

Abstract
Metabolic reprogramming is a hallmark of cancer which contributes to essential processes required for cell survival, growth, and proliferation. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer and its genomic classification has given rise to the design of therapies targeting tumors harboring specific gene alterations that cause aberrant signaling. Lung tumors are characterized with having high glucose and lactate use, and high heterogeneity in their metabolic pathways. Here we review how NSCLC cells with distinct mutations reprogram their metabolic pathways and highlight the potential metabolic vulnerabilities that might lead to the development of novel therapeutic strategies.
Keywords: Non-small cell lung cancer, metabolic rewiring, oncogenic drivers, therapeutic strategies
1. Introduction
Energy is generated inside the cell through the metabolism of carbon molecules in a series of catabolic reactions, while anabolic reactions enable the biosynthesis of complex biomolecules [1]. Cancer cells require both energy and increased formation of biomolecules to maintain their high proliferative rate and they have adapted their metabolism for this need [2]. Extensive research has focused on understanding the metabolic adaptations of cancer cells to potentially therapeutically target the alterations, for improved treatment of patients.
The observation of increased consumption of glucose in tumors was first documented by Otto Warburg in the 1920’s [3]. This discovery has been translated to the clinic, in the form of a diagnostic test using imaging of fluorine-labelled glucose analog, 18F-fluorodeoxygluoce (FDG) with positron emission tomography (PET) for cancer diagnosis and determination of cancer stage [4]. Decades of research has also focused around Warburg’s report of increased glycolysis and lactate excretion in the presence of oxygen in tumor cells, now known as the Warburg effect [5]. This observation has guided the curiosity of the field to explore other aspects of cancer metabolism and it is now appreciated that many metabolic adaptions are acquired to support the growth of tumor cell; summarized in Figure 1.
Figure 1: Metabolic pathways and signaling in cancer.
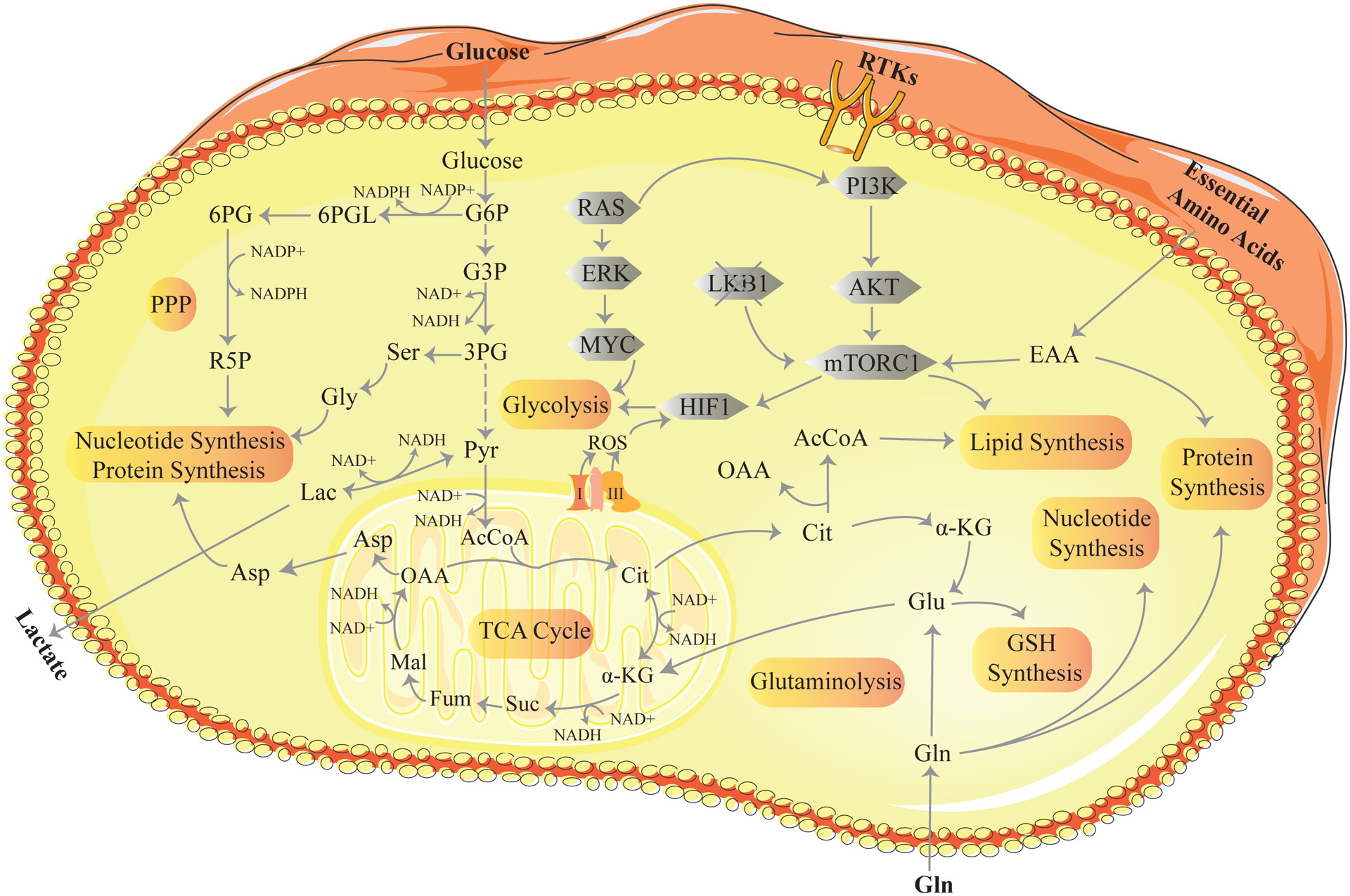
The key metabolic processes are highlighted in orange. Glycolysis is a 10-step process that converts glucose to pyruvate releasing energy in the form of ATP and NADH. The pentose phosphate pathway (PPP) generates NADPH used for the biosynthesis of fatty acids, cholesterol and reduced glutathione. In addition, the PPP generates ribose-5-phosphate required for the synthesis of purines, pyrimidines, nucleotides and nucleic acids. In the mitochondria, glycolytic-derived pyruvate is fed into the tricarboxylic acid (TCA) cycle. TCA cycle dehydrogenases consume NAD+ to produce NADH. Each turn in the cycle forms three NADH molecules. Nucleotide synthesis provides molecules that make up the nucleic acids RNA and DNA. Lipid synthesis is the synthesis of fatty acids from acetyl Co-A and malonyl-CoA precursors via fatty acid synthases. Glutaminolysis is a series of biochemical reactions that converts glutamine to α-ketoglutarate. This takes place outside and inside the mitochondria. GSH (glutathione) synthesis is the synthesis of GSH from glutamate, cysteine, and glycine. The grey shapes represent the signaling events that influence cancer metabolism. RAS activates the transcriptional program for metabolic enzymes through the activation of the RAF/MEK/ERK pathway leading to upregulation of the MYC transcription factor. MYC promotes metabolic phenotypes through transcriptional regulation of key metabolic genes. Receptor tyrosine kinases (RTKs) activate PI3K/AKT/mTORC1 signaling. Through a series of downstream effectors mTORC1 promotes anabolic growth by converting available nutrients for lipid, nucleotide and protein synthesis. Mutant LKB1 results in the hyper-activation of mTORC1 signaling and elevated HIF signaling and glycolysis. Reactive oxygen species (ROS) are generated by the electron transport chain (complex I and III) on the mitochondria membrane and activate HIF signaling. 6PG, 6-P-gluconate; R5P, Ribose-5-phosphate; Ser, Serine; Gly, Glycine; G6PL, glucose-6-phosphogluconolactone; G6P, glucose-6-phosphate; G3P, glyceraldehyde-3-phosphate; 3PG, 3-phosphoglycerate; Pyr, pyruvate; Lac, Lactate; Cit, citrate; α-KG, α-ketoglutarate; Suc, succinate; Fum, fumarate; Mal, malate; Asp, aspartate; OAA, oxaloacetate; Gln, glutamine; Glu, glutamate; EAA, essential amino acids.
In parallel, recent advances in biochemical and molecular biological tools, such as advances in mass spectrometry [6], unique in silico modelling [7] and technologies to measure metabolite levels in intact tissue [8], have reignited the metabolic field and offered an unprecedented opportunity to dissect cancer metabolism in more depth [9, 10]. For instance, an elegant study presented by Davidson and colleagues illustrates the influence of the microenvironment on metabolic dependences in tumors [11]. More recently, a study also demonstrates the discrepancies of T cell metabolism in vitro and in vivo [12]. These studies highlight the advantages of newly developed toolboxes, and the need for the research community to examine cancer metabolism within the context of the tumor microenvironment.
Moreover, the concept that metabolic phenotypes are tissue specific and cancer subtype specific is increasingly appreciated. For the purpose of this review, we will discuss the current understanding of metabolic rewiring and heterogeneity within non-small cell lung cancer (NSCLC), with a key focus on how oncogenic drivers influence these metabolic pathways.
2. Metabolic rewiring driven by oncogenic drivers in NSCLC
Lung cancer is the leading cause of cancer deaths worldwide and approximately 70% of all newly diagnosed patients present with local advanced or metastatic disease, requiring systemic chemotherapy [13]. NSCLC accounting for nearly 85% of all cases is the most common type of lung cancer. Based on histological and pathological features, this disease is classified as adenocarcinoma (LUAD), squamous cell carcinoma (LUSC) and large cell carcinoma [14]. Adenocarcinoma is the most frequent histological subtype and the main focus of this review.
Distinct metabolic phenotypes have been identified between adenocarcinoma and squamous cell carcinomas. In comparison to adenocarcinoma, squamous cell carcinoma display higher glucose uptake and glucose metabolism [15, 16] and are more susceptible to glucose deprivation [15]. Squamous cell carcinoma demonstrate a reliance on glutamine metabolism as a mechanism to overcome suppression of glycolysis [17]. Differences in metabolic pathways associated with tumor grade in lung cancer have also come to light in recent times. The absence of FDG uptake in early stage lung lesions has led to an association of a low requirement of glucose for these lesions [18, 19]. However, a recent study reveals that pulmonary premalignant lesions in fact utilize sodium- glucose transporter 2 (SGLT2) as an alternative to glucose transporters (GLUTs) to import glucose into tumors [19]. SGLT-mediated glucose uptake is not detected by FDG PET and hence why it was originally thought that glucose was not required at this early stage. Moreover, treatment with SCLT2 inhibitors in vivo delays lung adenocarcinoma development and growth, suggesting this as a potential therapeutic strategy for premalignant and early stage lung adenocarcinoma [19].
Recent advances in technology have allowed for the interrogation of metabolic reprogramming in human lung tumors in vivo [20, 21]. Hensley and colleagues conducted an informative study in which they analyze the fate of glucose in NSCLC patients by injecting 13C-glucose before and after surgery. This study reveals both inter- and intra-tumoral heterogeneity with regards to glucose oxidation and oxidation of multiple non-glucose nutrients in well-perfused areas of the tumor [20]. A later report demonstrates that lactate is in fact also used as a TCA cycle carbon source in human NSCLC tumors and this utilization is most evident in aggressive tumors [21].
The role of lung cancer mutations in driving specific metabolic pathways is also of intense interest to the field. For instance, mutations in kirsten rat sarcoma 2 viral oncogene homolog (KRAS) and epidermal growth factor receptor (EGFR) as well as liver kinase B1 (LKB1), among others, are commonly found in lung adenocarcinoma [22]. An unbiased assessment of the metabolism of a panel of over 80 genetically diverse lung cancer cell lines reveals major heterogeneity in their metabolic profiles [23]. This study and others highlight the metabolic diversity and heterogeneity within lung cancer (Figure 2). Here, we examine the metabolic reprogramming and vulnerabilities in subtypes of NSCLC that harbor the key oncogenic drivers.
Figure 2: Summary of the main metabolic alterations associated with the key oncogenic drivers in NSCLC.
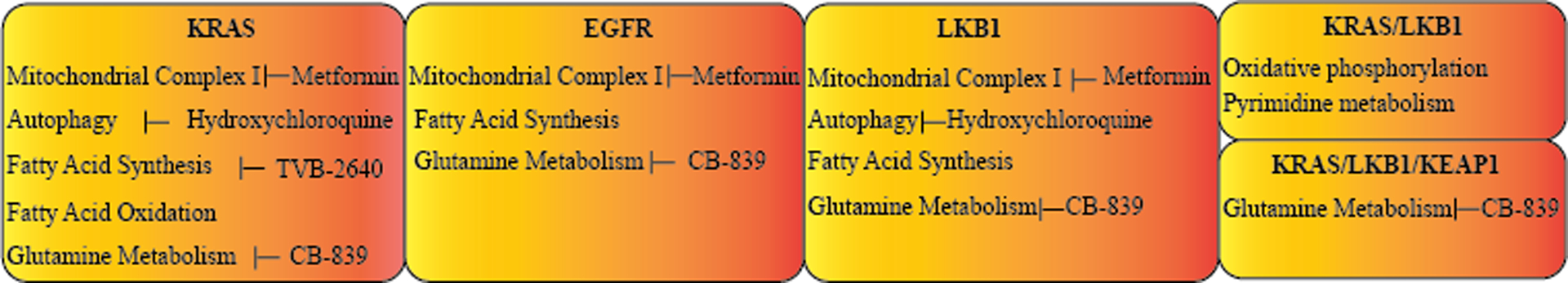
Each box shows the metabolic pathways which are altered in NSCLC harboring the named oncogenic driver. The therapeutic strategies that are currently available to target these pathways are also listed beside them.
2.1. KRAS
Approximately 30% of NSCLC patients with LUAD histology have mutations in the KRAS gene [24]. Decades of research provides comprehensive knowledge on the rewired metabolic programs driven by oncogenic KRAS, including glucose metabolism, fatty acid synthesis, glutamine metabolism, autophagy signaling and redox homeostasis.
Increased glucose uptake and the elevated flow of carbons derived from glucose into the TCA cycle are common features attributed to oncogenic KRAS signaling [11, 25, 26]. For instance, late stage gentically engineered mouse lung models (GEMMs) exhibit KRASG12D allelic enrichment when compared to early stage and normal murine lung tissue [27]. These tumors also have a distinct metabolic phenotype with increased glucose derived metabolities in the TCA cycle and they are more suspectibile to low levels of glucose and apoptosis induced by the glucose analogue, 2-deoxyglucose (2-DG) [26]. However, of note, utilization of glucose in the TCA cycle can be attributed to experimental conditions, as multiple studies now demonstrate that both mouse and human tumors oxidize glucose and lactate in the TCA cycle, independent of their mutational driver [20, 21, 25, 28]. Replenishment of the TCA cycle from glucose derived carbons requires the carboxylation of pyruvate to oxaloacetate via ATP-dependent pyruvate carboxylase (PC). Deletion of PC decreases cell proliferation in human NSCLC cell lines [25] and reduces tumor growth in murine KRAS driven GEMMs [11]. Indeed, early stage NSCLC patients infused with labelled glucose before resection show enhanced PC activity and PC expression is increased in cancerous tissue when compared to normal tissue [25]. A recent study by Rao and colleagues demonstrates that deletion of apoptosis-induced factor (AIF), in a KRAS driven mouse lung cancer model, induces a switch from oxidative phosphorylation (OXPHOS) to a more glycolytic phenotype [29]. Interestingly, lung cancer patients with high AIF expression have poor prognosis and AIF-regulated mitochondrial respiration and OXPHOS phenotype promotes the progression of tumors in mouse lung cancer models [29]. Oncogenic KRAS exerts its effects on metabolic rewiring mainly through regulating transcriptional programs of key metabolic enzymes. For instance, KRAS activation of the RAF/MEK/ERK pathway leads to upregulation of the MYC transcription factor. In turn, MYC increases the expression of glycolytic enzymes, such as Hexokinase 2 (HK2), an enzyme predominantly expressed in cancer cells catalyzing the first step in glucose metabolism and hence promoting glucose uptake and consumption [30]. HK2 is required for tumor initiation and maintenance in KRAS lung tumor models. Deletion of HK2 results in a reduction in glucose-derived carbon incorporation into the TCA cycle, leading to inhibition of ribonucleotide and serine biosynthesis in KRAS mutant NSCLC cells [30]. Collectively, it is evident that oncogenic KRAS plays a key role in driving glucose metabolism to fuel the demanding anabolic activities of lung cancer cells.
The amino acid glutamine acts as a major carbon source for the TCA cycle intermediates and much research suggests that there is an enhancemnt of glutamine metabolism in KRAS mutant lung cancer cells [11, 31, 32]. Oncogenic KRAS activates the PI3K/AKT pathway [32] and the NRF2/AKT pathway, leading to enhanced activating transcription factor 4 (ATF4) transcription and consequently an increase in glutamate uptake and metabolsim [31]. It is worth noting that much of this research is based around studies conducted in an in vitro setting, where NSCLC cell lines primarily use glutamine to feed the TCA cycle, independent of their driver mutations. Davidson and colleagues observe that while KRAS mutant lung cancer cells display a dependency on glutamine metabolism in vitro, neither the genetic deletion nor pharmacological inhibition of the mitochondrial enzyme glutaminase 1 (GLS1) alters the growth of KRAS-mutant lung tumors in vivo [11]. Moreover, early stage NSCLC patients have increased PC activity, and freshly resected lung tissue samples labelled with glutamine tracers have significantly less GLS activity, when compared to PC activity [25]. It has previously been reported that loss of the gene coding retinoblastoma protein (Rb1) promotes glutamine utilization in vitro [33]. Interestingly, a recent study demonstrated that Rb1 loss in KRAS driven lung tumors has no effect on glutamine carbon labelling of TCA cycle intermediates or on the expression of genes essential for glutamine utilization in vivo. Instead, loss of Rb1 results in an increase in glucose carbon incorporation to select glycolytic intermediates and an increase in the expression of key glycolytic genes in these tumors [34]. The discrepancy observed in the in vitro and in vivo settings warrants further investigation into glutamine utilization in KRAS driven lung cancer. The use of glutamine as a carbon source produces ammonia, however this is well tolerated in vitro due to high volumes of tissue culture media preventing ammonia accumulating to toxic levels. There is also difficulties associated with interpreting in vivo data due to difficulties in measuring single cell metabolic changes and also in deciphering the contribution of other cells in the microenvironment to the metabolic changes.
In addition to glutamine, other amino acids play an important role in the growth of KRAS mutant lung cancers. A study which compared the use of branched chain amino acids (BCAA) in KRAS driven pancreatic ductal adenocarcinoma (PDAC) and NSCLC found an increase in BCAA uptake and transamination in KRAS NSCLC tumors but not in KRAS PDAC tumors [35].This highlights the importance of tissue of origin, as well as oncogenic drivers, in the rewiring of metabolic pathways. Labelling of the amino acids demonstrates increased uptake of valine and leucine in tumor tissue compared to normal lung tissue and loss of the BCAA catabolic enzymes branched-chain amino acid aminotransferase 1 (BCAT1) and branched-chain amino acid aminotransferase 2 (BCAT2) impairs NSCLC tumor growth [35]. This increased uptake and transamination provides a nitrogen source for protein and nucleotide biosynthesis. The expression of enzymes responsible for the catabolism of BCAAs, such as SLC7A5, BCAT, and BCKDH, are also found to be upregulated in human NSCLC tumors [35]. Moreover, as mentioned above, oncogenic KRAS activates the AKT/NRF2 pathway which leads to the activation of ATF4 transcription to support amino acid homeostasis [31].
In order to meet the biosynthetic demands, cancer cells have acquired mechanisms to scavenge both intracellular and extracellular sources [36]. Autophagy is one such mechanism in which cells can capture, degrade and recycle the degraded products back into the cytosol to sustain metabolism and to enable survival during starvation [37]. KRAS tumors demonstrate high levels of basal autophagy and are highly dependent on autophagy for survival [36, 38–40]. More specifically, KRAS driven NSCLC tumors require autophagy for mitochondrial function, lipid metabolism, maintenance of nucleotide pools and growth [37, 38, 41]. The central autophagy gene, Atg7, is critical for KRAS lung tumor proliferation and deletion of Atg7, results in tumor regression to oncocytomas. These are a benign tumor associated with defective mitochondria, the accumlation of lipid stores, and increased sensitivity to fatty acid oxidation (FAO) inhibition [41, 42]. Moreover, KRAS mutant lung cancer cells with Atg7 knockdown display an increase in reactive oxygen species (ROS) production, lower energy charge and a dramatic reduction in nucleotide pools during stravation [37]. This effect can be rescued in the autophagy deficient cells by glutamine or glutamate or nucleosides, highlighting the importance of autophagy in providing substrates for nucleotide synthesis to maintain survival during starvation [37].
Mutant KRAS also promotes tumor growth through upregulation of anabolic pathways such as lipid biosynthesis [43–46]. Lung tumors with oncogenic KRAS drive a lipogeneic gene expression program which leads to de novo fatty acid synthesis in vitro and in vivo. Global gene expression profiling from KRAS mutant murine tumors reveals an enrichment in genes assoicated with fatty acid metabolism such as ATP citrate lyase (ACLY), fatty acid synthase (FASN) and acetyl coenzyme A carboxylase (ACC). Mutant KRAS promotes carbon flux through de novo fatty acid synthesis and treatment of KRAS lung tumors with fatty acid synthesis inhibitors reduces tumor growth [43]. It has been demonstrated that this activated lipogenesis in KRAS mutant lung adenocarcinoma is via induction of fatty acid synthases through the ERK2-medited pathway [44]. Moreover, mutant KRAS regulates FAO through the upregulation of the enzyme acyl-CoA synthetases long-chain family member 3 (ACSL3) [45]. ACSL3 suppression leads to inhibition of β-oxidation, depletion of cellular ATP, loss of mitochondrial potential and impaired tumor growth in vivo [45]. Indeed, FASN [43] and ACSL3 expression [45] is increased in lung cancer patients harboring mutant KRAS. Furthermore, a recent study demonstrated that regulated in development and DNA damage response 1 (REDD1) acts as a tumor suppressor in KRAS mutant GEMMs through its stress response function. REDD1 loss triggers enhanced uptake of lysophospholipids and lipid storage, coupled to augmented FAO and sustained ATP levels, providing a distinct growth advantage for these tumors [46].
The metabolic adaptions induced by oncogenic KRAS are essential for the generation of nutrients to supply the high demand of cancer cells, as well as maintenance of redox homeostasis. There is evidence that LUADs harboring KRAS mutations, are dependent on glutathione (GSH) metabolism [47, 48]. Cystine/glutamate antiporter SLC7A11 is overexpressed in LUAD patients with KRAS mutations [47]. Genetic abrogation or pharmacological inhibition of SLC7A11 selectively killed mutant KRAS cells in vitro and inhibited tumor growth in vivo. SLC7A11 inhibition reduces cystine uptake and GSH biosynthesis and hence leads to increased oxidative and ER stress resulting in apoptosis. Furthermore, lung cancer models with KRAS mutations are sensitive to histone H3K27 demethylase inhibitor GK-J4 [48]. Interestingly, treatment with GK-J4 upregulates the expression of glutamine/glutamate transport and metabolism genes. Subsequently, there is a reduction in GSH leading to reduced cell viability and supplementing with GSH protects KRAS-mutant cells from GSK-J4–mediated reductions in viability [48].
As is evident here NSCLC driven by oncogenic KRAS display a plastic metabolism and a reliance on autophagy to maintain anabolic and catabolic processes.
2.2. EGFR
Approximately 15–30% of NSCLC patients harbor mutations in EGFR, and this incidence is higher among Asian patients with no smoking history [49]. Research examining the metabolic alterations in EGFR mutant NSCLC highlights the dependency of these tumors on glucose metabolism, glutamine metabolism and fatty acid synthesis.
Many studies document the pivotal role of oncogenic EGFR in the regulation of glycolysis [50–54]. Mutant EGFR mediated enhancement of glycolysis is critical for EGFR stability. EGFR ablation in NSCLC cells reduces levels of glycolytic pathway intermediates via transcriptional regulation of glycolytic genes. Failure to sustain glucose derived ATP production triggers ROS mediated activation of JNK, and subsequently the induction of apoptosis via autophagy-induced EGFR degradation [50]. The activation of the glycolytic regulator, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) is required for glycolysis stimulation following EGFR activation and inhibition of PFKFB3 markedly reduces the EGF-mediated increase in glycolysis [55]. Treatment of LUAD cells with EGFR tyrosine kinase inhibitors (TKIs) reduces glucose derived metabolites in the glycolysis, phosphate pentose pathway (PPP) and pyrimidine biosynthesis pathway and this reduction in glucose utilization is associated with the downregulation of the GLUT3 [51]. Aberrant EGFR signaling via the PI3K/AKT/ rapamycin complex 1(mTORC) cascade regulates glycolysis by facilitating the localization of GLUT1 to the plasma membrane [52]. Aerobic glycolysis and PPP are reduced following ablation of GLUT1 in EGFR-mutated LUAD cells [52]. Moreover, there is an increase in GLUT1 expression and glucose uptake in TKI resistant NSCLC cells and ablation of GLUT1 re-sensitizes resistant cells to gefitinib [56]. Treatment of TKI resistant cells with the glucose analogue, 2-DG, improves the efficacy of the TKIs in EGFR mutated NSCLC models in vitro and in vivo. This effect is mediated through the activation of AMP-activated protein kinase (AMPK)/mTORC signaling due to intracellular ATP depletion [53].
Glutamine is utilized through several different pathways, including glutamate metabolism through TCA cycling via oxidative or reductive carboxylation and for GSH synthesis. Glutamine has been recognized as a source for bioenergetics and biosynthesis in NSCLC carrying mutant EGFR [57]. It has recently been documented that NSCLC cell lines harboring EGFR mutations have increased reductive carboxylation relative to other cell lines [23]. Treatment of EGFR mutant cells with the EGFR inhibitor, erlotinib, and the glutaminase inhibitor, CB839, induces severe energetic stress, and reduces cell viability in vitro, and rapid tumor regression in vivo [57]. This combination treatment reduces the expression of GLUT1, SLC1A5 and SLC38A1 in vivo which correlates with reduced glucose and glutamine uptake. Moreover, the combination of erlotinib and CB839 impairs GSH production compromising redox homeostasis [57]. Interestingly, NSCLC cells which are resistant to TKIs display increased utilization of glutamine to generate ATP and GSH, whereas TKI sensitive cells display ATP shortage and ROS accumulation leading to cell death [58]. A recent study, revealed that TKI resistance involves H3K9 demethylation upregulation of BCAT1. This epigenetic regulation of BCAA metabolic reprogramming leads to the elimination of ROS through the generation of GSH [59].
Aberrant lipid metabolism is described in the EGFR mutant NSCLC [60–62]. EGFR increases monosaturated fatty acid synthesis (MUFA) to facilitate lung tumor growth. Stearoyl-CoA desaturase-1 (SCD1) is a rate-limiting enzyme responsible for MUFA synthesis. The binding of EGFR to SCD1 and subsequent phosphorylation maintains SCD1 stability and EGFR stimulated cancer growth. Evaluation of patient samples reveals a positive correlation between EGFR activation and SCD1 protein expression and phosphorylation. Moreover, the level of phosphorylated SCD1 is found to be an independent prognostic factor for poor survival in patients [61]. Fatty acid metabolism gene program is increased with acquired TKI resistance in EGFR lung tumors. This includes a significant increase in FASN, which produces 16-C saturated fatty acid palmitate. Persistent signaling by mutated EGFR in TKI-resistant tumor cells is dependent on EGFR palmitoylation. Inhibition of FASN with orlistat suppresses tumor growth both in vitro and in vivo in TKI-resistant EGFR models [60]. Interestingly, sterol regulatory element binding proteins (SREBP) are a family of transcription binding proteins which regulate genes involved in fatty acid synthesis and inhibition of the SREBP driven lipogenic program increases sensitivity to gefitinib in vitro and in vivo [62].
Furthermore, recent work in preclinical models has shown that metabolic properties of NSCLC can reprogram stromal cells to induce resistance to EGFR inhibitors [54]. NSCLC cells export lactate which induces the neighboring fibroblasts to secrete hepatocyte growth factor (HGF), consequently activating its receptor tyrosine kinase c-MET on cancer cells and hence sustaining oncogenic signaling even in the presence of EGFR inhibitors [54]. This study highlights the need to consider the metabolic properties of specific tumor microenvironments when establishing treatment regimes.
In brief summary, EGFR mutations in NSCLC contribute to an increased reliance on glucose and glutamine metabolism and TKI resistance can be driven by enhanced glucose uptake.
2.3. LKB1
LKB1 is a serine-threonine kinase that acts as a tumor suppressor in many human cancers. Mutations in LKB1 are found in 20% of patients with NSCLC, and contribute significantly to lung cancer progression [63]. LKB1 is also linked to altered metabolism as it directly phosphorylates and activates the central metabolic sensor, AMPK [64]. In normal cells, AMPK activation leads to the stimulation of bioenergetic pathways and inhibition of ATP-consuming processes such as biosynthesis and proliferation, in part through the regulation of mammalian target of mTORC1 [65]. Upon detection of energy stress, LKB1 activates AMPK, which in turn inhibits fatty acid synthesis through inhibitory phosphorylation of acetyl-CoA carboxylase (ACC) and sterol regulatory element-binding proteins (SREBP). Subsequently, this prevents protein synthesis through inactivation of mTORC1 signaling, leading to a restriction in cell growth and proliferation [64].
Gene profiling of over 600 patient tumors found an upregulation of OXPHOS and mitochondria associated genes in lung adenocarcinoma samples with LKB1 deficiency [66]. LKB1 deletion in NSCLC cells also increases glucose uptake and lactate production [67]. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is a key enzyme in the glycolytic pathway, and deletion in LKB1-deficent NSCLC cells induces senescence as a result of compromised glycolysis and energy crisis [68]. Moreover, NSCLC cells deficient in LKB1 display enhanced flux of glucose and glutamine into the TCA cycle resulting in increased aerobic glycolysis and glutaminolysis [69, 70]. These events fuel cell growth and lipid biosynthesis [69]. This metabolic reprogramming is induced by increased hypoxia-inducible factor-1α (HIF-1α) protein stabilization under normoxia conditions and targeting HIF-1α impairs growth and survival of LKB1 deficient cells [69, 71]. LKB1 loss also leads to the upregulation of de novo fatty acid synthesis, and systemic inhibition of the rate limiting enzyme, ACC, is sufficient to block tumor growth in GEMMs of LKB1-deficient NSCLC [72].
While loss of LKB1 promotes cancer cell proliferation, it also renders cancer cells with a reduction in metabolic plasticity when dealing with energy crisis [70, 73]. As discussed earlier, autophagy is a protective process that helps cells maintain cellular energy homeostasis during nutrient deprivation. Deletion of Atg7 is synthetically lethal with LKB1 loss in lung tumors, leading to slower tumor initiation and a reduction in tumor growth. This is due to reduced availability of amino acids for mitochondrial energy production as a result of defective intracellular recycling. Consequently, there is a greater dependency on FAO causing a reduction in lipid reserve and ultimately energy crisis [74].
As already mentioned, LKB1 activates AMPK, and AMPK restores ATP levels while also maintaining NADPH levels to neutralize ROS levels. KRAS/LKB1 mouse models of adenocarcinoma accumulate ROS resulting from redox imbalance due to deregulation of PPP [75]. In contrast, KRAS/P53 adenocarcinoma mouse models have less ROS accumulation and LKB1 deletion in this model leads to an accumulation of ROS [75]. While LKB1 loss is advantageous for cancer growth and progression, it is clear that the induction of oxidative stress is a major vulnerability in LKB1 deficient tumors. Interestingly, analysis of TCGA lung adenocarcinoma reveal that the probability of a tumor harboring a kelch like ECH associated protein 1 (KEAP1) mutation significantly increases with LKB1 loss. This indicates a potential selective pressure existing for the activation of the antioxidant KEAP1/NRF2 pathway to compensate for oxidative stress induced by LKB1 loss [66]. This reduction in metabolic plasticity in LKB1 deficient tumors also results in hyper-sensitivity to pharmacological agents that induce energy stress [67, 70, 73, 76]. For example, in NSCLC cells, LKB1 loss leads to synthetic vulnerability to metabolic stress by the mitochondrial complex 1 inhibitor phenformin [70, 76], which further synergizes with mTOR1 inhibition to block cell growth [73, 76]. Similarly, LKB1-deficient NSCLC cells exhibit enhanced sensitivity to the energetic stress induced by erlotinib in vitro and in vivo [67]. Treatment with phenformin or erlotinib induces mitochondrial damage, impaired ATP homeostasis and an increase in ROS levels in LKB1-deficient cells [67, 76].
A dependency on pyrimidine metabolism is also evident in LKB1 deficient KRAS driven lung tumors [77, 78]. A high throughput RNA interference screen identified deoxythymidylate kinase (DTYMK) as synthetically lethal with LKB1 deficiency in mouse and human lung cancer cells. DTYMK catalyzes deoxythymidine triphosphate (dTTP) biosynthesis and depletion of DTYMK in NSCLC results in greater growth inhibition in LKB1 deficient tumors. Moreover, global metabolite profiling shows that LKB1-null cells have a significant decrease in multiple nucleotide pools when compared to wild type cells [77]. KRAS/LKB1 cells express the urea cycle enzyme carbamoyl phosphate synthetase-1 (CPS1), which produces carbamoyl phosphate in the mitochondria from ammonia and bicarbonate, initiating nitrogen disposal. Transcription of CPS1 is suppressed by LKB1 through AMPK and silencing of CPS1 in KRAS/LKB1 cells induces cell death and reduces tumor growth. Interestingly, this is as a result of pyrimidine depletion rather than ammonia toxicity and treatment with exogenous pyrimidines rescues growth [78].
Although AMPK has conventionally been viewed as the focal-point of tumor-suppressor activity downstream of LKB1, it has recently been demonstrated that AMPK deletion in KRAS-driven models of NSCLC does not mimic the effects of LBK1 loss [79]. Two independent studies have highlighted that the two understudied AMPKR kinases, SIK1 and SIK3, are critical targets in NSCLC [80, 81]. Both studies identified SIK1 and SIK3 as tumor suppressors, which cooperatively mediate the functions of LKB1 in KRAS-driven NSCLC models. Given the vast amount of literature linking LKB1 loss in lung cancer to metabolic vulnerabilities, it will also be worthwhile to further investigate the LKB1-SIK tumor suppressor axis.
LKB1 loss renders NSCLC tumors dependent on many metabolic pathways, while also reducing their metabolic plasticity and hence inducing a significant susceptibility to energy crisis.
2.4. Other mutations
Many other mutations common to lung cancer also play a role in modulating metabolic pathways such as KEAP1, anaplastic lymphoma kinase (ALK), BRAF and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA). Loss-of-function KEAP1 mutations occur in approximately 20–30% of lung adenocarcinoma [82, 83] and KEAP1 inactivation accelerates KRAS induced lung tumorigenesis in vivo [84]. KEAP1 normally associates with NRF2 and promotes the degradation of NRF2, thus preventing the antioxidant transcriptional program [75]. NRF2 induced transcription redirects the flow of bioenergetic mass towards antioxidant production, providing proliferative and survival advantages [85, 86]. Lung adenocarcinoma harboring KEAP1 mutations are dependent on PPP [84] and glutaminolysis [85]. KRAS/P53 tumors with KEAP1 mutations show a dramatic reduction in tumor growth rates when treated with glutaminase inhibitors [85]. Similarly, the loss of KEAP1 plays a critical role in elevating the redox stress observed in LKB1 deficient cells in a glutamine-dependent manner, leading to the sensitivity of KRAS/LKB1 cells to glutaminase inhibitors in vitro and in vivo [87]. The activation of NRF2 by KEAP1 mutation results in the system xc- dependent export of glutamate, leading to a depletion in intracellular glutamate [85, 88]. A recent study demonstrated that the limited glutamate availability reduces total non-essential amino acids (NEAA) pools and drives a dependency of KEAP1 mutant cells on multiple NEAAs such as serine, glycine and asparagine. This vulnerability can be targeted by enzymatic depletion of individual NEAAs or dietary restriction in KEAP1 mutant tumors [89]. Moreover, it has recently been shown that a KRAS/P53 lung adenocarcinoma model displays a KEAP1-mutant-specific dependency on solute carrier family 33 member 1 (SLC33A1) [90]. SLC33A1 is an ER-localized transmembrane protein and its loss leads to an induction of the unfolded protein response [90]. Increased GSH synthesis as a result of KEAP1 mutations renders KRAS/P53 cells highly sensitive to genetic perturbations of ER-localized proteins, such as SLC33A1 loss. This SLC33A1 dependency is rescued by inhibition of GSH synthesis [90].
Rearrangements in the ALK receptor tyrosine kinase are found in approximately 3–7% of NSCLC patients [91] and the impact of ALK rearrangements on metabolic alterations in NSCLC is poorly understood. Choi and colleagues used FDG PET/CT to compare the metabolic and metastatic features of lung adenocarcinoma patients with ALK and EGFR mutations. Interestingly, patients with ALK-rearrangements display higher glucose metabolism and a quicker rate of metastasis when compared to EGFR wild type and mutated tumors [92]. The most frequently observed ALK rearrangement is the EML4-ALK fusion [91] and a significant decrease in glycolysis is observed in EM4-ALK positive xenografts when treated with ALK inhibitors [93]. The upregulation of HK2 is essential for the proliferation of EML4-ALK NSCLC cells and HIF-1α is the key transcription factor in driving this upregulation [93].
The most common BRAF mutation is an alteration in the kinase domain (V600E) leading to constitutive activation and these occur in roughly 3% of lung cancer patients. Most research on the metabolic effect of oncogenic BRAF focuses around melanoma where it is demonstrated that BRAFV600E drives OXPHOS through its control of pyruvate metabolism, leading to increased oxidized pyruvate into the TCA cycle [94, 95]. In a mouse model of BRAFV600E-driven lung cancer, Atg7 deletion initially provides an advantage in tumor growth due to the induction of oxidative stress. However, at later stages of tumorigenesis, loss of autophagy leads to metabolic crisis resulting in defective mitochondria, reduced tumor growth and the conversion of adenomas and adenocarcinomas to oncocytomas [96].
Oncogenic PIK3CA signaling promotes glycolysis by increasing glucose uptake through regulation of GLUT1/4 [97–99] and this increased reliance on glucose metabolism is utilized to sustain proliferation [100]. Moreover, mutant PIK3CA in colorectal cancer (CRC) upregulates mitochondrial glutamate pyruvate transaminase 2 (GPT2), leading to enhanced conversion of glutamate to α-ketoglutarate to replenish the TCA cycle, and hence a dependency on glutamine metabolism [101, 102]. PIK3CA is mutated in 1% to 2% of lung cancers [103], however little is known about the specific metabolic consequences of this oncogenic signaling in the lung and future studies are needed to characterize its role in lung cancer.
3. Targeting metabolic vulnerabilities in NSCLC
The alterations observed in metabolism in lung cancer has led to the development of numerous compounds, some of which are being actively investigated in clinical trials (Table 1) [104, 105]. The glucose analogue, 2-DG, was one of the first compounds shown to inhibit glycolysis [106] and while early clinical testing showed some promise, adverse effects associated with hypoglycaemia resulted in limited clinical use [107]. A more recent clinical trial revisited the use of 2-DG at lower doses in various solid malignancies, including NSCLC, and in combination with docetaxel; however these reduced doses are insufficient to inhibit disease progression [108].
Table 1.
Ongoing Clinical Trials That Target Cancer Metabolism in NSCLC
| Drug | Target | Study | Phase | NCT Number | Metabolic Effect | Primary Outcome |
|---|---|---|---|---|---|---|
| Metformin | mitochondrial complex I | Pre-operative Metformin in Patients With Clinical Stage I - IIIA NSCLC Proceeding to Surgical Resection | 2 | NCT03086733 | Oxidative Phosphorylation | Tumor proliferation and apoptosis |
| Metformin | mitochondrial complex I | Sintilimab Combined With Metformin in First-Line Chemotherapy Refractory Advanced NSCLC Patients (SMART) | 2 | NCT03874000 | Oxidative Phosphorylation | Objective Response Rate (ORR) |
| Metformin | mitochondrial complex I | Nivolumab and Metformin Hydrochloride in Treating Patients With Stage III-IV Non-small Cell Lung Cancer That Cannot Be Removed by Surgery | 2 | NCT03048500 | Oxidative Phosphorylation | Objective Response Rate (ORR) |
| Metformin | mitochondrial complex I | Metformin Plus TKI Use in Patients With Non-Small Cell Lung Carcinoma | 2 | NCT03071705 | Oxidative Phosphorylation | Overall Survival |
| Metformin | mitochondrial complex I | Chemotherapy and Radiation Therapy With or Without Metformin Hydrochloride in Treating Patients With Stage III Non-small Cell Lung Cancer | 2 | NCT02186847 | Oxidative Phosphorylation | Progression-free survival |
| Metformin | mitochondrial complex I | The Addition of Metformin to Definitive Radiotherapy in Patients With Stage III NSCLC (RADFORMIN) | 2 | NCT04170959 | Oxidative Phosphorylation | Loco regional progression-free survival rate |
| Metformin | mitochondrial complex I | Metformin Plus/Minus Fasting Mimicking Diet to Target the Metabolic Vulnerabilities of LKB1-inactive Lung Adenocarcinoma (FAME) | 2 | NCT03709147 | Oxidative Phosphorylation | Progression-free survival |
| CB-839 | glutaminase inhibitor | Testing of the Anti Cancer Drugs CB-839 HCl (Telaglenastat) and MLN0128 (Sapanisertib) in Advanced Stage Non-small Cell Lung Cancer | 1 | NCT04250545 | Glutamine Metabolism | Maximum tolerated dose; Response rate; Median progression free survival |
| CB-839 | glutaminase inhibitor | Study CB-839 in Combination With Nivolumab in Patients With Melanoma, ccRCC and NSCLC | 1/2 | NCT02771626 | Glutamine Metabolism | Safety and Tolerability |
| CB-839 | glutaminase inhibitor | Glutaminase Inhibitor CB-839 Hydrochloride and Osimertinib in Treating Patients With EGFR-Mutated Stage IV Non-small Cell Lung Cancer | 1/2 | NCT03831932 | Glutamine Metabolism | Dose evaluation; Objective Response Rate (ORR) |
| CB-839 | glutaminase inhibitor | A Study of Telaglenastat (CB-839) in Combination With Palbociclib in Patients With Solid Tumors | 1/2 | NCT03965845 | Glutamine Metabolism | Safety and Tolerability; Dose evaluation |
| CB-839 | glutaminase inhibitor | A Study of Telaglenastat (CB-839) With Standard-of-Care Chemoimmunotherapy in 1L KEAP1/NRF2-Mutated, Nonsquamous NSCLC | 2 | NCT04265534 | Glutamine Metabolism | Progression-free survival; Safety and Tolerability; Dose evaluation |
| simvastatin | 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase | BIBW 2992 (an irreversible EGFR-TKI) Plus Simvastatin vs. BIBW 2992 in Previously Treated Patients With Advanced Non-adenocarcinomatous NSCLC | 2 | NCT01156545 | Inhibition of HMG-CoA reductase and de novo synthesis of cholesterol and isoprenoids | Objective Response Rate (ORR) |
| TVB-2640 | fatty acid synthase (FASN) | Study of TVB-2640 in KRAS Non-Small Cell Lung Carcinomas | 2 | NCT03808558 | Fatty Acid Metabolism | Preliminary disease control rate and response rate |
| Hydroxychloroquine | Lysozomes | Erlotinib With or Without Hydroxychloroquine in Chemo-Naive Advanced NSCLC and (EGFR) Mutations | 2 | NCT00977470 | Autophagy | Progression-free survival |
| NG-monomethyl-L-arginine (L-NMMA) | pan-nitric oxide synthase inhibitor | Safety of L-NMMA and Pembrolizumab in cancer | 1 | NCT03236935 | L-argine metabolism | Maximum tolerated dose |
| Trigriluzole | Glutamate | Trigriluzole With Nivolumab and Pembrolizumab in Treating Patients With Metastatic or Unresectable Solid Malignancies or Lymphoma | 2 | NCT03229278 | Glutamine Metabolism | Maximum tolerated dose |
| Vitamin C | Oxidative Stress | Pharmacological Ascorbate for Lung Cancer | 2 | NCT02420314 | Reactive Oxgen Species | Tumor response |
| Vitamin C | Oxidative Stress | Vitamin C and Tyrosine Kinase Inhibitor in Lung Cancer Patients With Epidermal Growth Factor Receptor Mutations | 1/2 | NCT03799094 | Reactive Oxgen Species | Progression-free survival |
| Vitamin C | Oxidative Stress | A Phase 2 Study Adding Ascorbate to Chemotherapy and Radiation Therapy for NSCLC (XACT-LUNG) | 2 | NCT02905591 | Reactive Oxgen Species | Progression rate |
| Vitamin C | Oxidative Stress | High Dose Vitamin C Intravenous Infusion in Patients With Resectable or Metastatic Solid Tumor Malignancies | 2 | NCT03146962 | Reactive Oxgen Species | Disease control rate |
| INCB001158 | Arginase | Arginase Inhibitor INCB001158 as a Single Agent and in Combination With Immune Checkpoint Therapy in Patients With Advanced/Metastatic Solid Tumors | 1/2 | NCT02903914 | L-argine metabolism | Safety and Tolerability; Dose evaluation |
Metformin is a mitochondrial drug which exhibits its function through the inhibition of Complex I of the respiratory chain and it is the most commonly administrated antidiabetic drug. Clinical and observational studies demonstrate that metformin improves response to chemotherapy [109, 110] and concurrent chemoradiotherapy (cCRT) [111] in lung cancer patients. Diabetic patients with advanced NSCLC undergoing metformin treatment at the time of lung cancer diagnosis, show longer overall survival in comparison to those not on metformin [112, 113]. Interestingly, a recent clinical study in a small cohort of lung cancer patients demonstrates that diabetic patients receiving metformin have a higher response rate and overall survival to immune checkpoint blockade inhibitors than non-diabetic patients not receiving metformin [114]. However, controversy exists in the field as there is also evidence to suggest the contrary. A recent study conducted on diabetic lung cancer patients using metformin and undergoing platinum based chemotherapy show no significant effect on overall survival [115]. Another clinical trial (NRG-LUOO1) in non-diabetic patients with nonresectable stage III NSCLC demonstrates that while the addition of metformin to cCRT is well tolerated, it does not improve clinical outcome for these patients [116].
The use of metformin in combination with EGFR TKIs has also been explored in the clinic for the treatment of NSCLC patients and similar contradictory findings are observed. A study examining data from NSCLC patients with diabetes receiving either EGFR-TKI plus metformin or EGFR-TKI plus an alternative hypoglycemic agent reveals an increase in progression free survival and overall survival in the patients receiving metformin [117]. Similarly, non-diabetic patients treated with metformin plus EGFR TKIs (erlotinib hydrochloride, afatinib dimaleate, or gefitinib) have significant benefit in terms of progression free survival and overall survival compared with those treated with EGFR TKI alone [118]. However, results from another recent phase 2 clinical trial (NCT01864681) demonstrate that there is no survival benefit when adding metformin to the TKI, gefitinib, in non-diabetic NSCLC patients harboring an EGFR mutation [119]. These contradictory results indicate that further studies with a larger cohort of patients need to be conducted to fully decipher the benefit of metformin for NSCLC patients. Moreover, given the differences observed in response to metformin; perhaps it will be interesting in future studies to focus on the metabolic discrepancies between lung cancer tumors of diabetic patients who have received long term exposure to metformin and non-diabetic patients. These comparisons may provide evidence of the metabolic liabilities which influence response of patients to mitochondrial drugs and hence provide information to allow other cancer types with similar vulnerabilities to benefit from these drugs.
Enhanced glutamine dependency is another common feature of NSCLC, creating a potential treatment option with glutamine inhibitors. Attempts to target this pathway in vivo led to the isolation of a compound from fermentation broth of a Streptomyces and subsequent development of the glutamine analog 6-diazo-5-oxo-L-norleucine (DON) [120]. This drug exerts its actions by inhibiting numerous enzymes that use glutamine as a substrate and although it is highly effective in killing cancer cells, it has severe side effects on the gastrointestinal tract [121]. Recently, a DON analog, JHU083, has been developed. It is a prodrug that circulates intact and inert and becomes activated in the tumor microenvironment upon enzymatic cleavage of the promoieties, thus improving the therapeutic window [122]. In syngeneic mouse models of colon cancer, lymphoma and melanoma, treatment with JHU083 produces robust antitumor immunity that results in tumor regression [122]. Interestingly, JHU083 treatment in tumor bearing mice suppresses oxidative and glycolytic metabolism of the cancer cells, while it surprisingly up-regulates oxidative metabolism in the effector T cells leading to long-lived highly activated T cells [122]. This discovery should lead the way to future studies examining differences in metabolic plasticity between cancer cells and immune cells. Such studies will help guide the design of better combination treatments and improve the therapeutic window for the use of metabolic inhibitors. Glutamine dependence has also been targeted with selective glutaminase inhibitors such BPTES, compound 968 and CB-839 [123–125]. There are numerous ongoing clinical trials investigating the use of CB839 in combination with various other agents for the treatment of NSCLC (NCT03831932, NCT02771626, NCT04250545, NCT03965845). As discussed earlier in this review, KEAP1 mutations in NSCLC results in a marked dependency on glutamine, hence a clinical trial has also been launched to examine the benefit of adding CB839 to the standard of care in KEAP1/NRF2 mutated NSCLC (NCT04265534).
Given the role of de novo lipogenesis in KRAS mutant NSCLC [43], there is an ongoing phase 2 clinical trial (NCT03808558) evaluating the benefits of the fatty acid synthases inhibitor, TVB-2640, as a single agent in KRAS mutant NSCLC patients. Moreover, autophagy addiction is a metabolic vulnerability associated with lung tumors and there has been a number of clinical trials conducted to evaluate the effects of the autophagy inhibitor, hydroxychloroquine (HCQ), in NSCLC patients (NCT01649947, NCT00728845, NCT00977470). Interestingly, recent work indicates that targeting autophagy appears to enhance antitumor immunity [126]. Surface major histocompatibility complex class I (MHC-I) is decreased in KRAS driven NSCLC and PDAC cell lines via autophagy-lysosome signaling and inhibiting this pathway via genetic deletion or pharmacological inhibition with chloroquine (CQ) restores MHC-I surface expression. Moreover, targeting the autophagy-lysosome pathway leads to enhanced T cell immunity and improves response to immune checkpoint blockade therapy in PDAC in vivo models [126]. Future clinical trials should explore the dependency of the autophagy-lysosome pathway as a metabolic liability in NSCLC to enhance anti-tumor immunity.
4. Concluding remarks
Despite the early concept of targeting metabolism to treat cancer and all the extensive studies since, to date, the development of metabolic therapies with broad and predictable efficacy in human cancers have been limited. One such issue arises from the traditional belief that tumors are thought to contain generalized metabolic vulnerabilities. However, as highlighted in this review, there are many different metabolic phenotypes driven by specific mutations which exist even within the context of NSCLC. Future studies need to focus on establishing the specific metabolic liabilities that arise within different cancers and their subtypes. This may require an understanding of the plasticity inbuilt into the metabolic network and perhaps a multi-nodal metabolic attack is required for effective treatment. Moreover, recent advances in technology to study metabolism in the context of the tumor microenvironment will shine new light on the inter metabolic dependencies between the cancer and surrounding cells. Finally, a better understanding of the metabolic changes in immune cells and the differences in metabolic plasticity between cancer cells and immune cells may help facilitate the development of combinational immunotherapy in lung cancer.
Acknowledgements
We would like to thank Dr. Kate ER Hollinshead for editing the figures.
Funding
This work was funded by grants from H2020 MSCA Global Fellowship (799724) (C.M.D). This work was supported by NIH grants CA233084-01 (K.K.W.), CA219670 (K.K.W), CA216188 (K.K.W.).
References
- 1.Vernieri C, et al. , Targeting cancer metabolism: dietary and pharmacologic interventions. Cancer discovery, 2016. 6(12): p. 1315–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D and Weinberg RA, Hallmarks of cancer: the next generation. cell, 2011. 144(5): p. 646–674. [DOI] [PubMed] [Google Scholar]
- 3.Warburg ОН PК and Negelein E, Uber den Stoffwechsel der Karzinomzelle. Biochem Z, 1924. 152: p. 309–44. [Google Scholar]
- 4.Almuhaideb A, Papathanasiou N, and Bomanji J, 18F-FDG PET/CT imaging in oncology. Annals of Saudi medicine, 2011. 31(1): p. 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warburg O, Wind F, and Negelein E, The metabolism of tumors in the body. The Journal of general physiology, 1927. 8(6): p. 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren J-L, et al. , Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC advances, 2018. 8(40): p. 22335–22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berndt N, et al. , Kinetic modelling of quantitative proteome data predicts metabolic reprogramming of liver cancer. British Journal of Cancer, 2020. 122(2): p. 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Björkblom B, et al. , Metabolic response patterns in brain microdialysis fluids and serum during interstitial cisplatin treatment of high-grade glioma. British Journal of Cancer, 2020. 122(2): p. 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faubert B, Solmonson A, and DeBerardinis RJ, Metabolic reprogramming and cancer progression. Science, 2020. 368(6487). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frezza C, Metabolism and cancer: the future is now. 2019, Nature Publishing Group. [Google Scholar]
- 11.Davidson SM, et al. , Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell metabolism, 2016. 23(3): p. 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma EH, et al. , Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity, 2019. 51(5): p. 856–870. e5. [DOI] [PubMed] [Google Scholar]
- 13.Ramalingam SS, Owonikoko TK, and Khuri FR, Lung cancer: New biological insights and recent therapeutic advances. CA: a cancer journal for clinicians, 2011. 61(2): p. 91–112. [DOI] [PubMed] [Google Scholar]
- 14.Majem B, Nadal E, and Muñoz-Pinedo C. Exploiting metabolic vulnerabilities of Non small cell lung carcinoma. in Seminars in Cell & Developmental Biology. 2020. Elsevier. [DOI] [PubMed] [Google Scholar]
- 15.Goodwin J, et al. , The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nature communications, 2017. 8(1): p. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schuurbiers OC, et al. , Glucose metabolism in NSCLC is histology-specific and diverges the prognostic potential of 18FDG-PET for adenocarcinoma and squamous cell carcinoma. Journal of Thoracic Oncology, 2014. 9(10): p. 1485–1493. [DOI] [PubMed] [Google Scholar]
- 17.Momcilovic M, et al. , The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell, 2018. 33(5): p. 905–921. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higashi K, et al. , Fluorine-18-FDG PET imaging is negative in bronchioloalveolar lung carcinoma. Journal of Nuclear Medicine, 1998. 39(6): p. 1016–1020. [PubMed] [Google Scholar]
- 19.Scafoglio CR, et al. , Sodium-glucose transporter 2 is a diagnostic and therapeutic target for early-stage lung adenocarcinoma. Science translational medicine, 2018. 10(467). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hensley CT, et al. , Metabolic heterogeneity in human lung tumors. Cell, 2016. 164(4): p. 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faubert B, et al. , Lactate metabolism in human lung tumors. Cell, 2017. 171(2): p. 358–371. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pikor LA, et al. , Genetic alterations defining NSCLC subtypes and their therapeutic implications. Lung cancer, 2013. 82(2): p. 179–189. [DOI] [PubMed] [Google Scholar]
- 23.Chen P-H, et al. , Metabolic diversity in human non-small cell lung cancer cells. Molecular Cell, 2019. 76(5): p. 838–851. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kempf E, et al. , KRAS oncogene in lung cancer: focus on molecularly driven clinical trials. European respiratory review, 2016. 25(139): p. 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sellers K, et al. , Pyruvate carboxylase is critical for non–small-cell lung cancer proliferation. The Journal of clinical investigation, 2015. 125(2): p. 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerr EM, et al. , Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature, 2016. 531(7592): p. 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Junttila MR, et al. , Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature, 2010. 468(7323): p. 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hui S, et al. , Glucose feeds the TCA cycle via circulating lactate. Nature, 2017. 551(7678): p. 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao S, et al. , AIF-regulated oxidative phosphorylation supports lung cancer development. Cell research, 2019. 29(7): p. 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patra KC, et al. , Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer cell, 2013. 24(2): p. 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gwinn DM, et al. , Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell, 2018. 33(1): p. 91–107. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caiola E, et al. , Different metabolic responses to PI3K inhibition in NSCLC cells harboring wild-type and G12C mutant KRAS. Oncotarget, 2016. 7(32): p. 51462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reynolds MR, et al. , Control of glutamine metabolism by the tumor suppressor Rb. Oncogene, 2014. 33(5): p. 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conroy LR, et al. , Loss of Rb1 Enhances Glycolytic Metabolism in Kras-Driven Lung Tumors In Vivo. Cancers, 2020. 12(1): p. 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayers JR, et al. , Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science, 2016. 353(6304): p. 1161–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimmelman AC, Metabolic dependencies in RAS-driven cancers. 2015, AACR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo JY, et al. , Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes & development, 2016. 30(15): p. 1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo JY and White E, Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRASG12D-driven lung tumors. Autophagy, 2013. 9(10): p. 1636–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryant KL, et al. , Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nature medicine, 2019. 25(4): p. 628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinsey CG, et al. , Protective autophagy elicited by RAF→ MEK→ ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nature medicine, 2019. 25(4): p. 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo JY, et al. , Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes & development, 2013. 27(13): p. 1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karsli-Uzunbas G, et al. , Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer discovery, 2014. 4(8): p. 914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh A, et al. , De novo lipogenesis represents a therapeutic target in mutant Kras non-small cell lung cancer. The FASEB Journal, 2018. 32(12): p. 7018–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gouw AM, et al. , Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proceedings of the National Academy of Sciences, 2017. 114(17): p. 4300–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Padanad MS, et al. , Fatty acid oxidation mediated by Acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell reports, 2016. 16(6): p. 1614–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiao S, et al. , REDD1 loss reprograms lipid metabolism to drive progression of RAS mutant tumors. Genes & development, 2020. 34(11–12): p. 751–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu K, et al. , Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. The Journal of Clinical Investigation, 2020. 130(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong B-J, et al. , Oncogenic KRAS Sensitizes Lung Adenocarcinoma to GSK-J4–Induced Metabolic and Oxidative Stress. Cancer Research, 2019. 79(22): p. 5849–5859. [DOI] [PubMed] [Google Scholar]
- 49.Gridelli C, et al. , Non-small-cell lung cancer. Nature reviews Disease primers, 2015. 1(1): p. 1–16. [DOI] [PubMed] [Google Scholar]
- 50.Kim JH, et al. , Enhanced glycolysis supports cell survival in EGFR-mutant lung adenocarcinoma by inhibiting autophagy-mediated EGFR degradation. Cancer research, 2018. 78(16): p. 4482–4496. [DOI] [PubMed] [Google Scholar]
- 51.Makinoshima H, et al. , Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. Journal of Biological Chemistry, 2014. 289(30): p. 20813–20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Makinoshima H, et al. , Signaling through the phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) axis is responsible for aerobic glycolysis mediated by glucose transporter in epidermal growth factor receptor (EGFR)-mutated lung adenocarcinoma. Journal of Biological Chemistry, 2015. 290(28): p. 17495–17504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim SM, et al. , Glycolysis inhibition sensitizes non–small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of mcl-1 by AMPK activation. Molecular cancer therapeutics, 2013. 12(10): p. 2145–2156. [DOI] [PubMed] [Google Scholar]
- 54.Apicella M, et al. , Increased lactate secretion by cancer cells sustains non-cell-autonomous adaptive resistance to MET and EGFR targeted therapies. Cell metabolism, 2018. 28(6): p. 848–865. e6. [DOI] [PubMed] [Google Scholar]
- 55.Lypova N, et al. , Increased 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase-3 activity in response to EGFR signaling contributes to non–small cell lung cancer cell survival. Journal of Biological Chemistry, 2019. 294(27): p. 10530–10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki S, et al. , Involvement of GLUT1-mediated glucose transport and metabolism in gefitinib resistance of non-small-cell lung cancer cells. Oncotarget, 2018. 9(66): p. 32667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Momcilovic M, et al. , Targeted inhibition of EGFR and glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell reports, 2017. 18(3): p. 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang L, et al. , Increased glutamine anabolism sensitizes non-small cell lung cancer to gefitinib treatment. Cell death discovery, 2018. 4(1): p. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y, et al. , Branched-chain amino acid metabolic reprogramming orchestrates drug resistance to EGFR tyrosine kinase inhibitors. Cell reports, 2019. 28(2): p. 512–525. e6. [DOI] [PubMed] [Google Scholar]
- 60.Ali A, et al. , Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non‐small cell lung cancer. EMBO molecular medicine, 2018. 10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, et al. , EGFR modulates monounsaturated fatty acid synthesis through phosphorylation of SCD1 in lung cancer. Molecular cancer, 2017. 16(1): p. 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J, et al. , Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells. Oncotarget, 2016. 7(32): p. 52392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding L, et al. , Somatic mutations affect key pathways in lung adenocarcinoma. Nature, 2008. 455(7216): p. 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shackelford DB and Shaw RJ, The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nature Reviews Cancer, 2009. 9(8): p. 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Laplante M and Sabatini DM, mTOR signaling in growth control and disease. Cell, 2012. 149(2): p. 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaufman JM, et al. , LKB1 Loss induces characteristic patterns of gene expression in human tumors associated with NRF2 activation and attenuation of PI3K-AKT. Journal of Thoracic Oncology, 2014. 9(6): p. 794–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whang YM, et al. , LKB1 deficiency enhances sensitivity to energetic stress induced by erlotinib treatment in non-small-cell lung cancer (NSCLC) cells. Oncogene, 2016. 35(7): p. 856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Phadke M, et al. , Accelerated cellular senescence phenotype of GAPDH-depleted human lung carcinoma cells. Biochemical and biophysical research communications, 2011. 411(2): p. 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Faubert B, et al. , Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proceedings of the National Academy of Sciences, 2014. 111(7): p. 2554–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parker SJ, et al. , LKB1 promotes metabolic flexibility in response to energy stress. Metabolic engineering, 2017. 43: p. 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shackelford DB, et al. , mTOR and HIF-1α-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proceedings of the National Academy of Sciences, 2009. 106(27): p. 11137–11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Svensson RU, et al. , Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nature medicine, 2016. 22(10): p. 1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Momcilovic M, et al. , Heightening energetic stress selectively targets LKB1-deficient non–small cell lung cancers. Cancer research, 2015. 75(22): p. 4910–4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhatt V, et al. , Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes & development, 2019. 33(3–4): p. 150–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li F, et al. , LKB1 inactivation elicits a redox imbalance to modulate non-small cell lung cancer plasticity and therapeutic response. Cancer cell, 2015. 27(5): p. 698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shackelford DB, et al. , LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer cell, 2013. 23(2): p. 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Y, et al. , Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer discovery, 2013. 3(8): p. 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim J, et al. , CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature, 2017. 546(7656): p. 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eichner LJ, et al. , Genetic analysis reveals AMPK is required to support tumor growth in murine Kras-dependent lung cancer models. Cell metabolism, 2019. 29(2): p. 285–302. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hollstein PE, et al. , The AMPK-related kinases SIK1 and SIK3 mediate key tumor-suppressive effects of LKB1 in NSCLC. Cancer discovery, 2019. 9(11): p. 1606–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murray CW, et al. , An LKB1–SIK axis suppresses lung tumor growth and controls differentiation. Cancer discovery, 2019. 9(11): p. 1590–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shen R, et al. , Harnessing clinical sequencing data for survival stratification of patients with metastatic lung adenocarcinomas. JCO precision oncology, 2019. 3: p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jaramillo MC and Zhang DD, The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes & development, 2013. 27(20): p. 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Best SA, et al. , Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nature communications, 2019. 10(1): p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Romero R, et al. , Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nature medicine, 2017. 23(11): p. 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sayin VI, LeBoeuf SE, and Papagiannakopoulos T, Targeting Metabolic Bottlenecks in Lung Cancer. Trends in cancer, 2019. 5(8): p. 457–459. [DOI] [PubMed] [Google Scholar]
- 87.Galan-Cobo A, et al. , LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer research, 2019. 79(13): p. 3251–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sayin VI, et al. , Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife, 2017. 6: p. e28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.LeBoeuf SE, et al. , Activation of oxidative stress response in cancer generates a druggable dependency on exogenous non-essential amino acids. Cell metabolism, 2020. 31(2): p. 339–350. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Romero R, et al. , Keap1 mutation renders lung adenocarcinomas dependent on Slc33a1. Nature Cancer, 2020: p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Katayama R, Lovly CM, and Shaw AT, Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. 2015, AACR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Choi H, et al. , Metabolic and metastatic characteristics of ALK-rearranged lung adenocarcinoma on FDG PET/CT. Lung Cancer, 2013. 79(3): p. 242–247. [DOI] [PubMed] [Google Scholar]
- 93.Ma Y, et al. , A causal link from ALK to hexokinase II overexpression and hyperactive glycolysis in EML4-ALK-positive lung cancer. Oncogene, 2016. 35(47): p. 6132–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaplon J, et al. , A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature, 2013. 498(7452): p. 109–112. [DOI] [PubMed] [Google Scholar]
- 95.Scortegagna M, et al. , Genetic inactivation or pharmacological inhibition of Pdk1 delays development and inhibits metastasis of Braf V600E:: Pten–/–melanoma. Oncogene, 2014. 33(34): p. 4330–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Strohecker AM, et al. , Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E–driven lung tumors. Cancer discovery, 2013. 3(11): p. 1272–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Taha C, et al. , Opposite Translational Control of glut1 and glut4 glucose transporter mrnas in response to insulin role of mammalian target of rapamycin, protein kinase b, and phosphatidylinositol 3-kinase in glut1 mRNA TRANSLATION. Journal of Biological Chemistry, 1999. 274(46): p. 33085–33091. [DOI] [PubMed] [Google Scholar]
- 98.Brachmann SM, et al. , Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Molecular and Cellular Biology, 2005. 25(5): p. 1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hu H, et al. , Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell, 2016. 164(3): p. 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ilic N, et al. , PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proceedings of the National Academy of Sciences, 2017. 114(17): p. E3434–E3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hao Y, et al. , Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nature communications, 2016. 7(1): p. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Feng X, Hao Y, and Wang Z, Targeting glutamine metabolism in PIK3CA mutant colorectal cancers. Genes & diseases, 2016. 3(4): p. 241–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Devarakonda S, Morgensztern D, and Govindan R, Genomic alterations in lung adenocarcinoma. The lancet oncology, 2015. 16(7): p. e342–e351. [DOI] [PubMed] [Google Scholar]
- 104.Hay N, Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nature Reviews Cancer, 2016. 16(10): p. 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao Y, Butler EB, and Tan M, Targeting cellular metabolism to improve cancer therapeutics. Cell death & disease, 2013. 4(3): p. e532–e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wick AN, et al. , Localization of the primary metabolic block produced by 2-deoxyglucose. J Biol Chem, 1957. 224(2): p. 963–969. [PubMed] [Google Scholar]
- 107.Landau BR, et al. , Certain metabolic and pharmacologic effects in cancer patients given infusions of 2-deoxy-D-glucose. Journal of the National Cancer Institute, 1958. 21(3): p. 485–494. [PubMed] [Google Scholar]
- 108.Raez LE, et al. , A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer chemotherapy and pharmacology, 2013. 71(2): p. 523–530. [DOI] [PubMed] [Google Scholar]
- 109.Tan BX, et al. , Prognostic influence of metformin as first‐line chemotherapy for advanced nonsmall cell lung cancer in patients with type 2 diabetes. Cancer, 2011. 117(22): p. 5103–5111. [DOI] [PubMed] [Google Scholar]
- 110.Marrone KA, et al. , A randomized phase II study of metformin plus paclitaxel/carboplatin/bevacizumab in patients with chemotherapy‐naïve advanced or metastatic nonsquamous non‐small cell lung cancer. The oncologist, 2018. 23(7): p. 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wink KC, et al. , Improved progression free survival for patients with diabetes and locally advanced non-small cell lung cancer (NSCLC) using metformin during concurrent chemoradiotherapy. Radiotherapy and Oncology, 2016. 118(3): p. 453–459. [DOI] [PubMed] [Google Scholar]
- 112.Lin JJ, et al. , Survival of patients with stage IV lung cancer with diabetes treated with metformin. American journal of respiratory and critical care medicine, 2015. 191(4): p. 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cao X, et al. , The clinical effect of metformin on the survival of lung cancer patients with diabetes: a comprehensive systematic review and meta-analysis of retrospective studies. Journal of Cancer, 2017. 8(13): p. 2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Afzal MZ, et al. , Clinical outcomes in non-small-cell lung cancer patients receiving concurrent metformin and immune checkpoint inhibitors. Lung Cancer Management, 2019. 8(2): p. LMT11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wen-Xiu X, et al. , Impact of metformin use on survival outcomes in non-small cell lung cancer treated with platinum. Medicine, 2018. 97(51). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tsakiridis T, et al. , Initial reporting of NRG-LU001 (NCT02186847), randomized phase II trial of concurrent chemoradiotherapy (CRT)+/−metformin in locally advanced Non-Small Cell Lung Cancer (NSCLC). 2019, American Society of Clinical Oncology. [Google Scholar]
- 117.Chen H, et al. , Synergistic effects of metformin in combination with EGFR-TKI in the treatment of patients with advanced non-small cell lung cancer and type 2 diabetes. Cancer letters, 2015. 369(1): p. 97–102. [DOI] [PubMed] [Google Scholar]
- 118.Arrieta O, et al. , Effect of Metformin Plus Tyrosine Kinase Inhibitors Compared With Tyrosine Kinase Inhibitors Alone in Patients With Epidermal Growth Factor Receptor–Mutated Lung Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA oncology, 2019. 5(11): p. e192553–e192553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li L, et al. , Combination of Metformin and Gefitinib as First-Line Therapy for Nondiabetic Advanced NSCLC Patients with EGFR Mutations: A Randomized, Double-Blind Phase II Trial. Clinical Cancer Research, 2019. 25(23): p. 6967–6975. [DOI] [PubMed] [Google Scholar]
- 120.Pinkus LM and Windmueller HG, Phosphate-dependent glutaminase of small intestine: localization and role in intestinal glutamine metabolism. Archives of biochemistry and biophysics, 1977. 182(2): p. 506–517. [DOI] [PubMed] [Google Scholar]
- 121.Lemberg KM, et al. , We’re not “DON” yet: optimal dosing and prodrug delivery of 6-Diazo-5-oxo-L-norleucine. Molecular cancer therapeutics, 2018. 17(9): p. 1824–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Leone RD, et al. , Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science, 2019. 366(6468): p. 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Elgogary A, et al. , Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proceedings of the National Academy of Sciences, 2016. 113(36): p. E5328–E5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang J-B, et al. , Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer cell, 2010. 18(3): p. 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gao M, et al. , Glutaminolysis and transferrin regulate ferroptosis. Molecular cell, 2015. 59(2): p. 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yamamoto K, et al. , Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature, 2020: p. 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]