZSF1 exhibits the best consensus with alteration observed in human samples (for summary see Table 2 ).
Table 2.
Summary of molecular alterations observed in human and animal skeletal muscle tissue
| Protein | Human | ZSF1 | DSS | TAC/DOCA |
|---|---|---|---|---|
| Anabolism | ||||
| MuRF1 | ↑ | ↑ | ↑ | = |
| MAFBx | ↑ | = | = | = |
| Ubiquitination | ↑ | ↑ | = | = |
| LC3‐I/LC3‐II | ↓ | ↓ | = | = |
| Mitochondria | ||||
| SDH | ↓ | ↓ | = | = |
| MDH | ↓ | ↓ | = | = |
| CK | ↑ | = | = | = |
| LDH | ↑ | = | = | = |
| Porin | ↑ | ↑ | = | = |
| PGC1‐alpha | ↑ | = | = | = |
| Structural protein | ||||
| MHC | = | = | = | = |
| Actin | = | = | ↑ | = |
| ROS/Inflammation | ||||
| Gp91phox | = | = | = | = |
| SOD1 | = | = | = | = |
| SOD2 | = | = | = | = |
| TNF‐alpha | ↑ | = | = | = |
DSS, Dahl salt‐sensitive; HFpEF, heart failure with preserved ejection fraction; LVEDP, left ventricular end‐diastolic pressure; LVEF, left ventricular ejection fraction; MHC, Myosin heavy chain; ROS, reactive oxygen species; TAC/DOCA, transverse aortic constriction/deoxycorticosterone acetate.
= no change between HFpEF and control; ↑ up‐regulation in HFpEF when compared with control; ↓ down‐regulation in HFpEF when compared with control; grey boxes mark changes, which are similar to changes observed in human skeletal muscle.
Heart failure with preserved ejection fraction in human subjects and animal models
The identification of authentic animal models of HFpEF is crucial to improve our understanding of underlying mechanisms towards the goal to develop new treatment strategies. For a proper comparison of alterations in the SM between human and animals, it is important to reliably verify the development of HFpEF. According to the ESC guidelines, 34 the patients included in our study were diagnosed as HFpEF due to diastolic dysfunction (assessed by elevated E/é ratio), increased levels of circulating NT‐pro brain natriuretic peptide and preserved LVEF (above 60%). We analysed these parameters in the three tested animal models, which develop heart failure based on either hypertension (DSS rat and TAC/DOCA mouse), diabetes/hypertension and obesity (ZSF1 rat model). All three animal models exhibited increased E/é ratios, increased LVEDP, myocardial hypertrophy (as evident by increased heart weight), and preserved LVEF compared with their respective controls. In addition, ZSF1‐obese rats, 35 DSS rats fed with high salt diet, 32 and mice subjected to TAC and DOCA‐pellet implantation 36 presented with a significantly lower exercise capacity compared with the respective control. Taken together, in all tested animal models, HFpEF was evident, and therefore, a comparability with human samples with respect to alterations in the SM was given.
Recently, an additional mouse model inducing HFpEF by feeding a high fat diet and giving L‐NAME in the drinking water has been reported. 37 This model also recapitulates numerous systemic and cardiovascular features of human HFpEF, including exercise intolerance. The molecular analysis of the SM revealed no change in mRNA expression of myosin heavy chain isoforms, but an in depth molecular analysis is warranted in the future to finally conclude that in this model exercise intolerance occurred in the absence of molecular changes in the SM.
Similarities and differences between animal models and patients
Proteins involved in muscle atrophy
Skeletal muscle atrophy is a hallmark of HFpEF 14 , 38 and HFrEF, 39 and the ubiquitin proteasome system, as well as autophagy, are the main protein degradation system activated in the SM14. Regarding UPS components and autophagy, our results reveal that among all three tested animal models, the ZSF1‐rat acts most comparable with the human SM. In ZSF1‐obese as well as in human HFpEF patients, MuRF1 is significantly upregulated, leading to an increase of ubiquitinylated proteins, which are subsequently recognized and degraded by the proteasome system. Regarding the MafBx expression, no difference was seen in all three animal models when compared with the respective controls, whereas a significant upregulation was observed in the human HFpEF samples. This is in accordance with our former study of DSS rats, 19 where no difference in MafBx expression was observed. Additionally to the observed activation of the UPS, the ratio of LC‐3 I/II, a marker of autophagy, is comparable between human biopsies and SM tissue of ZSF1 obese rats. The proven down regulation of the LC3 I/II ratio in SM tissue of ZSF1 obese rats is in accordance to the findings of Bowen and colleagues. 40 In summary, with respect to proteins involved in SM atrophy, ZSF1 HFpEF rats showed the best match to changes observed in the SM of HFpEF patients (for summary see Table 2 ).
Structural proteins
In our study, for structural proteins, like MHC, actin and telethonin, no significant difference was detected between human controls and HFpEF samples. In the three animal models, only the DSS HFpEF rat showed a slightly increased expression of these proteins. For SM tissue obtained from HFpEF patients, only a shift in fibre type composition and a reduction in capillary density has been reported. 41 Until now, no reports are available regarding the expression of structural proteins for human HFpEF SM. The situation seems to be quite different in HFrEF, where a clear reduction of MHC, 42 , 43 , 44 and actin expression 45 have been reported. Besides reduced actin expression level, posttranslational modifications resulting in a reduced functionality have been reported. 46 , 47 These differences between HFpEF and HFrEF suggest that SM alterations are rather exacerbated in HFrEF, as recently postulated by Seiler and colleagues. 38
Mitochondrial proteins
Mitochondria are central organelles in the SM as they act as important regulators of the energy supply and abnormalities can significantly contribute to impaired oxygen utilization and to exercise intolerance in HFpEF. 48 Direct assessment of mitochondrial function in a rat model of HFpEF 19 revealed an impaired coupling ratio between O2 consumption and ATP production. In the present study, it became evident that enzymes involved in mitochondrial energy production, like SDH and MDH are significantly reduced in SM of HFpEF patients. This reduction could also be detected in the SM of ZSF1 HFpEF animals, whereas no changes were detected in the DSS rat and TAC/DOCA model. Regarding the expression of porin, we showed increased amounts in human and ZSF HFpEF samples, which is in contrast to the current literature. Molina et al. 49 demonstrated significantly reduced porin expression levels in muscle biopsies obtained from HFpEF patients compared with age‐matched healthy controls. This discrepancy to our present study might go back to a compromised mitochondrial quality control, thereby leading to an accumulation of non‐functional mitochondria (reviewed in Hammerling and Gustafsson 50 ) and subsequently resulting in an increased porin expression. Furthermore, the enhanced generation of mitochondria could serve as a counter reaction of the SM to keep its important mitochondrial energy supply at a high level. At least for the human situation, this is supported by the increased expression of PGC1‐α, a key transcription factor for mitochondrial biogenesis. 51 However, further investigations will be necessary for that. Taken together, mitochondrial energy metabolism seems to be impaired in SM tissue of HFpEF patients and ZSF1 obese rats, whereas no changes were observed in the other two HFpEF animal models.
Reactive oxygen species generating enzymes and inflammation
Reactive oxygen species are constantly generated in the cell by different enzymes and mostly as a by‐product of mitochondrial oxidative phosphorylation. 52 In heart failure and especially in HFrEF, this homeostasis between ROS generation and elimination is shifted towards an increased generation of ROS. 38 For HFpEF the situation is less clear, and in the present study, a trend towards increased NADPH oxidase expression was detected in the SM of HFpEF patients. The ROS load might also be increased in the SM of ZSF1 HFpEF animals because SOD1, one of the main detoxifying enzymes, was significantly reduced. We found no alterations of enzymes involved in controlling the ROS in DSS samples. This is in accordance with an earlier observation from our group, when we compared alterations in the SM of HFrEF (left anterior descending artery [LAD] ligation model) and HFpEF (DSS rat model). At least in the human HFpEF situation the increased expression of TNFα might be a trigger for increased muscle atrophy, by stimulating the expression of MuRF1, and by elevating the expression of NADPH oxidase, thereby inhibiting mitochondrial oxidative phosphorylation. In the animal models investigated in the present study, no increased TNFα expression was observed. However, further investigations have to clarify whether other inflammatory cytokines are increased in the animal models, as seen for the DSS rat model. 38
Study limitation
Interpreting the results from the present comparison of human and animal models of HFpEF with respect to alterations of the peripheral SM we have to consider the following limitations.
First, an important limitation of the study is that the analysed animal models develop HFpEF based on one or two comorbidities like hypertension (DSS and TAC/DOCA) or metabolic syndrome (ZSF1 rat model). In the human situation, HFpEF is triggered by a variety of underlying comorbidities with different influence on SM alterations. This may be one reason why we did not found an animal model mimicking the human situation to 100% with respect to SM changes.
Second, the disease severity may differ between humans and the observed animal models, because the duration of comorbidities in patients before the onset of HFpEF might vary. Additionally, also in the animal models the time span from initiating the comorbidities until the harvest of SM tissue varies from 4 weeks (TAC/DOCA model) to 20 weeks (DSS model) and to 32 weeks (ZSF1 model). This different timing might also have an impact on the molecular alterations seen in the different models.
Third, the assessment of morphologic and functional features of mitochondria is missing in the present study. These measurements would have supported the importance of changes seen in mitochondrial enzyme activities especially in human and ZSF1 SM samples. Nevertheless, based on the current literature we have to assume that mitochondrial function is impaired in humans and animal models of HFpEF (for review refer to Adams et al. 14 ).
Conclusions
The present study is to our knowledge the first report comparing molecular alterations in SM tissue obtained from human HFpEF patients and three different HFpEF animal models. In summary, none of the three tested animal models mimics the situation in HFpEF patients completely. Still, among the models, the ZSF1 rat (ZSF1‐lean vs. ZSF1‐obese) shows the highest overlap to the human condition (see Table 2 ). Therefore, when studying therapeutic interventions to treat HFpEF and especially alterations in the SM, we suggest that the ZSF1 rat is a suitable model.
Conflict of interest
Ephraim Winzer reports personal fees from Boehringer‐Ingelheim, Novartis, and CVRx outside of this study.
Funding
We are grateful to the Foundation Leducq (network 13CVD04) for generous support. This study was supported by MSD Life Foundation, Public Interest Incorporated Foundation.
Acknowledgement
Open access funding enabled and organized by Projekt DEAL.
Goto, K. , Schauer, A. , Augstein, A. , Methawasin, M. , Granzier, H. , Halle, M. , Craenenbroeck, E. M. V. , Rolim, N. , Gielen, S. , Pieske, B. , Winzer, E. B. , Linke, A. , and Adams, V. (2021) Muscular changes in animal models of heart failure with preserved ejection fraction: what comes closest to the patient?. ESC Heart Failure, 8: 139–150. 10.1002/ehf2.13142.
References
- 1. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. New England Journal of Medicine 2006; 355: 251–259. [DOI] [PubMed] [Google Scholar]
- 2. Shah KS, Xu H, Matsouaka RA, Bhatt DL, Heidenreich PA, Hernandez AF, Devore AD, Yancy CW, Fonarow GC. Heart failure with preserved, borderline, and reduced ejection fraction: 5‐year outcomes. J Am Coll Cardiol 2017; 70: 2476–2486. [DOI] [PubMed] [Google Scholar]
- 3. Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A, Gong Y, Liu PP. Outcome of heart failure with preserved ejection fraction in a population‐based study. New England Journal of Medicine 2006; 355: 260–269. [DOI] [PubMed] [Google Scholar]
- 4. Nichols GA, Reynolds K, Kimes TM, Rosales AG, Chan WW. Comparison of risk of re‐hospitalization, all‐cause Mortality, and Medical Care Resource Utilization in Patients With Heart Failure and Preserved Versus Reduced Ejection Fraction. Am J Cardiol 2015; 116: 1088–1092. [DOI] [PubMed] [Google Scholar]
- 5. Steinberg BA, Zhao X, Heidenreich PA, Peterson ED, Bhatt DL, Cannon CP, Hernandez AF, Fonarow GC. Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction. Circulation 2012; 126: 65–75. [DOI] [PubMed] [Google Scholar]
- 6. Somaratne JB, Berry C, McMurray JJV, Poppe KK, Doughty RN, Whalley GA. The prognostic significance of heart failure with preserved left ventricular ejection fraction: a literature‐based meta‐analysis. Eur J Heart Fail 2009; 11: 855–862. [DOI] [PubMed] [Google Scholar]
- 7. Gevaert AB, Boen JRA, Segers VF, Van Craenenbroeck EM. Heart failure with preserved ejection fraction: a review of cardiac and noncardiac pathophysiology. Front Physiol 2019; 10: 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Solomon SD, Zile M, Pieske B, Voors A, Shah A, Kraigher‐Krainer E, Shi V, Bransford T, Takeuchi M, Gong J, Lefkowitz M, Packer M, McMurray JJ. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double‐blind randomised controlled trial. Lancet 2012; 380: 1387–1395. [DOI] [PubMed] [Google Scholar]
- 9. Edelmann F, Wachter R, Schmidt AG, Kraigher‐Krainer E, Colantonio C, Kamke W, Duvinage A, Stahrenberg R, Durstewitz K, Löffler M, Düngen HD, Tschöpe C, Herrmann‐Lingen C, Halle M, Hasenfuss G, Gelbrich G, Pieske B. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo‐DHF randomized controlled trial. JAMA 2013; 309: 781–791. [DOI] [PubMed] [Google Scholar]
- 10. Yamamoto K, Origasa H, Hori M. on behalf of theEffects of carvedilol on heart failure with preserved ejection fraction: the Japanese Diastolic Heart Failure Study (J‐DHF). Eur J Heart Fail 2013; 15: 110–118. [DOI] [PubMed] [Google Scholar]
- 11. Belardinelli R, Georgiou D, Cianci G, Purcaro A. Randomized, controlled trial of long‐term moderate exercise training in chronic heart failure. Circulation 1999; 99: 1173–1182. [DOI] [PubMed] [Google Scholar]
- 12. Jeng C, Yang MH, Chen PL, Ho CH. The influence of exercise tolerance on quality of life among patients with heart failure. Qual Life Res 2004; 13: 925–932. [DOI] [PubMed] [Google Scholar]
- 13. Chung CJ, Schulze PC. Exercise as a nonpharmacologic intervention in patients with heart failure. Phys Sportsmed 2011; 39: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adams V, Linke A, Winzer E. Skeletal muscle alterations in HFrEF vs. HFpEF. Curr Heart Fail Rep 2017; 14: 489–497. [DOI] [PubMed] [Google Scholar]
- 15. Duscha BD, Kraus WE, Keteyian SJ, Sullivan MJ, Green HJ, Schachat FH, Pippen AM, Brawner CA, Blank JM, Annex BH. Capillary density of skeletal muscle: a contributing mechanism for exercise intolerance in class II–III chronic heart failure independent of other peripheral alterations. J Am Coll Cardiol 1999; 33: 1956–1963. [DOI] [PubMed] [Google Scholar]
- 16. Erbs S, Höllriegel R, Linke A, Beck EB, Adams V, Gielen S, Möbius‐Winkler S, Sandri M, Kränkel N, Hambrecht R, Schuler G. Exercise training in patients with advanced chronic heart failure (NYHAIIIb) promotes restoration of peripheral vasomotor function, induction of endogenous regeneration, and improvement of left ventricular function. Circ Heart Fail 2010; 3: 486–494. [DOI] [PubMed] [Google Scholar]
- 17. Hambrecht R, Fiehn E, Yu J, Niebauer J, Weigl C, Hilbrich L, Adams V, Riede U, Schuler G. Effects of endurance training on mitochondrial ultrastructure and fiber type distribution in skeletal muscle of patients with stable chronic heart failure. J Am Coll Cardiol 1997; 29: 1067–1073. [DOI] [PubMed] [Google Scholar]
- 18. Linke A, Adams V, Schulze PC, Erbs S, Gielen S, Fiehn E, Möbius‐Winkler S, Schubert A, Schuler G, Hambrecht R. Antioxidative effects of exercise training in patients with chronic heart failure. Increase in radical scavenger enzyme activity in skeletal muscle. Circulation 2005; 111: 1763–1770. [DOI] [PubMed] [Google Scholar]
- 19. Bowen TS, Rolim NPL, Fischer T, Baekkerud FH, Medeiros A, Werner S, Bronstad E, Rognmo O, Mangner N, Linke A, Schuler G, Silva GJJ, Wisloff U, Adams V. on behalf of the Optimex Study GroupHeart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur J Heart Fail 2015; 17: 263–272. [DOI] [PubMed] [Google Scholar]
- 20. Gevaert AB, Shakeri H, Leloup AJ, Van Hove CE, De Meyer GRY, Vrints CJ, Lemmens K, Van Craenenbroeck EM. Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ Heart Fail 2017; 10: e003806. [DOI] [PubMed] [Google Scholar]
- 21. Adams V, Alves M, Fischer T, Rolim N, Werner S, Schütt N, Bowen TS, Linke A, Schuler G, Wisloff U. High‐intensity interval training attenuates endothelial dysfunction in a Dahl salt‐sensitive rat model of heart failure with preserved ejection fraction. J Appl Physiol 2015; 119: 745–752. [DOI] [PubMed] [Google Scholar]
- 22. Bowen TS, Brauer D, Rolim N, Bakkerud F, Kricke A, Ormbostad AM, Fischer T, Linke A, Silva GJJ, Wisloff U, Adams V. Exercise training reveals inflexibility of the diaphragm in an obesity‐driven HFpEF animal model. J Am Heart Assoc 2017; 6: e006416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmederer Z, Rolim NP, Bowen TS, Linke A, Wisloff U, Adams V. Endothelial function is disturbed in a hypertensive diabetic animal model of HFpEF: moderate continuous vs. high intensity interval training. Int J Cardiol 2018; 273: 147–154. [DOI] [PubMed] [Google Scholar]
- 24. Alex L, Russo I, Holoborodko V, Frangogiannis NG. Characterization of a mouse model of obesity‐related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol 2018; 315: H934–H949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Methawasin M, Strom JG, Slater RE, Fernandez V, Saripalli C, Granzier H. Experimentally increasing the compliance of titin through RNA binding motif‐20 (RBM20) inhibition improves diastolic function in a mouse model of heart failure with preserved ejection fraction. Circulation 2016; 134: 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suchy C, Massen L, Rogmo L, Van Craenenbroeck E, Beckers P, Kraigher‐Krainer E, Linke A, Adams V, Wisloff U, Pieske B, Halle M. Optimising exercise training in prevention and treatment of diastolic heart failure (OptimEx‐CLIN): rationale and design of a prospective, randomised, controlled trial. Eur J Prev Cardiol 2014; 21: 18–25. [DOI] [PubMed] [Google Scholar]
- 27. Gielen S, Sandri M, Kozarez I, Kratsch J, Teupser D, Thiery J, Erbs S, Mangner N, Lenk K, Hambrecht R, Schuler G, Adams V. Exercise training attenuates MuRF‐1 expression in the skeletal muscle of patients with chronic heart failure independent of age: the randomized Leipzig exercise intervention in chronic heart failure and aging (LEICA) catabolism study. Circulation 2012; 125: 2716–2727. [DOI] [PubMed] [Google Scholar]
- 28. Bücher T, Luh W, Pette D. Einfache und zusammengesetzte optische tests mit pyridinnucleotiden In Lang K., Lelnartz E., eds. Hoppe–Seyler Thierfelder Handbuch der Physiologisch‐und Pathologisch‐Chemischen Analyse. Berlin: Springer Verlag; 1964. p 293–339. [Google Scholar]
- 29. Mukherjee A, Srere PA, Frenkel EP. Studies of the mechanism by which hepatic citrate synthase activity increases in vitamin B12 deprivation. J Biol Chem 1976; 251: 2155–2160. [PubMed] [Google Scholar]
- 30. Vanderlinde RE. Measurement of total lactate dehydrogenase activity. Ann Clin Lab Sci 1985; 15: 13–31. [PubMed] [Google Scholar]
- 31. Dzeja PP, Pucar D, Redfield MM, Burnett JC, Terzic A. Reduced activity of enzymes coupling ATP‐generating with ATP‐consuming processes in the failing myocardium. Mol Cell Biochem 1999; 201: 33–40. [DOI] [PubMed] [Google Scholar]
- 32. Guazzi M, Brenner DA, Apstein CS, Saupe KW. Exercise intolerance in rats with hypertensive heart disease is associated with impaired diastolic relaxation. Hypertension 2001; 37: 204–208. [DOI] [PubMed] [Google Scholar]
- 33. Batista J, Rosa JC, Lopes RD, Lira FS, Martins J, Yamashita AS, Brum PC, Lancha J, Lopes AC, Seelaender M. Exercise training changes IL‐10/TNF‐[alpha] ratio in the skeletal muscle of post‐MI rats. Cytokine 2010; 49: 102–108. [DOI] [PubMed] [Google Scholar]
- 34. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, Gonzalez‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur J Heart Fail 2016; 18: 891–975. [DOI] [PubMed] [Google Scholar]
- 35. Miranda‐Silva D, Wüst RCI, Conceicao G, Goncalves‐Rodrigues P, Goncalves N, Goncalves A, Kuster DWD, Leite‐Moreira AF, van der Velden J, de Sousa Beleza JM, Magalhaes J, Stienen GJM, Falcao‐Pires I. Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk‐related rat model of heart failure with preserved ejection fraction. Acta Physiol 2019; 2019: e13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Slater RE, Strom JG, Methawasin M, Liss M, Gotthardt M, Sweitzer N, Granzier HL. Metformin improves diastolic function in an HFpEF‐like mouse model by increasing titin compliance. J Gen Physiol 2019; 151: 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, Hill TM, Mammen PPA, Huang J, Lee DI, Hahn VS, Sharma K, Kass DA, Lavandero S, Gillette TG, Hill JA. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019; 568: 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seiler M, Bowen TS, Rolim N, Dieterlen MT, Werner S, Hoshi T, Fischer T, Mangner N, Linke A, Schuler G, Halle M, Wisloff U, Adams V. Skeletal muscle alterations are exacerbated in heart failure with reduced compared with preserved ejection fraction: mediated by circulating cytokines? Circ Heart Fail 2016; 9: e003027. [DOI] [PubMed] [Google Scholar]
- 39. Martinez PF, Okoshi K, Carvalho RF, Oliveira Junior SA, Lima AR, Campos DH, Damatto RL, Padovani CR, Nogueira CR, Dal Pai‐Silva M, Okoshi MP. Chronic heart failure‐induced skeletal muscle atrophy, necrosis, and changes in myogenic regulatory factors. Med Sci Monit 2010; 16: BR374–BR383. [PubMed] [Google Scholar]
- 40. Bowen TS, Herz C, Rolim NPL, Berre AO, Halle M, Kricke A, Linke A, da Silva GJ, Wisloff U, Adams V. Effects of endurance training on detrimental structural, cellular, and functional alterations in skeletal muscles of heart failure with preserved ejection fraction. J Card Fail 2018; 24: 603–613. [DOI] [PubMed] [Google Scholar]
- 41. Kitzman DW, Nicklas B, Kraus WE, Lyles MF, Eggebeen J, Morgan TM, Haykowsky MJ. Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. Am J Physiol Heart Circ Physiol 2014; 306: H1364–H1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sullivan MJ, Duscha BD, Klitgaard H, Kraus WE, Cobb FR, Saltin B. Altered expression of myosin heavy chain in human skeletal muscle in chronic heart failure. Med Sci Sports Exerc 1997; 29: 860–866. [DOI] [PubMed] [Google Scholar]
- 43. Carvalho RF, Cicogna AC, Campos GER, De Assis JMF, Padovani CR, Okoshi MP, Pai‐Silva MD. Myosin heavy chain expression and atrophy in rat skeletal muscle during transition from cardiac hypertrophy to heart failure. Int J Exp Pathol 2003; 84: 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miller MS, VanBuren P, LeWinter MM, Lecker SH, Selby DE, Palmer BM, Maughan DW, Ades PA, Toth MJ. Mechanisms underlying skeletal muscle weakness in human heart failure: alterations in single fiber myosin protein content and function. Circ Heart Fail 2009; 2: 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simonini A, Massie BM, Long CS, Qi M, Samarel AM. Alterations in skeletal muscle gene expression in the rat with chronic congestive heart failure. J Moll Cell Cardiol 1996; 28: 1683–1691. [DOI] [PubMed] [Google Scholar]
- 46. Mangner N, Bowen TS, Werner S, Fischer T, Kullnick Y, Oberbach A, Linke A, Steil L, Schuler G, Adams V. Exercise training prevents diaphragm contractile dysfunction in heart failure. Med Sci Sports Exerc 2016; 48: 2118–2124. [DOI] [PubMed] [Google Scholar]
- 47. Vescovo G, Ravara B, Dalla Libera L. Skeletal muscle myofibrillar protein oxidation and exercise capacity in heart failure. Basic Res Cardiol 2007; 103: 285–290. [DOI] [PubMed] [Google Scholar]
- 48. Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation 2019; 139: 1435–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Molina AJA, Bharadwaj MS, Van Horn C, Nicklas BJ, Lyles MF, Eggebeen J, Haykowsky MJ, Brubaker PH, Kitzman DW. Skeletal muscle mitochondrial content, oxidative capacity, and Mfn2 expression are reduced in older patients with heart failure and preserved ejection fraction and are related to exercise intolerance. JACC Heart Fail 2016; 4: 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hammerling BC, Gustafsson AB. Mitochondrial quality control in the myocardium: cooperation between protein degradation and mitophagy. J Mol Cell Cardiol 2014; 75: 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Islam H, Edgett BA, Gurd BJ. Coordination of mitochondrial biogenesis by PGC‐1α in human skeletal muscle: a re‐evaluation. Metabolism 2018; 79: 42–51. [DOI] [PubMed] [Google Scholar]
- 52. Bowen TS, Schuler G, Adams V. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle 2015; 6: 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]