

SUMMARY
Biological processes are regulated by intermolecular interactions and chemical modifications that do not affect protein levels, thus escaping detection in classical proteomic screens. We demonstrate here that a global protein structural readout based on limited proteolysis-mass spectrometry (LiP-MS) detects many such functional alterations, simultaneously and in situ, in bacteria undergoing nutrient adaptation and in yeast responding to acute stress. The structural readout, visualized as structural barcodes, captured enzyme activity changes, phosphorylation, protein aggregation, and complex formation, with the resolution of individual regulated functional sites such as binding and active sites. Comparison with prior knowledge, including other ‘omics data, showed that LiP-MS detects many known functional alterations within well-studied pathways. It suggested distinct metabolite-protein interactions and enabled identification of a fructose-1,6-bisphosphate-based regulatory mechanism of glucose uptake in E. coli. The structural readout dramatically increases classical proteomics coverage, generates mechanistic hypotheses, and paves the way for in situ structural systems biology.
Keywords: structural proteomics, mass spectrometry, limited proteolysis, structural systems biology, protein aggregation, functional proteomics, metabolism, structural biology, yeast, E. coli
Graphical Abstract
Highlights
-
•
Dynamic structural proteomic screens detect functional changes at high resolution
-
•
Detect enzyme activity, phosphorylation, and molecular interactions in situ
-
•
Generate new molecular hypotheses and increase functional proteomics coverage
-
•
Enabled discovery of a regulatory mechanism of glucose uptake in E. coli
Introduction
Quantitative mass-spectrometry-based proteomics is used to profile proteome expression across different conditions (Aebersold and Mann, 2003). This approach has identified pathways regulated during cellular perturbations and disease development and has uncovered mechanisms of drug action and resistance (Boisvert et al., 2010; Costenoble et al., 2011; Ideker et al., 2001; Kolkman et al., 2006; Mertins et al., 2016; Ressa et al., 2018). However, many molecular events that result in protein functional changes do not involve changes in protein abundance. Proteins might undergo functional changes upon post-translational modification (PTM) (Ardito et al., 2017), binding to other molecules (Chubukov et al., 2014; Niphakis et al., 2015; Nussinov et al., 2013; Sahni et al., 2013), cleavage (Russell, 2014), or conformational changes induced by environmental changes (e.g., pH or temperature) (Damaghi et al., 2013; Robertson and Murphy, 1997). The regulation of many cellular processes, such as signaling cascades (Kolch, 2005; Shaul and Seger, 2007), relies solely on these types of events rather than on altered protein levels. Variants of the proteomics workflow such as phosphoproteomics (Batth et al., 2018; Humphrey et al., 2015), interactomics (Sowa et al., 2009; Wepf et al., 2009), and activity-based proteomics (Cravatt et al., 2008) can capture specific molecular events that affect protein function but typically report on only a single type of mechanism. The high-throughput, simultaneous analysis of diverse regulatory events on a proteome-wide scale is not practically feasible.
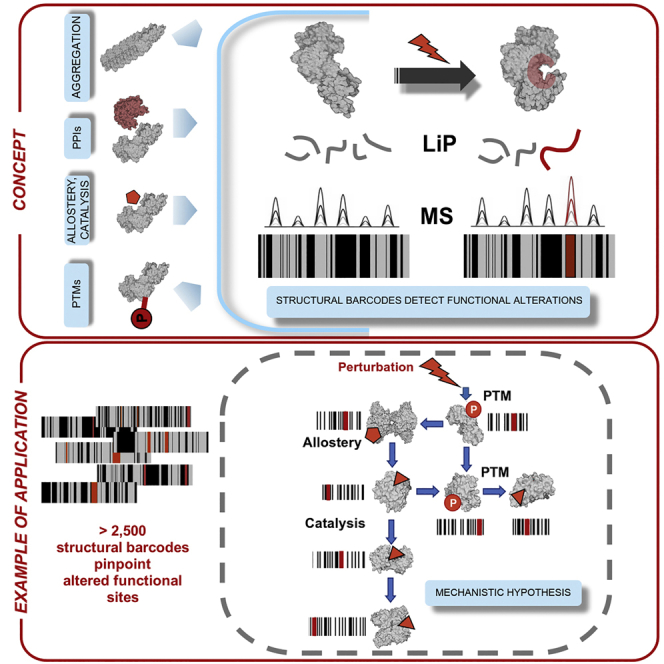
We speculated that global analysis of protein structures could serve as a quantitative readout to capture most events that alter protein functional states. It is dogma that the structure of a protein is intimately linked to its function (Pauling and Itano, 1949; White and Anfinsen, 1959). Protein structures integrate different types of molecular cues that result in functional alterations: binding of other molecules, protein-protein interactions, post-translational modifications (PTMs), mutations, aggregation, and conformational alterations due to changes in the cellular matrix all result in local or global structural alterations of proteins. We hypothesized that, by measuring altered protein structural states on a proteome-wide scale, we could detect protein functional changes of various types simultaneously, yielding a more detailed and nuanced picture than measurement of abundance changes alone.
We previously developed limited proteolysis-coupled mass spectrometry (LiP-MS) to monitor protein structural changes directly within complex biological extracts and on a proteome-wide scale (Feng et al., 2014). Comparison of structure-specific proteolytic fingerprints from different conditions identifies structurally altered proteins and can pinpoint structurally altered regions. LiP-MS and other structural proteomics approaches have been used in numerous protein structural studies (Aebersold and Mann, 2016; Huber et al., 2015; Leuenberger et al., 2017; Liu et al., 2018; Piazza et al., 2018; Savitski et al., 2014).
Here we test the idea that LiP-MS, and a global structural readout in general, can monitor functional changes, focusing on bacterial cells undergoing nutrient adaptation and yeast cells responding to acute stress. This structural approach captured enzyme activity changes, enzymatic substrate site occupancy, allosteric regulation, phosphorylation, and protein-protein interactions with a resolution that pinpoints single functional sites, thereby driving the generation of molecular hypotheses. We showed that LiP-MS detects a greater number of altered biological processes than do protein abundance measurements alone and captures information overlapping and complementary to metabolomics, flux analyses, and phosphoproteomics. We validated the interaction between an E. coli sugar phosphotransferase and the metabolite fructose-1,6-bisphosphate (FBP), suggesting a previously uncharacterized regulatory system of glucose uptake. In sum, this global structural approach reports on many functional events in situ and constitutes a powerful readout to detect molecular events underlying physiological and pathological phenotypes.
Results
Protein structural changes during the yeast response to acute stress
We used multiple experimental systems to test whether global protein structural data can detect functional alterations of proteins and protein networks (Figure 1A). First, we studied cellular responses activated on short timescales in yeast, which are typically independent of gene expression changes and thus less amenable to protein abundance screens. We applied a short osmotic or heat stress to exponentially growing yeast cultures, extracted the proteomes under native conditions, and applied the LiP-MS workflow, which monitors in parallel protein abundance and structural changes. We analyzed the resulting peptide mixtures by data-independent acquisition followed by label-free quantification and corrected data from LiP experiments for protein abundance changes to yield structure-specific proteolytic fingerprints for every detectable protein. We detected structural fingerprints for more than 2,700 proteins and monitored abundance changes for a similar number of proteins (Figure S1A; Table S1). Only 1% or less of the detected proteins varied in abundance upon stimulation (Figure 1B; Table S1), consistent with previous studies (Jarnuczak et al., 2018; Kanshin et al., 2015; Mackenzie et al., 2016; Soufi et al., 2009; Storey et al., 2020; Wallace et al., 2015). In contrast, 23% and 11% of the detected proteomes underwent structural alterations upon heat shock and osmotic shock, respectively (Figure 1B; Table S1; Table S2 for sequence coverage). Peptide intensities showed excellent correlation across replicates (Figures S2A–S2D) and replicates clustered by condition (data not shown); independent quality control (QC) analyses confirmed reproducibility (Figures S2E and S2F).
Figure 1.

Global protein structural and abundance changes during cellular responses in yeast and E. coli
(A) The experimental systems used in this work. We studied E. coli grown on eight different nutrient sources and yeast subjected to acute heat or osmotic shock. We monitored protein abundance and structural changes with LiP-MS and assessed the functional information content of both readouts.
(B) The number of proteins significantly changed (|log2FC| >1, q-value < 0.05) in abundance (green) or structure (yellow) in yeast subjected to heat shock or osmotic stress (two-sample t test with Storey method correction for multiple testing).
(C) Heat map of GO biological processes enriched among significantly changed proteins in yeast subjected to heat shock or osmotic stress. p values for the enrichment (gray scale) were determined with Fisher’s exact tests. Blank cells indicate biological processes that were not enriched significantly (i.e., p value > 0.01). Red and blue indicate categories expected to be enriched under heat and osmotic shock, respectively.
Figure S1.

Proteomic coverage and growth rate of bacteria in this study, and functional analysis of proteins that show structural and abundance changes during yeast response to acute stress, related to Figure 1
(A) The plot shows the number of yeast proteins detected by LC/MS-MS after digestion with trypsin only (which measures protein abundance, green bars) or upon limited proteolysis (which identifies structure-specific peptides, yellow bars), after the indicated stresses. (B) The plot shows the number of E. coli proteins detected by LC/MS-MS after digestion with trypsin only (which measures protein abundance, green bars) or upon limited proteolysis (which identifies structure-specific peptides, yellow bars) under the indicated conditions. (C) The plot shows the doubling time of E. coli in the seven different nutrient conditions used in this study. (D) The heat maps show functional categories (GO Biological Processes, Molecular Functions or Cellular Components) enriched among proteins that significantly change (|log2FC| >1, q-value < 0.05) in abundance (green) or structure (yellow) under the indicated stress conditions. P values for the enrichment (gray scale) were determined using Fisher’s exact test. Blank cells indicate molecular functions or cellular components that were not significantly enriched (i.e with p-value > 0.01) in a given condition. For the heat map showing Biological Processes, p-values were corrected for multiple hypothesis testing with the Benjamini-Hochberg method; blank cells indicate biological processes that were not significantly enriched (i.e with q-value > 0.05) in a given condition.
Figure S2.

Tests of LiP-MS reproducibility, related to STAR Methods
Correlation of replicate LiP-MS data sets in yeast responding to stress. (A-B) Reproducibility of the data set in yeast responding to osmotic stress. Correlation matrix of LiP peptide intensities between control conditions (C1-C3) and osmotic stress conditions (OS1-OS3; 10 min in 0.4M NaCl) after limited proteolysis (A) or in the trypsin-only control (B). (C-D) Reproducibility of the heat stress data set. Correlation matrix of LiP peptide intensities between control conditions (C1-C4) and heat stress conditions (HS1-HS4; 3 min at 42 degrees) after limited proteolysis (C) or in the trypsin-only control (D). The color scale indicates the Pearson correlation coefficient. (E-F) LiP-MS reproducibility across operators and replicates. LiP-MS experiments were conducted on unperturbed S. cerevisiae lysates by two different operators and in three replicates each. Shown is a principal component analysis of peptide intensities in all eight replicates, colored by operator (E). (F) The differential analysis shows the number of changing peptides between the two operators. 5 out of 16924 detected peptides change significantly (|log2FC| >1, q-value < 0.05).
Heat shock in yeast results in protein misfolding, activation of quality control mechanisms, translation inhibition, and formation of stress granules and protein aggregates (Verghese et al., 2012; Wallace et al., 2015). Osmotic stress activates the high omolarity glycerol (HOG) pathway and mitogen-activated protein (MAP) kinases, inducing allosteric events and flux alterations that lead to fast cytosolic accumulation of the osmoprotectant glycerol (Figure 2) (Brewster and Gustin, 2014; Hohmann, 2015). To ask whether the structural readout captured activation of these known processes, we performed a functional enrichment analysis of structurally altered proteins. For both perturbations, we found an enrichment of glucose metabolic pathways and translation (Figure 1C; Table S1) and of cytoplasmic stress granules and general cytosolic cellular components (Figure S1D). As expected, several categories related to regulation of translation and to protein folding, misfolding, and refolding were enriched specifically in the heat-stress dataset (Figures 1C and S1D; Table S1). Compatible with the known increase of glycerol production under osmotic stress, we detected enrichment of the “glycerol metabolic process” and the “NADPH regeneration” and “NADH metabolism” biological processes only after osmotic stress (Figure 1C); these gene ontology (GO) categories include glycerol biosynthetic enzymes. “Response to osmotic stress” was unexpectedly enriched in the heat-shock condition, probably because most altered proteins within this GO term are heat-shock proteins and proteins altered upon different stresses. Functional enrichment analysis of protein abundance data showed enrichment of very few GO terms, likely due to the low number of proteins that change abundance (Figure 1C).
Figure 2.

Structural changes capture multiple regulatory events in yeast responding to osmotic shock
A schematic of the yeast HOG-MAPK pathway and its links to glycolytic and glycerol biosynthesis pathways. Proteins undergoing significant structural alterations upon osmotic shock are indicated with orange labels (|log2FC| >1, q-value < 0.05, two-sample t test with Storey method correction for multiple testing). The barcodes represent the changes in proteolytic fingerprints from N to C terminus. Each vertical bar represents a peptide that could be detected in samples subject to LiP. Peptides that changed in intensity between conditions are indicated by yellow (|log2FC| >1, q-value < 0.05), peptides detected by MS but unchanged between conditions are in gray, and peptides not detected by MS are in black. The structural models show changed LiP peptides (orange) mapped onto the 3D protein structures of yeast protein-metabolite complexes or evolutionary conserved holo-complex structures obtained by homology modeling; metabolites positioned in allosteric or active sites are indicated in green. For Hog1 and Gpd1, phosphorylation sites are indicated in blue on protein sequences. The allosteric regulator Fructose 2, 6-bisphosphate (F2,6bP), is depicted in red. The models shown are based on available structures: Pfk1 (PDB: 3o8o), Fba1 (PDB: 3qm3), Ste20 (PDB: 4zlo), Hog1 (PDB: 5ci6), Tpi1 (PDB: 1nf0), Gpd1 (PDB: 6e9o), Gpp1 (PDB: 2qlt), Tdh2 (PDB: 3pym), Pgk1 (PDB: 1qpg), Gpm (PDB: 1qhf), Eno2 (PDB: 1ebh), and Pyk2 (PDB: 1a3x).
Structural changes capture multiple regulatory events in the response to osmotic stress
We next assessed whether the detected structural changes relate to events known to occur in the yeast response to osmotic stress (Brewster and Gustin, 2014; Hohmann, 2015). Indeed, we detected structural alterations for most proteins of the HOG1 and glycerol production pathways (Figure 2; Table S1). Structurally altered enzymes included kinases Ste20 and Hog1 of the MAPK-HOG1 signaling pathway, enzymes in the glycerol biosynthesis branch of the pathway (Gpd1 and Gpp1/2), and enzymes of both upper (Pfk1, Fba1, Tpi1, and Tdh1/2/3) and lower glycolysis (Pgk1, Gpm1, Eno2, Cdc19, and Pyk2).
To illustrate the richness of the structural information provided, we visualize the data as structural barcodes, representing proteolytic fingerprints along the sequence of a protein (Figure 2). The barcodes provide a concise visual summary of protein regions that showed structural alterations between conditions, were detected by MS but not structurally altered, and were not detected by the MS analysis. On average, only two LiP peptides were altered per enzyme, suggesting that structural alterations were confined to specific protein regions.
We mapped altered LiP peptides to structures of the relevant proteins in complex with substrates or allosteric regulators and then assessed their proximities to known functional sites with a threshold value of 6.4 Å as determined in previous work (Piazza et al., 2018). Strikingly, most altered LiP peptides corresponded to known functional sites (Figure 2; Table S1). For MAP kinases, regulated LiP peptides mapped either to the protein region embedding the known activating phosphorylation site (Hog1) or to the allosterically regulated catalytic site (Ste20). For the MAP kinase target Gpd1, one LiP peptide mapped to the active site, a second was adjacent to the downregulated phosphosites (Lee et al., 2012), and a third mapped to the C-terminal domain that undergoes an extensive conformational change upon substrate binding (Mydy et al., 2019). We also detected LiP peptides at sites bound by small-molecule allosteric regulators; one of the two altered peptides in the beta subunit of 6-phosphofructokinase Pfk1 mapped exactly to the allosteric binding site of fructose 2,6-bisphosphate (Banaszak et al., 2011; Dihazi et al., 2004; Sträter et al., 2011). The increase of Pfk1 activity during osmotic stress increases the flux of upper glycolysis through the two downstream enzymes Fba1 and Tpi1, and regulated peptides for Fba1 and Tpi1 mapped to their active sites, likely reporting on increased substrate site occupancy. Indeed, we previously showed in experiments with exogenously added metabolites that LiP peptides at metabolite binding sites are increasingly regulated with increasing occupancy of binding sites (Piazza et al., 2018). Regulated LiP peptides of Gpp1, which generates glycerol, are also in close proximity to its active site.
Most (79%) of the LiP peptides mapping to proteins of the glycolysis pathway were in the active sites of these enzymes, including all enzymes of lower glycolysis (Pgk1, Gpm1, Cdc19, Eno2, and Pyk2). An alteration in the occupancy of these sites is consistent with decreased flux through lower glycolysis upon acute osmotic stress, possibly as a result of most upper glycolytic flux being diverted to the glycerol biosynthetic branch during this response. Thus, LiP-MS captures multiple molecular events, including allostery, altered enzyme activity, site occupancy, and phosphorylation, during the yeast response to acute osmotic stress, with the resolution of single functional sites.
Structural changes capture phosphorylation events during the response to osmotic stress
Because several phosphorylation events occur in the MAPK pathway during the osmotic stress response, we compared the LiP-MS readout to a parallel phosphoproteomic analysis. Phosphoproteomics identified 11,078 phosphopeptides mapping to 2,022 proteins (Table S1). LiP-MS found 605 differentially phosphorylated proteins (|log2FC| >1, q-value < 0.05; Table S1) upon osmotic shock. Of the 316 phosphorylated proteins detected by both methods, 48 were structurally altered (|log2FC| >1, q-value < 0.05) upon stimulation (Table S1). Among these 48 proteins, 38% have LiP peptides overlapping or in close proximity (± 10 amino acids) to a phosphopeptide; thus, the structural readout detects a subset of phosphorylation events reported by phosphoproteomics.
As expected, differentially phosphorylated proteins included proteins of the HOG1 pathway, in particular MAPK kinases of the upstream osmotic response and plasma membrane osmosensors Figure S3B), in line with previous reports (Figure S3C) (Kanshin et al., 2015). In parallel, LiP-MS identified a set of 20 structurally altered proteins within the HOG1, glycolysis, and glycerol biosynthesis pathways (Figure S3A; Table S1). Six proteins showed both structural variations and differentially regulated phosphopeptides upon osmotic stress: Hog1, Ste20, Gpd1/2, Tdh3, and Fba1 (Figures S3A and S3B). Structural variations included peptides that were differentially phosphorylated (Hog1, Fba1), mapped to a sequence adjacent to the phosphosite (Gpd1) or to a region located in close proximity to the phosphosite in the protein 3D structure (Tdh3) (Figure S3D). In summary, the structural analysis detected a subset of proteins that not only became phosphorylated upon osmotic shock, as defined by phosphoproteomics, but also detected changes in additional proteins. The structural and phosphoproteomics analyses are thus complementary (Figure S3). Finally, based on our phosphoproteomics data and known kinase/substrate relationships, we identified 12 kinases or phosphatases with significantly altered activity upon osmotic stress. We detected structural changes and altered phosphorylation in 14 known target proteins of 11 of these enzymes (Figure 3A). Thus, these structural changes can be explained by altered upstream activity of specific kinases and phosphatases.
Figure S3.

LiP-MS detects phosphorylation events in yeast responding to acute osmotic stress, related to Figure 2
(A-C): The schematics depict the yeast HOG1-MAPK pathway, including its links to the glycolysis and glycerol biosynthesis pathways. Proteins with altered structure (A) and phosphorylation (B-C) upon acute osmotic stress are shown. Depicted are: proteins with significantly altered structure (|log2FC| >1, q-value < 0.05, two-sample t-test with Storey methods correction for multiple testing) as detected by LiP-MS (A, yellow) and proteins with significantly altered phosphorylation (|log2FC| >1, q-value < 0.05; empirical Bayes moderated t-test, P values adjusted for multiple testing using the Benjamini-Hochberg method) as detected by phosphoproteomics in our data (B, blue) and as reported by (Kanshin et al., 2015) (C, blue) during acute osmotic stress. (D) Examples of significant (as in panel A) structural alterations associated with phosphorylation. For Hog1 and Gpd1, the altered LiP peptide (yellow) is overlapping or near the known phosphorylation sites (blue) in the linear sequence. For Tdh3, the LiP peptide (orange) is near the phosphorylation site (green) in 3D space.
Figure 3.

Molecular events underlying structural changes in the yeast proteome upon osmotic and heat shock
(A) Network representation of deregulated kinase activities and their target phosphosites on proteins showing structural changes upon osmotic shock of yeast cells. Structurally altered proteins are indicated by gray circles, kinases by squares, and phosphatases by diamonds; phosphorylation sites are indicated. Kinase and phosphatase activities are reported as normalized enrichment scores (NES), and phosphosite abundance changes are reported as p value-associated z-scores.
(B) Venn diagrams of the numbers of proteins of the indicated categories that are significantly structurally altered (|log2FC| >1, q-value < 0.05; two-sample t test with Storey method correction for multiple testing) after heat stress (inner circle) in relation with all detected proteins in that category (outer circle).
(C) Specific chaperones that show significant structural alterations (|log2FC| >1, q-value < 0.05; two-sample t test with Storey method correction for multiple testing) in heat or osmotic stress labeled by subcellular location.
(D) Structural barcode indicating differences in proteolytic resistance of alpha-synuclein fibrils versus monomer. Red/blue vertical bars indicate regions that show an increase/decrease in proteolytic resistance between fibril and monomer based on peptide intensity (|log2FC| >1, q-value < 0.05; Welch modified two-sample t test, p values adjusted for multiple testing with the Benjamini-Hochberg method). Detected peptides that do not change between conditions and non-detected peptides are plotted as grey and black bars, respectively. The aggregation core (NAC) is indicated.
(E) Bar plot showing protease resistance for all superaggregators and aggregators that become insoluble upon heat shock. The clear and hatched regions of the histograms show peptides indicative of increased/decreased (red/blue) proteolytic resistance for the indicated comparisons. The number of changed LiP peptides is plotted for each protein; hues indicate average strength of the fold change. Structural barcodes (as in D) are shown for selected proteins with large fold changes upon heat shock. Red/blue bars in the barcodes represent protein regions that increase/decrease proteolytic resistance in either of the two shown comparisons.
(F and G) LiP peptides (orange) of Hsp104 in (F) supernatant S2 and (G) whole-cell lysate L1 that change in response to heat shock mapped to the Hsp104 hexameric structure (PDB: 6n8t). ATP molecules binding to the chaperone catalytic site are depicted in cyan.
Protein assemblies detected via structural alterations
We probed the ability of LiP-MS to report on protein-protein interactions, focusing on the heat-shock dataset. A previous yeast study based on centrifugation of cell extracts and MS analysis of the resulting pellets identified 177 proteins that become insoluble upon heat shock, likely as a consequence of aggregation, misfolding, or formation of protein/RNA complexes (Wallace et al., 2015). These proteins were referred to as aggregators. Seventeen of these, termed superaggregators, became insoluble within minutes of heat shock. In our data, structurally altered proteins upon heat shock of yeast were clearly enriched for aggregators (Fisher’s exact test, p value < 0.05), with 96 of 177 aggregators showing structural changes (Figure 3B). Among them, only four (Nug1, Faa4, Nog2, and Ett1) showed an abundance change.
Heat shock is also known to activate molecular chaperones (Mackenzie et al., 2016; O’Connell et al., 2014), which should engage in interactions with their clients (Balchin et al., 2016). We detected a significant enrichment for chaperones among structurally altered proteins after heat shock (Figure 3B; Fisher’s exact test, p value < 0.05) but not after osmotic stress (Figures 3B and 3C). Based on literature-curated data on chaperone interactors, 67 of 96 structurally altered aggregators are known to physically interact with a chaperone in which we also detected a structural change. These changes might therefore indicate chaperone-substrate interactions in response to heat-induced protein misfolding or aggregation.
To confirm that aggregates are insoluble and less accessible to limited proteolysis than the corresponding monomeric proteins as previously reported (Fontana et al., 2004; Leuenberger et al., 2017), we spiked into yeast lysates monomeric or fibrillar alpha-synuclein, which forms well-characterized aggregates implicated in Parkinson’s disease, and ultracentrifuged the lysates (L1). As expected, the fibrils were predominantly recovered in the pellet upon ultracentrifugation, whereas the monomer was depleted in this fraction (Figure S4B). Fibrillar alpha-synuclein in the bulk lysate was also more protease resistant than in the monomeric form, and the protease-protected region mainly corresponded to its known aggregation core (Figure 3D).
Figure S4.

LiP-MS detects multiple molecular events after yeast heat shock, related to Figure 3
(A) Structural barcodes for the 9 superaggregators or 6 aggregators detected in the insoluble fractions (P2) upon heat shock. The barcodes represent the change in proteolytic fingerprints along the sequence of each protein (N- to C-term) between conditions. Each vertical bar represents a potential LiP peptide, colored to show: peptides that increase/decrease in intensity between conditions (|log2FC| >1, q-value < 0.05) (red/blue), detected peptides that do not change between conditions (gray), and peptides that are not detected by MS (black). (B) Differential analysis of alpha-synuclein (a-syn) monomer (M) or fibril (F) upon ultracentrifugation. a-syn was spiked into yeast lysates either in monomeric or fibrillar form and the samples were ultracentrifuged to separate soluble and insoluble fractions. The whole lysate before centrifugation is referred to as L1 and the insoluble pellet after centrifugation is referred to as P2. The differential analyses compare different fractions (L1 or P2) with spiked-in monomer (M) or fibril (F) as indicated. Each dot represents a protein and a-syn is indicated (SNCA). The dotted lines indicate a log2FC of 2. We interpret the plots in the following way (left-to right): The L1F/L1M comparison shows that monomer and fibril have been spiked into the same levels in the lysate. The P2F/L1F comparison shows that a-syn fibrils are not substantially lost upon ultracentrifugation. The P2F/P2M comparison shows that the a-syn fibril is enriched in the insoluble pellet after ultracentrifugation. The P2M/L1M comparison shows that the a-syn monomer is enriched in the soluble supernatant after ultracentrifugation. (C) Differential analysis of protein abundance in the pelleted fraction of a yeast lysate in heat shocked versus control samples. Significantly upregulated (red) and downregulated (blue) peptides in heat shocked pellets are indicated (|log2FC| >1, q-value < 0.05). (D) Structural changes in the fraction of the ATPase chaperone Hsp104 that pellets upon ultracentrifugation after heat stress in yeast. LiP peptides that change during the response to heat shock (orange) are mapped to the Hsp104 structure (PDB ID: 6n8t). The hexameric structure is shown. ATP molecules binding to the chaperone catalytic site are depicted in cyan. The structural barcodes indicate changes in the proteolytic pattern of Hsp104 upon heat shock and are calculated as in Figure 3D. Each vertical bar represents a potential LiP peptide, colored to show: peptides that change in intensity upon heat shock irrespective of the direction of the change (|log2FC| >1, q-value < 0.05) (yellow), detected peptides that do not change between conditions (gray), and peptides that are not detected by MS (black). Structural barcodes are shown for Hsp104 in the P2, L1 and S2 fractions. Additional changes that appear in S2 and P2 may be due to increased coverage of the analysis once soluble and insoluble Hsp104 have been separated by centrifugation.
We used the same ultracentrifugation experiment to determine whether LiP-MS detects aggregation upon heat shock or rather detects unfolding or other structural changes prior to aggregation. In lysates of yeast cells, 34 aggregators identified by Wallace et al. (2015) were either enriched in the pellet fraction and/or were depleted from the supernatant fraction (S2) upon heat shock (Figure S4C; Table S1). This indicated that these proteins become insoluble after heat shock. In line with the previous study, this set included 12 of the 17 superaggregators and 22 other aggregators that likely form insoluble assemblies under our conditions.
Proteins that became insoluble due to aggregation upon heat shock should show increased protease protection in the pellet, whereas proteins that unfold should be highly accessible to proteases. We used LiP-MS to examine the set of superaggregators and aggregators in the insoluble fraction (P2) upon heat shock. These proteins were generally more protease resistant than the same proteins in the soluble (S2) fraction of cells not subjected to heat shock (Figures 3E and S4A) and in the bulk lysate (L1) upon heat shock (Figures 3E and S4A). This indicates that the centrifugation step enriches for insoluble, protease-resistant species and suggests that the structural changes we observed in the insoluble fraction of the proteome are due to aggregation. We note that our experiment cannot distinguish between homomeric aggregates and proteins trapped in a densely interacting insoluble protein network. Further, LiP-MS analyses of aggregators in the insoluble versus soluble fraction pinpoint putative aggregation interfaces (Figures 3D, 3E, and S4A), which could be used to identify mutations that modulate aggregation events.
We next looked more closely at the LiP patterns of the protein disaggregase ATPase Hsp104, which we detected in the total (L1), soluble (S2) and insoluble (P2) fractions (Figures 3F, 3G, and S4D). Interestingly, proteolytic patterns were distinct in these fractions, suggesting that LiP-MS captures structural changes in Hsp104 that reflect different molecular events. In the soluble fraction, three of the five altered peptides mapped to the ATP binding site or substrate channel of Hsp104 (Figure 3F). ATP binding is known to trigger substrate binding in Hsp104 (Gates et al., 2017), suggesting that LiP might capture the activation cycle of Hsp104 and its engagement in chaperone-client interactions induced by heat shock (Gates et al., 2017). In the insoluble fraction, four out of six altered peptides clustered around a large, solvent-exposed region, possibly indicating an interaction or aggregation interface (Figure S4D). In the unseparated lysate L1, altered peptides mapped to several of the above described regions (Figure 3G), suggesting that this sample contains a mixture of Hsp104 structural states. This example shows how coupling of LiP-MS with fractionation enabled us to deconvolve complex structural readouts indicative of the coexistence of different protein structural states.
In summary, in heat-shocked yeast, the structural readout captured protein aggregation, chaperone-client interactions, as well as potential allosteric regulation of chaperones.
Protein structural changes during nutrient adaptation in E. coli
For our second system, we studied E. coli grown in eight carbon sources: acetate, galactose, succinate, glycerol, pyruvate, fructose, glucose, and gluconate. We chose this model to leverage a recent analysis in which metabolite levels and fluxes through E. coli central carbon metabolism (CCM) were shown to be condition dependent under these same growth conditions (Gerosa et al., 2015). We reasoned that flux variations serve as a proxy for altered functional states of enzymes and used them to assess the capability of our structural readout to report on functional changes in the CCM.
We cultured E. coli in a medium containing each of the eight carbon sources, harvested cells in exponential phase (optical density [OD] = 0.8 ± 0.1), extracted the proteomes under native conditions, and analyzed samples by LiP-MS. We derived structural fingerprints for a minimum of 1,895 proteins (growth in galactose) to a maximum of 1,917 proteins (growth in gluconate) and measured abundance changes for a similar number of proteins (minimum 2,085 in glycerol and galactose to a maximum of 2,102 in pyruvate) (Figure S1B; Table S3). Differential analysis of protein structure and abundance in each growth condition compared to glucose showed that on average 365 proteins underwent structural alterations (15%–25% of identified proteins), and 190 proteins changed in abundance (3%–13% of identified proteins) (Figure 4A). As observed in our yeast experiments, a higher number of proteins underwent structural alterations than abundance changes. Replicates clustered together, and correlations of protein abundance changes with previous data were good (Schmidt et al., 2016, data not shown). Further, each growth condition resulted in the up-regulation of the expected nutrient transporters and uptake regulators (Figure 4B).
Figure 4.

Global protein structural and abundance changes during nutrient adaptation in E. coli
(A) Number of proteins significantly changed (|log2FC| >2, q-value < 0.05; p values adjusted for multiple testing with the Benjamini-Hochberg method) in structure (green) or abundance (yellow) under the indicated nutrient conditions in relation with glucose.
(B) Schematic of the known regulators for different nutrient sources (upper). Abundance differences of known nutrient transporters and uptake regulators under the indicated nutrient conditions in relation with growth in glucose (log2FC) (lower).
Global protein structural data are complementary to protein abundance information
To assess the overlap of information derived from protein abundance and structural measurements, we performed functional enrichment analyses on proteins with altered structure and/or abundance in different carbon sources. The structural and protein abundance readouts captured different sets of biological processes, although several GO terms overlapped (Figure S5A; Table S3). There was no enrichment of the glycolytic pathway in proteins that changed abundance (Figure S5A), consistent with previous observations that glycolytic fluxes are not primarily controlled at the transcriptional level (Gerosa et al., 2015). In contrast, proteins with structural alterations were enriched in glycolytic enzymes in all growth conditions (Figure S5A). Out of 20 identified glycolytic enzymes, 16 showed a structural alteration in multiple conditions, whereas only three changed abundance in at least one condition (Figure S5B). Similarly, seven out of nine enzymes from the pentose phosphate pathway were altered exclusively in their structure in multiple conditions. Other biological processes only enriched at the structural level were amino acid biosynthesis and tRNA aminoacylation for protein translation, particularly under conditions resulting in the slowest growth, and ATP biosynthesis (Figure S5A; Table S3). The tricarboxylic acid (TCA) cycle was regulated at both the abundance and the structural level, as were the glyoxylate cycle, transmembrane transport, and aerobic respiration (Figures S5A and S5B). Our data suggest that different regulatory mechanisms control different branches of the CCM in E. coli grown on different carbon sources. Glycolysis is controlled by regulatory mechanisms that affect protein structure and not gene expression, whereas the TCA and glyoxylate cycles show changes in expression levels in addition to other regulatory processes that affect protein structure.
Figure S5.

Functional analysis of proteins that show structural and abundance changes during nutrient adaptation in E. coli, related to Figures 4 and 5
(A) The plot shows functional categories (GO biological processes) enriched among proteins significantly changing (|log2FC| >2, q-value < 0.05; P values adjusted for multiple testing using the Benjamini-Hochberg method) in abundance (green) and structure (yellow) under the indicated nutrient conditions, relative to glucose. P values for the enrichment (gray scale) were determined using Fisher’s exact test. Blank cells indicate biological processes that were not significantly enriched (i.e with p-value > 0.01) in a given condition. (B) The heat maps show which E. coli CCM proteins significantly change (blue, |log2FC| >2, q-value < 0.05; P values adjusted for multiple testing using the Benjamini-Hochberg method) in either structure (left) or abundance (right) under the indicated nutrient conditions, relative to glucose. Proteins are arranged according to the CCM pathway to which they belong. (TCA= tricarboxylic acid cycle, GS = glyoxylate shunt, PPP = pentose phosphate pathway, ED = Entner-Doudoroff pathway). (C) The barcodes represent the change in proteolytic fingerprints along the sequence of Pgk (N- to C-term), comparing growth in the indicated carbon source relative to growth in glucose. Each vertical bar represents a peptide that could be detected in samples subject to LiP. The color code indicates: peptides that change in intensity (|log2FC| >1, q-value < 0.05) between galactose and glucose, correlate with flux across all conditions, and correlate with substrate levels in an in vitro LiP experiment (orange), peptides that change in intensity between galactose and glucose but do not meet the other two conditions (yellow), peptides detected by MS but that do not change between conditions (gray), and peptides that are not detected by MS (black).
Structural changes reflect functional alterations of metabolic enzymes
Changes in flux for a reaction catalyzed by a given enzyme could occur due to changes in enzyme activity (in response to allosteric interactions or PTM), in reactant concentrations, or in enzyme levels. We hypothesized that a change in flux due to altered enzyme activity or altered binding of reactants might affect protein structure, resulting in a LiP signal. To test this, we asked whether LiP-MS data, corrected for protein abundance changes, captured structural changes for enzymes known to catalyze the reactions that change flux. We calculated flux ratios in relation to glucose for 25 CCM reactions in E. coli grown under the different nutrient conditions (Gerosa et al., 2015). Between 18 (for acetate) and 25 (for gluconate) reactions significantly changed in flux in the different growth conditions (t test, adjusted p value < 0.05) (Table S3). LiP-MS detected a structural alteration for enzymes associated with the majority of flux changes (87%, Figure 5A), supporting the notion that a structural readout captures alterations in enzyme functional states. Protein abundance data detected changes in enzymes, mostly of the TCA and glyoxylate cycles, associated with only 39% of flux alterations (Figure 5A), confirming that only some flux changes are explained by altered concentrations of the associated enzymes.
Figure 5.

Structural changes reflect functional flux alterations of E. coli metabolic enzymes
(A) 13C-based metabolic flux maps for E. coli grown in indicated nutrient conditions reported in Gerosa et al. (2015). The thickness of the black arrows indicates the flux fold change in relation to growth in glucose. Proteins with significant changes (|log2FC| >2, q-value < 0.05; p values adjusted for multiple testing with the Benjamini-Hochberg method) in abundance (green), structure (yellow), or both are indicated.
(B) Schematic of glycolytic enzymes. Red circles indicate enzymes with correlations between LiP peptide levels and metabolic flux.
(C) Linear regression between levels of the indicated LiP peptides derived from pgk and relative flux values through pgk across all nutrient conditions.
(D) Level of the best correlating LiP peptide of recombinant pgk spiked into an E. coli lysate with increasing 3-phosphoglycerate (3PG) concentration. This peptide is almost identical to the one correlated with flux across growth conditions in vivo (in [C]).
(E) The two LiP peptides that correlate with flux (orange) mapped onto the structure of pgk (PDB: 1zmr). 3PG bound to pgk is indicated in cyan. The barcode represents the change in proteolytic fingerprint along the sequence of pgk in galactose in relation to glucose (for barcodes corresponding to all growth conditions, see Figure S5C). Orange indicates peptides that change in intensity (|log2FC| >1, q-value < 0.05) between galactose and glucose, correlate with flux across all conditions, and correlate with substrate levels in an in vitro LiP experiment; yellow indicates peptides that change in intensity between galactose and glucose but do not meet the other two conditions; gray indicates peptides detected by MS that do not change between conditions; and black indicates peptides that are not detected by MS.
See also Table S3.
Flux associated with an enzyme could be regulated by the same molecular event across growth conditions. Alternatively, different molecular events could regulate flux in different conditions. To distinguish between these two scenarios, we used linear regression to ask for which enzymes structural or abundance changes were quantitatively related to metabolic flux changes across the eight conditions. Of 11 glycolytic enzymes, one or multiple LiP peptides from gapA, pgk, and eno were linearly correlated with flux measurements over the set of growth conditions (R2 > 0.7 and adjusted p value < 0.05, Table S3; Figure 5B), suggesting that for these enzymes structural changes at specific sites are a quantitative predictor of fluxes. For the remaining enzymes in the network, there was no linear correlation between flux and a specific LiP peptide changing across at least four conditions, suggesting that fluxes for these enzymes are likely not regulated by the same molecular events in the different conditions, as previously suggested (Gerosa et al., 2015). The abundance of mdh was linearly correlated with flux, suggesting that mdh fluxes are regulated by enzyme abundance in these conditions (R2 > 0.7 and adjusted p value < 0.05, Table S3).
Of LiP peptides detected for glycolytic enzymes gapA, pgk, and eno, only 11% correlated with fluxes (Figure 5C; Table S3; for all correlation plots see https://doi.org/10.5281/zenodo.3964994). We mapped the LiP peptides that correlate with fluxes to available enzyme structures (Table S3). For the three glycolytic enzymes, a large fraction of LiP peptides that correlated with flux (2/2 for pgk, 11/14 for gapA, and 9/26 for eno) mapped to the enzyme active site or, in the case of gapA, to a known allosteric site (Table S3).
We speculated that active-site LiP peptides report on substrate occupancy and that they correlate with flux because substrate occupancy integrates events that affect flux. To confirm this, we performed LiP-MS on purified pgk in the presence of different amounts of its substrate 3-phosphoglycerate. To mimic as much as possible the in vivo experiment, we spiked purified pgk and substrate into an E. coli lysate cleared of endogenous metabolites. Remarkably, of the 180 peptides detected, the two LiP peptides that increased with added substrate in vitro covered exactly the same active-site region we detected for the endogenous enzyme across the different nutrient conditions (Figures 5C–5E; Table S3). This suggests that these LiP peptides report on pgk substrate occupancy in situ and that substrate occupancy monitored by LiP correlates with flux changes. Taken together, our data show that the structural readout captures functionally relevant changes of E. coli CCM enzymes in situ.
The structural readout identifies regulatory events in CCM
Metabolites can regulate enzyme activity by allosteric interactions (Chubukov et al., 2014). We reasoned that some of the structural alterations we detected for CCM enzymes could underlie cases of allosteric regulation. In these cases, levels of metabolite regulators should correlate with structural alterations at the allosteric site of the target enzyme. We used linear regression to test for a correlation between structural changes in enzymes of the CCM and relevant metabolite levels across the eight growth conditions. We found a linear correlation between metabolite concentration and LiP peptide abundance for 32 enzymes (Figures 6A and 6B; Table S3; for all correlation plots see https://doi.org/10.5281/zenodo.3965002). Among these metabolite-enzyme pairs, the allosteric interaction between FBP and pykF and catalytic interactions between FBP and pfkA, NAD and sucA, and alpha-ketoglutarate and gltA have been well characterized. Interactions between dihydroxyacetone phosphate and fbaA, NAD and eno, glucose-6-phosphate and gapA, FBP and pgk, citrulline and pta, FBP and ptsI, and NAD and pfkA were corroborated by recent physical interaction data (Diether et al., 2019; Piazza et al., 2018). Importantly, LiP peptides that correlated with metabolite levels were in close proximity to the binding sites identified by previous experiments (Piazza et al., 2018) (Table S3).
Figure 6.

Structural changes capture allosteric regulators of E. coli metabolic enzymes
(A) Depiction of E. coli CCM showing the 32 enzymes with significant correlations between LiP peptide levels and regulatory metabolite levels (gray dots) across all growth conditions. Red outlines indicate interactions supported by previous data. Metabolites are denoted by rectangular boxes, and the points of entry of different nutrient sources are shown.
(B) Correlations between levels of metabolites (rows) and CCM enzyme-derived LiP peptides (columns) in a linear regression analysis across all nutrient conditions. All metabolites with at least one significant correlation are plotted (|log2FC| >1, q-value < 0.05 in at least four conditions and for the regression analysis an adjusted p value <0.05 with R2 >0.7). The color scale indicates the correlation coefficient.
(C) LiP peptides of purified ptsI with significance level |log2FC| >2, q-value < 0.01 (two-sample t test with Storey methods correction for multiple testing) mapped onto the 3D structure of ptsI (PDB: 2xz7). Peptides in dark orange are positioned within the active site (< 6.4 Å), light orange peptides are outside the active site. A close-up of the ptsI active site is shown with PEP in cyan and Mg2+ in red. The structure shown is the only one for which a 3D structure with bound PEP was available.
(D) Binding mode of FBP (carbon atoms in cyan) to ptsI (gray, with carbon atoms in active-site side chains in green) as predicted by ligand docking and molecular dynamics simulations (PDB: 2xz7). A close-up of the ptsI active site is shown with FBP and the cofactor Mg2+ (blue sphere).
(E) ptsI in vitro activity assay. Bar plot of the fitted rate constants of the PEP-labeling reaction, which is a measure of ptsI activity. Rates are shown as means; error bars indicate the standard deviation (n=4).
We further validated the interaction between ptsI and the metabolite FBP (Figures S6A–S6C). In order to confirm that this interaction is not due to indirect effects, we repeated the LiP-MS analysis on purified ptsI in the presence and absence of FBP. Addition of FBP to the protein triggered alterations of the same region that was altered in vivo upon increasing intracellular concentrations of FBP (though not identical peptides), suggesting that this region corresponds to the ptsI-FBP binding site (Figures 6C and S6C). This site overlaps with the known binding site of the ptsI substrate phosphoenolpyruvate (PEP). In addition, possibly due to the higher coverage for purified protein, we detected four other altered peptides upon addition of FBP, two of which were located at the PEP binding site (Figure 6C). These data suggest that FBP could act as a competitive inhibitor of ptsI.
Figure S6.

Molecular events underlying structural changes in E. coli metabolic enzymes, related to Figure 6
(A-B) The plots show linear regressions between levels of the indicated LiP peptide derived from ptsI and levels of FBP, across all nutrient conditions. (C) LiP peptides that correlate with levels of fructose-bis-phosphate (FBP) in vivo (shown in A and B) are mapped (orange) onto the 3D structure of ptsI (PDB ID: 2xz7). Dark orange peptides indicate those positioned within the active site (< 6.4 Å) and light orange peptides indicate those outside the active site. A close-up of the ptsI active site is shown, with phosphoenolpyruvate (PEP) in cyan and the cofactor Mg2+ in red. The structure shown is the only one for which a 3D structure with bound PEP was available. (D-F) Controls for the ptsI activity assay. The gel (D) shows the purified protein used for the assay, in the indicated dilutions. The plots (E-F), show time-course data of the labeled fraction of phosphoenolpyruvate (PEP) in the presence (E) and absence (F) of 25 mM FBP. The colors indicate four independent assays; the solid line indicates the weighted non-linear least-squares regression using the following equation: L(t) = 0.45∗[1-exp(k∗t)] + c, where L denotes the fractional labeling of PEP, k the rate constant, t the time and c the intercept. (G) The table shows protein-metabolite interactions upon growth in each of six nutrient conditions versus growth in glucose. Proteins are shown in rows and metabolites are shown in columns. Plotted are interactions for which the metabolite showed an at least 3-fold change in any condition versus glucose (Gerosa et al., 2015) and which overlap with a dataset of interactions previous detected in vitro (Piazza et al., 2018). Colored cells indicate protein-metabolite interactions where the in vivo protein structural changes detected exactly match a previously determined in vitro structural change dependent on the same metabolite (see Methods). Gray cells indicate protein-metabolite pairs for which changes are detected in multiple conditions relative to glucose, other colors indicate condition-specific changes according to the following code: G/orange- galactose, F/green – fructose, S/dark blue – succinate, Y/pink – glycerol, A/peach – acetate, P/light blue – pyruvate. Triangles indicate whether the interaction is higher in glucose (apex down) or in the compared condition (apex up). Interactions marked in red text have been previously characterized.
To test this, we performed ligand docking and molecular dynamics simulations. These computational analyses provided strong evidence that FBP binds at the PEP binding site in ptsI (Figure 6D). The two phosphate groups of FBP coordinate the cofactor Mg2+, forming an octahedron coordination system together with the negatively charged side chains of Glu431 and Asp455 of ptsI and surrounding water molecules. These interactions were structurally stable along the simulations (Video S1). Finally, we performed in vitro activity assays and observed a strong inhibitory effect of FBP on ptsI activity (Figures 6E and S6D–S6F), thus confirming the hypothesis generated by our structural analysis.
Binding mode of FBP (carbon atoms in cyan) to enzyme I of the phosphoenolpyruvate-protein phosphotransferase system (PtsI) (gray, with carbon atoms in active site side chains in green) as predicted by ligand docking and molecular dynamics simulations. A close-up of the PtsI active site is shown, with FBP and the cofactor Mg2+ (blue sphere).
We next sought to systematically explain structural changes observed during the shift between pairs of conditions. Some regulatory metabolite-protein interactions have been proposed to be active only in the presence of specific nutrients (Gerosa et al., 2015) and would therefore not be captured by our correlation analysis across eight conditions. We identified, based on previous metabolomics data (Gerosa et al., 2015), metabolites that changed their concentration at least 3-fold in each condition in relation with glucose (Figure S6G; Table S3). For these altered metabolites, we examined a dataset of metabolite-protein interactions that we had previously generated (Piazza et al., 2018), where interactions had been induced by adding specific metabolites to cell extracts. That analysis had identified marker peptides at binding sites that showed altered levels upon binding of the metabolite, but we did not know whether the interactions identified in vitro would occur also in vivo.
We reasoned that interactions that occur in vivo should show changes in in vitro-defined marker peptides during metabolic transitions where the endogenous concentration of the specific metabolite is substantially altered. We identified 121 such metabolite-protein interactions (Figure S6G). Reassuringly, nine of these interactions were previously characterized in the literature; seven are enzyme-substrate relationships known to be relevant in the associated metabolic transitions (Table S3). This suggests that our approach identifies physiologically relevant interactions and that the remaining 112 metabolite-protein interactions could also occur in vivo. Moreover, our data suggest the specific metabolic transitions under which the interactions are regulated.
In summary, our structural approach suggested both transition-dependent and more broadly active metabolite-protein interactions in E. coli, in situ. We have validated a regulatory interaction between ptsI and FBP, showing that identification of structurally altered regions supports the generation of testable molecular hypotheses.
Discussion
We demonstrated that detecting dynamic alterations of protein structures on a proteome-wide scale provides a powerful global readout of protein functional alterations in situ. Our approach captured protein functional alterations due to different molecular events, including enzyme activity changes, altered enzyme active-site occupancy, PTMs, metabolite-driven allosteric events, substrate binding, and protein-protein interactions.
In all systems examined, the structural readout captured more altered proteins and processes than did a protein abundance readout, and data from both approaches were often complementary. This suggests that integrating structural and abundance-based proteomics will maximize detection of altered biological processes. Protein abundance information is also captured in the control step of LiP-MS experiments, allowing simultaneous probing of most types of functional molecular events in a single experiment (Figure S7). Although we focused our analyses on specific well-studied pathways, this rich dataset should yield biological insight beyond that presented here.
Figure S7.

LiP-MS data captures both abundance and structural changes, related to Figure 4
The plot shows functional categories (GO biological processes) enriched among proteins significantly changing (|log2FC| >2, q-value < 0.05; P values adjusted for multiple testing using the Benjamini-Hochberg method) in E. coli grown in the indicated nutrient conditions, relative to growth in glucose. Proteins showing only abundance changes, only structural changes (measured by normalizing LiP-MS data for proteins that also show abundance changes), both abundance and structure changes (consisting of the previous two categories added together) and proteins detected as changing based on the raw (i.e. non-normalized) LiP-MS data, are plotted separately. P values for the enrichment (gray scale) were determined using Fisher’s exact test. Blank cells indicate biological processes that were not significantly enriched (i.e with p-value > 0.01) in a given condition.
The LiP-MS structural readout provides data complementary to other ‘omic data. For instance, when conducted in parallel to phosphoproteomic analysis, the structural readout captured multiple types of molecular events known to regulate protein function; however, phosphoproteomics additionally revealed phosphorylation-associated functional alterations of low-abundance proteins, because the phosphopeptide enrichment step intrinsic to these analyses increases proteome coverage. For proteins analyzed by both approaches, LiP-MS detected structural alterations for 40% of the regions that changed phosphorylation state. The phosphoproteomic analysis is restricted to the phosphorylated fraction of protein molecules, whereas LiP-MS monitors the average structural state of both phosphorylated and non-phosphorylated protein pools. Thus, LiP-MS might not detect a structural change for a differentially phosphorylated protein region if the degree of phosphorylation is low.
In bacteria grown under different nutrient conditions, we detected structural alterations for almost all enzymes associated with flux changes, suggesting that LiP-MS is a good readout for enzyme functional alterations. In contrast, only about a third of flux changes were explained by enzyme abundances. The structural readout reports on different molecular events that can affect flux, including allosteric and phosphorylation events and changes in the relative levels of enzyme and/or substrate. For three enzymes, fluxes across metabolic conditions correlated with levels of LiP peptides at active sites, and we showed that these peptides likely report on site occupancy. For these enzymes, fluxes are likely regulated by relative levels of enzyme and reactants. For enzymes where we did not detect a correlation with flux across conditions, different types of molecular events presumably regulate fluxes across these conditions, as previously suggested (Gerosa et al., 2015).
LiP peptides located at functional sites can be used as markers to probe specific functional events in situ. For example, LiP peptides at the active site of pgk and at the metabolite binding sites of ptsI and eno reported on the occupancy of those sites both in vitro and in situ. Similarly, the abundance of a LiP peptide from Hog1 that contains a phosphorylation site changed concomitantly with phosphorylation. It could be possible to extract similar markers for many proteins by mapping structural proteomics data from perturbed proteomes to high-resolution structures and integrating prior knowledge on protein functional states. Structural proteomics could also be applied in a clinical context to identify structural biomarkers for disease.
Our approach does not directly inform on the causes of the detected structural alterations. However, LiP-MS detects structural changes with peptide-level resolution (i.e., the change can be pinpointed to stretches of around 10 amino acids), allowing us to relate effects of perturbations to specific functional sites. This supports the generation of testable molecular hypotheses, the design of follow-up biochemical experiments, and the design of mutations for functional studies, thus linking holistic and mechanistic approaches. Our method is particularly useful if a high-resolution structure of a protein is available, but sequence-based information on locations of functionally relevant sites or domains could be sufficient for hypothesis generation.
Based on our data we hypothesized that the metabolite FBP bound to the active site of the enzyme ptsI in E. coli. Computational analyses indicated that FBP likely acts as a competitive inhibitor of ptsI, and in vitro assays confirmed that FBP reduces ptsI activity. Previous work has shown that ptsI controls hexose uptake and regulates glycolytic flux (Doucette et al., 2011). Interestingly, FBP has been shown to act as an intracellular glycolytic flux sensor (Kochanowski et al., 2013). Thus, we hypothesize that the FBP-ptsI interaction serves as a negative feedback loop that prevents excessive glucose uptake when glycolytic intermediates are already abundant. In support of this, the in vitro inhibitory effect of FBP was observed only at high FBP concentrations (25 mM) (Figure 6E) but was negligible at around four times lower FBP levels (data not shown). These high FBP concentrations are in a physiological range for cells grown in glycolytic carbon sources (15 mM) (Bennett et al., 2009; Gerosa et al., 2015). Consistently, our in situ data indicate that the FBP-ptsI interaction becomes relevant when going from gluconeogenic (pyruvate, acetate, and succinate) to glycolytic (glucose, fructose, and glycerol) carbon sources. The low affinity of the FBP-ptsI interaction likely explains why it was not previously detected and indicates that our strategy does not only detect high-affinity interactions.
Structurally altered proteins identified in a global analysis could be followed up by high-resolution structural studies (e.g., by x-ray crystallography or cryo-electron microscopy) and visualized in cells and tissues (e.g., by cryo-electron tomography), thus potentially leading to innovative structural biology workflows. In principle, LiP-MS could also provide insight into protein organization more broadly, because it could detect changes in protein localization if this is accompanied by structural alterations. We note however that, because the protocol used here involves cell lysis prior to LiP, intracellular compartmentalization will be disrupted, so changes due to a different internal environment (e.g., pH of an organelle) are unlikely to be detected. An assessment of the ability of LiP-MS to monitor larger-scale proteome reorganization will require a more systematic study.
LiP-MS is not the only structural proteomics approach that can be used for the in situ detection of protein functional changes. Other techniques such as crosslinking mass spectrometry or surface footprinting (e.g., by fast photochemical oxidation of proteins) could, in principle, also be applied for this purpose. Although it is difficult to analyze complex proteomes with these approaches or to perform comparative analysis of differently treated samples, recent technical developments suggest that these techniques might be a promising direction for the dynamic analysis of structural and functional proteomes (Espino and Jones, 2019; Liu et al., 2015; Rinas et al., 2016). Thermal proteome profiling has also been used to identify protein-protein interaction and small molecule binding in complex samples (Savitski et al, 2014; Tan et al, 2018).
Pioneering computational biology studies have exploited information from static protein structures to assess the properties of specific biological systems, thus illustrating the potential of global structural data (Chang et al., 2013; Zhang et al., 2009). We propose that the incorporation of dynamic in situ structural data obtained for proteomes under different conditions, like those generated by our LiP-MS approach, will extend the potential of structural systems biology and link systems and reductionist approaches. The quantitative measurement of molecular events such as active-site occupancy should also support the development of frameworks for the modeling of biological systems. By linking dynamic and high-resolution structural data to function, our global structural approach brings us one step closer to a 3D model of the functioning of a cell.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| TCEP (tris(2-carboxyethyl)phosphine hydrochloride) | Pierce | Cat#20490; CAS#51805-45-9 |
| Iodoacetamide | Sigma-Aldrich | Cat#I1149; CAS#144-48-9 |
| Ammonium bicarbonate | Sigma-Aldrich | Cat#09830; CAS#1066-33-7 |
| Formic acid 98-100% | AppliChem | Cat#A38580500 |
| HEPES (4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-Hydroxyethyl)piperazine-N¢-(2-ethanesulfonic acid) | Sigma-Aldrich | Cat#H4034 |
| Sodium deoxycholate | Sigma-Aldrich | Cat#D6750; CAS #302-95-4 |
| Proteinase K (PK) from Engyodontium album | Sigma-Aldrich | Cat#P2308 |
| HRM calibration kit | Biognosys AG | Cat#Ki-3003 |
| Phospho(enol)pyruvic acid monopotassium salt | Sigma-Aldrich | Cat#860077; CAS#4265-07-0 |
| Sodium pyruvate | Sigma-Aldrich | Cat#P2256; CAS# 113-24-6 |
| Sodium pyruvate (13C3, 99%) | Cambridge Isotope Laboratories | Cat# CLM-2440-PK; 142014-11-7 |
| Fructose-bis-phosphate sodium salt | Sigma-Aldrich | CAT# 47810 |
| Potassium chloride | Merck | Cat#K41042236-032; CAS#64-18-6 |
| D-Glucose | Sigma-Aldrich | Cat #G8270; CAS#50-99-7 |
| D-Fructose | Sigma-Aldrich | Cat#F3510; CAS #57-48-7 |
| Sucrose | Sigma-Aldrich | Cat#84100 CAS #57-50-1 |
| Acetate (potassium acetate) | Sigma-Aldrich | Cat#60035 CAS #127-08-2 |
| Gluconate (potassium D-gluconate) | Sigma-Aldrich | Cat#G4500 CAS #299-27-4 |
| Glycerol (anhydrous) | PanReac AppliChem | Cat#A1123 Cas#56-81-5 |
| Galactose | Formedium | Cat#GAL03 |
| Sodium pyruvate | Sigma-Aldrich | Cat#P5280 Cas# 113-24-6 |
| Sodium chloride | Merck | Cat#1.06404 Cas# 7647-14-5 |
| Magnesium chloride hexahydrate | Fluka | Cat#63072; CAS#7791-18-6 |
| Alpha-synuclein | Purified in-house | Feng et al., 2014 |
| Critical Commercial Assays | ||
| BCA protein assay | Pierce | Cat: 23228 |
| His GraviTrap™ TALON® | GE Healthcare | Cat: GE29-0005-94 |
| Deposited Data | ||
| Raw and analyzed data | This paper | Pride: PXD022297 |
| LiP peptide correlations with flux | This paper | https://doi.org/10.5281/zenodo.3964994 |
| LiP peptide correlations with metabolite levels | This paper | https://doi.org/10.5281/zenodo.3965002 |
| Heat aggregating proteins | Wallace et al., 2015 | N/A |
| 13C-based metabolic fluxes | Gerosa et al., 2015 | N/A |
| Protein-metabolite interactions (LiP-SMAP) | Piazza et al., 2018 | N/A |
| S. cerevisiae Gene Ontology (GO) annotation | Gene Ontology Consortium | http://current.geneontology.org/annotations/sgd.gaf.gz |
| E. coli Gene Ontology (GO) annotation | Gene Ontology Consortium | http://current.geneontology.org/annotations/ecocyc.gaf.gz |
| Yeast chaperones | Gong et al., 2009 | N/A |
| Yeast chaperones physical interactors | BioGRID | https://thebiogrid.org/ |
| Kinase-substrate network (KSN) | BioGRID | https://downloads.thebiogrid.org/File/BioGRID/Release-Archive/BIOGRID-3.5.186/BIOGRID-PTMS-3.5.186.ptm.zip |
| Protein-metabolite interactions detected by ligand-detected NMR | Diether et al., 2019 | N/A |
| Pfk1 protein structure | Banaszak et al., 2011 | PDB: 3o8o |
| Fba1 protein structure | • https://doi.org/10.2210/pdb3QM3/pdb | PDB: 3qm3 |
| Ste20 protein structure | Karpov et al., 2015 | PDB: 4zlo |
| Hog1 protein structure | Wang et al., 2016 | PDB: 5ci6 |
| Tpi1 protein structure | Jogl, et al., 2003 | PDB: 1nf0 |
| Gpd1 protein structure | Leano et al., 2019 | PDB: 6e9o |
| Gpp1 protein structure | • https://doi.org/10.2210/pdb2QLT/pdb | PDB: 2qlt |
| Tdh2 protein structure | • https://doi.org/10.2210/pdb3PYM/pdb | PDB: 3pym |
| Pgk1 protein structure | McPhillips et al., 1996 | PDB: 1qpg |
| Gpm protein structure | Crowhurst et al., 1999 | PDB: 1qhf |
| Eno2 protein structure | Wedekind et al., 1995 | PDB: 1ebh |
| Pyk2 protein structure | Jurica et al., 1998 | PDB: 1a3x |
| Hsp104 protein structure | Lee et al., 2019 | PDB: 6n8t |
| pgk protein structure | Young et al., 2007 | PDB: 1zmr |
| ptsI protein structure | Navdaeva et al., 2011 | PDB: 2xz7 |
| Experimental Models: Organisms/Strains | ||
| E. coli: Strain background BW25113 | Baba et al., 2006 | N/A |
| S. cerevisiae BY4742: S288C isogenic yeast strain. Genotype: MATa his3D1 leu2D0 lys2D0 ura3D0 | Euroscarf | http://www.euroscarf.de/plasmid_details.php?accno=Y10000 |
| E. coli ASKA collection strain: ptsI, b2416 | Kitagawa et al., 2005 | JW2409-AP |
| E. coli ASKA collection strain: 6xHis-tagged Pgk | Kitagawa et al., 2005) | EcoCyc: EG10703 |
| Software and Algorithms | ||
| Rstudio | Rstudio | https://www.rstudio.com |
| R version v. 3.6.1 | The R Foundation | https://www.r-project.org/ |
| Python version v. 2.7, 3.0 | Python Software Foundation | https://www.python.org |
| Pandas library for python, 0.18.1 | NumFOCUS | https://pandas.pydata.org/ |
| Seaborn library for python v. 0.9.0 | Michael Waskom | https://seaborn.pydata.org/index.html# |
| PyMOL 2.4 | Schrödinger | https://pymol.org/2/ |
| Proteome discoverer v. 2.2 | ThermoFisher Scientific | https://www.thermofisher.com/us/en/home.html |
| Spectronaut v. 13 | Biognosys AG | https://biognosys.com/ |
| MaxQuant 1.5.2.8 | Max-Planck-Institute of Biochemistry | https://www.maxquant.org/ |
| Progenesis QI 2.0 | Nonlinear Dynamics | http://www.nonlinear.com/progenesis/qi-for-proteomics/ |
| SafeQuant 2.3.1 | Erik Ahrne | https://github.com/eahrne/SafeQuant |
| MSstats 3.1 | Choi et al., 2014 | https://www.bioconductor.org/packages/release/bioc/html/MSstats.html |
| proteusLabelFree | Marek Gierlinski | https://github.com/bartongroup/proteusLabelFree |
| Bioconductor | Huber et al., 2015 | https://www.bioconductor.org/about/ |
| topGO | Adrian Alexa, Jorg Rahnenfuhrer | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| viper | Mariano J Alvarez | http://bioconductor.org/packages/release/bioc/html/viper.html |
| Network analysis code | This paper | https://github.com/saezlab/conformationomic_yeast_picotti_2020.git |
| MatLab R2020a | MathWorks | https://www.mathworks.com |
| MarvinSketch 19.25 | ChemAxon | http://www.chemaxon.com |
| Maestro 11.5 | Schrödinger | https://www.schrodinger.com/freemaestro |
| AutoDock Vina 1.1.2 | Trott and Olson, 2010 | http://vina.scripps.edu |
| NAMD 2.13 | University of Illinois at Urbana-Champaign | https://www.ks.uiuc.edu/Research/namd |
| CHARMM 42b2 | Harvard University | https://www.charmm.org |
| Other | ||
| Amicon Desalting Columns 3 kDa MWCO | Merck | N/A |
| Freezer Mill, 6870 | SPEX SamplePrep | N/A |
| Ni-IMAC column | GE Biotech | N/A |
| Sep-Pak Vac, tC18 Cartridges | Waters | Cat: WAT054960 |
| Orbitrap Q Exactive Plus mass spectrometer | ThermoFisher Scientific | https://www.thermofisher.com/us/en/home.html |
| Orbitrap Fusion Lumos Tribrid mass spectrometer | ThermoFisher Scientific | https://www.thermofisher.com/us/en/home.html |
| Orbitrap Q Exactive HF mass spectrometer | ThermoFisher Scientific | https://www.thermofisher.com/us/en/home.html |
Resource Availability
Lead Contact
Further information and requests for resources and reagents may be directed to and will be fulfilled by Paola Picotti (picotti@imsb.biol.ethz.ch).
Materials Availability
This study did not generate new unique reagents.
Data and Software Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD022297.
The complete data set of LiP peptide correlations with flux are at https://doi.org/10.5281/zenodo.3964994
The complete data set of LiP peptide correlations with metabolite levels are at https://doi.org/10.5281/zenodo.3965002
Experimental Model and Subject Details
E. coli
All experiments were performed with the E. coli BW25113 wild-type in shake-flask cultures (Baba et al., 2006). Frozen glycerol stocks were used to inoculate Luria-Bertani (LB) complex medium. After 6 hours of incubation at 37°C under constant shaking at 220 rpm, LB cultures were used to inoculate 25 ml of M9 minimal medium pre-cultures supplemented with 5 g/L of the indicated carbon source (glucose, fructose, sucrose, acetate, gluconate, glycerol, galactose and pyruvate) for over-night culture. The next day, final cultures were inoculated 1:100 (v/v) in 500 ml of M9 minimal medium supplemented with the same carbon source and grown to exponential phase (OD600 = 0.8 ± 0.1) at 37°C under constant shaking at 220 rpm. Cells were then harvested by centrifugation at 4,200 x g for 15 min at 4 °C and washed twice with 25 ml ice-cold lysis buffer (LB: 20 mM Hepes, 150 mM KCl, 10 MgCl2, pH 7.5). Cell pellets were resuspended in 500 μl cold LB and mixed with the same volume of acid-washed glass beads (Sigma Aldrich) and disrupted at 4 °C by 4 consecutive rounds of beads-beating at 30 sec with 4 min pause between the runs in a FastPrep-24TM 5G Instrument (MP Biomedicals). E. coli lysates were centrifuged at 16,000 x g for 15 min at 4 °C to remove cellular debris, the supernatants were collected and transferred to a fresh 1.5 ml tube and the protein concentration was determined with the bicinchoninic acid assay (BCA Protein Assay Kit, Thermo Fisher Scientific). The protein extracts were flash frozen in liquid nitrogen and stored at - 80°C until use.
For the preparation of E. coli extracts used as background proteome in in vitro LiP-MS experiments, E. coli cells were grown in 500 ml M9 minimal medium supplemented with 5 g/L glucose at 37°C under shaking at 220 rpm and harvested in exponential phase (OD600 = 0.8 ± 0.1). Proteome extracts were prepared as described above for the different carbon sources. Endogenous metabolites and nucleic acids were removed by size-exclusion chromatography (Amicon Desalting Columns 3 kDa MWCO, Merck), protein concentration was determined with the bicinchoninic acid assay (BCA Protein Assay Kit, Thermo Fisher Scientific). The protein extracts were flash frozen in liquid nitrogen and stored at -80°C until use.
Saccharomyces cerevisiae
Single colonies of the BY4742 Saccharomyces cerevisiae strain picked from a fresh plate were inoculated in synthetic complete (SC, Cold Spring Harbor Protocols, 2016) medium and grown for 6 hours at 30°C under shaking at 180 rpm. The pre-cultures were inoculated into fresh SC medium cultures to a final OD600 of 0.0003 and grown overnight at 30°C under constant shaking. When cultures reached OD600 = 0.8±0.1 the liquid medium was removed by 1 min centrifugation at 1000 x g. For the heat shock experiment, cell pellets were resuspended in the same volume of 42°C pre-warmed SC medium and incubated at 42°C for 3 min under shaking at 180 rpm. As control, the same procedure was followed but cell pellets were resuspended with 30°C pre-warmed SC medium and cell cultures were incubated at 30°C. For the osmotic stress perturbation, cell pellets were resuspended in SC medium supplemented with 0.4 M NaCl and with an equivalent volume of SC medium in the control samples, and cell cultures were incubated for 10 min at 28°C under constant shaking at 180 rpm. Next, the liquid medium was removed by 1 min centrifugation at 1000 x g and cell pellets were resuspended in lysis buffer (100 mM HEPES, 1 mM MgCl2, 150 mM KCl, pH 7.5). Liquid-nitrogen frozen beads of cell suspensions were added to grinding vials and ground in a Freezer Mill (SPEX SamplePrep 6875). To remove cell debris, samples were centrifuged at 800 x g for 5 min at 4°C. The supernatant was collected and protein concentration determined with the bicinchoninic acid assay (Thermo Fisher Scientific).
For the analysis of differentially regulated phosphorylation sites during the acute osmotic perturbation, cells were prepared as described above using untreated cells as controls, and peptide mixtures were subjected to the enrichment step (see Phosphopeptide Enrichment section below). For the analysis of reproducibility of LiP, cells were prepared as described above, but Yeast Extract–Peptone–Dextrose (YPD) medium was used instead of SC medium. Additionally, cells were washed three times in Phosphate-buffered saline (PBS) buffer before resuspension in lysis buffer.
Method Details
Sedimentation analysis
The sedimentation analysis was used to separate high molecular weight protein assemblies, such as aggregates, from soluble protein assemblies. Yeast lysates were prepared as described above and centrifuged at 100.000 x g for 20 min at 4°C (Beckman Coulter Optima TLX). The supernatant was removed and protein concentration was determined as described above. The pellet was washed with 1 volume of lysis buffer and centrifuged again at 100.000 x g for 20 min at 4°C. The pellet was resuspended in lysis buffer by vortexing for 5min at RT. Subsequently, it was cleared of debris by centrifugation at 800 x g for 5 min at 4°C. Protein concentration was determined as described above.
To assess the effect of centrifugation on soluble proteins and aggregated proteins, heterologously expressed alpha-synuclein was spiked into non-treated yeast lysates. Monomeric alpha-synuclein and amyloid-like fibrils of alpha-synuclein were obtained as described before (Feng et al., 2014) and spiked into non-treated yeast lysates at 5pmol/ug lysate. The samples were further processed as described in the “Limited proteolysis (LiP)” section.
Recombinant protein purification
E. coli pgk: All purification steps were performed at 4°C, and protein concentration was determined spectrophotometrically at 280 nm. The Pgk expression strain was obtained from the ASKA collection (Kitagawa et al., 2005); pgk was expressed as an N-terminal His6 fusion protein. Briefly, 500 mL LB cultures containing 50 μg/mL chloramphenicol were inoculated with an aliquot of an LB overnight culture diluted 1:100. Cells were grown to a final OD600 of 0.5 at 37 °C under constant shaking at 220 rpm followed by induction for 2 h with 0.5 mM isopropyl b-d-thiogalactoside at 37°C. Cells were lysed in 20 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5mM phenylmethylsulfonyl, 0.5 mg/ml Lysozyme for 45 min on ice, followed by ultrasonication. The supernatant obtained after high-speed centrifugation (20,000 g, 40 min, 4 °C) was applied to an Ni-IMAC column (GE Biotech) equilibrated in 50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 10 mM imidazole, to capture the His6 fusion protein, followed by washing with 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 25 mM imidazole. The protein was eluted with 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 200 mM imidazole and the protein containing fractions were dialyzed against 20 mM Tris-HCl, pH 7.5, and 50 mM NaCl over-night at 4°C. Proteins were concentrated with 10 kDa MWCO (Milipore) concentrators, 5% glycerol was added, and fractions were flash frozen in liquid nitrogen and stored at -80 °C.
E. coli ptsI: 6 x His-tagged PtsI overexpression strain was obtained from the ASKA collection (Kitagawa et al., 2005) and purified following previously reported procedures (Sévin et al., 2017). Briefly, 1L LB cultures containing 200 μM IPTG and 20 μg/mL chloramphenicol were inoculated with an aliquot of an LB overnight culture diluted 1:200 and were grown at room temperature with stirring for 48 hours. Cells were harvested by centrifugation, washed twice with 0.9% NaCl, 2.5 mM MgCl2 and then lysed by three freeze-thaw cycles in 30 ml of 20 mM sodium phosphate pH 7.5, 500 mM NaCl, 20 mM imidazole, 2 mM MgCl2, 2 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, Lysoenzyme and DNAseI. Clear lysates were centrifuged for 30 min. at 4 °C (14000 g), and purified using His GraviTrap TALON columns (1 mL column volume, GE Healthcare) following manufacturer’s instructions. After elution with 20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, pH 7.5, pure protein was re-buffered four times with enzyme assay buffer using ultrafiltration columns with 10 kD cut-off (Millipore) and stored at 4°C. The enzyme assay buffer was 25 mM potassium-phosphate buffer pH 7.0 with 2.5 mM MgCl2.
Limited proteolysis (LiP)
Each proteome extract was split into a control sample, which was subjected to only tryptic digestion and used to measure protein abundance changes, and a LiP sample, containing information about protein structural changes, which was subjected to a double-protease digestion step with a nonspecific protease followed by complete digestion with trypsin. Both samples contained 100 μg of extracted proteome. Proteinase K from Tritirachium album (Sigma Aldrich) was added to the LiP samples at an enzyme/substrate (E:S) ratio of 1:100 (w/w) and incubated for 1 min (E.coli experiment) or 3 min (S. cerevisiae experiment) at 25°C. A corresponding volume of water was added to the control samples. Digestion reactions were stopped by heating LiP samples for 5 min at 98°C in a thermocycler followed by addition of sodium deoxycholate (Sigma Aldrich) to a final concentration of 5%. The same procedure was applied to control samples. Both LiP and control samples were then subjected to complete tryptic digestion in denaturing conditions as described below (tryptic digestion). The in vitro LiP experiment was performed by spiking in 10 μg purified E. coli pgk into 100 μg E. coli lysate cleared of endogenous metabolites, as described above (sample preparation). Cell lysates were incubated with the pgk substrate 3-phosphoglycerate (3PG) to a final concentration of 5mM, 10mM, 15mM, 20mM, and 25mM for 5 min at 25°C. As control, a cell lysate without metabolite addition was used. The metabolite solutions were freshly prepared from ultra-pure powders in 100mM HEPES, pH 7.5. After solubilization, pH was double-checked with pH strips. LiP experiments were carried out on both the lysate after metabolite addition and on control samples. For the analysis of reproducibility of LiP, samples were treated as described here, but the incubation with Proteinase K was prolonged to 5 min.
Tryptic digestion
Proteins fragments generated in the previous step were reduced by incubation of samples with tris(2-carboxyethyl)phosphine (Thermo Fisher Scientific) to a final concentration of 5 mM for 30 min at 37 °C. Next, the alkylation of free cysteine residues was achieved by adding iodoacetamide (Sigma Aldrich) to a final concentration of 40 mM for 30 min at 25°C in the dark. Samples were diluted with freshly prepared 0.1 M ammonium bicarbonate to a final concentration of 1% sodium deoxycholate. Samples were predigested with lysyl endopeptidase LysC (Wako Chemicals) at an enzyme/substrate ratio of 1:100. After 2 hours at 37°C, sequencing-grade porcine trypsin (Promega) was added to a final enzyme/substrate ratio of 1:100, and samples were incubated for 16 h at 37°C under shaking at 800 rpm. Protease digestion was quenched by lowering the reaction pH (< 3) The peptide mixtures were loaded onto Sep-Pak tC18 cartridges or 96 wells elution plates (Waters), desalted, and eluted with 80% acetonitrile, 0.1% formic acid. After elution from the cartridges, peptides were dried in a vacuum centrifuge, resolubilized in 0.1% formic acid, and analyzed by mass spectrometry. For the analysis of reproducibility of LiP, samples were treated as described here, but LysC and trypsin were added at the same time and samples incubated for 16 h.
Phosphopeptide enrichment
After the peptide clean-up step, each sample was diluted in 280 ml phtalic acid (PA) solution (86.7 mg/ml PA, 80% acetonitrile, 3.5% trifluoroacetic acid (Thermo Scientific)) by vortexing and sonicating for 5 min. The peptide solution was centrifuged at 16,000 x g for 5 min to remove solid debris. The peptide solution was transferred to Mobicol spin columns containing titaniumdioxide (TiO2) beads (GL Science) that had been washed with 280 ml of methanol by vortexing in short pulses and centrifuged at 800 x g, and equilibrated with 280 ml of PA solution by vortexing in short pulses and centrifuged at 800 x g. Transfer of peptide solution to the spin columns was achieved as follows: the bottom of the Mobicol columns was closed with a small plug and the columns were vortexed to mix the peptide solution with the TiO2 beads and incubated for 30 min at room temperature under end-over-end rotation. After incubation, the beads were washed twice with 280 ml of PA solution (load, vortex, spin down at 800 g) followed by two washing steps with 280 ml of 80% acetonitrile, 0.1% trifluoroacetic acid, two washing steps with 280 ml of 40% acetonitrile, 0.1% trifluoroacetic acid and two washing steps with 280 ml of 0.1% trifluoroacetic acid. For the elution, 280 ml of 0.3 M NH4OH were added to the beads, which were then incubated for 3 min at room temperature and centrifuged at 800 x g. The elution was performed a second time with the same parameters. The solution was acidified immediately after elution with 40 ml of 25% trifluoroacetic acid. The cleanup of phosphopeptides was performed as described in the “tryptic digestion” step using Sep-Pak tC18 cartridges (Waters). Dried phosphopeptides were re-solubilized with 30 μl 0.1% formic acid prior to analysis by mass spectrometry.
LC-MS/MS data acquisition
Peptide digests for LiP, trypsin-only control and phospho-enriched samples were analyzed on an Orbitrap Q Exactive Plus mass spectrometer (Thermo Fisher) equipped with a nanoelectrospray ion source and a nano-flow LC system (Easy-nLC 1000, Thermo Fisher). Peptide digests for LiP and trypsin-only control samples of heat stressed yeast cells in the sedimentation analysis, as well as peptide digests of the in vitro LiP-MS samples, were analyzed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher) equipped with a nanoelectrospray ion source and an UPLC system (ACQUITY UPLC M-Class, Waters).
For shotgun LC-MS/MS data dependent acquisition (DDA), 1 μl peptide digests from each biological replicate of LiP, trypsin-only control and phospho-enriched samples were injected independently at a concentration of 1 mg/ml. 1 μl of the same samples were also measured in data-independent acquisition (DIA) mode. Peptides were separated on a 40 cm x 0.75 μm i.d. column packed in-house with 1.9 μm C18 beads (Dr. Maisch Reprosil-Pur 120). For LC fractionation, buffer A was 0.1% formic acid and buffer B was 0.1% formic acid in 100% acetonitrile using a linear LC gradient from 5% to 25% or 5% to 35% acetonitrile, respectively, over 120 min and a flowrate of 300 nL/min and the column was heated to 50°C.
For DDA measurement on the Orbitrap Q Exactive Plus, MS1 scans were acquired over a mass range of 350-1500 m/z with a resolution of 70,000. The 20 most intense precursors that exceeded 1300 ion counts were selected for collision induced dissociation and the corresponding MS2 spectra were acquired at a resolution of 35000, collected for maximally 55 ms. All multiply charged ions were used to trigger MS-MS scans followed by a dynamic exclusion for 30 s. Singly charged precursor ions and ions of undefinable charged states were excluded from fragmentation.
For DIA measurements, 20 variable-width DIA isolation windows were recursively acquired. The DIA isolation setup included a 1 m/z overlap between windows, as described in (Piazza et al., 2018). DIA-MS2 spectra were acquired at a resolution of 17500 with a fixed first mass of 150 m/z and an AGC target of 1 x 106. To mimic DDA fragmentation, normalized collision energy was 25, calculated based on the doubly charged center m/z of the DIA window. Maximum injection times were automatically chosen to maximize parallelization resulting in a total duty cycle of approximately 3 s. A survey MS1 scan from 350 to 1500 m/z at a resolution of 70,000, with AGC target of 3 x 106 or 120 ms injection time was acquired in between the acquisitions of the full DIA isolation window sets.
For DDA measurement of the sedimentation analysis on the Orbitrap Fusion Lumos Tribrid, MS1 scans were acquired over a mass range of 350-1400 m/z with a resolution of 120,000. Survey spectra were scheduled for execution at least every 3 s, with the embedded control system determining the number of MS/MS acquisitions executed during this period. Precursors were selected for higher-energy collision dissociation and the corresponding MS2 spectra were acquired at a resolution of 30,000, collected for maximally 54 ms. All multiply charged ions were used to trigger MS-MS scans followed by a dynamic exclusion for 30 s. Singly charged precursor ions and ions of undefinable charged states were excluded from fragmentation. DIA measurements consisted of a survey MS1 scan from 300 to 2000 m/z at a resolution of 120,000, with a normalized AGC target of 200% or 100 ms injection time, followed by the acquisition of 41 DIA isolation windows spanning 16m/z. The DIA isolation setup included a 1 m/z overlap between windows.-DIA-MS2 spectra were acquired at a resolution of 30,000 with a fixed first mass of 358 m/z and a normalized AGC target of 200%. To mimic DDA fragmentation, normalized collision energy was 28, calculated based on the doubly charged center m/z of the DIA window. Maximum injection times was set at 54 ms.
For DDA measurements of the in vitro samples acquired with an Orbitrap Fusion Tribrid mass spectrometer, MS1 spectra were acquired from 300 to 1500 m/z at a resolution of 120,000. Survey spectra were scheduled for execution at least every 3 s, with the embedded control system determining the number of MS/MS acquisitions executed during this period. Precursors were selected for higher-energy collision dissociation and the corresponding MS2 spectra were acquired at a resolution of 30,000, collected for maximally 54 ms. All multiply charged ions were used to trigger MS-MS scans followed by a dynamic exclusion for 25 s. Singly charged precursor ions and ions of undefinable charged states were excluded from fragmentation. The DIA acquisition method for the in vitro samples acquired on the Orbitrap Fusion consisted of a survey MS1 scan from 300 to 1500 m/z at a resolution of 120,000, with AGC target of 4 x 105 or 50 ms injection time, followed by the acquisition of 20 variable-width DIA isolation windows. The DIA isolation setup included a 1 m/z overlap between windows.-DIA-MS2 spectra were acquired at a resolution of 30,000 with a fixed first mass of 150 m/z and an AGC target of 5 x 104. To mimic DDA fragmentation, normalized collision energy was 28, calculated based on the doubly charged center m/z of the DIA window. Maximum injection times were automatically chosen to maximize parallelization resulting in a total duty cycle of approximately 3 s.
The experiments for the analysis of reproducibility of LiP were acquired with a Q Exactive HF mass spectrometer (Thermo Fisher) equipped with a nanoelectrospray ion source and an UPLC system (ACQUITY UPLC M-Class, Waters). For DDA measurements, MS1 spectra were acquired from 300 to 1500 m/z at a resolution of 120,000. The 12 most intense precursors were selected for collision induced dissociation and the corresponding MS2 spectra were acquired at a resolution of 30000, collected for maximally 50 ms. All multiply charged ions were used to trigger MS-MS scans followed by a dynamic exclusion for 30 s.
Peptide and protein identification
The collected DDA spectra were searched against the E. coli (strain K12) Uniprot fasta database (version October 2017) and the S. cerevisiae (strain S288c) Uniprot fasta database (version November 2016) using the SEQUEST HT® database search engine (Thermo Fisher Scientific). Up to two missed cleavages were allowed, cleavage of KP and RP peptide bonds were excluded. For LiP samples, a semi-specific tryptic digestion rule type was applied. Cysteine carbamidomethylation (+57.0214 Da) and methionine oxidation (+15.99492) were allowed as fixed and variable modifications, respectively. In case of phosphopeptide search, the phosphorylation of serines, threonines and tyrosines (+79.966 Da) was defined as a variable modification. Monoisotopic peptide tolerance was set to 10 ppm, and fragment mass tolerance was set to 0.02 Da. The identified proteins were filtered using the high peptide confidence setting in Proteome Discoverer (version 2.2, Thermo Fisher Scientific), which correspond to a filter for 1% FDR on peptide level. For spectral library generation the software Spectronaut (Biognosys AG, version 13) was used with default settings. The spectral libraries contained normalized retention time iRT values for all peptides.
For the analysis of reproducibility of LiP, the collected DDA spectra were searched against a custom fasta database containing all open reading frames of Saccharomyces cerevisiae which was kindly provided by Rachel Brem. Andromeda and MaxQuant (Version 1.5.2.8) (Cox and Mann, 2008) were used to identify and quantify peptides using default settings, except for the activation of match between runs and digestion mode which was set to semi-specific.
PtsI activity assays
Approximately 8 μg of purified ptsI was incubated at 37 °C in 200 μL enzyme assay buffer as described previously (Doucette et al., 2011). Briefly, the enzyme assay was equilibrated for 30 min with 4 mM pyruvate and 1 mM phosphoenolpyruvate in the presence or absence of 25 mM fructose-bis-phosphate. All stock solutions were prepared in assay buffer and adjusted for pH. After addition of 4 mM U-13C pyruvate, 10 μl samples were sampled over time and mixed with 80 μl of ice-cold methanol to quench the reaction by enzyme denaturation. Relative reactant abundance was measured by time-of-flight mass spectrometry as described previously (Fuhrer et al., 2011). Each assay was repeated with four experimental replicates and data analysis was done with Matlab (The Mathworks, Natick) using functions embedded in the Bioinformatics and Statistics toolboxes as previously described (Fuhrer et al., 2011). Negatively charged ions were tentatively annotated as phosphoenolpyruvate and pyruvate based on accurate mass using 0.001 Da tolerance assuming simple deprotonation ([M-H]-) for monoisotopic pure mass and the fully labelled isotope mass ([M+3-H]-). Time-course data of labelled fraction of phosphoenolpyruvate was fitted by weighted non-linear least-squares regression in Matlab to estimate the rate constant of fully labelled phosphoenolpyruvate formation as described previously (Doucette et al., 2011).
Quantification and Statistical Analysis
LiP-MS data analysis
Targeted data extraction of DIA-MS acquisitions was performed with Spectronaut (Biognosys AG, version 13) with default settings, using spectral libraries generated as described above. Briefly, the dynamic mass tolerance strategy was applied to calculate the ideal mass tolerances for data extraction and no correction factor was applied (correction factor = 1). The local (non-linear) regression method was used for iRT calibration using the iRT kit peptides. The mutated decoy method was used to generate label-free decoys. Interference correction was enabled to exclude fragment ions with interferences from quantification across all runs but keeping at least three fragments for quantification. In the LiP samples only fully- and semi-tryptic peptides that were uniquely present in the sequence of one protein of the database (proteotypic peptides) were used for quantification, while in control samples only fully-tryptic proteotypic peptides were used for protein abundance. The false discovery rate (FDR) was estimated with the mProphet approach (Reiter et al., 2011) and set to 1% on peptide and protein level. Protein inference was performed using the implemented IDPicker algorithm to define protein groups (Zhang et al., 2007). Comparison analysis of protein (control samples) and peptide (LiP samples) levels was performed with the MSstats package (Choi et al., 2014). Spectronaut normalized peak areas were used as intensity values. Data were then processed with the “dataProcess” function which includes logarithm transformation with base 2 of intensities, median normalization, feature selection (all fragment ions in the dataset were selected) and imputation of missing values by AFT (accelerated failure time model). The “groupComparison” function using linear mixed-effects model was finally used to compare peptide and protein abundances between conditions. For each conditional comparison, MS stats provides model-based estimates of fold changes as well as p-values that are adjusted for multiple testing (q-values) using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). Significant protein abundance changes were used to correct LiP-peptide abundance changes (LiP samples) by dividing peptide-level abundance ratios by the significant abundance ratio of the respective protein. For proteins that did not significantly change abundance, a normalization factor of 1 was used (i.e., no correction). In the E. coli dataset, generated by exposing cells to the long-term metabolic perturbation which largely altered protein and peptide abundance levels, we used stringent cutoffs (|log2FC| >2, q-value < 0.05) to select for significant changes. In the S. cerevisiae datasets in contrast, where few proteins changed abundance due to the short perturbation time, the following cutoffs were applied: |log2FC| >1, q-value < 0.05).
The data for LiP reproducibility was analyzed in R (Version 3.6.1). The principle component analysis was based on consistently identified modified peptide sequences with centered and scaled intensities. The heatmap was generated using the pheatmap package (version 1.0.12) and was also based on consistently identified modified peptide sequences. Pearson correlation was used as distance measure for the LiP runs. The differential analysis used the proteusLabelFree package (Version 0.1.6; Gierlinski et al., 2018) which is based on limma (Smyth, 2004). Before comparing relative abundance, the data was median normalized and filtered for proteotypic peptides. Resulting p-values were finally adjusted by multiple testing using the Benjamin-Hochberg method (Benjamini and Hochberg, 1995). The output of this statistical analysis was filtered using the following cutoffs: q-value < 0.05 and |log2FC|>1.
Protease digestion accessibility analysis
Targeted data extraction of DIA-MS acquisitions was performed with Spectronaut (Biognosys AG, version 13) as described above. LiP samples and control samples were processed together and only fully tryptic peptides that were uniquely present in the sequence of one protein of the database (proteotypic peptides) were used for quantification. Spectronaut normalized peak areas were used as intensity values. Data were processed using the statistical software R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/) and the mean and standard deviation of peptides was calculated. The digestion accessibility was assessed as the ratio of the mean peptide intensity in LiP samples to the mean peptide intensity in trypsin-only control samples and was then compared between conditions using a Welch modified two-sample t-test. The resulting p-values were adjusted for multiple testing (q-values) using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) For the alpha-synuclein (a-syn) control (Figure 3D), we compared protease accessibility between alpha-synuclein monomer and fibrils spiked-in to yeast lysates. For aggregators (Figure 3E), we did two comparisons. We compared proteins recovered in the pellet (P2) after heat shock, relative to the soluble fraction (S2) without heat shock. We also compared proteins recovered in the pellet (P2) to proteins present in the bulk lysate (L1) prior to ultracentrifugation, both in the heat shocked sample. For all comparisons, the resulting p-values were adjusted for multiple testing (q-values) using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995).
Structural barcodes
To visualize our data as structural barcodes, we represent the change in proteolytic fingerprint along the sequence of a protein (N- to C-terminus) using the following color code: regions that show an increase/decrease in proteolytic resistance based on changing peptide intensity (|log2FC| >1, q-value < 0.05) in red, blue or yellow (see details below), regions where peptides are detected by MS but are not structurally altered in grey, and regions where peptides were not detected by MS in black. We show two types of barcodes in the manuscript. For proteins of the osmotic stress pathway (Figure 2), for pgk (Fig. 5E), for Hsp104 (Figure S5C), and for ptsI (Figure 6C), we depict peptides that change in intensity between conditions in yellow. For aggregators and the a-synuclein control in the ultracentrifugation study (Figures 3D–3E, S4 and S5C) we determined protease accessibility for each protein as described in the section “Protease digestion accessibility: peptide quantification and statistical analysis” using only fully tryptic peptides for this analysis and also introducing a color code (blue versus red) to pinpoint regions that become more versus less accessible to proteolysis, as this information is useful to identify aggregation interfaces.
Phosphopeptide data analysis
DDA data relative to phosphopeptide-enriched samples were analysed with Progenesis QI (Nonlinear Dynamics, version 2.0). Raw LC-MS/MS files were imported into Progenesis for MS1 feature alignment using the automatic alignment algorithm followed by manual revision and adjustment of the aligned chromatograms. Peak picking was then performed setting the maximum ions charge to 5. Peptide ion abundances were normalized using an automatically selected run as normalisation reference to allow comparisons across the different samples. An analysis of variance (ANOVA) was applied to all peptide ions. MS/MS spectra were exported in the .mgf format and searched against a yeast database using Proteome Discoverer (Thermo Fisher Scientific, version 2.2), as described above (Peptide and protein identification and spectral library generation). The resulted pepXML files containing peptides identified in the search were filtered with a false discovery rate (FDR) of 1% and imported into Progenesis. Peptide identifications from MS/MS spectra were mapped to the corresponding peptide ions detected in MS1 spectra, according to their accurate m/z and retention time and areas under the extracted ion chromatograms. The list of quantified peptide was exported in the .csv format. The R-framework based analysis tool SafeQuant (version 2.3.1) was used for statistical validation of differentially expressed phosphopeptides during osmotic stress. Peptide abundance values were used for statistical testing of differentially abundant phospho-peptides using an empirical Bayes moderated t-test as implemented in the R/Bioconductor limma package. Resulting p-values were finally adjusted by multiple testing using the Benjamin-Hochberg method (Benjamini and Hochberg, 1995). The output of this statistical analysis was filtered using the following cutoffs: q-value < 0.05 and |log2FC|>1. Moreover, phosphopeptides mapping to proteins for which we detected a significant change in abundance during osmotic stress (q-value < 0.05 and |log2FC|>1) were excluded from the analysis. Phosphorylation site abundances were calculated by grouping peptides reporting the same phospho modification and calculating fold change as the mean of all the peptide fold changes relative to the same modification. Phophorylation sites with a coefficient of variation (standard deviation to the mean) higher than 0.2 were filtered out. To statistically assess the combination of the qvalues, a Fisher's combined probability test has been applied to combine q-values using the “combine_pvalues” function of the open-source python-based Scipy library.
Functional Enrichment Analysis
We tested the proteins with significant changes in abundance or structure as defined above (in the “LiP-MS data analysis: peptide quantification and statistical analysis” section) for functional enrichments using the topGO-package in R (Alexa et al., 2006). We tested proteins with significant changes in abundance separately from those with structural changes, to be able to identify enrichments specific for each set. For the Escherichia coli but not the yeast data set we also tested for functional enrichments in the set of proteins that either change abundance or structure, and separately, for enrichments in proteins with at least one significant LiP-peptide reporting a structural change regardless of whether the trypsin-only control showed abundance changes. Each condition was tested separately. As seven conditions were measured for Escherichia coli, 28 enrichment analyses were performed per ontology for this species. For the Saccharomyces cerevisiae data set, we had measured the effects of two stressors on the proteome of Saccharomyces cerevisiae and therefore performed four enrichment analyses per ontology. We performed analyses in the ontologies Biological Process, Molecular Function, and Cellular Component.The enrichment analysis only considered those proteins for which abundance or structure was measured in the specific condition that was tested. Proteins with changes in abundance were tested against a background of proteins for which abundance was measured. Proteins with changes in structure were tested against a background of proteins for which structural signatures were measured.We downloaded current annotation files for S. cerevisiae (http://current.geneontology.org/annotations/sgd.gaf.gz, accessed May 11, 2020) and E. coli (http://current.geneontology.org/annotations/ecocyc.gaf.gz, accessed June 2 2020). To focus on the most informative terms, we tested for enrichments with Fisher’s exact tests using the elim-algorithm in topGO (Alexa et al., 2006). Here, genes that are annotated to a significantly enriched term are not included in the tests of parental terms. We tested terms with a minimum of 3 annotated genes in Escherichia coli and 5 annotated genes in Saccharomyces cerevisiae. Terms with more than 500 annotated genes were excluded for both species. The number of genes annotated to a term was determined separately for each enrichment analysis based on the measured proteins.
To be able to compare functional enrichments of terms across different analyses in an unbiased way, we removed redundant terms with the following procedure. First, all significant terms in an analysis were identified. For all figures showing functional enrichments other than Figure S6 (Biological Processes), all terms with an uncorrected p-value of 0.01 or smaller were considered significant. For Figure S6 (Biological Processes), we applied a significance cutoff of a corrected p-value of 0.1 or smaller. Then the relative overlap of these terms was computed based on all annotated proteins of the respective species. Next, we determined which pairs of terms were strongly overlapping. Terms were considered to strongly overlap if 70% of the genes annotated to the first term were also annotated to the second term and 70% of the genes annotated to the second term were also annotated to the first term for Escherichia coli. This threshold was changed to 50% for Saccharomyces cerevisiae, as more analyses were combined in the case of Escherichia coli. For these pairs of terms, we removed the term that was contained in the other term of the pair to a larger degree. Table S1 and Table S3 contains all significant terms for each enrichment analysis. The grey scale in the plots show p-values computed with Fisher’s exact tests using the classic algorithm, i.e. by using all annotated genes that were measured for the term.
We did not correct p-values generated with the elim algorithm from topGO for the following reasons. The elim algorithm conditions functional terms on significant child terms so that genes from these significant child terms are not considered when testing the parental terms. This is expected to result in inflated p-values for the parental terms, as the genes of insignificant child terms are still used. As recommended by the authors of the approach, we do not perform further corrections for multiple testing, as these p-values are viewed to be conservative [https://bioconductor.org/packages/release/bioc/vignettes/topGO/inst/doc/topGO.pdf]. However, to illustrate that our conclusions hold also when multiple testing correction is applied, we also show this for the biological processes ontology in Figure S1D. Here, we corrected p-values for multiple testing with the Benjamini-Hochberg method, as implemented in the p.adjust-function from the stats package in R (R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/). This correction was performed per enrichment analysis and ontology.
Enrichment analysis: chaperones & aggregators
We tested if chaperones proteins were over-represented in the S. cerevisiae datasets by performing a Fisher’s exact test using the “fisher_exact” function of the open-source python-based Scipy library, with the alternative parameter set to “greater”. The list of 63 yeast chaperones reported in (Gong et al., 2009) was used as reference set. The same analysis was repeated to test for the enrichment of aggregating or misfolded proteins in the heat shock dataset. The reference list of proteins aggregating or misfolding during heat shock was obtained from (Wallace et al., 2015). As in that paper, we defined as “aggregator” or “misfolded” a protein which was pelletable after heat shock. We finally assess the presence of chaperone-client relationships between the two groups of enriched proteins, chaperones and aggregators/misfolded proteins, using the list of yeast chaperone physical interactors retrieved from the BioGRID database (v3.5, https://thebiogrid.org/). We restricted the analysis to those interactions detected either by “affinity capture-MS” or by “affinity capture-western” experimental methods and performed the enrichment analysis as described above.
Network analysis of phosphoproteomics data
We linked structural changes upon yeast osmotic stress to the activity of regulated kinases or phosphatases (Figure 3A) in the following way. We used our yeast phosphoproteomics data and known kinase/substrate relationships to perform a kinase-substrate enrichment analysis, estimating the activity for the 58 kinases/phosphatases with at least three detected known targets and identifying kinases and phosphatases whose activities are increased or decreased upon osmotic stress. We estimate kinase activities following a variation of KSEA. The kinase-substrate network (KSN) was obtained from Biogrid (https://downloads.thebiogrid.org/File/BioGRID/Release-Archive/BIOGRID-3.5.186/BIOGRID-PTMS-3.5.186.ptm.zip) and the viper algorithm (Alvarez et al., 2016) was used with p value-associated z scores of phosphorylation sites as input. The "eset.filter" parameter of the viper function was set to false. When a phosphorylation site was present on multiple peptides, the peptide with the highest positive fold change was kept as a proxy of the phosphorylation site fold change. When building the network, we included kinases and phosphates that had an absolute normalised enrichment score (NES) over 1.7 standard deviation and at least three detected target phosphosites. These were connected to their measured phosphorylation site targets (according to Biogrid KSN). From those, phosphorylation site that were present on a protein that displayed at least one conformationally changing peptide with a Qvalue <= 0.05 were displayed. All code used for this analysis are available https://github.com/saezlab/conformationomic_yeast_picotti_2020.git
The analysis revealed a total of 12 kinases or phosphatases with significantly altered activity upon osmotic stress (as expected, this included Hog1 and two other MAP kinases of the HOG1 signaling pathway, which showed increased activity under these conditions). We then examined targets of these activated enzymes, identifying specifically those that displayed both altered phosphorylation and a structural change.
Curve fitting by regression analyses
A linear regression analysis was performed to investigate the relationship between metabolic fluxes and LiP peptide changes for all enzymes of the CCM for which 13C-based metabolic fluxes had been previous measured (Gerosa et al., 2015). First, we converted the estimated absolute fluxes to logarithmic fold changes, comparing each of the seven growth conditions to glucose, the carbon source we used as reference in the proteomics analysis. To identify significantly changing fluxes, absolute fluxes and their standard deviations were used to perform a t-test comparing each condition to glucose. A cutoff of 0.05 was applied to p-values adjusted for multiple testing (Benjamini and Hochberg, 1995). LiP-MS data were used for the regression analysis if q-value < 0.05 and |log2FC| > 1 in at least 4 conditions. A least-squares regression analysis was performed with the “stats.linregress” function of the open-source python-based Scipy library. The Wald test was finally applied to calculate the p-value for a hypothesis test whose null hypothesis is that the slope is zero. Calculated p-values were then adjusted for multiple test correction using the using the Benjamin-Hochberg correction method (Benjamini and Hochberg, 1995). Regression models were selected using a cutoff of 0.05 for the adjusted p-value and 0.7 for R-squared.
The same analysis was repeated to investigate the relationship between protein structural changes (LiP fold-changes) and the concentration of 26 metabolites (Table 8) known to regulate the CCM. Measurements of metabolites concentration over the 8 growth conditions was previously reported in (Gerosa et al., 2015). For the regression analysis, ratios to glucose were used. Peptides showing a linearity with metabolite concentrations where finally classified as “known” if the metabolite-protein (to which the peptide map) was reported in the EcoCyc database (https://ecocyc.org/) or in the BRENDA database (http://www.brenda-enzymes.org/). Moreover, we reported if the same interaction was identified through the LiP-SMap approach (Piazza et al., 2018) and/or ligand-detected NMR (Diether et al., 2019). Concentration-dependent structural effect curves for LiP peptides identified in the in vitro experiment were generated by plotting peptide abundance changes (log2FC) over the substrate concentration range. To investigate if LiP peptides followed a dose-response curve we selected peptides significantly changing (|log2FC| >2, q-value < 0.01) over at least 4 substrate concentrations. We then fit a higher-order polynomial regression using the “stats.linregress” function of the open-source python-based Scipy library with the parameter “order” set to 2 and select peptides following a hyperbolic curve. The lower limit of the peptide abundance change was set to zero to allow proper fitting.
All of the correlation analyses of LiP peptides versus flux and of LiP peptides versus metabolite levels can be accessed at https://doi.org/10.5281/zenodo.3964994 (for fluxes) and at https://doi.org/10.5281/zenodo.3965002 (for metabolite levels).
Metabolite-protein interaction analysis
To explain structural changes observed across pairs of metabolic conditions, we examined metabolite-protein interactions previously identified by (Piazza et al., 2018) and metabolomics data acquired under 8 metabolic conditions by (Gerosa et al., 2015). We asked for which of the interactions previously detected in vitro by exogenously adding metabolites we could see the identical structural change in vivo when levels of the same metabolites were physiologically regulated (Figure S7G), as follows. Based on the metabolomics data, we identified metabolites that changed their concentration at least 3-fold in each metabolic condition relative to growth in glucose. For these altered metabolites, we asked which proteins had been found by (Piazza et al., 2018) to interact with the metabolite. For these proteins we asked whether exactly the same marker peptides that changed upon addition of the metabolite to an E. coli lysate based on (Piazza et al., 2018) also changed upon the metabolic transition during which the concentation of the metabolite was regulated. We only retained peptides from (Piazza et al., 2018) that were regulated in the same direction at the two metabolite concentrations tested in that study. We further required that the in vivo change occurred in a direction consistent with both the in vitro observation and with the in vivo metabolite concentration change. For example, if addition of a metabolite to the E. coli lysate triggered the up-regulation of a given LiP peptide based on (Piazza et al., 2018), we required that the same peptide is up-regulated when the endogenous metabolite concentration is increased or that it is down-regulated when the endogenous metabolite concentration is decreased. We considered the interactions that passed these filters as interactions regulated in vivo across the considered pair of metabolic conditions. Using this approach, we identified 121 metabolite-protein interactions.
3D analysis of protein structural alterations
Selected LiP peptides from the different experiments were mapped to representative protein structures to investigate which regions of the different proteins (e.g. active site, allosteric site) were affected by the structural alterations. Structures of holocomplexes between protein and ligands were selected to position allosteric and active sites. For the yeast data dataset when only the apo- version of a protein was available homology models were built using holocomplexes from homologous proteins that have protein sequence similarity higher or equal to 30%. For those cases where experimental data was not available, we use the predicted active site residues as annotated in Uniprot for positioning metabolite binding. Using a custom-made PyMOL-Python script, we measured the minimal Euclidean distance in angstroms (Å) between all the atoms of the LiP peptide and those of the substrate or allosteric regulator, if present, or alternatively all the atoms of the peptide or amino acid defining the predicted active site (as reported in the Uniprot database). A peptide was assigned to a known functional site if the measured minimal distance was less than 6.4 Å, based on a previous study (Piazza et al., 2018).
Molecular Dynamics simulation
To determine the binding mode of FBP to the phosphoenolpyruvate-binding domain of enzyme I (EI) of the phosphoenolpyruvate-protein phosphotransferase system (encoded by ptsI and labeled as such hereafter), we first analyzed six ptsI structures from different bacterial species, namely, 2HWG (Teplyakov et al., 2006), 2BG5 (Oberholzer et al., 2005), 2HRO (Márquez et al., 2006), 2WQD (Oberholzer et al., 2009), 2XZ7 (Navdaeva et al., 2011), and 2XZ9. Our target protein is E. coli ptsI, but its structure 2HWG does not show a suitable space in the catalytic site for placing FBP because of the interplay between PEP-binding domain and His-domain. Based on our structural analysis, we chose the structure 2XZ7 as a surrogate for model building as only the PEP-binding domain is present. Although this structure is the T. tengcongesis ptsI, key residues in the active site are the same as in the ptsI of E. coli. This structure contains the necessary catalytic components, including PEP, a cofactor magnesium ion and its coordinated water molecules. Moreover, this structure shows a relatively open active site compared to others, which allows the initial placement of FBP in the active site.
Before docking FBP to the active site, PEP was removed, and all titratable residues were protonated to their correct states at pH 7. Because of the high conservation in the binding site between different species, we did not mutate any residue from the structure. The 3D structure of FBP was extracted from the X-ray structure 3D1R (Brown et al., 2009). We determined its protonation state with MarvinSketch (MarvinSketch 19.25, 2019, ChemAxon (http://www.chemaxon.com)), and added hydrogen atoms with Maestro (Maestro, Schrödinger, LLC, New York, NY, 2020.). The initial poses were obtained by the docking software AutoDock Vina 1.1.2 (Trott and Olson, 2010). The docking site was defined by a 303030 Å cubic grid box and centered on the PEP coordinates. We set the parameter “exhaustiveness” to 100 to enhance the configurational sampling of the binding poses and left the rest of parameters as the default. Finally, the ten best-ranked poses were saved for further analysis.
Two major binding modes emerged from a clustering of these ten best-ranked poses. Thus, we selected two representative docking poses for molecular dynamics (MD) based optimization. Each of the ptsI-FBP complex structures was solvated in an 80 Å rhombic dodecahedron TIP3P water box (Jorgensen et al., 1983), which ensured a 10 Å of buffer distance between protein atoms and the boundary of the water box. Sodium chloride (0.15 M) was added to the solvated systems to neutralize them and mimic physiological conditions. The CHARMM36 force field (Huang and MacKerell, 2013) was used for the ptsI protein and FBP was parametrized by the CGenFF force field (Vanommeslaeghe et al., 2010). Each complex was initially minimized by 10,000 steps of the conjugate gradient algorithm under a series of restraints and constraints to remove bad contacts and geometry. The minimized structure was heated to 300 K and equilibrated in an NVT condition (constant volume and temperature). Finally, the structure was further equilibrated in an NPT condition (constant pressure and temperature). The heating-up and equilibration phases lasted for 1 ns using the CHARMM program (version 42b2) (Brooks et al., 2009).
Production MD simulations were carried out in NPT conditions using NAMD 2.13 (Phillips et al., 2005). The pressure was controlled by the Nosé–Hoover Langevin piston method with a 200 ps piston period and 100 ps piston decay time (Feller et al., 1995; Martyna et al., 1994). The temperature was maintained at 300 K using the Langevin thermostat with a 5 ps friction coefficient. The integration time step was set to 2 fs by fixing all bonds connecting hydrogen atoms by the SHAKE algorithm. van der Waals energies were calculated using a switching function with a switching distance from 10 to 12 Å and long-range Lennard-Jones interactions was taken into account (Shirts et al., 2007). Electrostatic interactions were evaluated using the particle mesh Ewald summation (PME) method (Essmann et al., 1995) with a 1 Å of grid spacing. For each of the two starting poses, five independent 30-ns runs were carried out with different initial velocities. Thus, a cumulative sampling of 150 ns (7,500 snapshots) was collected from each of the two starting poses.
We downsampled the 7,500 snapshots of each system to 750 snapshots with an even spacing and then clustered them based on Root-Mean-Square Deviation (RMSD) matrix analysis. Specifically, we first mutually superposed the 750 snapshots to each other based on the ptsI active site atoms (5 Å around the docked FBP). We calculated the RMSD of FBP for each pair of snapshots, leading to a 750750 RMSD matrix. We then applied the clustering algorithm implemented in the routine “correl” of the CHARMM package to the RMSD matrix. As a result, 10 and 8 clusters were obtained for the MD sampling of the two starting poses, respectively. Finally, two representative poses were extracted from their respective largest clusters (one for each starting pose), and used as reference structure for the calculation of RMSD time series of FBP. One of the two poses was substantially more stable according to the RMSD time series, and was selected for Figure 6D.
Data visualization and processing
Data visualization, exploratory data analysis and processing were performed using Python (version 2.7 and 3.0) and the Python library Pandas (version 0.18.1). Heat-map diagrams, bar plots and regression model plots were created using the python data visualization library Seaborn (version 0.9.0).
Acknowledgments
We thank Judith Frydman (Stanford University), Matthias Heinemann (University of Groningen) Elad Noor and Uwe Sauer (ETH Zurich) for insightful discussions and Jan-Philipp Quast (ETH Zurich) and Franco Picotti for assistance with structural images. T.H., I.P., and L.M. were supported by long-term EMBO postdoctoral fellowships (ALTF 1240-2016, ALTF 846-2014, and ALTF 538-2016). Y.L. was supported by the International Postdoc Grant funded by the Swedish Research Council (VR 2019-00608). A.C. is funded by an Excellence grant of the Swiss National Science Foundation (SNSF grant 310030B-189363). This project received funding from the European Research Council (grant agreement no. 866004) and through the EPIC-XS Consortium (grant agreement no. 823839) both under the European Union’s Horizon 2020 research and innovation program. P.P. is also funded by a Personalized Health and Related Technologies (PHRT) grant (PHRT-506) and a Sinergia grant from the Swiss National Science Foundation (SNSF grant CRSII5_177195).
Author contributions
V.C. led bioinformatics analysis across the project. V.C., I.P., and T.H. designed and performed LiP screens and analyzed the data. T.H., L.G., and T.F. conducted in vitro experiments. M.P., L.M., P.B., and C.D. contributed to experiments. Y.Z. performed molecular dynamics simulations. J.G. and A.D. contributed to bioinformatics analyses. N.d.S., V.C., I.P., and P.P. wrote the manuscript with contributions from all authors. N.d.S. and P.P. coordinated the revision process. N.Z., A.C., J.S., and A.B. supervised parts of the project. P.P. conceived and supervised the project.
Declaration of interests
P.P. is a scientific advisor for the company Biognosys AG (Zurich, Switzerland) and an inventor of a patent licensed by Biognosys AG that covers the LiP-MS method used in this manuscript.
Published: December 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.12.021.
Supplemental Information
References
- Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Aebersold R., Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Alexa A., Rahnenführer J., Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- Alvarez M.J., Shen Y., Giorgi F.M., Lachmann A., Ding B.B., Ye B.H., Califano A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016;48:838–847. doi: 10.1038/ng.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardito F., Giuliani M., Perrone D., Troiano G., Lo Muzio L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review) Int. J. Mol. Med. 2017;40:271–280. doi: 10.3892/ijmm.2017.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2:0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin D., Hayer-Hartl M., Hartl F.U. In vivo aspects of protein folding and quality control. Science. 2016;353:aac4354. doi: 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- Banaszak K., Mechin I., Obmolova G., Oldham M., Chang S.H., Ruiz T., Radermacher M., Kopperschläger G., Rypniewski W. The crystal structures of eukaryotic phosphofructokinases from baker’s yeast and rabbit skeletal muscle. J. Mol. Biol. 2011;407:284–297. doi: 10.1016/j.jmb.2011.01.019. [DOI] [PubMed] [Google Scholar]
- Batth T.S., Papetti M., Pfeiffer A., Tollenaere M.A.X., Francavilla C., Olsen J.V. Large-Scale Phosphoproteomics Reveals Shp-2 Phosphatase-Dependent Regulators of Pdgf Receptor Signaling. Cell Rep. 2018;22:2784–2796. doi: 10.1016/j.celrep.2018.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Bennett B.D., Kimball E.H., Gao M., Osterhout R., Van Dien S.J., Rabinowitz J.D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert F.M., Lam Y.W., Lamont D., Lamond A.I. A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell. Proteomics. 2010;9:457–470. doi: 10.1074/mcp.M900429-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster J.L., Gustin M.C. Hog1: 20 years of discovery and impact. Sci. Signal. 2014;7:re7. doi: 10.1126/scisignal.2005458. [DOI] [PubMed] [Google Scholar]
- Brooks B.R., Brooks C.L., 3rd, Mackerell A.D., Jr., Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G., Singer A., Lunin V.V., Proudfoot M., Skarina T., Flick R., Kochinyan S., Sanishvili R., Joachimiak A., Edwards A.M. Structural and biochemical characterization of the type II fructose-1,6-bisphosphatase GlpX from Escherichia coli. J. Biol. Chem. 2009;284:3784–3792. doi: 10.1074/jbc.M808186200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang R.L., Andrews K., Kim D., Li Z., Godzik A., Palsson B.O. Structural systems biology evaluation of metabolic thermotolerance in Escherichia coli. Science. 2013;340:1220–1223. doi: 10.1126/science.1234012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M., Chang C.Y., Clough T., Broudy D., Killeen T., MacLean B., Vitek O. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics. 2014;30:2524–2526. doi: 10.1093/bioinformatics/btu305. [DOI] [PubMed] [Google Scholar]
- Chubukov V., Gerosa L., Kochanowski K., Sauer U. Coordination of microbial metabolism. Nat. Rev. Microbiol. 2014;12:327–340. doi: 10.1038/nrmicro3238. [DOI] [PubMed] [Google Scholar]
- Costenoble R., Picotti P., Reiter L., Stallmach R., Heinemann M., Sauer U., Aebersold R. Comprehensive quantitative analysis of central carbon and amino-acid metabolism in Saccharomyces cerevisiae under multiple conditions by targeted proteomics. Mol. Syst. Biol. 2011;7:464. doi: 10.1038/msb.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cravatt B.F., Wright A.T., Kozarich J.W. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- Crowhurst G.S., Dalby A.R., Isupov M.N., Campbell J.W., Littlechild J.A. Structure of a phosphoglycerate mutase:3-phosphoglyceric acid complex at 1.7 A. Acta Crystallogr. D Biol. Crystallogr. 1999;55:1822–1826. doi: 10.1107/s0907444999009944. [DOI] [PubMed] [Google Scholar]
- Damaghi M., Wojtkowiak J.W., Gillies R.J. pH sensing and regulation in cancer. Front. Physiol. 2013;4:370. doi: 10.3389/fphys.2013.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diether M., Nikolaev Y., Allain F.H., Sauer U. Systematic mapping of protein-metabolite interactions in central metabolism of Escherichia coli. Mol. Syst. Biol. 2019;15:e9008. doi: 10.15252/msb.20199008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dihazi H., Kessler R., Eschrich K. High osmolarity glycerol (HOG) pathway-induced phosphorylation and activation of 6-phosphofructo-2-kinase are essential for glycerol accumulation and yeast cell proliferation under hyperosmotic stress. J. Biol. Chem. 2004;279:23961–23968. doi: 10.1074/jbc.M312974200. [DOI] [PubMed] [Google Scholar]
- Doucette C.D., Schwab D.J., Wingreen N.S., Rabinowitz J.D. α-Ketoglutarate coordinates carbon and nitrogen utilization via enzyme I inhibition. Nat. Chem. Biol. 2011;7:894–901. doi: 10.1038/nchembio.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espino J.A., Jones L.M. Illuminating Biological Interactions with in Vivo Protein Footprinting. Anal. Chem. 2019;91:6577–6584. doi: 10.1021/acs.analchem.9b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- Feller S.E., Zhang Y.H., Pastor R.W., Brooks B.R. Constant-Pressure Molecular-Dynamics Simulation - the Langevin Piston Method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- Feng Y., De Franceschi G., Kahraman A., Soste M., Melnik A., Boersema P.J., de Laureto P.P., Nikolaev Y., Oliveira A.P., Picotti P. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 2014;32:1036–1044. doi: 10.1038/nbt.2999. [DOI] [PubMed] [Google Scholar]
- Fontana A., de Laureto P.P., Spolaore B., Frare E., Picotti P., Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim. Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- Fuhrer T., Heer D., Begemann B., Zamboni N. High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Anal. Chem. 2011;83:7074–7080. doi: 10.1021/ac201267k. [DOI] [PubMed] [Google Scholar]
- Gates S.N., Yokom A.L., Lin J., Jackrel M.E., Rizo A.N., Kendsersky N.M., Buell C.E., Sweeny E.A., Mack K.L., Chuang E. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science. 2017;357:273–279. doi: 10.1126/science.aan1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerosa L., Haverkorn van Rijsewijk B.R., Christodoulou D., Kochanowski K., Schmidt T.S., Noor E., Sauer U. Pseudo-transition Analysis Identifies the Key Regulators of Dynamic Metabolic Adaptations from Steady-State Data. Cell Syst. 2015;1:270–282. doi: 10.1016/j.cels.2015.09.008. [DOI] [PubMed] [Google Scholar]
- Gong Y., Kakihara Y., Krogan N., Greenblatt J., Emili A., Zhang Z., Houry W.A. An atlas of chaperone-protein interactions in Saccharomyces cerevisiae: implications to protein folding pathways in the cell. Mol. Syst. Biol. 2009;5:275. doi: 10.1038/msb.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann S. An integrated view on a eukaryotic osmoregulation system. Curr. Genet. 2015;61:373–382. doi: 10.1007/s00294-015-0475-0. [DOI] [PubMed] [Google Scholar]
- Huang J., MacKerell A.D., Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber K.V., Olek K.M., Müller A.C., Tan C.S., Bennett K.L., Colinge J., Superti-Furga G. Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat. Methods. 2015;12:1055–1057. doi: 10.1038/nmeth.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey S.J., Azimifar S.B., Mann M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol. 2015;33:990–995. doi: 10.1038/nbt.3327. [DOI] [PubMed] [Google Scholar]
- Ideker T., Thorsson V., Ranish J.A., Christmas R., Buhler J., Eng J.K., Bumgarner R., Goodlett D.R., Aebersold R., Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- Jarnuczak A.F., Albornoz M.G., Eyers C.E., Grant C.M., Hubbard S.J. A quantitative and temporal map of proteostasis during heat shock in Saccharomyces cerevisiae. Mol Omics. 2018;14:37–52. doi: 10.1039/c7mo00050b. [DOI] [PubMed] [Google Scholar]
- Jogl G., Rozovsky S., McDermott A.E., Tong L. Optimal alignment for enzymatic proton transfer: structure of the Michaelis complex of triosephosphate isomerase at 1.2-A resolution. Proc. Natl. Acad. Sci. USA. 2003;100:50–55. doi: 10.1073/pnas.0233793100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- Jurica M.S., Mesecar A., Heath P.J., Shi W., Nowak T., Stoddard B.L. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure. 1998;6:195–210. doi: 10.1016/s0969-2126(98)00021-5. [DOI] [PubMed] [Google Scholar]
- Kanshin E., Kubiniok P., Thattikota Y., D’Amours D., Thibault P. Phosphoproteome dynamics of Saccharomyces cerevisiae under heat shock and cold stress. Mol. Syst. Biol. 2015;11:813. doi: 10.15252/msb.20156170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpov A.S., Amiri P., Bellamacina C., Bellance M.H., Breitenstein W., Daniel D., Denay R., Fabbro D., Fernandez C., Galuba I. Optimization of a Dibenzodiazepine Hit to a Potent and Selective Allosteric PAK1 Inhibitor. ACS Med. Chem. Lett. 2015;6:776–781. doi: 10.1021/acsmedchemlett.5b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M., Ara T., Arifuzzaman M., Ioka-Nakamichi T., Inamoto E., Toyonaga H., Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Kochanowski K., Volkmer B., Gerosa L., Haverkorn van Rijsewijk B.R., Schmidt A., Heinemann M. Functioning of a metabolic flux sensor in Escherichia coli. Proc. Natl. Acad. Sci. USA. 2013;110:1130–1135. doi: 10.1073/pnas.1202582110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- Kolkman A., Daran-Lapujade P., Fullaondo A., Olsthoorn M.M., Pronk J.T., Slijper M., Heck A.J. Proteome analysis of yeast response to various nutrient limitations. Mol. Syst. Biol. 2006;2:0026. doi: 10.1038/msb4100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leano J.B., Batarni S., Eriksen J., Juge N., Pak J.E., Kimura-Someya T., Robles-Colmenares Y., Moriyama Y., Stroud R.M., Edwards R.H. Structures suggest a mechanism for energy coupling by a family of organic anion transporters. PLoS Biol. 2019;17:e3000260. doi: 10.1371/journal.pbio.3000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.J., Jeschke G.R., Roelants F.M., Thorner J., Turk B.E. Reciprocal phosphorylation of yeast glycerol-3-phosphate dehydrogenases in adaptation to distinct types of stress. Mol. Cell. Biol. 2012;32:4705–4717. doi: 10.1128/MCB.00897-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Roh S.H., Lee J., Sung N., Liu J., Tsai F.T.F. Cryo-EM Structures of the Hsp104 Protein Disaggregase Captured in the ATP Conformation. Cell Rep. 2019;26:29–36.e3. doi: 10.1016/j.celrep.2018.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuenberger P., Ganscha S., Kahraman A., Cappelletti V., Boersema P.J., von Mering C., Claassen M., Picotti P. Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science. 2017;355:355. doi: 10.1126/science.aai7825. [DOI] [PubMed] [Google Scholar]
- Liu F., Rijkers D.T., Post H., Heck A.J. Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods. 2015;12:1179–1184. doi: 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- Liu F., Lössl P., Rabbitts B.M., Balaban R.S., Heck A.J.R. The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes. Mol. Cell. Proteomics. 2018;17:216–232. doi: 10.1074/mcp.RA117.000470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie R.J., Lawless C., Holman S.W., Lanthaler K., Beynon R.J., Grant C.M., Hubbard S.J., Eyers C.E. Absolute protein quantification of the yeast chaperome under conditions of heat shock. Proteomics. 2016;16:2128–2140. doi: 10.1002/pmic.201500503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierlinski M., Gastaldello F., Geoffrey J., Barton G.J. Proteus: an R package for downstream analysis of MaxQuant output. bioRxiv. 2018 doi: 10.1101/416511. [DOI] [Google Scholar]
- Márquez J., Reinelt S., Koch B., Engelmann R., Hengstenberg W., Scheffzek K. Structure of the full-length enzyme I of the phosphoenolpyruvate-dependent sugar phosphotransferase system. J. Biol. Chem. 2006;281:32508–32515. doi: 10.1074/jbc.M513721200. [DOI] [PubMed] [Google Scholar]
- Martyna G.J., Tobias D.J., Klein M.L. Constant-Pressure Molecular-Dynamics Algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- McPhillips T.M., Hsu B.T., Sherman M.A., Mas M.T., Rees D.C. Structure of the R65Q mutant of yeast 3-phosphoglycerate kinase complexed with Mg-AMP-PNP and 3-phospho-D-glycerate. Biochemistry. 1996;35:4118–4127. doi: 10.1021/bi952500o. [DOI] [PubMed] [Google Scholar]
- Mertins P., Mani D.R., Ruggles K.V., Gillette M.A., Clauser K.R., Wang P., Wang X., Qiao J.W., Cao S., Petralia F., NCI CPTAC Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mydy L.S., Cristobal J.R., Katigbak R.D., Bauer P., Reyes A.C., Kamerlin S.C.L., Richard J.P., Gulick A.M. Human Glycerol 3-Phosphate Dehydrogenase: X-ray Crystal Structures That Guide the Interpretation of Mutagenesis Studies. Biochemistry. 2019;58:1061–1073. doi: 10.1021/acs.biochem.8b01103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navdaeva V., Zurbriggen A., Waltersperger S., Schneider P., Oberholzer A.E., Bähler P., Bächler C., Grieder A., Baumann U., Erni B. Phosphoenolpyruvate: sugar phosphotransferase system from the hyperthermophilic Thermoanaerobacter tengcongensis. Biochemistry. 2011;50:1184–1193. doi: 10.1021/bi101721f. [DOI] [PubMed] [Google Scholar]
- Niphakis M.J., Lum K.M., Cognetta A.B., 3rd, Correia B.E., Ichu T.A., Olucha J., Brown S.J., Kundu S., Piscitelli F., Rosen H., Cravatt B.F. A Global Map of Lipid-Binding Proteins and Their Ligandability in Cells. Cell. 2015;161:1668–1680. doi: 10.1016/j.cell.2015.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R., Tsai C.J., Ma B. The underappreciated role of allostery in the cellular network. Annu. Rev. Biophys. 2013;42:169–189. doi: 10.1146/annurev-biophys-083012-130257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J.D., Tsechansky M., Royal A., Boutz D.R., Ellington A.D., Marcotte E.M. A proteomic survey of widespread protein aggregation in yeast. Mol. Biosyst. 2014;10:851–861. doi: 10.1039/c3mb70508k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer A.E., Bumann M., Schneider P., Bächler C., Siebold C., Baumann U., Erni B. Crystal structure of the phosphoenolpyruvate-binding enzyme I-domain from the Thermoanaerobacter tengcongensis PEP: sugar phosphotransferase system (PTS) J. Mol. Biol. 2005;346:521–532. doi: 10.1016/j.jmb.2004.11.077. [DOI] [PubMed] [Google Scholar]
- Oberholzer A.E., Schneider P., Siebold C., Baumann U., Erni B. Crystal structure of enzyme I of the phosphoenolpyruvate sugar phosphotransferase system in the dephosphorylated state. J. Biol. Chem. 2009;284:33169–33176. doi: 10.1074/jbc.M109.057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L., Itano H.A. Sickle cell anemia a molecular disease. Science. 1949;110:543–548. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47(D1):D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza I., Kochanowski K., Cappelletti V., Fuhrer T., Noor E., Sauer U., Picotti P. A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication. Cell. 2018;172:358–372.e23. doi: 10.1016/j.cell.2017.12.006. [DOI] [PubMed] [Google Scholar]
- Reiter L., Rinner O., Picotti P., Hüttenhain R., Beck M., Brusniak M.Y., Hengartner M.O., Aebersold R. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods. 2011;8:430–435. doi: 10.1038/nmeth.1584. [DOI] [PubMed] [Google Scholar]
- Ressa A., Bosdriesz E., de Ligt J., Mainardi S., Maddalo G., Prahallad A., Jager M., de la Fonteijne L., Fitzpatrick M., Groten S. A System-wide Approach to Monitor Responses to Synergistic BRAF and EGFR Inhibition in Colorectal Cancer Cells. Mol. Cell. Proteomics. 2018;17:1892–1908. doi: 10.1074/mcp.RA117.000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinas A., Mali V.S., Espino J.A., Jones L.M. Development of a Microflow System for In-Cell Footprinting Coupled with Mass Spectrometry. Anal. Chem. 2016;88:10052–10058. doi: 10.1021/acs.analchem.6b02357. [DOI] [PubMed] [Google Scholar]
- Robertson A.D., Murphy K.P. Protein Structure and the Energetics of Protein Stability. Chem. Rev. 1997;97:1251–1268. doi: 10.1021/cr960383c. [DOI] [PubMed] [Google Scholar]
- Russell C.J. Acid-induced membrane fusion by the hemagglutinin protein and its role in influenza virus biology. Curr. Top. Microbiol. Immunol. 2014;385:93–116. doi: 10.1007/82_2014_393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni N., Yi S., Zhong Q., Jailkhani N., Charloteaux B., Cusick M.E., Vidal M. Edgotype: a fundamental link between genotype and phenotype. Curr. Opin. Genet. Dev. 2013;23:649–657. doi: 10.1016/j.gde.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M.M., Reinhard F.B., Franken H., Werner T., Savitski M.F., Eberhard D., Martinez Molina D., Jafari R., Dovega R.B., Klaeger S. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346:1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Schmidt A., Kochanowski K., Vedelaar S., Ahrné E., Volkmer B., Callipo L., Knoops K., Bauer M., Aebersold R., Heinemann M. The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 2016;34:104–110. doi: 10.1038/nbt.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sévin D.C., Fuhrer T., Zamboni N., Sauer U. Nontargeted in vitro metabolomics for high-throughput identification of novel enzymes in Escherichia coli. Nat. Methods. 2017;14:187–194. doi: 10.1038/nmeth.4103. [DOI] [PubMed] [Google Scholar]
- Shaul Y.D., Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim. Biophys. Acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Shirts M.R., Mobley D.L., Chodera J.D., Pande V.S. Accurate and efficient corrections for missing dispersion interactions in molecular simulations. J. Phys. Chem. B. 2007;111:13052–13063. doi: 10.1021/jp0735987. [DOI] [PubMed] [Google Scholar]
- Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Soufi B., Kelstrup C.D., Stoehr G., Fröhlich F., Walther T.C., Olsen J.V. Global analysis of the yeast osmotic stress response by quantitative proteomics. Mol. Biosyst. 2009;5:1337–1346. doi: 10.1039/b902256b. [DOI] [PubMed] [Google Scholar]
- Sowa M.E., Bennett E.J., Gygi S.P., Harper J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey A.J., Hardman R.E., Byrum S.D., Mackintosh S.G., Edmondson R.D., Wahls W.P., Tackett A.J., Lewis J.A. Accurate and Sensitive Quantitation of the Dynamic Heat Shock Proteome Using Tandem Mass Tags. J. Proteome Res. 2020;19:1183–1195. doi: 10.1021/acs.jproteome.9b00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sträter N., Marek S., Kuettner E.B., Kloos M., Keim A., Brüser A., Kirchberger J., Schöneberg T. Molecular architecture and structural basis of allosteric regulation of eukaryotic phosphofructokinases. FASEB J. 2011;25:89–98. doi: 10.1096/fj.10-163865. [DOI] [PubMed] [Google Scholar]
- Tan C.S.H., Go K.D., Bisteau X., Dai L., Yong C.H., Prabhu N., Ozturk M.B., Lim Y.T., Sreekumar L., Lengqvist J. Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science. 2018;359:1170–1177. doi: 10.1126/science.aan0346. [DOI] [PubMed] [Google Scholar]
- Teplyakov A., Lim K., Zhu P.P., Kapadia G., Chen C.C.H., Schwartz J., Howard A., Reddy P.T., Peterkofsky A., Herzberg O. Structure of phosphorylated enzyme I, the phosphoenolpyruvate:sugar phosphotransferase system sugar translocation signal protein. Proc. Natl. Acad. Sci. USA. 2006;103:16218–16223. doi: 10.1073/pnas.0607587103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A.D., Jr. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese J., Abrams J., Wang Y., Morano K.A. Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol. Mol. Biol. Rev. 2012;76:115–158. doi: 10.1128/MMBR.05018-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace E.W., Kear-Scott J.L., Pilipenko E.V., Schwartz M.H., Laskowski P.R., Rojek A.E., Katanski C.D., Riback J.A., Dion M.F., Franks A.M. Reversible, Specific, Active Aggregates of Endogenous Proteins Assemble upon Heat Stress. Cell. 2015;162:1286–1298. doi: 10.1016/j.cell.2015.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Qin X., Wu J., Deng H., Li Y., Yang H., Chen Z., Liu G., Ren D. Analysis of crystal structure of Arabidopsis MPK6 and generation of its mutants with higher activity. Sci. Rep. 2016;6:25646. doi: 10.1038/srep25646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedekind J.E., Reed G.H., Rayment I. Octahedral coordination at the high-affinity metal site in enolase: crystallographic analysis of the MgII--enzyme complex from yeast at 1.9 A resolution. Biochemistry. 1995;34:4325–4330. doi: 10.1021/bi00013a022. [DOI] [PubMed] [Google Scholar]
- Wepf A., Glatter T., Schmidt A., Aebersold R., Gstaiger M. Quantitative interaction proteomics using mass spectrometry. Nat. Methods. 2009;6:203–205. doi: 10.1038/nmeth.1302. [DOI] [PubMed] [Google Scholar]
- White F.H., Jr., Anfinsen C.B. Some relationships of structure to function in ribonuclease. Ann. N Y Acad. Sci. 1959;81:515–523. doi: 10.1111/j.1749-6632.1959.tb49333.x. [DOI] [PubMed] [Google Scholar]
- Young T.A., Skordalakes E., Marqusee S. Comparison of proteolytic susceptibility in phosphoglycerate kinases from yeast and E. coli: modulation of conformational ensembles without altering structure or stability. J. Mol. Biol. 2007;368:1438–1447. doi: 10.1016/j.jmb.2007.02.077. [DOI] [PubMed] [Google Scholar]
- Zhang B., Chambers M.C., Tabb D.L. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 2007;6:3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Thiele I., Weekes D., Li Z., Jaroszewski L., Ginalski K., Deacon A.M., Wooley J., Lesley S.A., Wilson I.A. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science. 2009;325:1544–1549. doi: 10.1126/science.1174671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Binding mode of FBP (carbon atoms in cyan) to enzyme I of the phosphoenolpyruvate-protein phosphotransferase system (PtsI) (gray, with carbon atoms in active site side chains in green) as predicted by ligand docking and molecular dynamics simulations. A close-up of the PtsI active site is shown, with FBP and the cofactor Mg2+ (blue sphere).
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD022297.
The complete data set of LiP peptide correlations with flux are at https://doi.org/10.5281/zenodo.3964994
The complete data set of LiP peptide correlations with metabolite levels are at https://doi.org/10.5281/zenodo.3965002