

Abstract
Tetralogy of Fallot (TOF) is the most common cyanotic congenital heart defect. It involves anatomical abnormalities that change the normal flow of blood through the heart resulting in low oxygenation. Although not all of the underlying causes of TOF are completely understood, the disease has been associated with varying genetic etiologies including chromosomal abnormalities and Mendelian disorders, but can also occur as an isolated defect. In this report, we describe a familial case of TOF associated with a 1.8 Mb deletion of chromosome 10p11. Among the three genes in the region one is Neuropilin1 (NRP1), a membrane co-receptor of VEGF that modulates vasculogenesis. Hemizygous levels of NRP1 resulted in a reduced expression at the transcriptional and protein levels in patient-derived cells. Reduction of NRP1 also lead to decreased function of its activity as a co-receptor in intermolecular VEGF signaling. These findings support that diminished levels of NRP1 contribute to the development of TOF, likely through its function in mediating VEGF signal and vasculogenesis.
Keywords: chromosomal deletion, congenital heart disease, neuropilin 1 (NRP1), prenatal ultrasound, tetralogy of fallot (TOF)
1. INTRODUCTION
Congenital heart disease (CHD) affects approximately 1% of live births and is the most common life-threatening birth defect in the United States. Among these congenital conditions are conotruncal heart defects (CTDs) that consist of structural abnormalities of the outflow tracts of the heart, and account for 20–30% of all CHD (Hobbs et al., 2014). The three major types of CTDs include truncus arteriosus, transposition of the great arteries, and Tetralogy of Fallot (TOF). TOF is the most common cyanotic congenital heart defect, occurring in 1 per 2,518 live births (Parker, Abt, & Parr, 1980). The classic form of TOF consists of four anatomic defects: ventricular septal defect (VSD), obstruction of the right ventricular outflow tract (pulmonary stenosis), overriding aorta and right ventricular hypertrophy (Dabbagh, Conte, & Lubin, 2017).
Historically, TOF was considered a disorder of multigenic etiology. However, population studies have demonstrated a recurrence risk ratio of 11.7 for conotruncal defects (Øyen et al., 2009), suggesting that Mendelian inheritance may account for a portion of the cases. A comprehensive genotype-phenotype analysis in 230 patients with TOF showed that it is often part of the constellation of findings in many genetic syndromes including Mendelian disorders and aneuploidy (Rauch et al., 2010). At this time, the most common identifiable cause of TOF is chromosome 22q11 deletion syndrome, a relatively common aneuploidy characterized by congenital heart disease, palatal abnormalities, hypocalcemia, characteristic facial features, learning difficulties, and immunodeficiency (Burn et al., 1993; Driscoll et al., 1993; Momma, Kondo, Ando, Matsuoka, & Takao, 1995). Supporting chromosome 22q11 as TOF susceptibility region, TOF has also been reported in rare cases of 22q11 microduplication syndrome (Hu et al., 2011). The second most common currently identifiable cause of TOF is down syndrome due to trisomy for chromosome 21 (OMIM 190685) (Baraona, Gurvitz, Landzberg, & Opotowsky, 2013; CASE RECORDS of the Massachusetts General Hospital, 1957; Ghosh & Biswas, 1955), followed by other chromosomal aberrations and copy number changes (Greenway et al., 2009; Lammer et al., 2009; Rauch et al., 2010; Silversides et al., 2012). A lesser percentage of TOF results from its occurrence in Mendelian disorders. For example, it is the characteristic congenital heart defect in Alagille (OMIM 118450) and Kabuki (OMIM 147920) syndromes, due to heterozygosity for mutations in JAG or NOTCH2 (OMIM 118450, OMIM 610205) and KMT2D (OMIM 147920), respectively (Krantz et al., 1999; McDaniell et al., 2006; Ng et al., 2010; Rauch et al., 2010). Although many TOF cases are associated with phenotypically complex syndromes, TOF can occur as an isolated non-syndromic defect. However, only a small percentage of isolated TOF cases have a recognized genetic etiology and result from heterozygosity for mutations in the genes TBX1 (MIM 602054), JAG1 (MIM 601920), NKX2.5 (MIM 600584), ZFPM2 (MIM 603693), TBX5 (MIM 601620), GATA6 (MIM 601656), and GATA4 (MIM 600576) (Benson et al., 1999; Burn et al., 1998; Eldadah et al., 2001; Maitra, Koenig, Srivastava, & Garg, 2010; Pizzuti et al., 2003; Rauch et al., 2010; Schott et al., 1998; Tomita-Mitchell, Maslen, Morris, Garg, & Goldmuntz, 2007).
Genome-wide association studies (GWAS) have been employed to identify risk alleles associated with TOF and have suggested a susceptibility locus on chromosome 10p11 (Cordell et al., 2013; Xu et al., 2014), however no specific gene in the region is yet shown to be causative. We identified a fetus and his affected father who both had TOF and harbored a chromosomal 10p11.22-p11.21 deletion. Within the region are three genes, including Neuropilin 1, NRP1 (MIM 602069). NRP1 is a receptor for semaphorins and vascular endothelial growth factor (VEGF). It has been demonstrated to function as a co-receptor of VEGFR2 to modulate cardiovascular development and vasculogenesis (Lampropoulou & Ruhrberg, 2014; Lee et al., 2002). Affected patient derived cells show diminished levels of NRP1 cDNA and protein, misregulation of VEGF signaling, and support a novel association between defective NRP1 levels and the development of TOF.
2. MATERIALS AND METHODS
2.1. Clinical information
Patients and their family members were assessed under a University of California at Los Angeles approved human subjects protocol. Clinical information and imaging studies were reviewed to determine the phenotypic findings in the affected individuals. Amniocytes were collected from proband R14-031A. No parental cells were available for this study.
2.2. SNP array
SNP microarray analysis was performed using the Affymetrix Cytoscan HD platform. DNA was processed and hybridized to the Affymetrix Cytoscan HD GeneChip. Data was analyzed using Chromosome Analysis Suite
2.3. Sequence analyses and epigenetic studies
DNA was isolated from blood or cultured fibroblasts via a standard manufacturer's protocol (QIAGEN, Hilden, Germany). Sanger sequence analysis of the remaining hemizygous allele of NRP1 in the proband was performed. Whole cDNA NRP1 sequence derived from amniocyte RNA was analyzed to identify other potential transcriptional defects. Complete sequencing of cDNA, including the 5′ UTR to account for possible non-coding variants in family members, was performed from blood DNA. Bi-allelic expression was studied by sequencing human NRP1 cDNA from amniocytes, endothelial cells, and striated muscle searching for heterozygosity for several high frequency variants (Ensembl.org: rs2229935, 0.35 MAF frequency; rs2229934, 0.35 MAF frequency; COSM146924 0.5 MAF frequency). cDNA derived from mouse heart was sequenced to identify polymorphisms between strains C57BL/6J and C3H/HeJ (Nrp1 SNP352). Primers are shown in Supplementary Table S1.
2.4. Quantitative expression analysis and genomic DNA
Total RNA was extracted from cultured amniocytes at approximately 80% confluence using TRIzol (Invitrogen, Carlsbad, CA), treated with DNAase (Invitrogen) and underwent reverse transcription (Thermo Scientific, Waltham, MA) according to the manufacturer's instructions. Quantitative PCR was performed with a real-time PCR detection system (Stratagene, La Jolla, CA, MX3005P) using SYBR green PCR master mix (Thermo Scientific) and optimized thermocycler conditions. qPCR was performed in three independent experiments. Each sample was analyzed using two housekeeping genes to normalize for RNA/cDNA input amounts and to determine relative quantifications. Levels of transcripts in controls were set at 1. Melting curve analysis showed a single, sharp peak with the expected temperature melting for all samples. DNA quantitative PCR was also analyzed with primers within and outside the deletion region to assess potential hemizygosity in other family members. Primers were designed with pairs from multiple different exons (Supplementary Table S1).
2.5. Western blot
For Western blot analyses, proteins lysates were separated by electrophoresis through a 10% SDS-polyacrylamide gel, transferred to PVDF membranes, blocked in 5% milk and probed with primary antibodies (anti-NRP1 antibody, 1:2000 Proteintech, Chicago, IL; anti-GAPDH 1:2000 Cell Signaling, Danvers, MA, 2118S). Peroxidase-conjugated secondary antibodies (Cell Signaling 7071 and 7072) were used and immuno-complexes identified by using the ECL detection reagent (Cell Signaling 7003). FIJI was used to quantify bands following Gel Analysis recommendations from ImageJ (Gassmann, Grenacher, Rohde, & Vogel, 2009) and the Mann–Whitney test was performed for statistical analysis using Prism software. Experiments were replicated three times.
2.6. Amniocytes and endothelial cells co-culture
Human umbilical vein endothelial cells (EC) (100,000 cells) were cultured alone or with control versus patient amniocytes (100,000 cells) in MCDB-131 with 10% FBS for at least 48 hr. The cells were incubated in media without serum overnight, treated with 200 μM Na3VO4 in MCDB-131 for 5 min at 37 °C, then VEGF (100 ng/ml) in MCDB 131 for 5 min 37 °C. Cells were washed with PBS then lysed in modified RIPA (mRIPA) buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycolate, 1 mM EDTA, 1 mM Na3VO4, 10 mM β-glycerophosphate, and protease inhibitor cocktail (Roche, Pleasanton, CA). Lysates were agitated at 4 °C for 30 min, then centrifuged for 10 min at 4 °C to remove debris. Proteins were separated by SDS-PAGE and immunoblot analyses were performed with antibodies against total VEGFR2 and phospho-VEGFR2 Y1175 (1:1000 Cell Signaling), NRP1 (1:2000 Proteintech), smooth muscle actin (SMA, an amniocyte marker, 1:1000 Cell Signaling), and GAPDH as a loading control (1:1000 Cell Signaling). Densitomery analysis was performed using ImageLab software (Bio-Rad) and the signal of phospho-VEGFR2 relative to total VEGFR2 was determined. This experiment was performed one time because the patient's amniocytes failed additional passages.
3. RESULTS
3.1. Clinical findings
The proband, patient reference number R14-031A was the offspring of a 32 year-old G2P1 mother and 37 year-old father. Fetal ultrasound at 16 weeks showed findings consistent with TOF that included narrowing of the pulmonic vessel at the level of the pulmonic valve and a ventral septal defect (VSD). No abnormalities were detected in other organ systems. Continued fetal evaluation by fetal echocardiography showed at 26 weeks moderate valvular/subvalvular pulmonary stenosis and moderately hypoplastic pulmonary arteries as well as a double aortic arch (Figure 1a). Postnatal echocardiogram showed the aforementioned anomalies and an overriding aorta (Figure 1b). At birth the child had no other appreciable abnormalities. The patient underwent surgical correction of the TOF at the age of 2 months.
FIGURE 1.

Cardiac defects in patient R14-031A. (a) 3D rendering of fetal heart showing VSD (thin arrow) and double aortic arch (thick arrows). RV = Right ventricle. LV = Left ventricle. Asc Ao = Ascending aorta. (b) Postnatal ECHO showing VSD and overriding aorta (Arrow).
On further questioning of the family at the time of the proband's 16 week fetal ultrasound the familial history revealed that the patient's father fatigued easily as a child and workup at age 4 years identified TOF and he underwent an uncomplicated surgical correction. The father also had unilateral equinovarus at birth that responded to physical manipulations and surgical correction was not necessary. As an adult, a horse-shoe kidney and fused thoracic vertebrae were noted by MRI. The father also reported unilateral congenital deafness. The patient's paternal uncle (Figure 2, II-3) has unilateral renal agenesis and there are two healthy paternal aunts (Figure 2, II-1 and 2). The remainder of the paternal and maternal family history was negative for cardiac defects, renal anomalies, skeletal malformations, or congenital deafness.
FIGURE 2.

Incomplete penetrance of the deletion 10p11. (a) Pedigree of R14-031 family with TOF patients (filled in black), carriers for the deletion 10p11 without TOF (dot inside symbol). Other members of the family were tested for the 10p11 deletion and were negative (*).
(b) Diagram of the chromosome 10 with details of the deleted region 10p11.22-p11.21 including the genes ITGB1, NRP1, and PARD3.
3.2. Genetic analyses
Because of the identification of the fetal TOF, amniocentesis was performed at 16 weeks (Figure 2a, III-3). SNP chromosomal microarray analysis from amniocyte derived DNA showed a 1.81 MB interstitial deletion at 10p11.22-p11.21 with estimated breakpoints at 32,686,908 to 34,498,716 base pairs (GRCh38/hg38: chr10:32686908–34498716). No other chromosomal abnormalities or rearrangements were detected by karyotype or microarray. Microarray analyses from DNA derived from peripheral blood in the parents revealed the same deletion in the proband's father (Figure 2a, II-4) and no abnormalities were noted in this region in the maternal sample (Figure 2a, II-5). FISH analysis was performed on blood samples from the paternal grandparents, revealed that the paternal grandfather also carried the deletion (Figure 2a, I-2). Further evaluation of the paternal family showed that the paternal uncle with the unilateral renal agenesis did not carry the deletion (Figure 2, II-3), though one unaffected aunt did (Figure 2, II-2). There are three genes localized in this region: ITGB1 (MIM135630), NRP1 (MIM 602069), and PARD3 (MIM 606745) (Figure 2b).
3.3. Deletion results in reduced expression of NRP1
As the deleted region contains three genes, ITGB1, NRP1, and PARD3, we determined the effects of the deletion on these loci by studying the expression of the three genes by quantitative RT-PCR on amniocytes derived from the patient R14-031A (Figure 2a, III-3). Among the three genes, only NRP1 showed reduced transcriptional levels, while ITGB1 and PARD3 cDNA levels were normal when compared with control amniocytes (Figure 3a-c). Interestingly, NRP1 expression was below the predicted 0.5-fold compared to control cells, suggesting perhaps an additional factor reducing its expression other than the deletion. Data obtained from DECIPHER and ExAC databases indicate intolerance of NRP1 to loss of function (pLi = 1.00) and low Haploinsufficiency score (HI index = 2.53%), suggesting that haploinsufficiency for NRP1 may produce pathogenicity. To determine if transcriptional changes corresponded with reduced levels of proteins we performed Western blot analysis. These results showed that similar to cDNA levels, there was markedly decreased NRP1 protein in amniocytes from patient, compared to control (Figure 3d), lending to the conclusion that NRP1 haploinsufficiency due to the deletion contributed to reduced levels of NRP1 protein in patient derived amniocytes.
FIGURE 3.

Transcriptional levels for the three genes found in the deletion 10p11. (a) Expression levels of NRP1 in amniocytes from the patient R14-031A showed a significant decreased compared to the control. ITGB1 and PARD3 expression (b,c) did not show changes due to the deletion 10p11. (d) Protein levels on NRP1 were decreased in R14-031A amniocytes more than 0.5-fold. Reduction under 50% of NRP1 levels did not correspond with epigenetic changes as shown by bi-allelic expression of the NPR1 mRNA in human and mouse tissues (e,f).
We then asked whether there was a sequence alteration on the maternally inherited NRP1 allele that contributed to reduced cDNA and protein levels below the 50% predicted for haploinsufficiency, as well as perhaps uncovering a recessive mode of inheritance leading to TOF. Sanger sequence analysis of the coding exons of NPR1 on the remaining maternal allele from affected amniocytes did not reveal any coding changes or silent polymorphisms. Furthermore, we asked if NRP1 could be an imprinted gene or that other epigenetic changes that could account for marked reduced expression contributing to the phenotype. No database showed NRP1 as an imprinting gene. We also performed cDNA sequencing to check for bi-allelic expression of three common synonymous variants in the NRP1 coding sequence (Ensembl.org: rs2229935, 0.35 MAF frequency; rs2229934, 0.35 MAF frequency; COSM146924 0.5 MAF frequency) in amniocytes, endothelial cell lines and striated muscle derived from four control individuals. For each individual and in multiple tissues, there was bi-allelic expression of these variants (Figure 3e, rs2229935), discarding the possibility that NRP1 is maternally imprinted. Due to the lack of human cardiac samples we could not test potential epigenetic changes in the heart, however, we were able to analyze the expression of Nrp1 in mouse heart. Mouse Nrp1 coding sequence contains a polymorphism variant between strains C57BL/6J and C3H/HeJ. RNA sequencing from heart samples of C57BL/6J-C3H/HeJ heterozygous males and females showed biallelic expression of the variant, suggesting no significant difference in allelic expression of Nrp1 in mouse heart. (Figure 3f, SNP352). These studies suggest that the phenotype was not recessive due to haploinsufficiency plus a nonsilent sequence change, or through loss of NRP1 through an epigenetic mechanism.
3.4. Decreased NRPlevels correlate with decreased NRP1 function
We then asked if reduced NRP1 protein levels observed in the patient amniocytes altered NRP1 function. NRP1 can modulate signaling by VEGFR2 by acting as a co-receptor for VEGF. While NRP1 by itself does not directly signal in response to VEGF, it alters the responsiveness of VEGFR2 to VEGF ligand. It has been reported that NRP1 on non-endothelial cells can be presented to VEGFR2 on endothelial cells in trans and suppress angiogenesis by reducing VEGFR2 internalization and signaling (Koch et al., 2014). We tested whether reduced expression of NRP1 on the patient amniocytes resulted in differences in trans signaling to VEGFR2. After co-culturing amniocytes with endothelial cells for 2 days, we challenged the cells with VEGF and assayed VEGFR2 signaling by measuring the phosphorylation of VEGFR2 relative to total receptor levels. We observed a 3.4-fold change when endothelial cells were co-cultured with patient amniocytes, while co-culture with control amniocytes had minimal effect by comparison (Figure 4), suggesting together with information from published findings that reduced the NRP1 levels resulted in less NRP1-VEGFR2 complexes in trans and loss of inhibition of VEGFR2 phosphorylation. Unfortunately, limited material did not allow for further studies. These findings suggest that increased VEGFR2 signaling due to reduced NRP1 inhibition may contribute to the phenotype through dysregulation of angiogenesis at a critical stage of development.
FIGURE 4.
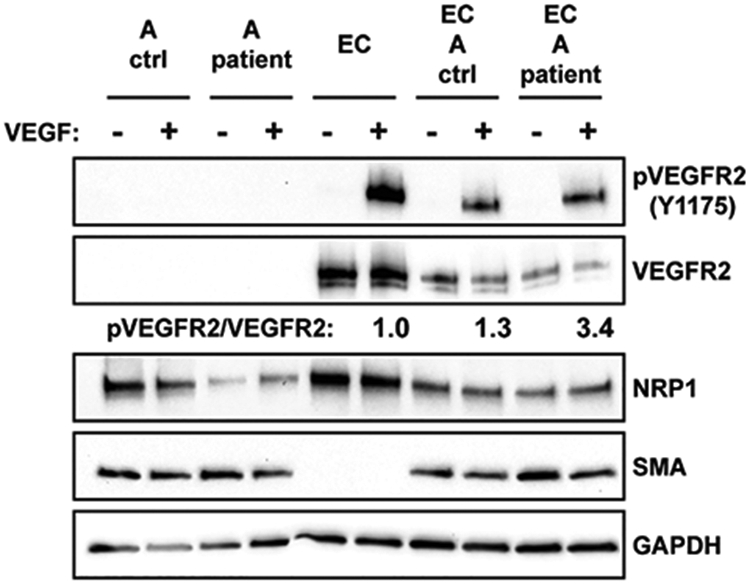
Altered levels of VEGFR2 phosphorylation from patient amniocytes (A), endothelial cells (EC) and co-cultured amniocytes and endothelial cells (EC+A). Ratio of phosphorylated (p) VEGFR2 at tyrosine 1175 versus total VEGFR2 were altered in endothelial cell (EC) when co-cultured with patient cells (3.4-fold) compared to endothelial cells co-cultured with control cells (1.3). SMA is an amniocyte marker.
4. DISCUSSION
NRP1 is a transmembrane protein, which serves as a co-receptor for both vascular endothelial growth factor (VEGF; 192240) and semaphorin (SEMA3A; 603961) family members (Fantin et al., 2014; Lee et al., 2002). NRP1 plays a role in angiogenesis, axon guidance, cell survival, migration, and invasion. It is expressed in the digestive tract, urinary tract, adrenal glands, cardiac muscle, and CNS. A zebrafish knockout model demonstrated that NRP1 regulated angiogenesis through a VEGF dependent pathway (Lee et al., 2002). Knock-out mice for Nrp1 die during development due to severe cardiovascular defects (Kawasaki et al., 1999; Kitsukawa et al., 1997). Targeted knockout mice have shown abnormalities of the cardiovascular system including persistent truncus arteriosus and abnormal vestibulocochlear development (Gu et al., 2003). Endothelial disruption of Nrp1 in mice caused a DiGeorge syndrome-like phenotype that included septal defects (Zhou, Pashmforoush, & Sucov, 2012). Additionally, a genetic variant at 10p11 has been associated with increased risk of TOF in a Chinese population study (Xu et al., 2014). Specifically, Xu et al. suggested that the presence of a variant in the NRP1 coding sequence (rs2228638) increases the risk of developing TOF. In a large scale study that included syndromic and nonsyndromic congenital heart defects, Sifrim et al, found a de novo rare variant in NRP1, but no specific details were reported (Sifrim et al., 2016). Finally, Shaheen, Hashem, and Alkuraya (2015), reported on a consanguineous family with three affected individuals that presented with recurrent truncus arteriosus due to homozygosity for a truncating splicing mutation in NRP1 (p.Asp25Gfs*25) (Shaheen et al., 2015). These findings support NRP1's association with cardiac defects, including TOF, making NRP1 the most likely candidate to cause the heart defects in patients with 10p11 deletion syndrome.
The family case presented in this study showed that two of four members of the family with the 10p11 deletion had TOF (Figure 2), suggesting that deletion of this region at 10p11 has incomplete penetrance for the risk of developing TOF. Incomplete penetrance for TOF has been reported for chromosome 5q33 deletion syndrome (Starkovich, Lalani, Mercer, & Scott, 2016), GATA4 missense mutations (Chen et al., 2016), low frequency variants in ROCK1 (Palomino et al., 2013), and JAG1 (Eldadah et al., 2001). The genetic or non-genetic etiologies that contribute to incomplete penetrance remain unresolved. In this work, other potential mechanisms were addressed. No coding variations were detected in the remaining NRP1 maternally inherited allele. Imprinting of the NRP1 gene was discarded because two brothers with the deletion showed both absence and presence of TOF. Additionally, several control human cell lines showed biallelic NRP1 expression, suggesting that alleles are not selectively silenced. One study showed that the NRP1 had a lower methylation level in myocardial tissue in patients with TOF (Sheng et al., 2014) suggesting that methylation state of NRP1 can be responsible for the cardiac defect, however, neither that study nor our results had evidence for altered NRP1 expression levels. Our analysis included human amniocytes, endothelial cells, muscle, and murine heart, however, we cannot rule out a very localized epigenetic control in the development of the cardiac outflow tract at the arterial pole of the heart (Buckingham, Meilhac, & Zaffran, 2005) that could affect development. Other genetic changes that could explain the incomplete penetrance of the NRP1 deletion are undetected variants in the remaining NRP1 promoter, non-coding changes affecting NRP1 regulatory elements or digenic variant influences where more than one gene could be involved in the control of NRP1 expression influencing development. Interestingly, the levels of NRP1 cDNA and protein derived from affected amniocytes showed levels that were more than 50% reduced, the amount expected based on the deletion, perhaps suggesting that the regulation of NRP1 is more complex than previously appreciated. Two other genes were deleted in the region, ITGB1 and PARP3. Both of these genes have roles in development (Hirose et al., 2006; Wu et al., 2009), however mouse knockout models did not manifest with congenital heart disease and neither gene showed decreased levels of cDNA expression in affected amniocytes. Thus, NRP1 remains the most likely candidate gene in the 10p11 deletion to contribute to TOF.
NRP1 was first discovered in 1987 as a cell adhesion molecule in xenopus neurogenesis (Takagi, Tsuji, Amagai, Takamatsu, & Fujisawa, 1987). It serves as a receptor for a class of semaphorin (SEMA3), a secreted ligand that regulates axon guidance (He & Tessier-Lavigne, 1997; Kolodkin & Ginty, 1997), and well as a receptor for specific isoforms of vascular endothelial growth factor A (VEGF) (Soker, Fidder, Neufeld, & Klagsbrun, 1996; Soker, Takashima, Miao, Neufeld, & Klagsbrun, 1998). Additionally, NRP1 can modulate PDGF, TGFβ, and integrin signaling (Cao et al., 2010; Evans et al., 2011; Pellet-Many et al., 2011; Valdembri et al., 2009; Yaqoob et al., 2012), thus the tissue dependent roles of NRP1 are complex. For example, conditional ablation of NRP1 in endothelial cells caused cardiovascular development defects, while directed mutagenesis for NRP1 semaphorin binding sites only showed defective neuronal development, but not cardiovascular abnormalities. (Gu et al., 2003; Vieira, Schwarz, & Ruhrberg, 2007). VEGF ligand binding induces complex formation between NRP1 and VEGFR2 enhancing VEGFR2 signaling during endothelial cell migration in vitro (Evans et al., 2011; Soker, Miao, Nomi, Takashima, & Klagsbrun, 2002; Wang, Zeng, Wang, Soker, & Mukhopadhyay, 2003) and as well as arteriogenesis in vivo (Lanahan et al., 2013). VEGF is a known modifier for TOF as low VEGF expression haplotypes have been observed to increase risk of non-syndromic TOF (Lambrechts et al., 2005), however, although the role of NRP1-VEGF in cardiovascular defects while well described in animal models, its role in human cardiac defects is poorly understood. Our data demonstrated that decreased NRP1 due to the deletion presented in trans from amniocytes to endothelial cells abnormally increased the internalization of the NRP1-VEGF-VEGFR demonstrating a functional role for the 10p11 deletion. In conclusion, our data shows that chromosome 10p11 deletion can produce TOF with incomplete penetrance and negative impacts VEGF signaling.
Supplementary Material
ACKNOWLEDGMENTS
We thank the family for the generosity of its participation in this study. This study was supported by grants from the National Institutes of Health (RO1 AR062651) and funds from the Orthopaedic Institute for Children and the March of Dimes Foundation.
Footnotes
CONFLICTS OF INTEREST
None.
REFERENCES
- Baraona F, Gurvitz M, Landzberg MJ, & Opotowsky AR (2013). Hospitalizations and mortality in the United States for adults with down syndrome and congenital heart disease. American Journal of Cardiology, 111, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, … Kugler JD (1999). Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. The Journal of Clinical Investigation, 104, 1567–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, Meilhac S, & Zaffran S (2005). Building the mammalian heart from two sources of myocardial cells. Nature Reviews. Genetics, 6, 826–835. [DOI] [PubMed] [Google Scholar]
- Burn J, Brennan P, Little J, Holloway S, Coffey R, Somerville J, … Hunter AS (1998). Recurrence risks in offspring of adults with major heart defects: Results from first cohort of British collaborative study. The Lancet, 351, 311–316. [DOI] [PubMed] [Google Scholar]
- Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, … Goodship J (1993). Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. Journal of Medical Genetics, 30, 822–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Szabolcs A, Dutta SK, Yaqoob U, Jagavelu K, Wang L, … Mukhopadhyay D (2010). Neuropilin-1 mediates divergent R-Smad signaling and the myofibroblast phenotype. Journal of Biological Chemistry, 285, 31840–31848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASE RECORDS of the Massachusetts General Hospital; case 43401. (1957). CASE RECORDS of the massachusetts general hospital; case 43401. The New England Journal of Medicine, 257, 672–677. [DOI] [PubMed] [Google Scholar]
- Chen J, Qi B, Zhao J, Liu W, Duan R, & Zhang M (2016). A novel mutation of GATA4 (K300T) associated with familial atrial septal defect. Gene, 575, 473–477. [DOI] [PubMed] [Google Scholar]
- Cordell HJ, Töpf A, Mamasoula C, Postma AV, Bentham J, Zelenika D, … Goodship JA (2013). Genome-wide association study identifies loci on 12q24 and 13q32 associated with tetralogy of Fallot. Human Molecular Genetics, 22, 1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabbagh A, Conte AH, & Lubin L (2017). Congenital heart disease in pediatric and adult patients (p. 1). New York City, NY: Springer. [Google Scholar]
- Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, & Emanuel BS (1993). Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: Implications for genetic counselling and prenatal diagnosis. Journal of Medical Genetics, 30, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, & Dietz HC (2001). Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Human Molecular Genetics, 10, 163–169. [DOI] [PubMed] [Google Scholar]
- Evans IM, Yamaji M, Britton G, Pellet-Many C, Lockie C, Zachary IC, & Frankel P (2011). Neuropilin-1 signaling through p130Cas tyrosine phosphorylation is essential for growth factor-dependent migration of glioma and endothelial cells. Molecular and Cellular Biology, 31, 1174–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin A, Herzog B, Mahmoud M, Yamaji M, Plein A, Denti L, … Zachary I (2014). Neuropilin 1 (NRP1) hypomorphism combined with defective VEGF-A binding reveals novel roles for NRP1 in developmental and pathological angiogenesis. Development, 141, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann M, Grenacher B, Rohde B, & Vogel J (2009). Quantifying western blots: Pitfalls of densitometry. Electrophoresis, 30, 1845–1855. [DOI] [PubMed] [Google Scholar]
- Ghosh SC, & Biswas ML (1955). The incidence and nature of congenital cardiac anomaly in mongols, with a case report of Fallot's tetralogy in a mongol. Journal of the Indian Medical Association, 25, 472–475. [PubMed] [Google Scholar]
- Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, … Seidman CE (2009). De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nature Genetics, 41, 931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, … Ginty DD (2003). Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Developmental Cell, 5, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, & Tessier-Lavigne M (1997). Neuropilin is a receptor for the axonal chemorepellent semaphorin III. Cell, 90, 739–751. [DOI] [PubMed] [Google Scholar]
- Hirose T, Karasawa M, Sugitani Y, Fujisawa M, Akimoto K, Ohno S, & Noda T (2006). PAR3 is essential for cyst-mediated epicardial development by establishing apical cortical domains. Development, 133, 1389–1398. [DOI] [PubMed] [Google Scholar]
- Hobbs CA, Cleves MA, Macleod SL, Erickson SW, Tang X, … Li J (2014). Conotruncal heart defects and common variants in maternal and fetal genes in folate, homocysteine, and transsulfuration pathways. Birth Defects Research Part A: Clinical and Molecular Teratology, 100, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Ji X, Yang C, Zhang J, Lin Y, Cheng J, … Xu Z (2011). 22q11.2 microduplication in a family with recurrent fetal congenital heart disease. European Journal of Medical Genetics, 54, e433–e436. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, & Fujisawa H (1999). A requirement for neuropilin-1 in embryonic vessel formation. Development, 126, 4895–4902. [DOI] [PubMed] [Google Scholar]
- Kitsukawa T, Shimizu M, Sanbo M, Hirata T, Taniguchi M, Bekku Y, … Fujisawa H (1997). Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron, 19, 995–1005. [DOI] [PubMed] [Google Scholar]
- Koch S, van Meeteren LA, Morin E, Testini C, Weström S, Björkelund H, … Claesson-Welsh L (2014). NRP1 presented in trans to the endothelium arrests VEGFR2 endocytosis, preventing angiogenic signaling and tumor initiation. Developmental Cell, 28, 633–646. [DOI] [PubMed] [Google Scholar]
- Kolodkin AL, & Ginty DD (1997). Steering clear of semaphorins: Neuropilins sound the retreat. Neuron, 19, 1159–1162. [DOI] [PubMed] [Google Scholar]
- Krantz ID, Smith R, Colliton RP, Tinkel H, Zackai EH, Piccoli DA, … Spinner NB (1999). Jagged1 mutations in patients ascertained with isolated congenital heart defects. American Journal of Medical Genetics, 84, 56–60. [PubMed] [Google Scholar]
- Lambrechts D, Devriendt K, Driscoll DA, Goldmuntz E, Gewillig M, Vlietinck R, … Carmeliet P (2005). Low expression VEGF haplotype increases the risk for tetralogy of Fallot: A family based association study. Journal of Medical Genetics, 42, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammer EJ, Chak JS, Iovannisci DM, Schultz K, Osoegawa K, Yang W, … Shaw GM (2009). Chromosomal abnormalities among children born with conotruncal cardiac defects. Birth Defects Research Part A: Clinical and Molecular Teratology, 85A, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampropoulou A, & Ruhrberg C (2014). Neuropilin regulation of angiogenesis. Biochemical Society Transactions, 42, 1623–1628. [DOI] [PubMed] [Google Scholar]
- Lanahan A, Zhang X, Fantin A, Zhuang Z, Rivera-Molina F, Speichinger K, … Simons M (2013). The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Developmental Cell, 25, 156–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Goishi K, Davidson AJ, Mannix R, Zon L, & Klagsbrun M (2002). Neuropilin-1 is required for vascular development and is a mediator of VEGF-dependent angiogenesis in zebrafish. Proceedings of the National Academy of Sciences of the United States of America, 99, 10470–10475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra M, Koenig SN, Srivastava D, & Garg V (2010). Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatric Research, 68, 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, & Spinner NB (2006). NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. The American Journal of Human Genetics, 79, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momma K, Kondo C, Ando M, Matsuoka R, & Takao A (1995). Tetralogy of Fallot associated with chromosome 22q11 deletion. American Journal of Cardiology, 76, 618–621. [DOI] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, … Shendure J (2010). Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature Genetics, 42, 790–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomino Doza J, Töpf A, Bentham J, Bhattacharya S, Cosgrove C, Brook JD, … Keavney B (2013). Low-frequency intermediate penetrance variants in the ROCK1 gene predispose to Tetralogy of Fallot. BMC Genetics, 14, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker ME, Abt AB, & Parr GV (1980). Fallot's tetralogy. Its occurrence with absent pulmonary valve and sinus of Valsalva aneurysm in an adult. Archives of Pathology and Laboratory Medicine, 104, 597–598. [PubMed] [Google Scholar]
- Pellet-Many C, Frankel P, Evans IM, Herzog B, Jünemann-Ramírez M, & Zachary IC (2011). Neuropilin-1 mediates PDGF stimulation of vascular smooth muscle cell migration and signalling via p130Cas. Biochemical Journal, 435, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzuti A, Sarkozy A, Newton AL, Conti E, Flex E, Digilio MC, … Dallapiccola B (2003). Mutations of ZFPM2/FOG2 gene in sporadic cases of tetralogy of Fallot. Human Mutation, 22, 372–377. [DOI] [PubMed] [Google Scholar]
- Rauch R, Hofbeck M, Zweier C, Koch A, Zink S, Trautmann U, … Rauch A (2010). Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. Journal of Medical Genetics, 47, 321–331. [DOI] [PubMed] [Google Scholar]
- Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, … Seidman JG (1998). Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science, 281, 108–111. [DOI] [PubMed] [Google Scholar]
- Shaheen R, Hashem A, Alghamdi A, Seidahmad MH, Wakil MZ, Dagriri SM, Keavney K, … Alkuraya FS (2015). Positional mapping of PRKD1, NRP1 and PRDM1 as novel candidate disease genes in truncus arteriosus. Journal of Medical Genetics, 52, 322–329. [DOI] [PubMed] [Google Scholar]
- Sheng W, Qian Y, Zhang P, Wu Y, Wang H, Ma X, … Huang G (2014). Association of promoter methylation statuses of congenital heart defect candidate genes with Tetralogy of Fallot. Journal of Translational Medicine, 12, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifrim A, Hitz M-P, Wilsdon A, Breckpot J, Turki SHA, Thienpont B, … Hurles ME (2016). Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nature Genetics, 48, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silversides CK, Lionel AC, Costain G, Merico D, Migita O, Liu B, … Bassett AS (2012). Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genetics, 8, e1002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soker S, Fidder H, Neufeld G, & Klagsbrun M (1996). Characterization of novel vascular endothelial growth factor (VEGF) receptors on tumor cells that bind VEGF via its exon 7-encoded domain. The Journal of Biological Chemistry, 271, 5761–5767. [DOI] [PubMed] [Google Scholar]
- Soker S, Miao H-Q, Nomi M, Takashima S, & Klagsbrun M (2002). VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. Journal of Cellular Biochemistry, 85, 357–368. [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, & Klagsbrun M (1998). Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell, 92, 735–745. [DOI] [PubMed] [Google Scholar]
- Starkovich M, Lalani SR, Mercer CL, & Scott DA (2016). Chromosome 5q33 deletions associated with congenital heart defects. American Journal of Medical Genetics, 170, 3338–3342. [DOI] [PubMed] [Google Scholar]
- Takagi S, Tsuji T, Amagai T, Takamatsu T, & Fujisawa H (1987). Specific cell surface labels in the visual centers of Xenopus laevis tadpole identified using monoclonal antibodies. Developments in Biologicals, 122, 90–100. [DOI] [PubMed] [Google Scholar]
- Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, & Goldmuntz E (2007). GATA4 sequence variants in patients with congenital heart disease. Journal of Medical Genetics, 44, 779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdembri D, Caswell PT, Anderson KI, Schwarz JP, König I, Astanina E, … Serini G (2009). Neuropilin-1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biology, 7, e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira JM, Schwarz Q, & Ruhrberg C (2007). Selective requirements for NRP1 ligands during neurovascular patterning. Development, 134, 1833–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zeng H, Wang P, Soker S, & Mukhopadhyay D (2003). Neuropilin-1-mediated vascular permeability factor/vascular endothelial growth factor-dependent endothelial cell migration. The Journal of Biological Chemistry, 278, 48848–48860. [DOI] [PubMed] [Google Scholar]
- Wu W, Kitamura S, Truong DM, Rieg T, Vallon V, Sakurai H, … Nigam SK (2009). Beta1-integrin is required for kidney collecting duct morphogenesis and maintenance of renal function. American Journal of Physiology Renal Physiology, 297, F210–F217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lin Y, Si L, Jin G, Dai J, Wang C, … Wang X (2014). Genetic variants at 10p11 confer risk of tetralogy of fallot in chinese of nanjing. PLoS ONE, 9, e89636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaqoob U, Cao S, Shergill U, Jagavelu K, Geng Z, Yin M, … Shah VH (2012). Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer Research, 72, 4047–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Pashmforoush M, & Sucov HM (2012). Endothelial neuropilin disruption in mice causes DiGeorge syndrome-like malformations via mechanisms distinct to those caused by loss of Tbx1. PLoS ONE, 7, e32429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Øyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PKA, & Melbye M (2009). Recurrence of congenital heart defects in families. Circulation, 120, 295–301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.