

Abstract
Aims
Amyloidogenic transthyretin (ATTR) amyloidosis is a fatal disease characterized by progressive cardiomyopathy and/or polyneuropathy. AKCEA‐TTR‐LRx (ION‐682884) is a ligand‐conjugated antisense drug designed for receptor‐mediated uptake by hepatocytes, the primary source of circulating transthyretin (TTR). Enhanced delivery of the antisense pharmacophore is expected to increase drug potency and support lower, less frequent dosing in treatment.
Methods and results
AKCEA‐TTR‐LRx demonstrated an approximate 50‐fold and 30‐fold increase in potency compared with the unconjugated antisense drug, inotersen, in human hepatocyte cell culture and mice expressing a mutated human genomic TTR sequence, respectively. This increase in potency was supported by a preferential distribution of AKCEA‐TTR‐LRx to liver hepatocytes in the transgenic hTTR mouse model. A randomized, placebo‐controlled, phase 1 study was conducted to evaluate AKCEA‐TTR‐LRx in healthy volunteers (ClinicalTrials.gov: NCT03728634). Eligible participants were assigned to one of three multiple‐dose cohorts (45, 60, and 90 mg) or a single‐dose cohort (120 mg), and then randomized 10:2 (active : placebo) to receive a total of 4 SC doses (Day 1, 29, 57, and 85) in the multiple‐dose cohorts or 1 SC dose in the single‐dose cohort. The primary endpoint was safety and tolerability; pharmacokinetics and pharmacodynamics were secondary endpoints. All randomized participants completed treatment. No serious adverse events were reported. In the multiple‐dose cohorts, AKCEA‐TTR‐LRx reduced TTR levels from baseline to 2 weeks after the last dose of 45, 60, or 90 mg by a mean (SD) of −85.7% (8.0), −90.5% (7.4), and −93.8% (3.4), compared with −5.9% (14.0) for pooled placebo (P < 0.001). A maximum mean (SD) reduction in TTR levels of −86.3% (6.5) from baseline was achieved after a single dose of 120 mg AKCEA‐TTR‐LRx.
Conclusions
These findings suggest an improved safety and tolerability profile with the increase in potency achieved by productive receptor‐mediated uptake of AKCEA‐TTR‐LRx by hepatocytes and supports further development of AKCEA‐TTR‐LRx for the treatment of ATTR polyneuropathy and cardiomyopathy.
Keywords: Transthyretin, Amyloidosis, Cardiomyopathy, Polyneuropathy, Antisense, Ligand‐conjugated
Introduction
Amyloidogenic transthyretin (ATTR) amyloidosis is a progressive and fatal disease manifested by the buildup of misfolded transthyretin (TTR) protein in major organ systems. There are two types of ATTR amyloidosis: one determined by genetic variation and the other by age. 1 , 2 The genetic form, hereditary ATTR (hATTR), is caused by pathogenic variants in the TTR gene that destabilize the normal tetrameric structure of the TTR oligomers, leading to the formation of insoluble, extracellular amyloid deposits in multiple organ systems. 3 Wild‐type TTR can also misfold and deposit as amyloid, causing wild‐type ATTR (wtATTR) amyloidosis in the elderly, which typically manifests primarily as a restrictive cardiomyopathy. 4 ATTR amyloidosis is a complex disease of mixed phenotypes. The most common phenotypes are polyneuropathy (PN) and cardiomyopathy (CM), but other tissues and organs can be affected, including the kidneys, gastrointestinal tract, and eyes, causing symptoms in multiple organs. 5 , 6 , 7
RNase H and RNA interference (RNAi) are two antisense RNA‐degradation mechanisms broadly recognized for development of RNA‐targeted drugs today. 8 , 9 Both mechanisms have recently been validated as an approach for treatment of patients with hATTR‐PN in the pivotal phase 3 studies of inotersen 10 (NEURO‐TTR) and patisiran 11 (APOLLO). Both drugs act through sequence‐specific binding to the target TTR mRNA through Watson‐Crick base pairing, to form an antisense : sense duplex. Once formed, the duplex is a substrate for an endoribonuclease (RNase H1 or argonaute 2 [Ago2] for RNAi), which degrades the mRNA at the site where the antisense strand is bound. 8 , 9 Degradation of the target mRNA reduces the amount of transcript (mRNA) available for translation, leading to subsequent reduction in synthesis of the protein product. In each respective pivotal phase 3 study, reduction of serum TTR protein levels by either antisense mechanism led to both clinical benefit in composite neurologic assessment and improved quality of life in patients with hATTR‐PN.
Inotersen is a single‐strand 2′‐O‐methoxyethyl (2′‐MOE) modified full phosphorothioate (PS) antisense oligonucleotide (ASO) that supports RNase H1 activity with a central ‘gap’ of ten 2′‐deoxynucleotides. 12 Inotersen includes five 2′‐MOE modified ribonucleotides at each terminus to increase the affinity for the target TTR mRNA as well as to enhance drug stability. 13 The PS substitutions throughout the ASO backbone provide resistance to nuclease activity and increase plasma protein binding to facilitate broad tissue distribution and cell uptake. 14 Patisiran, on the other hand, is a double‐stranded small interfering RNA (siRNA) that supports Ago2 activity and requires formulation in a lipid nanoparticle to provide sufficient stability, effective tissue distribution, and productive cellular uptake upon systemic intravenous delivery. 15
AKCEA‐TTR‐LRx (ION‐682884) is an ASO of similar design and identical sequence as inotersen that is conjugated to a triantennary N‐acetyl galactosamine (GalNAc3) moiety. This moiety acts as a ligand for productive receptor‐mediated uptake by the high‐capacity asialoglycoprotein receptors (ASGR) expressed by hepatocytes, 16 , 17 which are the principal source of systemically circulating TTR. After internalization into cells by the ASGR1‐mediated endocytic pathway, the GalNAc3 cluster is metabolized to release ‘free ASO’ inside the hepatocyte for binding to the target mRNA. 18 The ligand‐conjugated antisense (LICA) technology has provided a 20‐fold to 30‐fold increase in potency for targets expressed by liver hepatocytes. 19 , 20 Additionally, targeted receptor‐mediated delivery eliminates the need for a full PS‐modified backbone to facilitate tissue distribution and cell uptake. Based on prior clinical experience with GalNAc3‐conjugated ASOs, AKCEA‐TTR‐LRx is expected to support lower, less frequent dosing and achieve an improved safety and tolerability profile compared with inotersen. 19 , 21 , 22 , 23 , 24 Here, we present the preclinical comparison of inotersen and AKCEA‐TTR‐LRx pharmacologic activity in human hepatocyte cell culture and a human TTR transgenic mouse model, and the results from first‐in‐human dosing with AKCEA‐TTR‐LRx in a phase 1 study in healthy volunteers.
Methods
AKCEA‐TTR‐LRx and inotersen are 2′‐MOE‐modified ASOs that target the 3′ untranslated region (3′UTR) of the human TTR pre‐messenger RNA (NM_000371.3; base pairs 618 to 637; 5′‐TCTTGGTTACATGAAATCCC‐3′). The target sequence has no known hATTR pathogenic mutations and was identified through an extensive ASO screening process described previously. 25 Both ASOs are 20 nucleotides in length with five 2′‐MOE‐modified ribonucleotides at each terminus and a central region of ten 2′deoxynucleotide residues. AKCEA‐TTR‐LRx utilizes a LICA technology for targeted delivery to hepatocytes through conjugation of a triantennary cluster of GalNAc sugars on the 5′ end. 26 The conjugated form of the ASO has six phosphodiester linkages in the 2′MOE modified ends, as compared with the full number of PS linkages contained in inotersen. Decreasing the PS content of single‐stranded oligonucleotides has been found to attenuate sequence‐dependent proinflammatory effects 27 and, in the conjugated form, shows a similar or slightly enhanced increase in potency. 26
Detailed methods for the preclinical model studies are provided in the Supporting Information, Data S1 .
Phase 1 study
AKCEA‐TTR‐LRx was evaluated in a randomized, double‐blind, placebo‐controlled, dose‐ascending phase 1 study at a single site in Canada from December 2018 to February 2020. The study was approved by Institutional Review Board Services (Ontario, Canada) and conducted in compliance with the World Medical Association Declaration of Helsinki (October 2002), Good Clinical Practice (GCP) guidelines, and all national, state, and local laws of the appropriate regulatory authorities. All participants provided written informed consent prior to participation in the study. Participants, monitors, study centre personnel, and the sponsor were blinded to treatment throughout the study. Only the pharmacist who allocated the drug was unblinded to treatment.
Healthy male and non‐pregnant, non‐lactating female participants between the ages of 18 and 65 years with a body mass index <32 kg/m2 and a willingness to take vitamin A supplements were eligible for the study. Women of childbearing potential were excluded. Participants with clinically significant abnormalities in medical history, physical examinations, vital signs, electrocardiogram (ECG), or laboratory tests were also excluded.
Eligible participants were randomized 10:2, active to placebo, in one single‐dose cohort of 120 mg and three multiple‐dose cohorts of 45, 60, or 90 mg. Study drug was administered by SC injection. Participants in the multiple‐dose cohorts received one dose of the study drug once every 4 weeks on Days 1, 29, 57, and 85 (Supporting Information, Figure S1 ).
Phase 1 endpoints
Safety assessments included adverse events (AEs), physical examination findings, ECG results, vital signs, and routine laboratory tests for blood chemistry, coagulation, complement, haematology, and urinalysis.
Pharmacokinetic (PK) parameters were determined based on nominal times from intensive PK sampling following SC injection in the multiple‐dose (Days 1 and 85) and single‐dose (Day 1) cohorts. Concentrations of total full‐length ASO in human plasma were measured using a validated hybridization‐based assay with electrochemiluminescence (ECL) detection, a variation on the method reported previously. 28 Total full‐length ASO included full and partial (one‐, two‐, and three‐sugar deletions) conjugated forms and the unconjugated form of AKCEA‐TTR‐LRx. The assay was validated for precision, accuracy, selectivity, sensitivity, dilution linearity, anti‐drug antibody interference, and stability of AKCEA‐TTR‐LRx quantitation prior to analysis of the plasma samples. Equivalent mass concentrations (μg eq./mL) were determined using AKCEA‐TTR‐LRx standard curves. Plasma sample analyses were conducted at PPD Inc. and were performed based on the principles and requirements described in 21 CFR Part 58. The assay conducted with synthesized putative shortened oligonucleotide metabolite standards showed no measurable cross‐reactivity, confirming the assay's specificity for the parent full‐length oligonucleotides. The quantitation range was 0.129–86.1 ng/mL, with the low and high ends of the range defining the lower and upper limits of quantitation, respectively. Samples anticipated to be above the upper limits of quantitation were diluted prior to the hybridization‐ECL analysis.
Pharmacodynamic (PD) assessments evaluated the per cent change from baseline in serum TTR and retinol binding protein 4 (RBP4) concentration. Laboratory tests were performed by a central laboratory (Medpace Reference Laboratories). Serum TTR and RBP4 concentrations were determined using ELISA.
Phase 1 analysis
The sample size was based on previous experience with ASOs in the same chemical class to adequately assess the safety, tolerability, PK, and pharmacodynamics of AKCEA‐TTR‐LRx while minimizing unnecessary participant exposure.
Safety and pharmacodynamic populations included all participants who were randomized and received at least one dose of study drug. The baseline for pharmacodynamic analysis was defined as the average of the pre‐dose measurement closest to Day 1 and the pre‐dose measurement on Day 1, where Day 1 was the time of the first dose of study drug. Participants assigned to placebo treatment in the multiple‐dose cohorts were pooled for analysis.
Noncompartmental PK analysis of AKCEA‐TTR‐LRx was carried out on each individual subject data set (Phoenix WinNonlin version 8.1). Plasma PK parameters for total full‐length ASOs (ION‐682884 equivalent) included peak plasma concentration (Cmax) and time to Cmax (tmax), area under the concentration‐time curve from 0 to 24 h (AUC0‐24h), AUCτ after multiple doses, AUC0‐∞ and clearance (CL/F) for the single‐dose cohort only, clearance at steady‐state following multiple doses (CLss/F), and apparent terminal elimination half‐life (t1/2λz).
Per cent change from baseline in serum TTR and RBP4 levels was compared between each AKCEA‐TTR‐LRx treatment group and pooled placebo at each post‐baseline visit using one‐way ANOVA. If data departed substantially from normality, the Wilcoxon rank sum test was applied. Statistical tests were conducted using two‐sided tests with a 5% type 1 error rate (SAS version 9.4).
Results
Preclinical studies
In vitro pharmacodynamics of AKCEA‐TTR‐LRx
In HepatoPac cell culture, inotersen and AKCEA‐TTR‐LRx both exhibited dose‐dependent reductions in wild‐type TTR mRNA expression. AKCEA‐TTR‐LRx had a median EC50 of 0.059 μM compared with 3.01 μM for inotersen (Figure 1 A ). AKCEA‐TTR‐LRx showed greater reductions in TTR mRNA at the same drug concentrations and was approximately 50‐fold more potent than inotersen.
Figure 1.
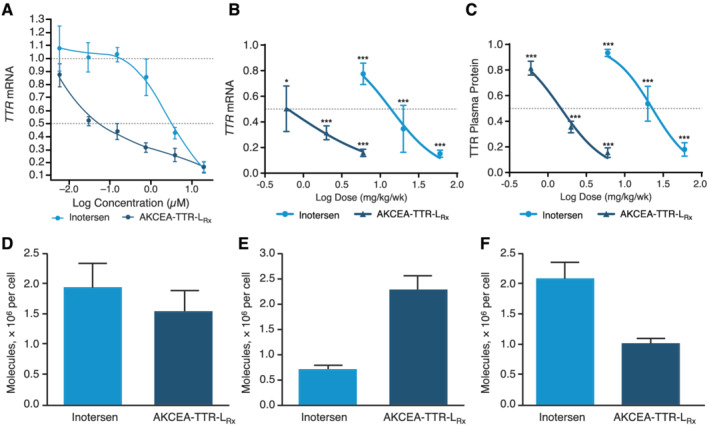
AKCEA‐TTR‐LRx produced dose‐dependent reductions in TTR mRNA in human hepatocytes and human TTR mRNA and protein levels in hTTR‐Tg mice, with preferential distribution to liver hepatocytes in hTTR‐Tg mice. (A) Mean percentage TTR mRNA levels relative to untreated primary human hepatocytes after culture with AKCEA‐TTR‐LRx at concentrations ranging from 0.0064 to 40 μM. (B) Mean liver human TTR mRNA levels relative to that of phosphate buffered saline‐treated hTTR‐Tg mice. (C) Mean plasma hTTR protein levels relative to baseline pre‐treatment levels in hTTR‐Tg mice. (D–F) Number of ASO molecules per cell in whole liver (D), hepatocytes (E), and non‐parenchymal cells (F) in hTTR‐Tg mice treated with equimolar amounts of inotersen or AKCEA‐TTR‐LRx. *P < 0.05; **P < 0.01; ***P < 0.001.
In vivo pharmacodynamics of AKCEA‐TTR‐LRx in hTTR‐Tg mice
The in vivo pharmacologic activity of inotersen and AKCEA‐TTR‐LRx was compared in transgenic mice expressing the human genomic TTR transgene containing the Ile84Ser pathogenic mutation. Three once‐weekly doses of inotersen or AKCEA‐TTR‐LRx resulted in dose‐dependent reductions of hepatic hTTR mRNA levels with a maximum target reduction of 85% for both drugs at the highest doses (60 and 6 mg/kg/week, respectively) compared with PBS‐treated controls (Figure 1 B ). The ED50 was 0.5 mg/kg/week for AKCEA‐TTR‐ LRx and 13.9 mg/kg/week for inotersen, representing a 28‐fold improvement in the potency for liver hTTR mRNA knockdown. Reductions in liver hTTR mRNA correlated with reductions in hTTR plasma protein (Figure 1 C ). AKCEA‐TTR‐LRx had an ED50 of 1.5 mg/kg/week compared with 22.9 mg/kg/week for inotersen, representing a 15‐fold improvement in TTR plasma protein reduction.
Following administration of a single dose of equal molar concentration of ASO (1.4 mmol/kg) to hTTR‐Tg mice, inotersen distributed preferentially to nonparenchymal cells by a mean (SD) of 2.09 (0.52) molecules per nonparenchymal cell, and 0.7 (0.19) molecules per hepatocyte cell, see Figure 1 D–F . Conversely, AKCEA‐TTR‐LRx preferentially accumulated in hepatocytes by a mean (SD) of 2.27 (0.60) molecules per hepatocyte cell and 1.18 (0.12) molecules per nonparenchymal cell, leading to an approximate three‐fold increase of drug delivered to hepatocytes.
Phase 1 trial
Forty‐seven eligible adults (20 women and 27 men) were assigned to one of three multiple‐dose cohorts (N = 36) or a single‐dose cohort (N = 11). Subjects were then randomized within each cohort to receive a total of four doses of either 45 mg (n = 10), 60 mg (n = 10), or 90 mg (n = 10) AKCEA‐TTR‐LRx, or placebo (n = 6, pooled), SC once every 4 weeks in the multiple dose cohorts; or one dose of 120 mg AKCEA‐TTR‐LRx (n = 9), or placebo (n = 2), in the single‐dose cohort (Figure 2 ). Across treatment groups, mean age ranged from 43.4 to 51.6 years and mean BMI ranged from 24.7 to 30.4 kg/m2 (Table 1 ). All participants completed dosing and were included in the pharmacodynamic and safety analyses.
Figure 2.
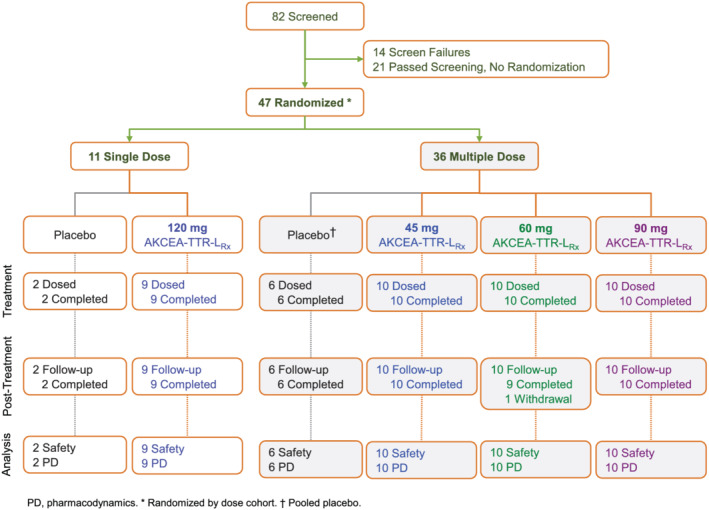
Flow of study participants in phase 1 trial of AKCEA‐TTR‐LRx.
Table 1.
Demographics and baseline characteristics of phase 1 study participants by treatment groups
| Single‐dose cohort | Multiple‐dose cohorts | |||||
|---|---|---|---|---|---|---|
| AKCEA‐TTR‐LRx | AKCEA‐TTR‐LRx | |||||
| Characteristic | Placebo | 120 mg | Placebo a | 45 mg | 60 mg | 90 mg |
| n | 2 | 9 | 6 | 10 | 10 | 10 |
| Age, years | 57.0 (53, 61) | 43.4 (26, 60) | 49.2 (39, 57) | 51.6 (28, 61) | 51.6 (23, 65) | 51.1 (28, 62) |
| Gender (M : F) | 0:2 | 8:1 | 2:4 | 7:3 | 4:6 | 6:4 |
| Race, n (%) | ||||||
| White | 2 (100.0%) | 3 (33.3%) | 4 (66.7%) | 6 (60.0%) | 4 (40.0%) | 6 (60.0%) |
| Black | 0 (0.0%) | 3 (33.3%) | 1 (16.7%) | 3 (30.0%) | 3 (30.0%) | 1 (10.0%) |
| Asian | 0 (0.0%) | 3 (33.3%) | 1 (16.7%) | 1 (10.0%) | 3 (30.0%) | 3 (30.0%) |
| BMI, kg/m2 | 30.4 (0.2) | 25.2 (2.4) | 24.7 (2.5) | 26.3 (2.6) | 26.2 (3.7) | 25.4 (2.8) |
| TTR, mg/L | 237.4 (10.7) | 317.8 (70.7) | 324.3 (84.5) | 316.0 (99.3) | 303.5 (53.5) | 304.6 (79.2) |
| RBP4, mg/L | 29.59 (3.09) | 32.14 (9.67) | 34.045 (6.73) | 37.95 (10.08) | 25.35 (4.39) | 29.93 (7.42) |
BMI, body mass index; RBP4, retinol binding protein 4; SD, standard deviation; TTR, transthyretin.
Values shown for BMI, TTR, and RBP4 are the mean (standard deviation) and for age are the mean (minimum, maximum).
Pooled placebo.
Pharmacokinetics
Following SC administration, AKCEA‐TTR‐LRx was absorbed rapidly into the systemic circulation with median tmax values of one to 6 h (Table S1 ). Similar tmax values were observed at all dose levels. Mean Cmax and total exposure (AUC) were dose‐dependent and near dose‐proportional over the studied SC dose range of 45 to 120 mg. After reaching Cmax, mean plasma concentrations of AKCEA‐TTR‐LRx declined over time in a biphasic fashion, with an initial rapid distribution phase that dominated the plasma clearance, followed by a slower elimination phase with an apparent terminal half‐life (t1/2λz) of 2 to 4 weeks. This result was consistent with the slow elimination of AKCEA‐TTR‐LRx observed in monkeys and the comparatively long half‐lives observed for this chemical class. 20 There was little or no increase in mean plasma Cmax or AUC after four repeated SC injections once every 4 weeks as compared with the first dose, consistent with the expected result of little or no plasma accumulation and time‐invariant plasma kinetics.
Pharmacodynamics
Four SC doses of AKCEA‐TTR‐LRx once every 4 weeks produced dose‐dependent and prolonged decreases in TTR levels over time compared to placebo (Figure 3 ). Mean per cent changes from baseline at 2 weeks after the fourth dose were statistically significant compared with pooled placebo (−5.9%) in each AKCEA‐TTR‐LRx multiple‐dose group of 45 mg (−85.7%, P < 0.001), 60 mg (−90.5%, P < 0.001), and 90 mg (−93.8%, P < 0.001) (Table 2 ) and at all observations from 1 week after the first dose (Day 8) to 3 months after the last dose (Day 176) (Table S2 ). Dose‐dependent and prolonged reductions in RBP4 levels also occurred, with a mean per cent change from baseline to 2 weeks after last dose of −77.3%, −79.2%, and −84.0% in the 45, 60, and 90 mg AKCEA‐TTR‐LRx multiple‐dose groups, respectively (Figure S2 , Table 2 ).
Figure 3.
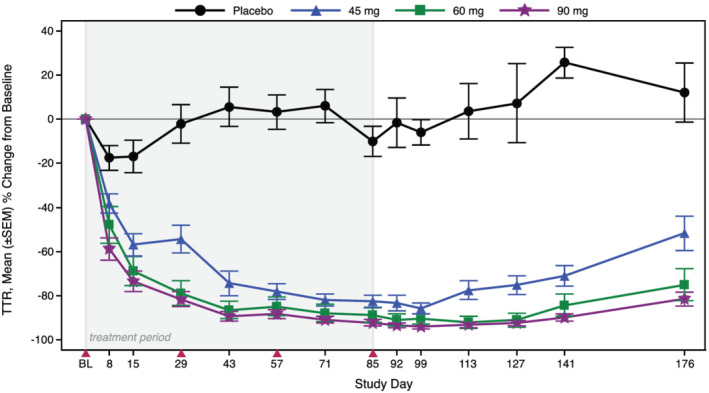
Mean per cent change from baseline in TTR over time in the multiple‐ascending dose cohorts. Subjects were administered four SC doses of 45, 60, or 90 mg AKCEA‐TTR‐LRx or placebo, once every 4 weeks. Shaded region represents treatment period and solid red triangles indicate dosing days.
Table 2.
Mean per cent change in serum TTR and RBP4 levels from baseline to 2 weeks after the last dose in the multiple dose groups
| AKCEA‐TTR‐LRx | ||||
|---|---|---|---|---|
| % change from baseline | Placebo | 45 mg | 60 mg | 90 mg |
| TTR | ||||
| n | 6 | 10 | 10 | 10 |
| Mean (SD) | −5.9 (14.0) | −85.7 (8.0) | −90.5 (7.4) | −93.8 (3.4) |
| Min, Max | −28.5, 10.0 | −93.5, −71.7 | −97.2, −71.8 | −98.4, −87.0 |
| P‐value [A] | <0.001 | <0.001 | <0.001 | |
| RBP4 | ||||
| n | 6 | 10 | 10 | 10 |
| Mean (SD) | −0.2 (13.3) | −77.3 (12.5) | −79.2 (5.5) | −84.0 (4.6) |
| Min, Max | −15.5, 21.4 | −89.7, −51.1 | −89.0, −70.2 | −90.2, −74.6 |
| P‐value [T] | 0.006 | 0.006 | 0.006 | |
[A] ANOVA test P‐value, [T] Wilcoxon rank sum t approximation test two‐sided P‐value. Max, maximum; Min, minimum; RBP4, retinol binding protein 4; SD, standard deviation; TTR, transthyretin.
A single dose of 120 mg AKCEA‐TTR‐LRx produced a rapid mean (SD) reduction in TTR levels of −60.4% (14.0) 1 week after dosing, with a maximum mean (SD) reduction from baseline of −86.3% (6.5) on Day 29 (Figure S3 ). RBP4 showed a similar profile over time, with a mean (SD) per cent change from baseline of −53.6% (12.1) 1 week after dosing and maximum mean (SD) reduction of −77.4% (6.2) on Day 29 in the single‐dose group.
Safety and tolerability
No serious AEs occurred in this study, and no dose discontinuations occurred due to an AE. AEs that occurred in two or more participants included headache in two subjects of the 120 mg single‐dose group, alanine aminotransferase (ALT) increase in four subjects in the 90 mg multiple‐dose group, and blood creatine phosphokinase (CK) increase in one placebo‐treated subject and four subjects in the 90 mg multiple‐dose group (Table 3 ). The increase in ALT levels of two of the four subjects was three‐fold above the upper limit of normal (ULN) and reversed after the last dose without sequelae. There were no abnormalities in laboratories values of bilirubin, international normalized ratio (INR), or alkaline phosphatase in these subjects. CK increases were transient, and subjects were asymptomatic. Two of the four subjects in the 90 mg group who had a mild transient increase in CK levels on treatment also had a transient increase prior to dosing.
Table 3.
Treatment‐emergent adverse events reported in two or more participants: (A) single‐dose cohort and (B) multiple‐dose cohorts
| (A) | |||||
| AKCEA‐TTR‐LRx | |||||
| Preferred term a , n | Placebo (2) | 120 mg (9) | |||
| Headache | 0 | 2 | |||
| (B) | AKCEA‐TTR‐LRx | ||||
| Preferred term b , n | Placebo (6) | 45 mg (10) | 60 mg (10) | 90 mg (10) | Total (30) |
| Alanine aminotransferase increased | 0 | 0 | 0 | 4 | 4 |
| Blood creatine phosphokinase increased | 1 | 0 | 0 | 4 | 4 |
One subject had an event considered mild in severity; a second subject had an event of moderate severity.
All events were considered mild in severity.
No local cutaneous injection site reactions, defined as injection site erythema, pain, swelling, or pruritis that persisted for two or more days post‐injection, were reported in this study. Influenza‐like illness was experienced by one subject in the 120 mg single‐dose group. No significant abnormalities were observed in either renal or haematologic parameters, such as increased serum creatinine levels or reduced platelet counts, respectively (Table S3 ).
Discussion
AKCEA‐TTR‐LRx is under development for treatment of both hATTR and wtATTR. In healthy volunteers, AKCEA‐TTR‐LRx produced an overall reduction of approximately 90% in TTR levels after four SC doses of 45, 60, or 90 mg once every 4 weeks. The mean reduction in serum TTR at the lowest dose tested was 86% from baseline under the pre‐steady state conditions of this dose regimen. As predicted by the PK properties of this class of LICA drugs, serum TTR reductions were maintained between doses and up to 3 months after the last dose in the multi‐dose treatment groups, supporting infrequent dosing. Except for one moderate AE of headache at the highest dose tested, all other reported AEs were mild in severity and resolved spontaneously without sequelae. AKCEA‐TTR‐LRx was well tolerated at all doses tested in phase 1, with all participants receiving the full number of planned doses.
The pharmacodynamics of AKCEA‐TTR‐LRx observed in healthy volunteers was supported by an approximately 50‐fold reduction in EC50 in human hepatocyte cell culture and a 28‐fold reduction in ED50 (<1 mg/kg) in the Ile84Ser hTTR‐Tg mouse model, upon head‐to‐head comparison with inotersen at the target TTR mRNA level. Preferential uptake of the ligand‐conjugated drug to hepatocytes was also demonstrated in hTTR‐Tg mice, further confirming the basis of the increase in potency across species. These results are consistent with other drugs in this LICA class, showing an improved pharmacodynamic profile compared with the unconjugated parent ASO across species 16 , 19 , 20 , 21 , 22 , 23 , 29 , 30 and support the potential for an increase in safety margin upon longer‐term exposure in the clinic. 24
GalNAc3‐conjugated siRNAs have also been investigated in the clinic. The first that advanced to late‐stage clinical development in patients with hATTR‐CM was revusiran in the pivotal phase 3 study ENDEAVOUR. 31 , 32 This trial was halted early, however, due to an unfavourable imbalance in mortality rate between the revusiran and placebo arms. 33 Further to this outcome, treatment‐emergent peripheral neuropathy and severe hepatic events were only reported in the revusiran arm, and four revusiran‐treated patients had elevations in liver transaminase accompanied by increases in total bilirubin. 34 Consequently, the revusiran development program was terminated.
Vutrisiran, a next‐generation siRNA of identical sequence as revusiran, is now in phase 3 investigations in patients with ATTR amyloidosis. This siRNA and its predecessor, revusiran, target the same site of the 3′‐UTR of the TTR mRNA as AKCEA‐TTR‐LRx and inotersen but differ in structure, chemistry, and the enzyme‐mediated mechanism of target mRNA reduction. Vutrisiran differs from revusiran in its chemical modifications, 35 through incorporation of two PS modifications at the 5′ terminus of each of the two strands for increased stability 36 and reduction in the number of 2′‐fluoro‐modified sugar residues (from 22 to 9 residues) to potentially minimize hybridization‐dependent off‐target effects 37 and lessen exposure to 2′‐fluoro nucleoside metabolites. 38 Outcomes from phase 3 assessments will provide further insight on the effect of these modifications.
An alternative approach to treating ATTR amyloidosis is to stabilize the functional TTR tetramer to prevent its dissociation to the monomeric form, a catalyst for formation of amyloidogenic fibrils. 39 Tafamidis is a TTR stabilizer and the first medicine to be approved for the treatment of patients with ATTR‐CM. Though tafamidis was found to attenuate disease progression in patients with ATTR‐CM, including morbidity and mortality rate, consistent worsening in patients' functional capacity and quality of life were observed over time despite continued treatment. 40 Thus, there remains an unmet medical need for more effective treatments.
The increased potency of AKCEA‐TTR‐LRx demonstrated first preclinically in human hepatocytes and hTTR‐Tg mice supports and predicts an equivalent pharmacologic effect to inotersen despite lower and less frequent dosing in humans. Based on the PK and PD findings for AKCEA‐TTR‐LRx and the absence of potential tolerability or safety issues, such as the increases in liver transaminase levels observed in the upper dose range of the current phase 1 trial, the 45 mg dose SC once every 4 weeks was chosen for the phase 3 studies.
The lower, less frequent SC dose is expected to improve the drug safety profile by reducing total systemic exposure and to provide a more convenient and tolerable dosing regimen for patients compared with inotersen. 10 , 41 , 42 Additionally, based on the absorption, distribution, metabolism, and excretion of this class of drugs, 20 , 43 combination with tafamidis or medications such as statins, antihypertensives, or diuretics is not expected to be contraindicated due to drug–drug interactions. 44 Finally, because AKCEA‐TTR‐LRx is designed to reduce both wild‐type and mutant TTR mRNA, treatment of both forms of ATTR is possible.
AKCEA‐TTR‐LRx is currently under investigation in two phase 3 trials, NEURO‐TTRansform in patients with hATTR‐PN (ClinicalTrials.gov, NCT04136184) and CARDIO‐TTRansform in patients with either hATTR‐CM or wtATTR‐CM (NCT04136171). Prior experience with dosing in the inotersen trials and the predictable antisense platform effect, as well as modelling, provide confidence that the reduction in circulating TTR levels will be replicated in patients with either hATTR or wtATTR amyloidosis. Notably though, TTR amyloidosis affects persons who are older in age than the participants in the current phase 1 trial and particularly those with the wild‐type form. Thus, a broader characterization of the safety profile of this investigational antisense drug awaits further evaluation in the phase 3 trials, which allow patients up to 82 and 90 years of age, respectively, as well as those with mild to moderate renal impairment and elevated hepatic transaminase levels.
The efficacy and safety of 45 mg AKCEA‐TTR‐LRx SC once every 4 weeks are being investigated in both pivotal, phase 3 trial. An approximate 30‐fold increase in potency was observed under pre‐steady state conditions at this dose of AKCEA‐TTR‐LRx in healthy volunteers, relative to 300 mg weekly SC dose of the unconjugated 2′‐MOE‐modified ASO inotersen. 10 , 25 Based on the profile of AKCEA‐TTR‐LRx and the LICA platform overall, AKCEA‐TTR‐LRx is expected to produce greater efficacy compared with existing RNA‐targeted medicines with an attractive safety profile and the convenience of a once monthly injection.
Conflict of interest
All authors are employees of Ionis Pharmaceuticals, Inc. M. D. B. is a consultant for Ionis Pharmaceuticals, Inc.
Funding
This study is funded by Ionis Pharmaceuticals, Inc., Carlsbad, CA.
Supporting information
Data S1. Methods ‐ Preclinical Models
Figure S1. Phase 1 study design in healthy volunteers
Figure S2. Mean percent change from baseline in RBP4 over time with multiple doses
Figure S3. Mean percent change from baseline in TTR and RBP4 after a single dose
Table S1. Plasma pharmacokinetics of AKCEA‐TTR‐LRx in healthy subjects
Table S2. Summary of percent change from baseline in plasma TTR overtime
Table S3. Clinical laboratory measurements
Acknowledgements
The authors thank Tracy Reigle of Ionis Pharmaceuticals, Inc., for providing graphic art support and Autumn Kelly of AHK Communications for editorial and administrative support.
Viney, N. J. , Guo, S. , Tai, L.‐J. , Baker, B. F. , Aghajan, M. , Jung, S. W. , Yu, R. Z. , Booten, S. , Murray, H. , Machemer, T. , Burel, S. , Murray, S. , Buchele, G. , Tsimikas, S. , Schneider, E. , Geary, R. S. , Benson, M. D. , and Monia, B. P. (2021) Ligand conjugated antisense oligonucleotide for the treatment of transthyretin amyloidosis: preclinical and phase 1 data. ESC Heart Failure, 8: 652–661. 10.1002/ehf2.13154.
References
- 1. Coelho T, Maurer MS, Suhr OB. THAOS ‐ The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild‐type transthyretin amyloidosis. Curr Med Res Opin 2013; 29: 63–76. [DOI] [PubMed] [Google Scholar]
- 2. Cuddy SAM, Falk RH. Amyloidosis as a systemic disease in context. Can J Cardiol 2020; 36: 396–407. [DOI] [PubMed] [Google Scholar]
- 3. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 2019; 15: 387–404. [DOI] [PubMed] [Google Scholar]
- 4. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, Steidley DE, Ventura H, Murali S, Silver MA, Jacoby D, Fedson S, Hummel SL, Kristen AV, Damy T, Plante‐Bordeneuve V, Coelho T, Mundayat R, Suhr OB, Waddington Cruz M, Rapezzi C, Investigators T . Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol 2016; 68: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease‐modifying treatments. J Neurol Neurosurg Psychiatry 2015; 86: 1036–1043. [DOI] [PubMed] [Google Scholar]
- 6. Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, Berk JL, Plante‐Bordeneuve V, Schmidt HHJ, Merlini G. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol 2015; 66: 2451–2466. [DOI] [PubMed] [Google Scholar]
- 7. Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S, Lewis WD, Obici L, Plante‐Bordeneuve V, Rapezzi C, Said G, Salvi F. Guideline of transthyretin‐related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 2013; 8: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bennett CF. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med 2019; 70: 307–321. [DOI] [PubMed] [Google Scholar]
- 9. Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA‐targeted therapeutics. Cell Metab 2018; 27: 714–739. [DOI] [PubMed] [Google Scholar]
- 10. Benson MD, Waddington‐Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Plante‐Bordeneuve V, Barroso FA, Merlini G, Obici L, Scheinberg M, Brannagan TH 3rd, Litchy WJ, Whelan C, Drachman BM, Adams D, Heitner SB, Conceicao I, Schmidt HH, Vita G, Campistol JM, Gamez J, Gorevic PD, Gane E, Shah AM, Solomon SD, Monia BP, Hughes SG, Kwoh TJ, McEvoy BW, Jung SW, Baker BF, Ackermann EJ, Gertz MA, Coelho T. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 22–31. [DOI] [PubMed] [Google Scholar]
- 11. Adams D, Gonzalez‐Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Plante‐Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH 3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 11–21. [DOI] [PubMed] [Google Scholar]
- 12. Wu H, Lima WF, Crooke ST. Properties of cloned and expressed human RNase H1. J Biol Chem 1999; 274: 28270–28278. [DOI] [PubMed] [Google Scholar]
- 13. Teplova M, Minasov G, Tereshko V, Inamati GB, Cook PD, Manoharan M, Egli M. Crystal structure and improved antisense properties of 2′‐O‐(2‐methoxyethyl)‐RNA. Nat Struct Biol 1999; 6: 535–539. [DOI] [PubMed] [Google Scholar]
- 14. Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol 2009; 5: 381–391. [DOI] [PubMed] [Google Scholar]
- 15. Kulkarni JA, Witzigmann D, Chen S, Cullis PR, van der Meel R. Lipid nanoparticle technology for clinical translation of siRNA therapeutics. Acc Chem Res 2019; 52: 2435–2444. [DOI] [PubMed] [Google Scholar]
- 16. Prakash TP, Graham MJ, Yu J, Carty R, Low A, Chappell A, Schmidt K, Zhao C, Aghajan M, Murray HF, Riney S, Booten SL, Murray SF, Gaus H, Crosby J, Lima WF, Guo S, Monia BP, Swayze EE, Seth PP. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N‐acetyl galactosamine improves potency 10‐fold in mice. Nucleic Acids Res 2014; 42: 8796–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanowitz M, Hettrick L, Revenko A, Kinberger GA, Prakash TP, Seth PP. Asialoglycoprotein receptor 1 mediates productive uptake of N‐acetylgalactosamine‐conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res 2017; 45: 12388–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shemesh CS, Yu RZ, Gaus HJ, Greenlee S, Post N, Schmidt K, Migawa MT, Seth PP, Zanardi TA, Prakash TP, Swayze EE, Henry SP, Wang Y. Elucidation of the biotransformation pathways of a Galnac3‐conjugated antisense oligonucleotide in rats and monkeys. Mol Ther Nucleic Acids 2016; 5: e319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crooke ST, Baker BF, Xia S, Yu RZ, Viney NJ, Wang Y, Tsimikas S, Geary RS. Integrated Assessment of the clinical performance of GalNAc3‐conjugated 2′‐O‐methoxyethyl chimeric antisense oligonucleotides: I. Human Volunteer Experience. Nucleic Acid Ther 2019; 29: 16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y, Yu RZ, Henry S, Geary RS. Pharmacokinetics and clinical pharmacology considerations of GalNAc3‐conjugated antisense oligonucleotides. Expert Opin Drug Metab Toxicol 2019; 15: 475–485. [DOI] [PubMed] [Google Scholar]
- 21. Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, Crooke ST, Witztum JL, Stroes ES, Tsimikas S. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double‐blind, placebo‐controlled, dose‐ranging trials. Lancet 2016; 388: 2239–2253. [DOI] [PubMed] [Google Scholar]
- 22. Graham MJ, Lee RG, Brandt TA, Tai LJ, Fu W, Peralta R, Yu R, Hurh E, Paz E, McEvoy BW, Baker BF, Pham NC, Digenio A, Hughes SG, Geary RS, Witztum JL, Crooke RM, Tsimikas S. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med 2017; 377: 222–232. [DOI] [PubMed] [Google Scholar]
- 23. Alexander VJ, Xia S, Hurh E, Hughes SG, O'Dea L, Geary RS, Witztum JL, Tsimikas S. N‐acetyl galactosamine‐conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J 2019; 40: 2785–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsimikas S, Karwatowska‐Prokopczuk E, Gouni‐Berthold I, Tardif JC, Baum SJ, Steinhagen‐Thiessen E, Shapiro MD, Stroes ES, Moriarty PM, Nordestgaard BG, Xia S, Guerriero J, Viney NJ, O'Dea L, Witztum JL, Investigators AK‐A‐LS . Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med 2020; 382: 244–255. [DOI] [PubMed] [Google Scholar]
- 25. Ackermann EJ, Guo S, Benson MD, Booten S, Freier S, Hughes SG, Kim TW, Jesse Kwoh T, Matson J, Norris D, Yu R, Watt A, Monia BP. Suppressing transthyretin production in mice, monkeys and humans using 2nd‐generation antisense oligonucleotides. Amyloid 2016; 23: 148–157. [DOI] [PubMed] [Google Scholar]
- 26. Prakash TP, Yu J, Migawa MT, Kinberger GA, Wan WB, Ostergaard ME, Carty RL, Vasquez G, Low A, Chappell A, Schmidt K, Aghajan M, Crosby J, Murray HM, Booten SL, Hsiao J, Soriano A, Machemer T, Cauntay P, Burel SA, Murray SF, Gaus H, Graham MJ, Swayze EE, Seth PP. Comprehensive structure‐activity relationship of triantennary N‐acetylgalactosamine conjugated antisense oligonucleotides for targeted delivery to hepatocytes. J Med Chem 2016; 59: 2718–2733. [DOI] [PubMed] [Google Scholar]
- 27. Vollmer J, Jepsen JS, Uhlmann E, Schetter C, Jurk M, Wader T, Wullner M, Krieg AM. Modulation of CpG oligodeoxynucleotide‐mediated immune stimulation by locked nucleic acid (LNA). Oligonucleotides 2004; 14: 23–31. [DOI] [PubMed] [Google Scholar]
- 28. Yu RZ, Baker B, Chappell A, Geary RS, Cheung E, Levin AA. Development of an ultrasensitive noncompetitive hybridization‐ligation enzyme‐linked immunosorbent assay for the determination of phosphorothioate oligodeoxynucleotide in plasma. Anal Biochem 2002; 304: 19–25. [DOI] [PubMed] [Google Scholar]
- 29. Yu RZ, Graham MJ, Post N, Riney S, Zanardi T, Hall S, Burkey J, Shemesh CS, Prakash TP, Seth PP, Swayze EE, Geary RS, Wang Y, Henry S. Disposition and pharmacology of a GalNAc3‐conjugated ASO targeting human lipoprotein (a) in mice. Mol Ther Nucleic Acids 2016; 5: e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu RZ, Gunawan R, Post N, Zanardi T, Hall S, Burkey J, Kim TW, Graham MJ, Prakash TP, Seth PP, Swayze EE, Geary RS, Henry SP, Wang Y. Disposition and pharmacokinetics of a GalNAc3‐conjugated antisense oligonucleotide targeting human lipoprotein (a) in monkeys. Nucleic Acid Ther 2016; 26: 372–380. [DOI] [PubMed] [Google Scholar]
- 31. Zimmermann TS, Karsten V, Chan A, Chiesa J, Boyce M, Bettencourt BR, Hutabarat R, Nochur S, Vaishnaw A, Gollob J. Clinical proof of concept for a novel hepatocyte‐targeting GalNAc‐siRNA conjugate. Mol Ther 2017; 25: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. ENDEAVOUR: Phase 3 multicenter study of revusiran (ALN‐TTRSC) in patients with transthyretin (TTR) mediated familial amyloidotic cardiomyopathy (FAC). In: ClinicalTrials.gov Identifier: NCT02319005. (July 2019).
- 33. Garber K. Alnylam terminates revusiran program, stock plunges. Nat Biotechnol 2016; 34: 1213–1214. [DOI] [PubMed] [Google Scholar]
- 34. Judge DP, Kristen AV, Grogan M, Maurer MS, Falk RH, Hanna M, Gillmore J, Garg P, Vaishnaw AK, Harrop J, Powell C, Karsten V, Zhang X, Sweetser MT, Vest J, Hawkins PN. Phase 3 multicenter study of revusiran in patients with hereditary transthyretin‐mediated (hATTR) amyloidosis with cardiomyopathy (ENDEAVOUR). Cardiovasc Drugs Ther 2020; 34: 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zimmermann T, Chan A, Jadhav VR, Maier MA, Rajeev KG, inventors; Alnylam Pharmaceuticals, Inc., assignee. Transthyretin (TTR) iRNA compositions and methods of use thereof for treating or preventing TTR‐associated diseases patent United States patent 10,683,501. 2020. June 16, 2020.
- 36. Nair JK, Attarwala H, Sehgal A, Wang Q, Aluri K, Zhang X, Gao M, Liu J, Indrakanti R, Schofield S, Kretschmer P, Brown CR, Gupta S, Willoughby JLS, Boshar JA, Jadhav V, Charisse K, Zimmermann T, Fitzgerald K, Manoharan M, Rajeev KG, Akinc A, Hutabarat R, Maier MA. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc‐siRNA conjugates. Nucleic Acids Res 2017; 45: 10969–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Janas MM, Schlegel MK, Harbison CE, Yilmaz VO, Jiang Y, Parmar R, Zlatev I, Castoreno A, Xu H, Shulga‐Morskaya S, Rajeev KG, Manoharan M, Keirstead ND, Maier MA, Jadhav V. Selection of GalNAc‐conjugated siRNAs with limited off‐target‐driven rat hepatotoxicity. Nat Commun 2018; 9: 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Janas MM, Zlatev I, Liu J, Jiang Y, Barros SA, Sutherland JE, Davis WP, Liu J, Brown CR, Liu X, Schlegel MK, Blair L, Zhang X, Das B, Tran C, Aluri K, Li J, Agarwal S, Indrakanti R, Charisse K, Nair J, Matsuda S, Rajeev KG, Zimmermann T, Sepp‐Lorenzino L, Xu Y, Akinc A, Fitzgerald K, Vaishnaw AK, Smith PF, Manoharan M, Jadhav V, Wu JT, Maier MA. Safety evaluation of 2′‐deoxy‐2′‐fluoro nucleotides in GalNAc‐siRNA conjugates. Nucleic Acids Res 2019; 47: 3306–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudiniere R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A 2012; 109: 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C, Investigators A‐AS . Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007–1016. [DOI] [PubMed] [Google Scholar]
- 41. Tegsedi [prescribing information]. Boston, MA: Akcea Therapeutics; October 2019. In.
- 42. Narayanan P, Curtis BR, Shen L, Schneider E, Tami JA, Paz S, Burel SA, Tai LJ, Machemer T, Kwoh TJ, Xia S, Shattil SJ, Witztum JL, Engelhardt JA, Henry SP, Monia BP, Hughes SG. Underlying immune disorder may predispose some transthyretin amyloidosis subjects to inotersen‐mediated thrombocytopenia. Nucleic Acid Ther 2020; 30: 94–103. [DOI] [PubMed] [Google Scholar]
- 43. Yu RZ, Grundy JS, Geary RS. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin Drug Metab Toxicol 2013; 9: 169–182. [DOI] [PubMed] [Google Scholar]
- 44. Shemesh CS, Yu RZ, Warren MS, Liu M, Jahic M, Nichols B, Post N, Lin S, Norris DA, Hurh E, Huang J, Watanabe T, Henry SP, Wang Y. Assessment of the drug interaction potential of unconjugated and GalNAc3‐conjugated 2′‐MOE‐ASOs. Mol Ther Nucleic Acids 2017; 9: 34–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Methods ‐ Preclinical Models
Figure S1. Phase 1 study design in healthy volunteers
Figure S2. Mean percent change from baseline in RBP4 over time with multiple doses
Figure S3. Mean percent change from baseline in TTR and RBP4 after a single dose
Table S1. Plasma pharmacokinetics of AKCEA‐TTR‐LRx in healthy subjects
Table S2. Summary of percent change from baseline in plasma TTR overtime
Table S3. Clinical laboratory measurements