

Abstract
The novel SARS-CoV-2 is the etiological agent causing the Coronavirus disease 2019 (COVID-19), which continues to become an inevitable pandemic outbreak. Over a short span of time, the structures of therapeutic target proteins for SARS-CoV-2 were identified based on the homology modelled structure of similar SARS-CoV transmission of 2003. Since the onset of the disease, the research community has been looking for a potential drug lead. Out of all the known resolved structures related to SARS-CoV, Main protease (Mpro) is considered an attractive anti-viral drug target on the grounds of its role in viral replication and probable non-interactive competency to bind to any viral host protein. To the best of our knowledge, till date only one compound has been identified and tested in-vivo as a potent inhibitor of Mpro protein, addressed as N3 (PubChem Compound CID: 6323191) and is known to bind irreversibly to Mpro suppressing its activity. Using computational approach, we intend to identify a probable natural fungal metabolite to interact and inhibit Mpro. After screening various small molecules for molecular docking and dynamics simulation, we propose Pyranonigrin A, a secondary fungal metabolite to possess potent inhibitory potential against the Main protease (Mpro) expressed in SARS-CoV-2 virus.
Keywords: Main protease (Mpro), SARS-CoV-2 novel corona virus, Fungal metabolites, Docking, Molecular dynamics simulation
Graphical abstract

Highlights
-
•
Searching inhibitor for Main Protease (Mpro) of SARS-CoV2
-
•
Purpose to block viral replication and packaging in host
-
•
Mpro interaction with fungal metabolite- Pyranonigrin A
-
•
Studies involving docking and molecular dynamics
1. Introduction
World Health Organization (WHO) announced the outbreak of coronavirus disease 2019 (COVID-19) to be pandemic on 11 March 2020. This outbreak of COVID-19 is the third major outbreak of Corona virus inflicting severe disease and death on a global scale. The Corona viral pathogen (so named because of the ‘crown’ or ‘wreath’ like surface appearance) first appeared in the year 2002 as Severe Acute Respiratory Syndrome (SARS) and later as Middle East Respiratory Syndrome (MERS) in 2012 as an epidemic affecting 26 countries resulting in more than 8000 reported cases to cause severe infections although lacked an ability to spread from person to person. What makes the novel SARS-CoV-2 infection even more dangerous, is its ability of being transmissible and a higher affinity to bind to the human Angiotensin Converting Enzyme-2 (ACE2) receptor surpassing the presently known SARS-CoV and MERS-CoV infections [[1], [2], [3]]. The novel Corona virus responsible for COVID-19 shares 89.1% genetic similarity with SARS-CoV and is hence, named as SARS-CoV-2, possessing a very strong potency to spread in humans which has not be seen in SARS-CoV and MERS-CoV infections back then [4].
The first diagnostic case of COVID-19 was reported on 17 November 2019 in Wuhan, China as severe clusters of pneumonia had been observed and since then the disease has spread globally with little over a 50% increase in the prevalence of the infection, and lowered ascertainment rate of less than 9.2% [[2], [3], [4]]
Since the inception of this virus, the researchers are trying to find drug leads that can be used to stop its replication in host, and currently there is no established therapy to overcome COVID-19. Recently, Non-structural group of SARS-CoV-2 proteins have been identified, namely: Main protease (Mpro), Papain-like protease, Non-structural protein 13 (nsp13, helicase), Non-structural protein 12 (nsp12), RNA-dependent RNA polymerase, (RdRp), Non-structural protein 14 (nsp14, N-terminal exoribonuclease and C-terminal guanine-N7 methyl transferase), Non-structural protein 15 (nsp15, Uridylate-specific endoribonuclease), Non-structural protein 16 (nsp16, 2’-O-methyltransferase) and Non-structural protein 10. These proteins are found to be responsible for replication and packing of the viral genomic content [4,5]. Blocking any of the proteins involved in this process will hinder successful replication or packing of its viral genomic content into the protein coat. Similarly, other group of proteins called the Structural proteins are also identified as drug likely leads, including; Spike protein (S protein), S2 of S protein, Envelop small membrane protein (E protein), Membrane protein (M protein) and Nucleocapsid protein (N protein) [5].
Replicase gene of SARS-CoV-2 codes polyproteins namely, pp1a and pp1ab protein which is required for viral replication and transcription. These two polyproteins in active form are released after proteolytic processing by a 33.8-kDa Mpro, also referred to as the 3C-like protease. Mpro can digest polyprotein at 11 or more conserved sites, that is initiated by autolytic cleavage of this enzyme itself from pp1a and pp1ab8 [4,6,7]. The quintessential role of Mpro in viral replication and development, with no similarity to any human protein makes Mpro an attractive antiviral drug target [[8], [9], [10], [11]]. In order to accelerate the Efforts are extensively been made using Computer-Aided Drug Design (CADD) to develop Mpro blockers with stabilised covalent bonding and the success of only one compound ‘N-[(5-METHYLISOXAZOL-3-YL)CARBONYL]ALANYL-L-VALYL-N~1~-((1R,2Z)-4-(BENZYLOXY)-4-OXO-1-([(3R)-2-OXOPYRROLIDIN-3-YL]METHYL)BUT-2-ENYL)-L-LEUCINAMIDE’ (PubChem Compound CID: 6323191) abbreviated as ‘N3’ has shown potentials to effectively supress the activity backed up by in-vivo trials [4]. However, this compound is synthetically designed and before it reaches the commercial setup, it must undergo prolonged clinical trials, thus, inviting natural alternatives as lead molecules as target against Mpro.
This study was designed to identify potential inhibitor of Mpro from the pool of fungal metabolites. Fungal metabolites have proved as a boon in past by making up more than 90% of naturally occurring antibiotics and possess compounds that may possess anti-viral capabilities [12]. Of about hundred secondary fungal metabolites were computationally docked to Mpro keeping N3 as positive control compound. The best compound found, in this case was Pyranonigrin A (PubChem Compound CID: 16756786) which could make equivalent H-bonds as that made by N3 with Mpro. To validate the docking results, molecular dynamics (MD) simulation study was carried out to screen the behaviour of ligand-target interactions under simulated physiological conditions. Moreover, the drug likeliness prediction and ADMET (drug absorption, distribution, metabolism, excretion, and toxicity) profiles of Pyranonigrin A were also undertaken and compared with that of N3.
2. Materials and methods
2.1. Preparation of receptor and ligands
The protein Mpro was retrieved from Protein databank (PDB), ID 6 LU7. The protein was co-crystallized with N3. The co-ordinates of N3 binding site on Mpro was determined using UCSF Chimera. Prior to docking studies, all the co-crystallized residues were removed in UCSF Chimera. The protein structure was then prepared by assigning the hydrogen atoms, charges and energy minimization using DockPrep tool [13]. The charges were assigned as per the AM1-BCC method which quickly and efficiently generates high-quality atomic charges for protein and the charges were computed using ANTECHAMBER algorithm [14]. The energy minimization was performed using 500 steepest descent steps with 0.02 Å step size and an update interval of 10. All the steps mentioned were performed in UCSF Chimera.
All the ligands used for the in-silico interaction assays were fungal secondary metabolites and were retrieved from PubChem. Before performing the molecular docking of ligand and receptor, the ligands were optimized by addition of hydrogen and energy minimization using Gasteiger algorithm [15] in structure editing wizard of UCSF Chimera, which works on the chemoinformatic principle of electronegativity equilibration and the files were saved in mol2 format.
2.2. Molecular docking
Receptor-ligand docking analysis was performed using AutoDock Vina [16] and the program was executed as an add-on in UCSF Chimera. The ligand binding site in Mpro was chosen based on the crystallized ligand (N3) attached in the original pdb file and the co-ordinates were recorded for docking of fungal ligands. Further, the hydrophobic cavity of Mpro. The coordinates of hydrophobic cavity of the active site was used in the docking of N3 (as control) and fungal metabolites.
In the AutoDock Vina algorithm, the following parameters were set as: (i) number of binding modes- 10; (ii) exhaustiveness of search- 8 and (iii) maximum energy difference- 3 kcal/mol. Out of all the possible poses suggested by AutoDock Vina, the pose showing maximum hydrogen bonds and minimum binding free energy change (kcal/mol) as represented in the ViewDock window were chosen. They were further analysed in Biovia Discovery Studio (DS) visualizer for hydrogen bond formation by the functional groups of ligands with amino acids. DS also suggested other supporting hydrophobic interactions made by the N3 and fungal metabolites. The metabolite compound making highest number of H-bonds, showing capability to form covalent interaction with Mpro and showed highest binding affinity was chosen for further molecular dynamics simulation analysis.
2.3. Molecular dynamics (MD) simulation
The simulation of the Mpro in the presence of N3 and Pyranonigrin A were performed in two sets of experiments using GROMACS 2019 software [[17], [18], [19]]. Pyranonigrin A has best docking score when compared to other fungal metabolites. Simulations were carried out for the Pyranonigrin A-Mprokeeping N3-Mpro docked complex as a control. The topology of the ligand was generated using SwissParam, which provides topology and parameters for small organic molecules compatible with the CHARMM all atoms force field, for use with CHARMM and GROMACS [20]. Whereas, the topology of the protein was created using GROMACS utilities using CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins) with the water model set to TIP 3-point. The structure (Ligand-protein complex) were defined with unit cell box under periodic boundary conditions using 1.0 nm distance from the protein to the box faces with triclinic shape and was filled with water [21]. This was then followed by neutralization by Cl− or Na+ counter ions. Steepest descent energy minimization was performed, and the systems were equilibrated under NVT (constant number of particles, volume and temperature) conditions for 50 ns at 300 K. Once the NVT run was completed the system was proceeded with NPT (constant number of particles, pressure, and temperature) simulation and MD run was performed for 50 ns. All the covalent bonds were constrained using the LINCS (Linear Constraint Solver) algorithm [22]. The electrostatic interactions were treated using the Particle Mesh Ewald (PME) method. The cut-off radii for Coulomb and van der Waals interactions were set to 10.0 and 14.0 Å, respectively. On completion of NVT and NPT simulation, the potential of each trajectory produced were analysed. Trajectories were analysed for root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg) and for the number of H-bonds formed between the ligand and proteins using ‘gmx rms’,’gmx rmsf’, ‘gmx gyrate’ and ‘gmx hbond’ of GROMACS utilities [17,18]. Ligand–protein stability was determined by the dynamics of hydrogen bonds between ligand and protein with respect to time. XMgrace tool was used to prepare the graphs [23].
2.4. ADMET analysis
The pkCSM - pharmacokinetics server [24] was used to predict the ADMET properties of the N3 and Pyranonigrin A. It predicted both physiochemical and pharmacological properties. SMILES (Simplified Molecule Input Line Entry Specification) of the compounds were retrieved from PubChem and uploaded to pkCSM - pharmacokinetics server. It computed in-vivo Absorption parameters like, Water solubility in buffer system (SK atomic types, mg/L), in-vivo Caco2 cell permeability (Human colorectal carcinoma), Human intestinal absorption (HIA, %), in-vivo P-glycoprotein inhibition and in-vivo skin permeability (logKp, cm/h). Metabolic parameters where determined using in-vivo Cytochrome P450 2C19 inhibition, in-vivo Cytochrome P450 2C9 inhibition, in-vivo Cytochrome P450 2D6 inhibition, in-vivo Cytochrome P450 2D6 substrate, in-vivo Cytochrome P450 3A4 inhibition and in-vivo Cytochrome P450 3A4 substrate, Distribution property included tests like, Blood-Brain Barrier (BBB) penetration, Lipinskis Rule (Rule of Five), Cental Nervous System (CNS) permeability. While, toxicity properties covered a range of important endpoints including, Acute algae toxicity, Ames test, 2 years carcinogenicity bioassay in mouse, 2 years carcinogenicity bioassay in rat, in-vivo Ames test result in TA100 strain (Metabolic activation by rat liver homogenate) were computed to access the toxicity of compounds under study. Excretion again is a very important parameter and many drugs were withdrawn due to their poorer renal clearance. In this study we included Total Renal clearance and Renal OCT2 Substrate to identify Excretion efficacy of the proposed metabolite.
3. Results
3.1. Analysing Mpro and its interaction with N3
The structure analysis of SARS-CoV2 Mpro shows that Domains I extend from residues 8 to 101 and Domain II ranges from residues 102 to 184 comprise of antiparallel β-barrel structure. Domain residues are formed from residue 201 to 303 and possess five α-helices arranged into a largely antiparallel globular cluster, which is connected to domain II means of a long loop region extending from residue 185 to 200. COVID-19 virus Mpro has a Cys-His catalytic dyad, and the substrate-binding site is in a cleft between Domain I and II. Binding of N3 with Mpro shows that it binds in the substrate binding pocket as shown in (Fig. 1 ). N3 is relatively a large molecule where its backbone forms an antiparallel sheet with residues 164 to168 (His164, Glu166, Met165, Leu167, Pro168) of Mpro and also interacting with residues 189 to191 (Gln189, Thr190, Ala191) of the loop that links Domain II and III.
Fig. 1.

(a) Orientation and position of N3 in the binding cleft of Mpro (PDB ID: 6LU7) of SARS CoV-2 is shown in 3D representation where ligand (N3) in red is representing co-crystallized orientation and in blue-cyan is the orientation of same ligand obtained after performing Docking. (b) Interaction of N3 in the binding cleft of Mpro shown in 3D representation and 2D representation describing ligands interactions by formation of various H-bonds and hydrophobic interactions with protein. 6LU7 (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Briefly the inhibitor N3 makes six hydrogen bonds (Gly143, Phe140, His163, His164, Glu166 and Thr190), one Amide Pi-stacked interaction (with Leu141), five Pi-Alkyl interactions (His41, Met49, Leu167, Pro168 and Ala191) two carbon hydrogen bonds (Met165 and His172) and one Van der Waals interactions (Asn142) with Mpro.
3.2. Docking of fungal metabolites with Mpro
For the docking of fungal metabolites with Mpro, the coordinates of N3 binding was used to assess the binding affinity of various fungal metabolites with Mpro. On performing the docking of all the metabolites one by one with Mpro, it was observed that only five molecules could make three hydrogen bonds or more with Mpro at the binding cleft of N3.
Out of all 100 screened fungal metabolites, Pyranonigrin A interacted in the ligand binding cleft of Mpro making eight hydrogen bonds in total (with Leu141, Gly143, Ser144, Cys145, His163, Glu166 and Gln189), one carbon hydrogen bond (Asn142) and the docking energy as predicted by AutoDock Vina was −7.3 Kcal/mol for the best pose (Fig. 2a). As shown previously N3 showed interaction with residues 164 to168 (His164, Glu166, Met165, Leu167, Pro168) of Mpro along with interacting with residues 189 to191 (Gln189, Thr190, Ala191) of the loop that links Domain II and III, which are quite similar to what Pyranonigrin A has shown upon interacting with Mpro. All these properties make Pyranonigrin A, a natural fungal metabolite as the best choice to be the most suitable inhibitor against Mpro. (See Fig. 3.)
Fig. 2.

6LU7(a) Interaction of Pyranonigrin A in the binding cleft of Mpro (PDB ID: 6LU7) of COVID-19 shown in 3D representation and 2D representation describing ligands interactions by formation of various H-bonds and hydrophobic interactions with protein. (b) Interaction of second ranked ligands in the binding cleft of Mpro.
Fig. 3.

3D image representing binding of N3 and Pyranonigrin A at same cleft of Mpro (PDB ID: 6LU7) of COVID-19.
From all the docked compounds, those which were ranked second to Pyranonigrin A include Tensyuic acid A, Asprtnigrin A, Asperic acid, Aurosperone B and Carboxymethyl-3hexylmaleic acid (Fig. 2b). First one, Tensyuic acid A is shown to make five hydrogen bonds (Leu141, Gly143, Ser144, Cys145 and Glu166), while, Asperic acid showed to form four hydrogen bonds (Leu141, Gly143, Ser144 and Cys145) along with one alkyl interaction (Cys145), the docking energy as predicted by AutoDock Vina was −6.4 kcal/mol. Another fungal metabolite, Aspernigrin A was able to interact with Mpro by making three hydrogen bonds (Phe140, His163 and His164), one Pi-sulphur bond (Cys145), one Carbon‑hydrogen bond (His172,) a Pi-Pi T-shaped bond with His41 and Pi-Alkyl bond along with Met165, the docking energy as predicted by AutoDock Vina was −6.6 kcal/mol for the best pose. While, Aurosperone B interacted with Mpro making three hydrogen bonds (Gly143, Ser144 and Cys145), two Pi-Alkyl bond (Met49 and Cys145) and one amino acid (His163) formed unfavourable Donor interaction. The docking energy in this case was poor which valued −5.8 kcal/mol for the best pose. In contrast, Carbomymethyl-3-hexylmaleic acid, another fungal metabolite showed better efficacy to interact with Mpro by making six hydrogen bonds (Gly143, Ser144, Cys145, His163, His 164 and Glu166), one carbon hydrogen bond (Met165) and one Pi-Alkyl interaction (Leu27) with the docking score of −7.0 kcal/mol for the best pose.
As we overlay the structures of the best docked pose of SARS-CoV-2 virus Mpro protein, it showed that N3 and Pyranonigrin A bind to Mpro in a similar mode and exactly at the same N3 binding cleft (Fig. 3). Furthermore, the docking analysis suggested that Pyranonigrin A interacted with almost all the same amino acids as done by N3. To validate the molecular docking results, a comparative MD simulation was performed in two pairs of experiments studying (i) N3-Mpro and (ii) Pyranonigrin A-Mpro individually and the obtained results were compared. The basic structural and chemical characteristics of N3 and Pyranonigrin is represented Table 1, Table 2 respectively.
Table 1.
Structural and chemical characteristics of N3.
| Compound | N3 |
|---|---|
Molecule depiction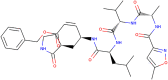
| |
| Descriptor | Value |
| Molecular weight | 680.803 |
| LogP | 2.08362 |
| #Rotatable bonds | 17 |
| #Acceptors | 9 |
| #Donors | 5 |
| Surface area | 286.079 |
Table 2.
Structural and chemical characteristics of Pyranonigrin A.
| Compound | Pyranonigrin A |
|---|---|
Molecule depiction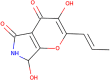
| |
| Descriptor | Value |
| Molecular weight | 223.184 |
| LogP | 0.1128 |
| #Rotatable bonds | 1 |
| #Acceptors | 5 |
| #Donors | 3 |
| Surface area | 89.827 |
Pyranonigrin being small by 1/3rd proportions with a surface area of 3.2 times smaller than that of N3 (Table 1, Table 2) makes almost same number of H-bonds, therefore the ratio of H-bond to surface area is much greater than that of N3 suggesting very strong interaction by pyranonigrin A with that of Mpro in the binding cleft of N3. Pyranonigrin A, therefore, by making sufficient H-bonds to settle and block the space in the cleft renders the protein ineffective and this is the rationale behind pyranonigrin A's mode of action. Both the compound may have different hydrophobicity (Table 1, Table 2), but the binding cleft of Mpro is huge, so Pyranonigrin A can bind to the region in the cleft where it forms more favourable H-bonds than hydrophobic interactions.
3.3. Molecular dynamics simulation
Simulations are conducted for the protein–ligand complexes (i) N3-Mpro and (ii) Pyranonigrin A-Mpro individually and their results were compared. This provided a better picture of the overall stability of the Mpro in presence of N3 and Pyranonigrin A, also the stability of both the ligands N3 and Pyranonigrin A while interacting with Mpro was obtained and compared. Root-mean-square-deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg) and H-bonds are used to check the stability of the model system.
The RMSD is a crucial parameter to analyse the equilibration of MD trajectories. RMSD of the protein backbone atoms are plotted as a function of time to check the stability of each system throughout the simulation. The RMSD values of the Mpro backbone with N3 and Pyranonigrin A was calculated against the simulation time scale (0–50 ns) and results are shown in Fig. 4 .
Fig. 4.
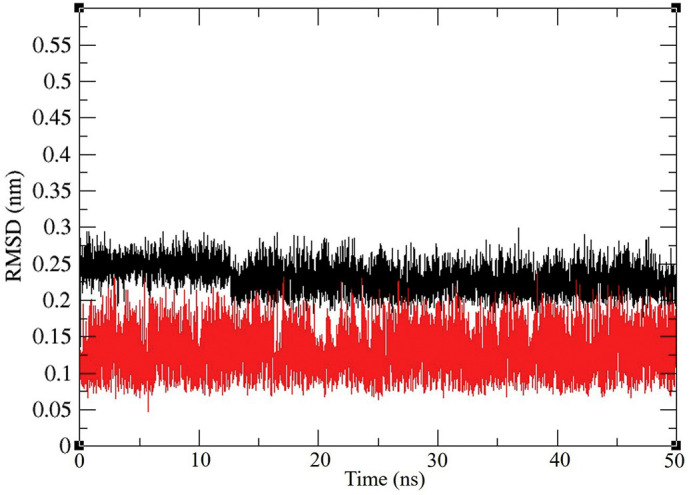
6LU7Representation of ligand RMSD of Mpro backbone RMSD during interaction with N3 (black) and Pyranonigrin A (red) during their interaction with Mpro (PDB ID: 6LU7) of COVID-19 derived from NVT Simulation at 300 K. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The RMSD values of two trajectories have values 0.1–0.3 nm during simulation for the backbone of proteins in presence of both the ligands, the Mpro was found to be more stable in presence of Pyranonigrin A as and when compared to N3 (Fig. 4).
The RMSF with respect to the average MD simulation conformation reflects as a means of portraying flexibility differences amongst residues. The RMSF of the backbone atoms of each residues of (i) N3-Mpro and (ii) Pyranonigrin A-Mpro individually is calculated to reveal the flexibility of the backbone structure in presence of both the ligands. This shows how the protein behaves in presence of different ligands. The high RMSF value indicates more flexibility whereas the low RMSF value indicates limited movements during simulation in relation to its average position. The RMSF of the residues are shown in Fig. 5 .
Fig. 5.

Representation of Molecular Dynamics (a) RMSF values of Mpro backbone during its interaction with N3, (b) RMSF values of Mpro backbone during its interaction with Pyranonigrin A (c) ligand N3's RMSF values during Simulation, and (d) ligand Pyranonigrin A's RMSF values during simulation.
The RMSF of the protein backbone Mpro in presence of both the ligands N3 and Pyranonigrin A were almost identical suggesting similar stability of protein not being altered by Pryanonigrin A binding with the protein. If the binding is poor than the protein can have higher RMSF values, which is not the case here (Fig. 5a, b). While the RMSF of the ligands N3 and Pyranonigrin A is shown in Fig. 5c and d respectively. As both the ligands are differing in size and shapes, comparing their RMSF doesn't hold any importance.
The inter molecular hydrogen bonding between the protein and the ligand plays an essential role in stabilizing the protein–ligand complexes. The stability of the hydrogen bond network formed between (i) N3-Mpro and (ii) Pyranonigrin A-Mpro were calculated throughout the simulation at 300 K for the ligated system and the results are depicted in Fig. 6 . The N3-Mpro complex exhibited seven hydrogen H-bonds formation during the course of simulation. While for Pyranonigrin A-Mpro, there were consistently six hydrogen bonds being formed throughout the simulation. Overall scenario suggests both ligands fits in to the binding cleft making appropriate hydrogen bonds. Pyranonigrin A being small by 1/3rd proportions with a surface area of 3.2 times smaller than that of N3 (Table 1, Table 2) makes almost same number of H-bonds, therefore the ratio of H-bond to surface area is much greater than that of N3 suggesting very strong interaction by pyranonigrin A with that of Mpro in the binding cleft of N3. Pyranonigrin A, therefore, by making sufficient H-bonds to settle and block the space in the cleft renders the protein ineffective and this is the rationale behind pyranonigrin A's mode of action.
Fig. 6.

Hydrogen Bonds formed by (a) N3 and (b) Pyranonigrin A with Mpro of COVID-19 during simulation.
For the MD analysis it can be said that the protein behaves identical in presence of both the ligands, N3 and Pyranonigrin A both forms similar hydrogen bonds with Mpro and hence the efficacy of Pyranonigrin A to interact with Mpro can be depicted to be at par with N3.
3.4. ADMET analysis
All the ADMET properties of both the compounds is shown in Table 3 . One of the most important challenges for an oral drug is its movement across the intestinal epithelial barrier that determines the rate and extent of human absorption and ultimately affects its bioavailability. As per Absorption is considered, The Caco2 permeability predicts the absorption of orally administered drugs, the value >8 × 10−6 cm/s suggests high permeability. Both the compounds have values less than the suggested value suggesting poor permeability. Intestinal absorption (human) value predicts absorption of the drug from orally administered solution. Both the compound under study has this value above 30% suggesting good absorption. P-glycoprotein is a component of ATP-binding cassette (ABC) transporter, the positive value by N3 suggests it can get transported across cell membrane by ABC transporter. On the contrary, N3 is also predicted to be a P-glycoprotein I inhibitor which can inhibit its transport. For, skin permeability, if the logKp > −2.5, drug cannot be skin permeable, in this context the both the compounds under study show better skin permeability.
Table 3.
ADMED values for N3 and Pyranonigrin A.

The volume of Distribution (VDss) suggests the total volume of drug needed to be uniformly distributed in blood. The value below −0.15 logVDss stands for a low VDss value and above 0.45 Log VDss, stands for considerably high VDss value. In this context, N3 can be considered to be a low VDss compound. Blood Brain barrier (BBB) permeability gives an account on compound's ability to reach brain or not. It is presumed that compounds with logBB >0.3 can pass BBB therefore, logBB values of both the compounds show their poor capability to pass BBB. Similarly, both the compounds also show poor Central Nervous System (CNS) permeability as for both the compounds their logPS values is smaller than −2.0 which is good as only the drugs for the nervous system disease should pass through the BBB and not others. Metabolism prediction suggested that Pyranonigrin A does not affect cytochrome functioning while N3 inhibits only one type of Cytochrome P that is, CYP3A4. Renal excretion of both the compounds differ greatly. The important criteria for toxicity assessment is Maximum Tolerated Dose (MRTD), this value less than 0.44 log(mg/kg/day) is considered low, and high if greater than 0.477 log(mg/kg/day) thus, MRTD for N3 is considered to be low and for Pyranonigrin A MRTD is considered high as per their log(mg/kg/day) values. Moreover, N3 shows hepatotoxicity, while Pyranonigrin A does not show this type of toxicity. The hERG I and II are potassium channels encoded by hERG, and their inhibition can cause QT syndrome (QT refers to the peaks of heart electrocardiogram) which affects repolarization of the heart after a heartbeat. Here, N3 has is predicted to be hERG inhibitor and shows tendency to induce QT syndrome. The OCT2 was the first step in the renal secretion of many cationic drugs as blockers and inhibitors may result nephrotoxicity.
4. Discussion
Coronaviruses (CoVs) are enveloped, positive stranded RNA viruses. They belong to the genus Coronavirus of the family Coronaviridae. All the species of CoVs are bifurcated in to 3 major groups based on their genome sequences and serological reactions. Different CoVs have potency to infect humans while other are specific to infect animals and cause variety of highly prevalent and severe diseases. For instance, strains of human coronavirus (HCoV) responsible for respiratory tract infections with different severity include 229E (HCoV-229E), NL63 (HCoV-NL63), OC43 (HCoVOC43), and HKU1 (HCoV-HKU1). They create respiratory complications such as common colds, bronchiolitis, and pneumonia. Well identified animal CoVs are Avian Infectious Bronchitis Virus (IBV), Turkey Coronavirus (TCV), Porcine Transmissible Gastroenteritis Virus (TGEV), Porcine Hemagglutinating Encephalomyelitis Virus (HEV), canine coronavirus (CCoV), Feline Infectious Peritonitis Virus (FIPV) And Bovine Coronavirus (BCV) [[25], [26], [27]]. Cross infectivity of these viruses to other animals is rare [28,29]. However, the outbreak of SARS-CoV in the year 2002 was unique in sense the virus's true host was Bat and instead cross infected humans leading to higher mortality than that of HCoVs. Another similar case there MERS-CoV infected humans in the year 2005 was known to have Camel as its native host. Such cross infectivity caused by CoV has prompted a new health alert. Recently COVID-19 outbreak in 2019 is the most severe outbreaks caused by Corona viruses and again, its origin is thought to be from Bats and the virus causing this pandemic condition has very high similarity with previous 2002 SARS-CoV and hence is named SARS-CoV2 [4,5] Strategies to control CoVs, there are two main ventures (i) discover inhibitors to block virus entry into the host cells, and (ii) discover bioactive compounds that prevent viral replication and transcription. In total of twelve proteins that are identified as a target to control CoVs, Mpro is identified as one of the most vital targets due to its pivotal role in mediating viral replication and transcription [4,5]. The Mpro is a chymotrypsin-like cysteine protease (~33 kDa) and is termed the Main protease because of its dominant role in processing replicase polyproteins and genes. The first Mpro structure to be solved was that of TGEV at 1.96 Å in the year 2002 [25,26].
Due to the high degree of similarity with amongst the Mpro's of different CoVs, the inhibitor designed previously for MERS or SARS can also work effectively against SARS-CoV-2. An inhibitor named N2 previously designed to inhibit Mpro of SARS-CoV and MERS-CoV was also tested for SARS-CoV-2 and showed promising results [4]. The Mpro of SARS-CoV2 is in fact co-crystalized with N3 and is deposited in PDB (ID: 6LU7). For this reason, under present study, N3 was taken as a positive control to access potency of other inhibitors using computational approach. Moreover, the structure of N3 was artistically developed by CADD approach. Other compounds that are previously designed for SARS-CoV and MERS-CoV include, Hesperetin, Calmidazolium, Cinanserin, Aza-peptide epoxides, FL-166, 8c, 2a and 4o [25,26]. For short discovery of inhibitors for Mpro of SARS-CoV-2, these previously identified compounds can also be tested. Recently the efforts are been made to construct efficient inhibitors using CADD [10]. For rapid commercialization of drug to inhibit SARS-CoV2 targets, the possible approach is to identify bioactive compounds already approved by FDA using computational approach of docking and molecular dynamics. Such approach was recently perused by Arya et al. [30] where that proposed Procainamide, Tetrahydrozoline, Levamisole to interfere the action of papain-like protease of SARS-CoV2. Thus, molecular docking and molecular dynamics can successfully provide insights to the potency of drugs towards the targets of SARS-CoV2 with relative accuracy and in short duration of time. Here we have therefore used the same approach to identify bioactive compounds from fungal origin that involved the use of docking followed by molecular dynamics. The group of scientists who co-crystalized this protein with N3 also suggests strong covalent bond formed between Cys145 of Mpro with N3 of 1.8 Å length making the interaction with of this inhibitor bind irreversibly with the protein [4]. However, when we performed the docking, we cannot make the deduction as docking doesn't provide such information on covalent bond formation. In past Chloropyridine was known to show moderate interaction with Mpro of previously identified SARS-CoV [31]. There are several efforts made to find inhibitors of other corona viruses that have known to causef threat to humans in past, but there limited experiments performed to identify one for SARS-CoV2 [32]. ADMET analysis suggested that, LD50 levels of Pyranonigrin A to be much lower than N3, the possible reason could be the larger size of N3 and its peptide like properties. However, the toxicity of 1.979 mol/kg of Pyranonigrin A is accepted in the medical community and is found to be like several drugs. The classic example would be penicillin, exhibiting LD50 toxicity of 1.8 mol/kg. (https://www.drugbank.ca/drugs/DB00417). Thus, molecules possessing such levels of LD50 values are acceptable.
Reflecting in the history, fungal metabolites have been a boon for the mankind, starting from antibiotics to flavoring agents to food preservatives. Fungi are known to produce large amounts of secondary metabolites and by chance that might interact with Mpro, with this rationale the fungal metabolites were looked for pursuing this research. Previously, the potentials of fungal metabolites as anti-viral agents are explored and the success was promising. The anti-viral compounds from fungal origin is vividly described by Linnakoski et al. [12] where the promising anti-viral compounds portrayed were belong to the chemical class of Indole alkaloids, Non-ribosomal peptides, Polyketides, Terpenoids. The computational experiments and calculations performed virtually screening hundreds of different fungal metabolites; Pyranonigrin A was identified as a potent inhibitor of Mpro. The comparative analysis of its result with N3, this fungal metabolite could make seven hydrogen bonds at par with N3 and is also predicted to form covalent bond with Mpro making it a promising compound that could be seen for. A polyketide synthase and nonribosomal peptide synthetase hybrid gene cluster from the genome of Penicillium thymicola and expression of this cluster leads to the production of Pyranonigrin A is reported [33]. Last but not the least this compound is extensively studied for its antioxidant property [34].
With efforts been made to control the pandemic of SAR-CoV2 in the form of novel Coronavirus, to find a lead compound against the target protein that can suppress its replication in host. We here are proposing Pyranonigrin A to interact with one of the important target proteins of SAR-CoV2, Mpro and block its function as deduced using docking and molecular dynamics.
Compliance with ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Author statement
PR and AS Conseptulized the work, performed Data curation. AS and DG Wrote the manuscript. PR and DG Conceptualize idea. Review & Editing was performed by AS and PR. Supervision was conducted by RR, BV and MS.
Author approvals
All authors have seen and approved the manuscript, and that it hasn't been accepted or published elsewhere.
Declaration of Competing Interest
The authors declare that they have no conflict of interest.
Acknowledgements
Authors are thankful to Gujarat University for providing necessary facilities to perform experiments.
References
- 1.de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14(8):523. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., Yuan M.L., Zhang Y.L., Dai F.H., Liu Y., Wang Q.M., Zheng J.J., Xu L., Holmes E.C., Zhang Y.Z. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. 2020. Structure of Mpro From COVID-19 Virus and Discovery of Its Inhibitors. [DOI] [PubMed] [Google Scholar]
- 5.Kong R., Yang G., Xue R., Liu M., Wang F., Hu J., Guo X., Chang S. COVID-19 docking server: an interactive server for docking small molecules, peptides and antibodies against potential targets of COVID-19. arXiv:2003.00163. 2020 doi: 10.1093/bioinformatics/btaa645. (preprint) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang F., Chen C., Yang K., Xu Y., Liu X., Gao F., Liu H., Chen X., Zhao Q., Liu X., Cai Y., Yang H. Michael acceptor-based peptidomimetic inhibitor of Main protease from porcine epidemic diarrhea virus. J. Med. Chem. 2017;60(7):3212–3216. doi: 10.1021/acs.jmedchem.7b00103. [DOI] [PubMed] [Google Scholar]
- 7.Xue X., Yang H., Shen W., Zhao Q., Li J., Yang K., Chen C., Jin Y., Bartlam M., Rao Z. Production of authentic SARS-CoV Mpro with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 2007;366(3):965–975. doi: 10.1016/j.jmb.2006.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui W., Cui S., Chen C., Chen X., Wang Z., Yang H., Zhang L.J.B. The crystal structure of main protease from mouse hepatitis virus A59 in complex with an inhibitor. J. Biochem. Biophys. Res. Commun. 2019;511(4):794–799. doi: 10.1016/j.bbrc.2019.02.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.John S.E.S., Tomar S., Stauffer S.R., Mesecar A.D. Targeting zoonotic viruses: Structure-based inhibition of the 3C-like protease from bat coronavirus HKU4—The likely reservoir host to the human coronavirus that causes Middle East Respiratory Syndrome (MERS) Bioorg. Med. Chem. 2015;23(17):6036–6048. doi: 10.1016/j.bmc.2015.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macchiagodena M., Pagliai M. Inhibition of the main protease 3CL-pro of the coronavirus disease 19 via structure-based ligand design and molecular modeling. arXiv. 2020 doi: 10.1016/j.cplett.2020.137489. (preprint arXiv: 09937) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang F., Chen C., Liu X., Yang K., Xu X., Yang H. Crystal structure of feline infectious peritonitis virus main protease in complex with synergetic dual inhibitors. J. Virol. 2016;90(4):1910–1917. doi: 10.1128/JVI.02685-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linnakoski R., Reshamwala D., Veteli P., Cortina-Escribano M., Vanhanen H., Marjomäki V. Antiviral agents from fungi: diversity, mechanisms and potential applications. Front. Microbiol. 2018;9:2325. doi: 10.3389/fmicb.2018.02325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krivov G.G., Shapovalov M.V., Dunbrack R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77(4):778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J., Wang W., Kollman P.A., Case D.A. Antechamber: an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001;222:U403. [Google Scholar]
- 15.Gasteiger J., Jochum C. An algorithm for the perception of synthetically important rings. J. Chem. Inform. 1979;19(1):43–48. [Google Scholar]
- 16.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bekker H., Berendsen H., Dijkstra E., Achterop S., van Drunen R., der Spoel D., BEKKER H., Dijkstra E., Van Der Spoel D., Sijbers A. 1993. Gromacs: A Parallel Computer for Molecular Dynamics Simulations. [Google Scholar]
- 18.Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D.J.B. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H. 26(16) 2005. GROMACS: Fast, Flexible, and Free; pp. 1701–1718. [DOI] [PubMed] [Google Scholar]
- 20.Zoete V., Cuendet M.A., Grosdidier A., Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011;32(11):2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 21.Berendsen H.J., Postma J.P., van Gunsteren W.F., Hermans J. Intermolecular Forces. Springer; 1981. Interaction models for water in relation to protein hydration; pp. 331–342. [Google Scholar]
- 22.Hess B., Bekker H., Berendsen H.J., Fraaije J.G. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18(12):1463–1472. [Google Scholar]
- 23.Turner P., Land-Margin Research, O.G.I.o.S., Technology, B., OR . 2005. XMGRACE, Version 5.1; p. 19. [Google Scholar]
- 24.Pires D.E., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58(9):4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H., Bartlam M., Rao Z.J.C.p.d. Drug design targeting the main protease, the Achilles' heel of coronaviruses. Curr. Pharm. Des. 2006;12(35):4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 26.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3(10):e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003;100(23):13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H. An overview of Severe Acute Respiratory Syndrome–Coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59(14):6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., Rao Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an "Achilles' heel": current effective inhibitor targeting a 3C-like protease. Protein Cell. 2013;4(4):248. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arya R., Das A., Prashar V., Kumar M. 2020. Potential Inhibitors Against Papain-like Protease of Novel Coronavirus (SARS-CoV-2) From FDA Approved Drugs. [Google Scholar]
- 31.Niu C., Yin J., Zhang J., Vederas J.C., James M.N. Molecular docking identifies the binding of 3-chloropyridine moieties specifically to the S1 pocket of SARS-CoV Mpro. Bioorg. Med. Chem. 2008;16(1):293–302. doi: 10.1016/j.bmc.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C.J. 2020. Structure of M pro From SARS-CoV-2 and Discovery of its Inhibitors; pp. 1–5. [DOI] [PubMed] [Google Scholar]
- 33.Tang M.C., Zou Y., Yee D., Tang Y. Identification of the pyranonigrin A biosynthetic gene cluster by genome mining in Penicillium thymicola IBT 5891. AICHe J. 2018;64(12):4182–4186. doi: 10.1002/aic.16324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riko R., Nakamura H., Shindo K. Studies on pyranonigrins–isolation of pyranonigrin E and biosynthetic studies on pyranonigrin A. J. Antibiot. 2014;67(2):179–181. doi: 10.1038/ja.2013.91. [DOI] [PubMed] [Google Scholar]