

Abstract
Cyclotides are plant-derived peptides found within five families of flowering plants (Violaceae, Rubiaceae, Fabaceae, Solanaceae, and Poaceae) that have a cyclic backbone and six conserved cysteine residues linked by disulfide bonds. Their presence within the Violaceae species seems ubiquitous, yet not all members of other families produce these macrocyclic peptides. The genus Palicourea Aubl. (Rubiaceae) contains hundreds of neotropical species of shrubs and small trees; however, only a few cyclotides have been discovered hitherto. Herein, five previously uncharacterized Möbius cyclotides within Palicourea sessilis and their pharmacological activities are described. Cyclotides were isolated from leaves and stems of this plant and identified as pase A–E, as well as the known peptide kalata S. Cyclotides were de novo sequenced by MALDI-TOF/TOF mass spectrometry, and their structures were solved by NMR spectroscopy. Because some cyclotides have been reported to modulate immune cells, pase A–D were assayed for cell proliferation of human primary activated T lymphocytes, and the results showed a dose-dependent antiproliferative function. The toxicity on other nonimmune cells was also assessed. This study reveals that pase cyclotides have potential for applications as immunosuppressants and in immune-related disorders.
Cyclotides are plant-derived peptides with a molecular weight range of 2.8 to 3.9 kDa, characterized by a head-to-tail cyclized backbone that is stabilized by three disulfide bonds forming a cyclic cystine knot (CCK) motif.1−3 This structural topology makes cyclotides exceptionally resistant to thermal, chemical, or enzymatic degradation.4 Cyclotides have been classified into three subfamilies differing by either the presence (Möbius) or absence (bracelet) of a cis proline bond in the backbone and by their size and amino acid composition. A cis Pro residue gives the Möbius subfamily a conceptual backbone twist, likening it to a Möbius strip, compared to a conventional bracelet of trans peptide bonds in bracelet cyclotides.5 The third subfamily, termed trypsin inhibitor cyclotides, contains a CCK motif but differs markedly in sequence compared to Möbius and bracelet cyclotides.6,7 In general, all subfamilies are tolerant to sequence variability in the “loops” around the cystine knot core;8 however, the bracelet subfamily contains a higher number of cationic residues than the Möbius subfamily.9 Based on discovery studies thus far, bracelets are more common than Möbius cyclotides.10 The three-dimensional structure of the prototypic Möbius cyclotide, kalata B1, isolated from O. affinis was reported in 1995, and details of the cyclic cystine knot topology were established in 2003.11,12
Cyclotides have a wide range of bioactivities that make them interesting for biological or therapeutic applications, including anti-HIV,13−15 inhibition of cell migration,16 modulation of cell signaling,17,18 uterotonic,11,19−21 anthelmintic,22−24 insecticidal,25−27 molluscicidal,28 cytotoxic,29−31 antimicrobial,32,33 hemolytic,34,35 protease inhibition,6,36 and immunosuppressive properties.37,38 The immunosuppressive cyclotide, kalata B1 mutant [T20K], recently completed preclinical evaluation as a potential drug for multiple sclerosis (MS).39
Multiple sclerosis is a disease of the central nervous system (brain and spinal cord) characterized as an autoimmune disorder mediated primarily by activated T cells,40 and more than 2.3 million people suffer from it worldwide.40,41 After a breach of self-tolerance toward myelin and other central nervous system antigens, peripheral activation of autoreactive T cells promotes an immune attack.41,42 T20K is known to act on T-cell proliferation by downregulation of IL-2 release as well as IL-2R/CD25 surface expression and effector activity performance.43,44 Synthetic mutants of kalata B1, including T8K, V10K, V10A, G18K, and N29K, have been studied; the mutations for T8K, V10K, and V10A resulted in loss of immunosuppressive activity,43 which reinforced the notion that the “bioactive face” is the hydrophilic area, centered around the residue Glu7.43,45 However, changes to the surface-exposed hydrophobic patch formed by residues Leu2, Pro3, Val4, Val10, Trp23, Pro24, and Val25 did not significantly alter the activity.45
Although Brazil has the largest biodiversity in the world and is thus an ideal “playground” for phytochemists to discover peptides from plants, there are still few studies describing the isolation of cyclotides from Brazilian plants.16,46−51 The relative lack of study of cyclotides in Brazilian plants probably reflects an earlier focus among Brazilian researchers on nonpeptidic natural products based on available training and equipment, but with opportunities for international internships in cyclotide-specialist laboratories, as occurred in this project, cyclotide research is expanding in Brazil. CyBase, a database that showcases cyclic protein sequences and structures, reports approximately 500 natural cyclotides discovered from five plant families, i.e., Rubiaceae, Fabaceae, Poaceae, Solanaceae, and Violaceae.10 Earlier work estimated that at least 50 000 novel cyclotides may be discovered within the Rubiaceae, but a recent study demonstrated that Violaceae and Rubiaceae plants could exhibit a much higher cyclotide diversity and consist of >150 000 individual cyclotides.52,53
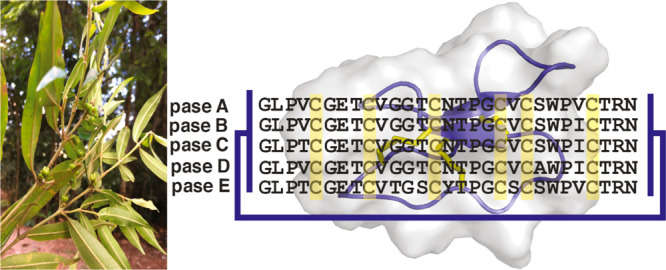
Cyclotides seem to be ubiquitously present within the Violaceae, but their distribution within other families such as the Rubiaceae remains unclear. The genus Palicourea Aubl. (Rubiaceae) contains hundreds of neotropical species of shrubs and small trees;54 however, only four cyclotides have been discovered from three different species: palicourein (P. condensata),55 parigidin-br1-3 (P. rigida),49−51 and paltet-1 (P. tetragona).21 The first cyclotide described for this genus was palicourein, one of the largest members of this macrocyclic plant peptide family (Figure 1).55
Figure 1.

Sequences and structure of Palicourea cyclotides. (A) palicourein, the first cyclotide isolated from the genus Palicourea (P. condensata).55 Cysteine residues and disulfide bonds are shown in yellow. (B) Three-dimensional structure of palicourein (PDB ID: 1R1F). Loop 3 forms an alpha helical segment and is highlighted in red. Disulfides are shown as yellow sticks. (C) Sequence alignment of cyclotides and acyclotides isolated from the genus Palicourea. Yellow boxes show the conserved cysteine residues; the connectivity between the disulfide bonds is shown by black lines, and backbone cyclization is represented by a blue line.
To explore the diversity and activity of Palicourea-type cyclotides, Palicourea sessilis (Vell.) C. M. Taylor (homotypic synonyms: Coffea sessilis Vell., Psychotria vellosiana Benth., non Psychotria sessilis Vell.),54 commonly known as “café do mato”56 was studied. This small tree, 2.5–4 m tall, grows in humid Atlantic forests at 700–1800 m stretching from eastern Brazil to central Paraguay and is also found in wet forests at 950–2250 m on sandstone mountains in the Guyana Highlands in western Guyana and southeastern Venezuela.54 Phytochemically, studies on the isolation of a range of small molecules including squalene, stigmasterol, sitosterol, lupeol, scopoletin, coumarin, and monoterpene indole alkaloids have been reported.56,57
The current study aimed to characterize new cyclotides isolated from P. sessilis (Rubiaceae), because plant species from the Rubiaceae tribe Palicoureeae have been previously examined to contain cyclotides.53 This included the taxonomically related species Palicourea coriacea and Palicourea rigida, and more distant species from the tribe Psychotrieae such as Psychotria longipes.17 Generally, species from the tribes Palicoureeae/Psychotrieae are widely distributed in Brazil. The leaves and stems of P. sessilis (Rubiaceae) were investigated allowing the identification and the characterization of five new Möbius cyclotides (pase A–E). The similarities of pase peptides with T20K prompted us to investigate their immunosuppressant activity on human peripheral blood mononuclear cells (PBMCs). Knowing that single amino acid replacements can have detrimental effects on activity as well as toxicity,43 we also explored such effects of natural modifications of pase peptides and other immortalized nonimmune cells, i.e., human umbilical vein endothelial cells (HUVECs), human colorectal adenocarcinoma (HT-29), and red blood cells (RBCs).
Results and Discussion
Extraction and Purification of Cyclotides from P. sessilis
Dried leaves and stems of P. sessilis were extracted as described in the Experimental Section. After we obtained the peptide-rich fraction for each tissue, LC-MS profiles (Figure 2) were acquired for the extracts. Previous studies have shown that the HPLC elution profile for cyclotides is around 25–55% of CH3CN.53,58 All signals in the cyclotide-elution region that had masses between 2000 and 4000 Da (after deconvolution) were analyzed individually. As shown in Figure 2, it is evident that few major cyclotides were present in the two P. sessilis extracts.
Figure 2.

Identification and purification of cyclotides from P. sessilis. (A) Base peak ion chromatogram profile (LC-MS) of cyclotides identified in the leaves and stems. A linear gradient of 35–45% B in 30 min was applied: Buffer A (H2O/0.1% FA) and Buffer B (90% CH3CN/0.1% FA). (B) HPLC traces showing the purity of isolated cyclotides. A linear gradient 40–70% B in 15 min was applied; Buffer A (H2O/0.1% TFA) and Buffer B (90% CH3CN/0.08% TFA). In both experiments, a Kromasil column (250 mm × 4.6 mm, 5 μm; flow rate = 1 mL per min), 300 Å, UV 220 nm was used.
The presence of disulfide bonds in putative peptides was confirmed through reduction and S-carbamidomethylation of cysteine residues. All peptide signals displayed a mass shift of 348 Da, indicating the presence of six cysteines, with each S-alkylated cysteine residue increasing the molecular mass by 58 Da. For the purification of these cyclotides, successive elution using preparative analysis was employed on peptide-rich fractions from leaves and stems. Purity profiles and the monoisotopic masses of compounds were verified by analytical RP-HPLC (Figure 2) and MALDI-TOF/TOF-MS, respectively. Six peptides were isolated from the leaves and stem of this plant, namely, pase A (m/z 2889), pase B (m/z 2903), pase C (m/z 2905), pase D (m/z 2887), pase E (m/z 2983), and kalata S (m/z 2877) (Figure 2).
The amounts of individual cyclotides from each tissue are listed in Table S1 (Supporting Information). Notably, the amount of cyclotides in the leaves was typically higher than in the stems. For example, the concentration of pase A was approximately 0.21 mg/g of dry plant material in the leaves and 0.07 mg/g dry weight in the stem. Variation in cyclotide expression pattern has been reported previously and may reflect seasonal variation and the exposure of different tissues to different types as well as different levels of biotic stresses.59,60
De Novo Sequencing of Pase A–E
After reduction and alkylation, cyclotides were sequenced by de novo MS/MS; the peptides were enzymatically hydrolyzed by targeting the conserved Glu residue in loop 1 using endoproteinase GluC (endo-GluC), which, for cyclotides, commonly results in a single fragment corresponding to the linearized cyclotide. This also confirmed the presence of a cyclic backbone, with an observed mass increase of 18 Da (in total the observed mass increased by 366 Da compared to the native form). Mass spectra were carefully examined for each peptide, and the sequence was determined based on the presence of both b- and y-ion series (N- and C-terminal fragment ions).
After we used endo-GluC and trypsin enzymes, digested pase A provided a precursor ion at m/z 3255, indicating ring-opening of the cyclotide backbone. MS/MS of this precursor ion from endo-GluC digestion only allowed for annotation of the sequence of a y-ion series fragment VCSWPVCTR (Figure 3). Additionally, the MS/MS of this precursor from tryptic digestion allowed for the identification of the full sequence as cyclo-NGI/LPVCGETCVGGTCNTPGCVCSWPVCTR, based on the b- and y-ion series. As Leu and Ile are difficult to distinguish by mass spectrometry alone, additional digestion studies were done. Chymotrypsin can hydrolyze amide bonds after Leu but not Ile and is widely used to differentiate Leu/Ile. However, if Leu is followed by a Pro, this cleavage does not occur, and hence, the two isobaric residues, i.e., Leu2 and Ile,11 could only be determined by NMR analysis. Observed b- and y-ions from both enzymatic digestions together with two-dimensional NMR data allowed the unambiguous characterization of Leu and Ile in the sequence of a Möbius cyclotide, named pase A (sequence = cyclo-GLPVCGETCVGGTCNTPGCVCSWPVCTRN). The same protocol was applied to characterize the sequences of the other four cyclotides, pase B–E, and confirm the presence of the known sequence of kalata S. Sequence fragments used for MS/MS analyses are shown in Table S2 (Supporting Information). Overall, MS-MS data from endoGlu-C digestions of pase A–D resulted in poor fragmentation patterns, and analyses of MS-MS spectra obtained from tryptic digests were more elucidative. To assemble the final sequence, MS/MS sequencing data and two-dimensional (TOCSY and NOESY) NMR experiments were used.
Figure 3.

De novo sequencing of pase A. (A) MALDI-TOF/TOF-MS spectra of native pase A and (B) after reduction and alkylation; (C) MALDI-TOF/TOF-MS/MS of the peak at m/z 3255.3 corresponding to a linearized cyclotide obtained from tryptic digestion.
Structure Determination by NMR Spectroscopy
Secondary αH shifts of the backbone protons were calculated using measured chemical shifts and random coil values.61,62 Peptides that have secondary αH shift values of <−0.1 ppm for three consecutive amino acids are likely to have a propensity to form an α-helix. In contrast, if consecutive values of >+0.1 are observed, the peptide sequence could consist of a β-strand structure. As illustrated in Figure 4, it is apparent that pase cyclotides contain a β-sheet motif as described previously for other Möbius cyclotides such as kalata B1. The αH signals of Gly11 (pase C), Asn15 (pase B–D), Asn16 (pase B–C), Ser22 (pase E), Trp23 (pase B and pase E), and Asn29 (pase C) were not observed at the temperature and pH tested. However, even in the absence of these values, there is no evidence for the presence of α-helical structure elements. This absence of helices is consistent with what is commonly found in most cyclotides. Detailed chemical shift data are reported in Tables S3–S7 (Supporting Information).
Figure 4.

Structural characterization of pase cyclotides. (A) Secondary αH shift comparison for pase A–E. The data are represented as a bar plot. A deviation of ±0.1 ppm shift from random coil values61 is indicated by a dashed line. Cysteines are highlighted with a yellow box. (B) Superposition of the 15 lowest energy structures of pase A and (C) cartoon representation of pase A (PDB ID: 7K7X, BMRB ID: 50464). Disulfide bonds are highlighted in yellow, and cysteine residue numbers labeled.
The configuration of proline residues was assigned based on comparison with αH and βH chemical shifts of kalata B1. Based on similar values for pase A–E, Pro24 was assigned as cis and Pro3 and Pro25 as trans. Thus, the NMR data and amino acid sequencing from mass spectrometry confirmed that pase A–E contain the unique CCK motif and are structurally similar to kalata B1.
3D Structure of Pase A
As the most abundant cyclotide in P. sessilis, the pase A structure was determined using torsion angle dynamics in the program CYANA, and the 15 lowest energy structures were chosen to represent the 3D fold. This run involved the calculation of 204 distance restraints, 50 dihedral angle restraints, and 8 pairs of hydrogen bonds (Table S8, Supporting Information). From these input data, the 15 lowest energy structures were calculated for pase A and showed that the major structural element is a β-hairpin involving residues 19–21 and 26–28 (Figure 4).
Amino Acid Diversity in Pase Cyclotides
Möbius cyclotides exhibit a wide sequence variation around the six cysteine residues and the conserved Pro residue in loop 5. To date, approximately 160 Möbius and 350 bracelet cyclotides have been reported in Cybase.10 All published Möbius cyclotides were compared to pase A–E cyclotides using Clustal Omega software,63 and a multiple sequence alignment was established (Figure S1, Supporting Information). Based on the results from the sequence logo, it is evident that Möbius cyclotides display a great degree of conservation in loops 1, 3, and 4, with loop 6 having the highest variability in size and sequence (Figure 5). Among all Möbius cyclotides reported hitherto, only seven (∼3%) have been reported to have a valine in loop 4, and herein an additional four cyclotides (i.e., pase A–D) are reported to contain this unusual structural feature. The absence of a hydroxy group in loop 4 and resulting loss of potential hydrogen bonding capability may lead to locally increased flexibility. While such changes may not be immediately apparent in the overall structure, effects on bioactivity can be significant. For example, kalata B12, a natural variant having a substitution of a highly conserved glutamic acid in loop 1, has an overall similar structure to kalata B1 but does not exhibit hemolytic activity.64 It is important to highlight that loops 1 and 4 are the backbone loops that form the ring of the cystine knot.5 Thus, changes in either structure or flexibility of these loops could significantly impact biological activities.
Figure 5.

Sequence Logo of cyclotides. (A) All Möbius cyclotides described in CyBase and (B) pase A–E and kalata S, showing the variability found in each amino acid position. Conserved cysteine residues are colored yellow, and conserved proline residues are shown in blue. The logos were generated using the application WebLogo version 2.8.2.65
The percentage of sequence identity was based on kalata B1 as the model Möbius cyclotide. For the top scoring full length matches, pase A showed 96.5% (S20 V) (serine position 20 of kalata B1 was replaced by valine in pase A), pase B showed 93.1% (S20 V, V25I), pase C (V4T, S20 V, V25I) and pase D showed 89.7% (S20 V, S22A, V25I), and pase E showed 82.8% (G11T, N15Y) sequence identity.63
Effects of Pase Cyclotides on Cell Proliferation of Primary Activated Human Lymphocytes
After we considered the high homology between the immunomodulatory cyclotide T20K43 and pase cyclotides, it was of interest to examine the bioactivity of these peptides on human immune cells. Thus, purified lymphocytes from the blood of healthy donors were treated with pase A–D. The results demonstrated a well-defined concentration-dependent proliferation inhibition for all tested cyclotides. As indicated in Figure 6, pase D has the strongest activity and pase C has the lowest immunosuppressive activity levels on human primary cells, denoted by IC50 values for pase A (4.5 ± 2.65 μM), pase B (2.3 ± 1.39 μM), pase C (7.1 ± 3.56 μM), and pase D (1.6 ± 1.78 μM). These data demonstrated similar potency to kalata B1 and its active analogs (including the clinical candidate cyclotide T20K),39,44 which have IC50 values between 1.9 and 4.4 μM.43
Figure 6.

Effect of pase cyclotides on proliferation of primary human lymphocytes. Human T lymphocytes were left unstimulated and untreated (negative control; NC); all other cells were stimulated with CD3/CD28 mAbs (100 ng/mL) and cultured with medium (positive control; PC), cyclosporin A (CsA; 5 μg/mL), camptothecin (CPT; 300 μM), and different concentrations (1–30 μM) of cyclotides. Cell division analyses were performed by flow cytometry after incubation for 72 h. Data are expressed as the means ± standard deviation (SD) of four independent experiments and were related to untreated stimulated control (PC = 100%). The asterisk (**p < 0.01, ***p < 0.001) represents significant differences from untreated stimulated cells; n.d. means not determined.
All tested cyclotides contain 8–11 hydrophobic amino acid residues (Ala, Val, Leu, Ile, Pro, Phe, and Trp) and have a similar ratio of these residues in the sequences. They also present a hydrophilic face, described previously as the “bioactive face” formed around the residue Glu7 and adjacent residues.45,66 For example, the natural mutation T20 V in pase A showed a higher IC50 value, but the concomitant mutations in T20 V and V25I (pase B) and T20 V, V25I, and S22A (pase D) showed increased inhibition of cell proliferation. Pase D was as active as T20K, which may point toward the importance of Ala22 for this activity. Pase C, with T20 V, V25I, and V4T mutations, lost considerable activity. This was expected, because residue 4 is a part of the hydrophobic face; an insertion of a polar residue such as threonine can produce a functional perturbation in the hydrophobic patch, which is important to membrane binding and consequently activity. This reinforces the notion that even minimal sequence changes can have significant influences on activity.
On comparing these results to previous investigations with kalata B1, T20K and natural mutants of the data demonstrate the potential of pase cyclotides for the development of new immunosuppressive drugs. Defined mode-of-action experiments must be performed to see if the pase cyclotides have a comparable impact on interleukin-2 signaling pathways as well as on cell function, as has been done in the past with the lead molecule T20K and native kalata B1.
Effects of Pase Cyclotides on Non-Immune Cells
Some cyclotides can have toxic activities, and hence, it is imperative to also assess their potential unspecific toxic and/or hemolytic effects. Thus, we investigated pase cyclotides on immune cells (i.e., PBMCs), red blood cells (RBCs), and umbilical vein endothelial cells (i.e., HUVECs) and cancer (i.e., HT-29) cells. All pase cyclotides were screened in a hemolytic assay along with the control peptide melittin (Table 2). The highest hemolytic activity was displayed by pase B, with a CC50 of 14.6 μM, whereas pase C showed the lowest hemolysis, with a CC50 of 62.8 μM. The reasons for the differences observed in hemolytic activity between all pase cyclotides are not clear, but it is possible that they are correlated to hydrophobicity. Based on the calculated GRAVY score obtained from the Expasy Web site (https://web.expasy.org/protparam/), pase B and pase C have GRAVY scores of 0.331 and 0.162, respectively. These two cyclotides differ only by one residue at position four (i.e., Val to Thr) in loop 6. A more positive GRAVY value indicates an increase in hydrophobicity.
Table 2. Cytotoxicity of HT-29, HUVEC in CC50 (μM) ± SD, and Hemolytic Activities of RBC in HC50 (μM) ± SD of Cyclotides against Different Cell Lines.
| cell
line; CC50 (μM) ± SDa |
|||
|---|---|---|---|
| cyclotide | HT-29 | HUVEC | RBC |
| pase A | 5.5 ± 0.04 | 5.2 ± 0.06 | 39.5 ± 0.02 |
| pase B | >10 | 7.5 ± 0.11 | 14.6 ± 0.02 |
| pase C | 8.4 ± 0.10 | >10 | 62.8 ± 0.06 |
| pase D | 6.1 ± 0.04 | 5.3 ± 0.06 | 33.5 ± 0.02 |
| pase E | 5.6 ± 0.10 | >10 | 51.7 ± 0.08 |
| kalata S | 7.3 ± 0.03 | >10 | 49.6 ± 0.02 |
| kalata B1 | >10 | 5.1 ± 0.08 | |
| melittin | 0.2 ± 0.01 | ||
Cytotoxic concentration required to kill 50% of the cells (CC50). Data represent the means ± standard deviation (SD) and have been normalized and analyzed using Graph Pad Prism, n = 3.
In general, Möbius cyclotides are less toxic compared to other subfamilies of cyclotides.67 The cytotoxicity data obtained for pase A–E with HT-29 cells and HUVEC endothelial cells showed CC50 values between 5 and 10 μM, approximately, showing a low toxicity and effectiveness to inhibit proliferation of lymphocytes.
In summary, a suite of cyclotides from P. sessilis including five previously uncharacterized sequences (pase A–E) and one known (kalata S)1,68 sequence are reported (Figure 7), increasing the total number of cyclotides from the genus Palicourea to 11. It was demonstrated that pase cyclotides can mitigate and stop the cell proliferation of lymphocytes, with low cytotoxicity and no discernible hemolytic effects at the tested concentrations. These results open new avenues to assay pase cyclotides in the future for use as immunosuppressive lead molecules.
Figure 7.

Sequence alignment of pase cyclotides isolated from the genus Palicourea, kalata B1, and T20K. Yellow boxes show the conserved cysteine residues; the connectivity between the disulfide bonds is shown by black lines, and backbone cyclization is represented by a blue line. *Isolated in this work.
Experimental Section
General Experimental Procedures
Analytical and semipreparative HPLC analyses were carried out on a Shimadzu (Tokyo, Japan) Prominence LC-20A instrument, with a LC-20AT pump, a DAD SPD-M20A detector, a SIL-20A autosampler, a CBM-20A controller and a DGU 20-A3 degassing unit, and a CTO-20A oven, with analytical and semipreparative Kromasil 300-5-SIL-C18 columns (250 mm × 4.6 mm and 250 mm × 10 mm, respectively). UV absorbance was recorded at 220 and 280 nm. Both analyses were performed using a linear gradient of 40–70% B in 15 min and 30–50% B in 80 min, respectively. Solvents are Buffer A (H2O/0.1% TFA) and Buffer B (90% CH3CN/0.08% TFA). The LC-MS traces were obtained on a Shimadzu (Tokyo, Japan) chromatograph coupled to an amaZon-SL ion trap Bruker Daltonics, LC-20AD solvent pump, CTO-20A column oven, DGU-20A3R online degasser, CBM-20A system controller, and SPD-M20A (190–800 nm) PDA detector and SIL-20A HT autosampler. MS and tandem MS analyses were performed in the positive-ion reflector mode on a 5800 Proteomics Analyzer (AB/Sciex, Foster City, CA, USA). The spectra were recorded for a mass window set to 1000–4500 Da using positive ion reflectron mode. To record NMR spectra, a Bruker ARX 600 (Karlsruhe, Germany) high-resolution NMR spectrometer with a shielded gradient unit was used. TOCSY and NOESY experiments were obtained with 80 and 200 ms mixing times, respectively. The water suppression was achieved using a WATERGATE (water suppression by gradient-tailored excitation) sequence.
Plant Material
Leaves and stem of P. sessilis were collected in Itatitaia (22°25′44.7″S 44°37′11.7″W), Rio de Janeiro, Brazil, during September 2015. The samples were collected by Dr. Marcelo Trovo and identified by Dr. Mario Gomes, and a voucher specimen has been deposited at Rio de Janeiro Botanical Garden with the collection number RB 640066, under harvest authorization SisGenAF5C371.
Extraction, Purification, and Isolation
A total of 175 and 360 g of dried and pulverized leaves and stems of P. sessilis, respectively, were extracted with 3000 mL of CH3OH/H2O (6:4, v/v) over 24 h at room temperature. The extracts were partitioned with CH2Cl2/CH3OH/H2O (1:1:1, v/v/v) (4 times), and the combined aqueous phases were concentrated on a rotary evaporator and lyophilized. The crude extracts were redissolved in CH3CN/H2O (1:9; v/v) and loaded onto Polygoprep 60–50 C18 silica gel reversed-phase (50 μm, 60 Å, 70 mg). The silica gel was activated with CH3OH and subsequently equilibrated with H2O/0.1% TFA (2 × 500 mL). After application of the extracts, the column was washed with 20 and 80% Buffer B. The 80% B fractions were considered peptide-rich fractions and named 80%-C18 leaves (1325 mg) and 80%-C18 stems (333 mg). The fraction of 80%-C18 stem (103 mg) was resuspended, filtered through a 0.45 μm membrane filter, loaded onto a RP-HPLC column, and separated using a linear gradient from 30 to 50% Buffer B in Buffer A over 80 min. Fractions were collected manually and lyophilized. Similarly, 80%-C18 leaves (198 mg) was resuspended in 3 mL of Buffer B and 12 mL of Buffer A. All obtained samples were analyzed by analytical HPLC, yielding six pure cyclotides pase A–E and kalata S (Table S1, Supporting Information), and subjected to enzymatic digestion.
LC-MS Analysis
The dried extract as well as peptide-rich fractions were resuspended in H2O/CH3CN/FA, 90:10:0.1% (v/v/v), and analyzed by RP-HPLC at 30 °C on Kromasil 300-5-SIL-C18 columns (250 mm × 4.6 mm i.d., 5 μm particle size, 300 Å pore size) at a flow rate of 1 mL/min with a linear gradient: 35–45% B in 30 min (A = H2O/0.1% FA and B = 90% CH3CN/0.1% FA). Absorbance was monitored on a PDA detector, set up at 220 and 280 nm. The mass spectra were obtained in the positive-ion mode with a mass range set to 400–2000 m/z. The mass spectrometer source parameters were as follows: capillary voltage at 3.0 kV. Nitrogen was used as the nebulizing and drying gas (7 psi, 4 L/min, 230 °C). Data were processed using Bruker Compass Data Analysis 4.1.
Sequencing of Palicourea Cyclotides
Cyclotides were characterized by manual de novo peptide sequencing using enzymatic digestion, MALDI-TOF/TOF mass spectrometry, and sequence comparison using CyBase tools (www.cybase.org.au). Data were analyzed in Data Explorer software Version 4.3. For acquisition, the dried compounds were dissolved in 0.1% TFA and mixed at a ratio of 1:1 (v/v) with a matrix solution of saturated α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich, St. Louis, MO, USA) in H2O/CH3CN/TFA, 50:50:0.1% (v/v/v). An aliquot (1 μL) of the sample/matrix mixture was directly spotted onto the MALDI target plate and air-dried. For sequencing, the peptides were reduced, alkylated, and digested using individual endoGlu-C, trypsin, and chymotrypsin enzymes, as described previously.43 The peptides obtained from enzymatic digestion were desalted using C18 ZipTips (Millipore, Billerica, MA, USA) and reconstituted in 80% Buffer B.
Secondary Structure Analysis of Pase A–E Using NMR
All peptides were dissolved in 550 μL (90% H2O/10% D2O) at pH ∼ 4.0, and 10 μL (1 mg/mL) of 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) was added as a chemical shift reference for spectral calibration. One-dimensional NMR (1H spectrum) and two-dimensional spectra (TOCSY and NOESY) were acquired. TOCSY and NOESY spectra were acquired with mixing times of 80 and 200 ms, respectively. All spectra were recorded on a Bruker Avance 600 MHz spectrometer at 298 K and were processed using TOPSIN 2.1 (Bruker) program. NMR data were analyzed using CCPNMR program version win32 2.3.1.54,69 and the sequences were assigned according to Wuthrich.70 The 3D structure was calculated from interproton distance restraints derived from cross-peaks in NOESY spectra. The cross-peaks were analyzed and integrated within the program CCPNMR, and structures were calculated with CYANA. Once a complete set of input restraints (distance and dihedral angle restraints) was determined using the program TALOS-N,71 structures were calculated with a simulated annealing protocol using CNS.72 A set of 50 structures was calculated, and the 15 lowest energy structures were selected for further analysis. The stereochemical quality of final structures was evaluated using MolProbity.73 The programs MolMol74 and PyMol75 were used to display the structural ensembles.
Hemolytic Assay
The cytotoxicity of cyclotides on red blood cells was evaluated through a hemolytic assay as described in the literature.76 Human red blood cells was collected from healthy donors following protocols approved by the Human Research Ethics Department at The University of Queensland. The approval number is 2013000582. Briefly, red blood cells were prepared by adding an aliquot of blood (4–5 drops) into Eppendorf tubes filled with 1 mL of phosphate-buffered saline (PBS) and mixed gently. The samples were centrifuged at 4000 rpm for 1 min, and the supernatant was carefully removed. An aliquot of suspended red blood cells was diluted in PBS to achieve a concentration of 0.25% (v/v). Peptide samples were prepared in water and serially diluted in a 96 well round-bottom plate obtaining a total of eight concentrations (0.187–24 μM). Melittin was used as a control peptide and prepared at the highest concentration of 20 μM. PBS and 1% Triton-X were used as negative and positive controls, respectively. Cells were incubated at 37 °C for one h in a final volume of 100 μL. The plate was then centrifuged at 1000 rpm, and the supernatant was transferred into a flat-bottom plate. The absorbance of the supernatants was measured at 415 nm. The percentage of cells hemolyzed was determined based on the log (agonist) vs response (variable slope), and the CC50 (peptide concentration required to lyse 50% of red blood cells) was calculated using GraphPad Prism software.
Cell Culture
Human umbilical vein endothelial cells (HUVECs) were cultured in EGM-2 BulletKit supplemented with SingleQuots (supplements = growth factors, cytokines, antibiotics; Lonza) and 10% FBS (catalog no. FFBS-500; Scientifix). Human colorectal adenocarcinoma cells (HT-29) were cultured in 10% FBS/DMEM (Dulbecco’s Modified Eagle Medium) with 1% penicillin–streptomycin (5000 U/mL; Life Technologies). The cells were maintained at 37 °C in a humidified incubator with a 5% CO2/95% air atmosphere.
Isolation of Human Peripheral Blood Mononuclear Cells (PBMCs)
Blood of healthy adult donors were obtained from the Blood Transfusion Centre (University Medical Centre Freiburg, Freiburg, Germany), and all experiments conducted on human material were approved by the ethics committee of the University of Freiburg (55/14). PBMC were isolated by centrifuging venous blood on a LymphoPrep gradient (density = 1.077 g/cm3, 20 min, 500 × g, 20 °C; Progen, Heidelberg, Germany). Cells were washed twice and resuspended in cold phosphate buffer saline (PBS; GE Healthcare, Munich, Germany) and used directly. The cell viability as well as concentration were determined using the trypan blue exclusion test.
Analysis of Proliferation Capacity of Activated Lymphocytes by CFSE Staining
To perform carboxyfluorescein diacetate succinimidyl ester (CFSE; 5 mM; Sigma-Aldrich, St. Louis, MO, USA) staining to determine T cell proliferation, 5 × 106 cells/mL were stained with CFSE (1:1000 dilution) and incubated for 10 min at 37 °C. Cells were washed twice with complete medium to stop the staining reaction. Afterward, stained cells were stimulated with antihuman CD3 (clone OKT3) and antihuman CD28 (clone 28.6) mAbs (each 100 ng/mL; both from eBioscience, Frankfurt, Germany). Furthermore, stimulated cells were treated with medium alone (positive control; PC), cyclosporin A (CsA; 5 μg/mL, purity ≥ 99%, Sandimmun 50 mg/mL, Novartis Pharma, Basel, Switzerland), camptothecin (CPT; 300 μM; purity > 98%; Tocris, Bristol, UK), or in the presence of different concentrations (1–30 μM) of cyclotides (pase A–D) for 72 h at 37 °C in a humidified incubator with a 5% CO2/95% air atmosphere. Cell division progress was analyzed by flow cytometric analysis using a FACS Calibur analyzer (BD Bioscience, Becton Dickinson, Franklin Lakes, NJ, USA). Data were analyzed using FlowJo software. Data were processed with Microsoft Excel and SPSS software (IBM, Version 22.0, Armonk, NY, USA). Values are represented as means ± standard deviation (SD) for the indicated number of independent experiments. Statistical significance was determined by a one-way ANOVA followed by Dunnett’s post hoc pairwise comparisons. Asterisks (*p < 0.05, **p < 0.01, ***p < 0.001) represent significant differences from the respective control.
MTT Assay on HUVECs and Human Colorectal Adenocarcinoma HT-29 Cells
Cell cytotoxicity assays were performed using similar methods to those described previously.77 For both cell lines, 5.0 × 103 cells/well (100 μL) were used. All peptides including kalata B1 (as a control) were used in both assays (10 μL, at final concentrations ranging from 0.78–100 μM). Triton X-100 (0.1% (v/v); 10 μL) was used as a positive control. Cells were treated with fresh medium the day after plating, before the addition of peptides, followed by a subsequent 2 and 48 h incubation for cell cytotoxicity. After different incubation periods, 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) (10 μL; 5 mg/mL in PBS) was added, and cells were incubated for a further 3 h. The supernatant was then removed, and 100 μL of DMSO was added to solubilize formazan salts. Experiments were performed in triplicate. Cell numbers were recorded at 600 nm. Results were evaluated with log (Inhibitor) vs normalized response (variable slope), using GraphPad Prism Version 6 software, for means ± SD from triplicate experiments.
Acknowledgments
The authors are thankful to grant no. 2017/17098-4 and grant no. 2019/04381-5, São Paulo Research Foundation (FAPESP), for scholarships to M.E.F.P. and to CEPID-FAPESP grant no. 2013/07600-3, and INCT-CNPq grant no. 465637/2014-0, for financial support. This work was supported by Australian Research Council grant DP150100443 and by access to the facilities of the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012). D.J.C. is an Australian Research Council Australian Laureate Fellow (FL150100146). L.Y.C. was supported by the Advance Queensland Women’s Academic Fund (WAF-6884942288). C.W.G. is supported by the Austrian Science Fund (FWF) through project P32109.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.0c01069.
Obtained amounts of pase cyclotides, sequence fragments generated through enzymatic digestion and characterization using MALDI-TOF/TOF-MS/MS, 1H chemical shifts (ppm) of all pase cyclotides, and multiple sequence alignment of Möbius cyclotides (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): C.W.G. is shareholder and chief scientific advisor of Cyxone AB.
Supplementary Material
References
- Craik D.; Daly N.; Bond T.; Waine C. J. Mol. Biol. 1999, 294, 1327–1336. 10.1006/jmbi.1999.3383. [DOI] [PubMed] [Google Scholar]
- Göransson U.; Burman R.; Gunasekera S.; Strömstedt A. A.; Rosengren K. J. J. Biol. Chem. 2012, 287, 27001–27006. 10.1074/jbc.R111.300129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik D.; Anderson M.; Barry D.; Clark R.; Daly N.; Jennings C.; Mulvenna J. Lett. Pept. Sci. 2001, 8, 119–128. 10.1007/BF02446507. [DOI] [Google Scholar]
- Colgrave M. L.; Craik D. J. Biochemistry 2004, 43, 5965–5975. 10.1021/bi049711q. [DOI] [PubMed] [Google Scholar]
- Rosengren K. J.; Daly N. L.; Plan M. R.; Waine C.; Craik D. J. J. Biol. Chem. 2003, 278, 8606–8616. 10.1074/jbc.M211147200. [DOI] [PubMed] [Google Scholar]
- Hernandez J. F.; Gagnon J.; Chiche L.; Nguyen T. M.; Andrieu J. P.; Heitz A.; Trinh Hong T.; Pham T. T.; Le Nguyen D. Biochemistry 2000, 39, 5722–5730. 10.1021/bi9929756. [DOI] [PubMed] [Google Scholar]
- Felizmenio-Quimio M. E.; Daly N. L.; Craik D. J. J. Biol. Chem. 2001, 276, 22875–22882. 10.1074/jbc.M101666200. [DOI] [PubMed] [Google Scholar]
- Camarero J. A.; Campbell M. J. Biomedicines 2019, 7, 31. 10.3390/biomedicines7020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland D. C.; Colgrave M. L.; Craik D. J. Biochem. J. 2006, 400, 1–12. 10.1042/BJ20060627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. K. L.; Kaas Q.; Chiche L.; Craik D. J. Nucleic Acids Res. 2007, 36, D206–D210. 10.1093/nar/gkm953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saether O.; Craik D. J.; Campbell I. D.; Sletten K.; Juul J.; Norman D. G. Biochemistry 1995, 34, 4147–4158. 10.1021/bi00013a002. [DOI] [PubMed] [Google Scholar]
- Göransson U.; Craik D. J. J. Biol. Chem. 2003, 278, 48188–48196. 10.1074/jbc.M308771200. [DOI] [PubMed] [Google Scholar]
- Gustafson K. R.; Sowder R. C. I.; Henderson L. E.; Parsons I. C.; Kashman Y.; Cardellina J. H. I.; McMahon J. B.; Buckheit R. W. J.; Pannell L. K.; Boyd M. R. J. Am. Chem. Soc. 1994, 116, 9337–9338. 10.1021/ja00099a064. [DOI] [Google Scholar]
- Daly N. L.; Koltay A.; Gustafson K. R.; Boyd M. R.; Casas-Finet J. R.; Craik D. J. J. Mol. Biol. 1999, 285, 333–345. 10.1006/jmbi.1998.2276. [DOI] [PubMed] [Google Scholar]
- Hallock Y. F.; Sowder R. C. I.; Pannell L. K.; Hughes C. B.; Johnson D. G.; Gulakowski R.; Cardellina J. H.; Boyd M. R. J. Org. Chem. 2000, 65, 124–128. 10.1021/jo990952r. [DOI] [PubMed] [Google Scholar]
- Pinto M. E. F.; Najas J. Z. G.; Magalhães L. G.; Bobey A. F.; Mendonça J. N.; Lopes N. P.; Leme F. M.; Teixeira S. P.; Trovó M.; Andricopulo A. D.; Koehbach J.; Gruber C. W.; Cilli E. M.; Bolzani V. S. J. Nat. Prod. 2018, 81, 1203–1208. 10.1021/acs.jnatprod.7b00969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherup K. M.; Bogusky M. J.; Anderson P. S.; Ramjit H.; Ransom R. W.; Wood T.; Sardana M. J. Nat. Prod. 1994, 57, 1619–1625. 10.1021/np50114a002. [DOI] [PubMed] [Google Scholar]
- Fahradpour M.; Keov P.; Tognola C.; Perez-Santamarina E.; McCormick P. J.; Ghassempour A.; Gruber C. W. Front. Pharmacol. 2017, 8, 616. 10.3389/fphar.2017.00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gran L. Lloydia 1973, 36, 174–178. 10.4043/1817-MS. [DOI] [PubMed] [Google Scholar]
- Grain L. Lloydia 1973, 36, 207–208. [PubMed] [Google Scholar]
- Koehbach J.; O’Brien M.; Muttenthaler M.; Miazzo M.; Akcan M.; Elliott A. G.; Daly N. L.; Harvey P. J.; Arrowsmith S.; Gunasekera S.; Smith T. J.; Wray S.; Göransson U.; Dawson P. E.; Craik D. J.; Freissmuth M.; Gruber C. W. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 21183–21188. 10.1073/pnas.1311183110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgrave M. L.; Kotze A. C.; Huang Y. H.; O’Grady J.; Simonsen S. M.; Craik D. J. Biochemistry 2008, 47, 5581–5589. 10.1021/bi800223y. [DOI] [PubMed] [Google Scholar]
- Colgrave M. L.; Kotze A. C.; Ireland D. C.; Wang C. K.; Craik D. J. ChemBioChem 2008, 9, 1939–1945. 10.1002/cbic.200800174. [DOI] [PubMed] [Google Scholar]
- Colgrave M. L.; Kotze A. C.; Kopp S.; McCarthy J. S.; Coleman G. T.; Craik D. J. Acta Trop. 2009, 109, 163–166. 10.1016/j.actatropica.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Jennings C. V.; Rosengren K. J.; Daly N. L.; Plan M.; Stevens J.; Scanlon M. J.; Waine C.; Norman D. G.; Anderson M. A.; Craik D. J. Biochemistry 2005, 44, 851–860. 10.1021/bi047837h. [DOI] [PubMed] [Google Scholar]
- Jennings C.; West J.; Waine C.; Craik D.; Anderson M. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 10614–10619. 10.1073/pnas.191366898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeta B. L.; Marshall A. T.; Gillon A. D.; Craik D. J.; Anderson M. A. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1221–1225. 10.1073/pnas.0710338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plan M. R.; Saska I.; Cagauan A. G.; Craik D. J. J. Agric. Food Chem. 2008, 56, 5237–5241. 10.1021/jf800302f. [DOI] [PubMed] [Google Scholar]
- Lindholm P.; Gullbo J.; Claeson P.; Goransson U.; Johansson S.; Backlund A.; Larsson R.; Bohlin L. J. Biomol. Screening 2002, 1, 365–369. 10.1177/108705710200700405. [DOI] [PubMed] [Google Scholar]
- Svangård E.; Göransson U.; Hocaoglu Z.; Gullbo J.; Larsson R.; Claeson P.; Bohlin L. J. Nat. Prod. 2004, 67, 144–147. 10.1021/np030101l. [DOI] [PubMed] [Google Scholar]
- Herrmann A.; Burman R.; Mylne J.; Karlsson G.; Gullbo J.; Craik D.; Clark R.; Goransson U. Phytochemistry 2008, 69, 939–952. 10.1016/j.phytochem.2007.10.023. [DOI] [PubMed] [Google Scholar]
- Tam J. P.; Lu Y. A.; Yang J. L.; Chiu K. W. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 8913–8918. 10.1073/pnas.96.16.8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pranting M.; Loov C.; Burman R.; Goransson U.; Andersson D. J. Antimicrob. Chemother. 2010, 65, 1964–1971. 10.1093/jac/dkq220. [DOI] [PubMed] [Google Scholar]
- Schöpke T.; Hasan Agha M. I.; Kraft R.; Otto A.; Hiller K. Sci. Pharm. 1993, 61, 145–153. [Google Scholar]
- Chen B.; Colgrave M. L.; Wang C.; Craik D. J. J. Nat. Prod. 2006, 69, 23–28. 10.1021/np050317i. [DOI] [PubMed] [Google Scholar]
- Hellinger R.; Koehbach J.; Puigpinos A.; Clark R. J.; Tarrago T.; Giralt E.; Gruber C. W. J. Nat. Prod. 2015, 78, 1073–1082. 10.1021/np501061t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thell K.; Hellinger R.; Schabbauer G.; Gruber C. W. Drug Discovery Today 2014, 19, 645–653. 10.1016/j.drudis.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellinger R.; Koehbach J.; Fedchuk H.; Sauer B.; Huber R.; Gruber C. W.; Gründemann C. J. Ethnopharmacol. 2014, 151, 299–306. 10.1016/j.jep.2013.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründemann C.; Stenberg K. G.; Gruber C. W. Int. J. Pept. Res. Ther. 2019, 25, 9–13. 10.1007/s10989-018-9701-1. [DOI] [Google Scholar]
- Yamout B. I.; Alroughani R. Semin Neurol. 2018, 38, 212–225. 10.1055/s-0038-1649502. [DOI] [PubMed] [Google Scholar]
- Multiple Sclerosis International Federation Atlas of MS Database https://www.msif.org/about-ms/what-is-ms/ (accessed 2020-05-05).
- Selter R. C.; Hemmer B. ImmunoTargets Ther. 2013, 2, 21–30. 10.2147/ITT.S31813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründemann C.; Thell K.; Lengen K.; Garcia-Käufer M.; Huang Y.-H.; Huber R.; Craik D. J.; Schabbauer G.; Gruber C. W. PLoS One 2013, 8, e68016 10.1371/journal.pone.0068016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thell K.; Hellinger R.; Sahin E.; Michenthaler P.; Gold-Binder M.; Haider T.; Kuttke M.; Liutkevičiutė Z.; Göransson U.; Gründemann C.; Schabbauer G.; Gruber C. W. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 3960–3965. 10.1073/pnas.1519960113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. H.; Colgrave M. L.; Clark R. J.; Kotze A. C.; Craik D. J. J. Biol. Chem. 2010, 285, 10797–10805. 10.1074/jbc.M109.089854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura H. N.; Poth A. G.; Yendo A. C. A.; Fett-Neto A. G.; Craik D. J. J. Nat. Prod. 2016, 79, 3006–3013. 10.1021/acs.jnatprod.6b00492. [DOI] [PubMed] [Google Scholar]
- Bernardino K.; Pinto M. E. F.; Bolzani V. S.; de Moura A. F.; Batista Junior J. M. Chem. Commun. 2017, 53, 7337–7340. 10.1039/C7CC02333B. [DOI] [PubMed] [Google Scholar]
- Bobey A. F.; Pinto M. E. F.; Cilli E. M.; Lopes N. P.; Bolzani V. S. Planta Med. 2018, 84, 947–952. 10.1055/a-0632-2204. [DOI] [PubMed] [Google Scholar]
- Pinto M. F. S.; Fensterseifer I. C. M.; Migliolo L.; Sousa D. A.; de Capdville G.; Arboleda-Valencia J. W.; Colgrave M. L.; Craik D. J.; Magalhaes B. S.; Dias S. C.; Franco O. L. J. Biol. Chem. 2012, 287, 134–147. 10.1074/jbc.M111.294009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha N. B. d.; Barbosa A. E. A. d. D.; de Almeida R. G.; Porto W. F.; Maximiano M. R.; Álvares L. C. S.; Munhoz C. B. R.; Eugênio C. U. O.; Viana A. A. B.; Franco O. L.; Dias S. C. Biopolymers 2016, 106, 784–795. 10.1002/bip.22938. [DOI] [PubMed] [Google Scholar]
- Pinto M. F. S.; Silva O. N.; Viana J. C.; Porto W. F.; Migliolo L.; da Cunha N. B.; Gomes N.; Fensterseifer I. C. M.; Colgrave M. L.; Craik D. J.; Dias S. C.; Franco O. L. J. Nat. Prod. 2016, 79, 2767–2773. 10.1021/acs.jnatprod.6b00270. [DOI] [PubMed] [Google Scholar]
- Hellinger R.; Koehbach J.; Soltis D. E.; Carpenter E. J.; Wong G. K.-S.; Gruber C. W. J. Proteome Res. 2015, 14, 4851–4862. 10.1021/acs.jproteome.5b00681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber C. W.; Elliott A. G.; Ireland D. C.; Delprete P. G.; Dessein S.; Göransson U.; Trabi M.; Wang C. K.; Kinghorn A. B.; Robbrecht E.; Craik D. J. Plant Cell 2008, 20, 2471–2483. 10.1105/tpc.108.062331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C. M. Novon 2015, 24, 55–95. 10.3417/2015001. [DOI] [Google Scholar]
- Bokesch H. R.; Pannell L. K.; Cochran P. K.; Sowder R. C. 2nd; McKee T. C.; Boyd M. R. J. Nat. Prod. 2001, 64, 249–250. 10.1021/np000372l. [DOI] [PubMed] [Google Scholar]
- Moreno B. P.; Ricardo Fiorucci L. L.; do Carmo M. R. B.; Sarragiotto M. H.; Baldoqui D. C. Biochem. Syst. Ecol. 2014, 56, 80–82. 10.1016/j.bse.2014.04.013. [DOI] [Google Scholar]
- Klein-Júnior L. C.; Cretton S.; Allard P.-M.; Genta-Jouve G.; Passos C. S.; Salton J.; Bertelli P.; Pupier M.; Jeannerat D.; Heyden Y. V.; Gasper A. L.; Wolfender J.-L.; Christen P.; Henriques A. T. J. Nat. Prod. 2017, 80, 3032–3037. 10.1021/acs.jnatprod.7b00681. [DOI] [PubMed] [Google Scholar]
- Koehbach J.; Attah A. F.; Berger A.; Hellinger R.; Kutchan T. M.; Carpenter E. J.; Rolf M.; Sonibare M. A.; Moody J. O.; Wong G. K.-S.; Dessein S.; Greger H.; Gruber C. W. Biopolymers 2013, 100, 438–452. 10.1002/bip.22328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabi M.; Craik D. J. Plant Cell 2004, 16, 2204–2216. 10.1105/tpc.104.021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabi M.; Svangard E.; Herrmann A.; Göransson U.; Claeson P.; Craik D. J.; Bohlin L. J. Nat. Prod. 2004, 67, 806–810. 10.1021/np034068e. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D.; Richards F. M. Biochemistry 1992, 31, 1647–1651. 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- Merutka G.; Dyson H. J.; Wright P. E. J. Biomol. NMR 1995, 5, 14–24. 10.1007/BF00227466. [DOI] [PubMed] [Google Scholar]
- Madeira F.; Park Y. M.; Lee J.; Buso N.; Gur T.; Madhusoodanan N.; Basutkar P.; Tivey A. R. N.; Potter S. C.; Finn R. D.; Lopez R. Nucleic Acids Res. 2019, 47, W636–W641. 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. K.; Clark R. J.; Harvey P. J.; Rosengren K. J.; Cemazar M.; Craik D. J. Biochemistry 2011, 50, 4077–4086. 10.1021/bi2004153. [DOI] [PubMed] [Google Scholar]
- Crooks G. E.; Hon G.; Chandonia J. M.; Brenner S. E. Genome Res. 2004, 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen S. M.; Sando L.; Rosengren K. J.; Wang C. K.; Colgrave M. L.; Daly N. L.; Craik D. J. J. Biol. Chem. 2008, 283, 9805–9813. 10.1074/jbc.M709303200. [DOI] [PubMed] [Google Scholar]
- Burman R.; Herrmann A.; Tran R.; Kivela J. E.; Lomize A.; Gullbo J.; Göransson U. Org. Biomol. Chem. 2011, 9, 4306–4314. 10.1039/c0ob00966k. [DOI] [PubMed] [Google Scholar]
- Claeson P.; Göransson U.; Johansson S.; Luijendijk T.; Bohlin L. J. Nat. Prod. 1998, 61, 77–81. 10.1021/np970342r. [DOI] [PubMed] [Google Scholar]
- Vranken W. F.; Boucher W.; Stevens T. J.; Fogh R. H.; Pajon A.; Llinas M.; Ulrich E. L.; Markley J. L.; Ionides J.; Laue E. D. Proteins: Struct., Funct., Genet. 2005, 59, 687–696. 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Wüthrich K.NMR of Proteins and Nucleic Acids; Wiley-Interscience: New York, 1986; p 292. [Google Scholar]
- Shen Y.; Bax A. J. Biomol. NMR 2013, 56, 227–241. 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A. T. Nat. Protoc. 2007, 2, 2728–2733. 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi R.; Billeter M.; Wüthrich K. J. Mol. Graphics 1996, 14, 51–55. 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.PyMOL: An Open-Source Molecular Graphics Tool, 2002. [Google Scholar]
- Chan L. Y.; Wang C. K.; Major J. M.; Greenwood K. P.; Lewis R. J.; Craik D. J.; Daly N. L. J. Nat. Prod. 2009, 72, 1453–1458. 10.1021/np900174n. [DOI] [PubMed] [Google Scholar]
- Chan L. Y.; Craik D. J.; Daly N. L. Sci. Rep. 2016, 6, 35347. 10.1038/srep35347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.