

Abstract
Background
Coronavirus disease 2019 (COVID-19) primarily affects the lungs, but evidence of systemic disease with multi-organ involvement is emerging. Here, we developed a blood test to broadly quantify cell-, tissue-, and organ-specific injury due to COVID-19.
Methods
Our test leverages genome-wide methylation profiling of circulating cell-free DNA in plasma. We assessed the utility of this test to identify subjects with severe disease in two independent, longitudinal cohorts of hospitalized patients. Cell-free DNA profiling was performed on 104 plasma samples from 33 COVID-19 patients and compared to samples from patients with other viral infections and healthy controls.
Findings
We found evidence of injury to the lung and liver and involvement of red blood cell progenitors associated with severe COVID-19. The concentration of cell-free DNA correlated with the World Health Organization (WHO) ordinal scale for disease progression and was significantly increased in patients requiring intubation.
Conclusions
This study points to the utility of cell-free DNA as an analyte to monitor and study COVID-19.
Funding
This work was supported by NIH grants 1DP2AI138242 (to I.D.V.), R01AI146165 (to I.D.V., M.P.C., F.M.M., and J.R.), 1R01AI151059 (to I.D.V.), K08-CA230156 (to W.G.), and R33-AI129455 to C.Y.C., a Synergy award from the Rainin Foundation (to I.D.V.), a SARS-CoV-2 seed grant at Cornell (to I.D.V.), a National Sciences and Engineering Research Council of Canada fellowship PGS-D3 (to A.P.C.), and a Burroughs-Wellcome CAMS Award (to W.G.). D.C.V. is supported by a Fonds de la Recherche en Sante du Quebec Clinical Research Scholar Junior 2 award. C.Y.C. is supported by the California Initiative to Advance Precision Medicine, and the Charles and Helen Schwab Foundation.
Keywords: SARS-CoV-2, COVID-19, cell-free DNA, methylation, liquid biopsy, tissue injury, disease severity
Graphical abstract
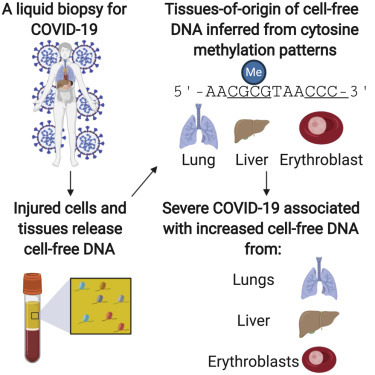
Context and significance
COVID-19 is a systemic disease with multi-organ involvement. Here, the authors report a blood test to quantify cell-, tissue-, and organ-specific injury due to COVID-19. This is accomplished by profiling methylation marks within circulating cell-free DNA (cfDNA) to trace their tissues of origin and to quantify tissue-specific injury due to COVID-19. The authors assessed the utility of this test to identify subjects with severe disease in two independent, longitudinal cohorts of hospitalized patients and report evidence of injury to the lung and liver and involvement of red blood cell progenitors associated with severe COVID-19. These results support the utility of cfDNA profiling as a prognostic tool for the early detection and monitoring of cell and tissue injury due to COVID-19.
COVID-19 affects the entire body. Cheng et al. describe a DNA-based blood test to quantify cell and tissue injury due to COVID-19. They show that severe COVID-19 is associated with increased lung, liver, and erythroblast DNA in blood and that the concentration of cell-free DNA correlates with disease progression.
Introduction
The coronavirus disease 2019 (COVID-19) pandemic is a major global health crisis. COVID-19 is a complex disease with diverse clinical features, ranging from asymptomatic infection to acute respiratory distress syndrome (ARDS) and multi-organ dysfunction. There is an urgent need for predictive biomarkers of COVID-19 severity detectable early in disease onset and improved understanding of the pathogenesis of COVID-19. Here, we have investigated the utility of circulating cell-free DNA (cfDNA) in blood as an analyte to (1) broadly monitor cell, tissue, and organ injury due to COVID-19, (2) assess disease severity and predict disease outcomes, and (3) elucidate the multi-organ involvement that characterizes COVID-19.
Autopsy studies indicate a broad organotropism for the severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) virus beyond the lungs.1 , 2 Detection of the virus in the kidneys, heart, liver, brain, and blood of many patients has been reported.2 , 3 The significant viral burden in the kidney seen in some patients may help explain the increased risk of acute kidney injury in patients with COVID-19. Damage to endothelial cells may contribute to COVID-19 coagulopathy and prothrombic state.4, 5, 6, 7, 8
Initial reports have primarily described COVID-19 as a disease affecting tissues expressing angiotensin-converting enzyme 2 (ACE-2).9 However, there are emerging data that SARS-CoV-2 infection may also be accompanied by hematological derangements.10, 11, 12, 13 In addition, a dysregulated immune response to SARS-CoV-2 can occur, contributing to the development of ARDS, systemic tissue injury, and multi-organ failure.14 A strong association between increased cytokine profiles and the severe deterioration of some patients has been observed.15 In children, a multisystem inflammatory syndrome linked to recent SARS-CoV-2 infection is reported.16 Given the disparate clinical manifestations and potential complications of COVID-19, there is an urgent need for tests that can quantify injury to multiple tissues simultaneously to monitor patients, analyze disease pathogenesis, predict clinical outcomes, and guide clinical management in patients with COVID-19.
Since the advent of cfDNA-based non-invasive prenatal testing, myriad applications of cfDNA in diagnostic medicine have been established.17, 18, 19 These short fragments of circulating DNA are the debris of dead cells from across the body. The value of cfDNA as a quantitative marker of tissue and organ injury was first recognized in solid-organ transplantation, in which the level of transplant donor-derived cfDNA in the blood is now widely used as a marker of transplant rejection.20, 21, 22 More recently, several approaches have been developed to quantify the tissues of origin of cfDNA and thus monitor injury to any cell, tissue, or organ type.23, 24, 25, 26, 27 This is achieved by profiling epigenetic marks within cfDNA by quantitative molecular measurement technologies such as DNA sequencing. Here, we tested the hypothesis that cfDNA tissues of origin profiling enables the identification of specific tissue or cell types that are directly or indirectly targeted and injured throughout COVID-19 pathogenesis. We studied two independent patient cohorts and found evidence of significant injury to the liver, lung, and kidney associated with COVID-19. We further observed a striking increase, both in terms of proportion and total abundance, of cfDNA derived from red blood cell (RBC) precursors when compared to patients infected with other RNA viruses and healthy controls. Last, the total burden of cfDNA correlated with the World Health Organization (WHO) ordinal scale for disease progression, with an increase in cfDNA being strongly associated with admission to the intensive care unit (ICU) and need for mechanical ventilation. Thus, cfDNA can provide a marker of disease severity and a prognostic tool that is straightforward to adopt.
Results
We tested the utility of cfDNA to quantify cell-, tissue-, and organ-specific injury associated with COVID-19 in two independent patient cohorts from two different hospitals in North America (Figure 1 A, patient characteristics in Tables S1 and S2). We assayed a total of 104 plasma samples from 33 patients across these cohorts. We performed shotgun DNA sequencing after bisulfite treatment to determine the tissues of origin of cfDNA isolated from all plasma samples by methylation profiling. We obtained 62 ± 35 million (means ± standard deviations) paired-end reads per sample, leading to a per-base genome coverage of 1.3 ± 0.8. We verified that we achieved a high bisulfite conversion efficiency for all of the samples (0.996 ± 0.005, Method Details). To determine the cell, tissue, and organ types that contribute cfDNA to the mixture in blood (Method Details), we analyzed plasma cfDNA methylation profiles against a reference set of 147 cell, tissue, and organ types using previously described bioinformatic approaches (Figures 1B and 1C; Data S1; Method Details).25
Figure 1.
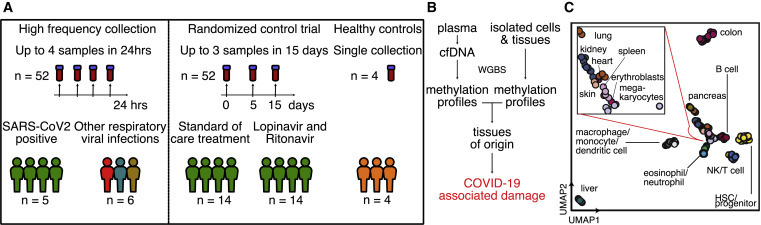
Study design
(A) Two independent cohorts were used in our study. First, a high-frequency collection cohort with 5 SARS-CoV-2 patients (n = 52 samples) and 6 SARS-CoV-2-negative, RNA virus-positive patients (n = 6 samples). Second, a randomized controlled trial of 28 SARS-CoV-2 patients with plasma at serial time points (n = 52 samples). Four healthy individuals volunteered plasma for cell-free DNA analysis.
(B) Experimental workflow. cfDNA is extracted from plasma, and whole-genome bisulfite sequencing is performed. In parallel, methylation profiles of cell and tissue genomes are obtained from publicly available databases. cfDNA methylation profiles are compared to those of cell and tissue references to infer relative contributions of tissues to the cfDNA mixtures.
(C) UMAP of differentially methylated regions for isolated cell and tissue types used as a reference.
Temporal dynamics of cfDNA tissues of origin in plasma of COVID-19 patients
We first assayed 52 serial samples collected at short time intervals from 5 adult patients with COVID-19 who were treated at University of California, San Francisco (UCSF) Medical Center (median of 8 samples per patient [range 6–18]). These plasma samples were residual from clinical testing and were collected from this group of patients over a treatment period of up to 14 days, with up to 4 samples collected within 24 h (median time between consecutive collections of 13 h [range 5–64]). These samples allowed us to study dynamic changes in cfDNA profiles in patients diagnosed with and treated for COVID-19 (Figure 2 A). One patient did not require oxygen at the time of hospital admission, 2 patients required oxygen by mask (n = 2), and 2 patients required mechanical ventilation (n = 2). Two patients presented with ARDS (Table S1). Treatments included standard of care (n = 2), remdesivir (n = 1), hydroxychloroquine (n = 1), or a combination of remdesivir, hydroxychloroquine, azithromycin, and tocilizumab (n = 1). In addition to plasma from COVID-19 patients, we performed cfDNA tissues of origin profiling for 6 samples collected from patients with other respiratory viral infection treated at the same hospital, including influenza B (n = 2), metapneumovirus (n = 1), coronavirus HKU1 (n = 1), coronavirus NL63 (n = 1), and respiratory syncytial virus B (n = 1) (Figure 2B).
Figure 2.
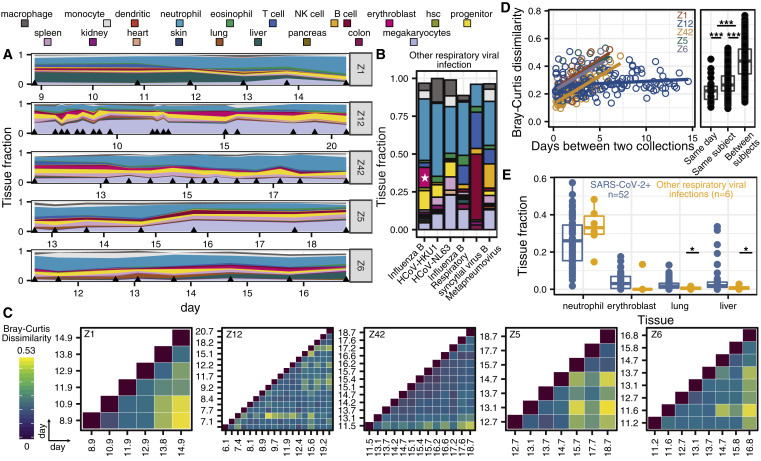
High-Frequency sample collection cohort at UCSF
(A and B) Patient-specific relative tissue contributions for SARS-CoV-2 patients (A) and other RNA virus infection patients (B). Triangles (A) indicate sampling times and the star (B) represents the erythroblast fraction of an influenza B patient who was being treated for recurrent stage IV diffuse large B cell lymphoma.
(C) Heatmaps of Bray-Curtis dissimilarity.
(D) Scatterplot of patient-specific Bray-Curtis dissimilarity (left) and boxplot of Bray-Curtis dissimilarity between cfDNA tissue proportions from samples collected from either the same day (within 24 h), the same person (but not within 24 h), or from all of the patients (right).
(E) Comparison of tissue fraction of 4 cell and tissue types (neutrophil, erythroblast, lung, and liver) between SARS-CoV-2-positive patients and other RNA virus-positive patients. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 (p values calculated using a Wilcoxon test)
We plotted the relative abundance of cfDNA derived from different cell, tissue, and organ types and found that differences in cfDNA profiles between individuals were larger than differences within individuals over the sampling period. For subjects Z1, Z5, Z6, and Z42, but not Z12, we observed gradual changes in the tissues of origin profiles over sampling periods of 6–7 days. We used the Bray-Curtis dissimilarity to quantify the inter- and intra-individual differences in cfDNA profiles (Figures 2C and 2D). This analysis confirmed the visual appearance of the tissues of origin profiles in Figure 2A and demonstrated that the largest differences in cfDNA were found for samples collected from different individuals. Within subjects, smaller differences were observed for samples collected on the same day (Figure 2D). Last, the Bray-Curtis dissimilarity increased with time intervals between samples for patients Z1, Z5, Z6, and Z42, but not for Z12. We verified that these observations were not affected by differences in the plasma volume of different samples (Figure S1A). These analyses indicate that cfDNA profiles are subject specific and that changes in cfDNA tissues of origin profiles occur gradually over days and not hours; therefore, adequate longitudinal data can be collected every few days.
We next compared the cfDNA tissues of origin profiles associated with COVID-19 versus those associated with respiratory infection with other viruses (Figure 2E; Table S3). We found significant increases in the relative proportion of lung-specific cfDNA in the blood of COVID-19 patients, which was likely related to COVID-19-associated tissue injury (2.5% versus 0.6%, p = 0.019, Wilcoxon). We found a similar association with liver-derived cfDNA (5.0% versus 0.9%, p = 0.025, Wilcoxon), and this was validated by the elevated liver function tests in 4 of 5 COVID-19 patients. Strikingly, we also observed an increase in the relative proportion of cfDNA derived from erythroblasts in the blood of COVID-19 patients compared to the control group (75% versus 17% samples with an erythroblast fraction >0, p = 0.003, 2-sample proportions test; Figures 2E and S1B). Erythroblasts are nucleated cells typically in the adult bone marrow from which RBCs develop and have been shown to contribute circulating cfDNA to plasma.28 The increase in cfDNA derived from red blood progenitor cells seen here may be an indirect consequence of the hypoxemia and/or cytokine-mediated anemia that characterize severe COVID-19, or it may indicate a more direct involvement of coronavirus with RBC precursors. We note that erythroblast cfDNA was elevated in a single patient in the control group, who was being treated for recurrent stage IV diffuse large B cell lymphoma (Figures 2B and 2E).
Randomized clinical trial cohort
To test the robustness of these initial observations, we assayed an additional 52 samples collected from 28 patients who were recruited into a randomized controlled trial at the McGill University Health Centre in Montreal, Canada. Patients were assigned to either an experimental antiviral therapy consisting of a combination of lopinavir and ritonavir or to the standard of care. Of these patients, 14 were treated with the lopinavir/ritonavir and 14 received the standard of care. At the time of hospital admission, patients either did not require oxygen (n = 9), required oxygen by mask (n = 10), were intubated (without ARDS, n = 5; with ARDS or requiring vasopressors, n = 2), or required mechanical ventilation and vasopressors, dialysis, or extracorporeal membrane oxygenation (ECMO, n = 2). Of the 28 patients, 22 were discharged after treatment and 6 patients died. Serial samples were collected from these patients at 3 predetermined time points: days 1, 5, and 15 after enrollment in the clinical trial, provided they remained hospitalized on the days of collection (Figure 3 A). We determined the relative abundance of tissue-specific cfDNA using the approaches described above. In addition, we quantified the absolute concentration of tissue-specific cfDNA by multiplying the proportion of tissue-specific cfDNA with the concentration of total cfDNA (Method Details).
Figure 3.
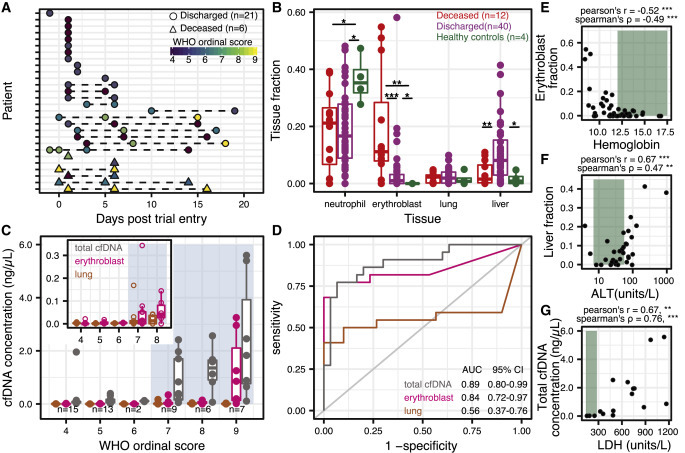
Randomized controlled trial cohort from MUHC
(A) Patient sample collection map by day of enrollment in the study.
(B) Relative proportion of cfDNA derived from 4 cell and tissue types (neutrophil, erythroblast, lung, liver) by hospitalization status (p values calculated using a Wilcoxon test).
(C) Absolute cfDNA concentrations compared to the WHO ordinal scale for COVID progression. Blue shading indicates ordinal scores requiring admittance to the intensive care unit (ICU).
(D) Receiver operating characteristic analysis of the performance of absolute cfDNA concentration of different tissues (lung, erythroblast, and total) in distinguishing patients presenting with ordinal scales from 4 to 6 (hospitalized) and 7 to 9 (hospitalized in the ICU).
(E–G) Scatterplot comparisons between relative proportions of erythroblast cfDNA fraction and hemoglobin (E), liver cfDNA fraction and alanine aminotransferase (ALT) (F), and total cfDNA concentration and lactase dehydrogenase (LDH) (H). Green shading indicates normal levels. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
We compared the cfDNA tissues of origin profiles measured for these patients with the tissues of origin profiles for four healthy subjects (Figures 3B and S2A). We found that 62% of samples from patients with COVID-19 had a higher concentration of lung cfDNA than the highest concentration measured for a healthy individual (p = 0.017, 2-sample proportions test). In addition, hospitalized patients with COVID-19 had both an elevated relative and absolute burden of cfDNA derived from the liver (liver fraction 9.1 versus 1.6%, p = 0.054, and 0.051 ng/μL versus 0.00029 ng/μL, p = 0.010, Wilcoxon). In addition to these tissue-specific features, we again observed a significant increase in cfDNA derived from erythroblast cells for COVID-19 patients compared to healthy controls (7.7% versus 0%, p = 0.027, Wilcoxon; 65% versus 0% of samples showing an erythroblast fraction >0, p = 0.0099, 2-sample proportions test; Figure 3B). We evaluated the temporal dynamics of the contribution of different cell and tissue types to the mixture in plasma of COVID-19 patients and observed a slow recovery in tissue injury and a slow increase in the contribution of cfDNA derived from erythroblasts (Figures S2B and S2C). To assess the prognostic value of the burden of erythroblast-derived cfDNA, we calculated the odds ratios of eventual mortality versus non-mortality across different erythroblast fractions for two different sample groups: (1) the earliest available sample per patient and (2) for all of the samples. Using a cutoff value of 6% erythroblast-derived cfDNA, we obtained odds ratios of 105 (p = 3.53 × 10−4, Fisher’s exact test) and 28 (p = 2.52 × 10−5, Fisher’s exact test) for each group, respectively (Figures S2D–S2G). Last, we found that an elevated proportion of erythroblast cfDNA was associated with decreased survival probability (p = 0.0052, log-rank test, high and low defined as ≥6% erythroblast-derived cfDNA or <6%, respectively, Kaplan-Meier; Figures S2D–S2G).
We next compared cfDNA signatures for COVID-19 patients as function of disease severity and found that erythroblast cfDNA proportions at any time point are predictive of in-hospital mortality (19.6% versus 4.1%, p = 0.0004, Wilcoxon). Receiver operating characteristic (ROC) analysis of the performance of the relative proportion of erythroblast-derived DNA to predict COVID-19 mortality yielded an area under the curve (AUC) of 0.83 (95% confidence interval [CI] 0.69–0.98 [deceased n = 12; discharged n = 40]). In addition, our analysis revealed that kidney cfDNA was significantly elevated in COVID-19 patients who eventually died (1.8% versus 0.5% versus 0.005% between deceased, non-deceased, and healthy controls, respectively; p = 0.0018 between deceased and non-deceased COVID-19 patients).
We then compared the cfDNA tissues of origin profiles to the 10-point WHO clinical status ordinal scale for COVID-1929 (Figure 3C). We found a strong association between the total cfDNA concentrations isolated from plasma and the WHO clinical progression scores (Figures 3C and 3D). Notably, a clinical score of ≥7 (indicating the need for admission to the ICU and invasive mechanical ventilation) was associated with a sharp increase in the total burden of cfDNA (Figures 3C and 3D, mean 1.5 ng/μL versus 0.16 ng/μL, between clinical scores from 7 to 9 and 4 to 6, respectively; p = 1.5 × 10−6, Wilcoxon, odds ratio = 62, p = 5.00 × 10−7, Fisher’s exact test; Figures S2D–S2G). ROC analysis of cfDNA concentrations to predict ordinal scores revealed AUCs of 0.89 (95% CI 0.80–0.99), 0.84 (95% CI 0.72–0.97), and 0.56 (95% CI 0.37–0.76) for total, erythroblast, and lung cfDNA, respectively. Furthermore, samples taken from patients with a clinical score of 9 (use of ECMO) had significantly higher erythroblast-derived cfDNA than patients with a clinical score of 7–8 (1.23 ng/μL versus 0.06 ng/μL, p = 0.006, Wilcoxon). Patients receiving ECMO tend to bleed and require additional blood volumes, which may contribute to the increased erythroblast signal. However, erythroblast-derived cfDNA was significantly increased in patients with a clinical score of ≥7 as well (Figures 3C and 3D; mean 0.43 ng/μL versus 0.003 ng/μL, p = 1.83 × 10−5, Wilcoxon).
Erythroblast and liver cfDNA contributions correlated with clinical metrics for anemia and liver damage, respectively (Figures 3E–3G). We observed significant negative correlations between the proportion of erythroblast cfDNA and hematocrit and hemoglobin (Hgb; Pearson’s R = −0.51, Spearman’s ρ = −0.37 and R = −0.52, ρ = −0.49, respectively). Similarly, we found positive correlations between the proportion of liver-derived cfDNA and alanine aminotransferase (ALT) and aspartate transaminase (AST; R = 0.63, ρ = 0.47 and R = 0.76, ρ = 0.24, respectively). We did not observe a correlation between kidney-derived cfDNA and serum creatinine (R = 0.05, ρ = 0.09). We found similar results when comparing the tissue-derived cfDNA concentration to these clinical markers (erythroblast cfDNA concentration versus hematocrit and Hgb: R = −0.42, ρ = −0.32 and R = −0.38, ρ = −0.45, respectively; liver cfDNA concentration versus ALT and AST: R = 0.84, ρ = 0.52 and R = 0.20, ρ = 0.23, respectively; kidney cfDNA concentration versus creatinine: R = 0.56, ρ = 0.20).
Recent papers from Zhou et al.30 and Yan et al.31 identified lactate dehydrogenase (LDH) as a strong predictor of COVID-19 outcome. LDH is found in virtually all cells and is a commonly used biomarker for tissue damage and hemolysis.32, 33, 34 We found a significant correlation between LDH and the proportion of erythroblast-derived cfDNA (R = 0.64, ρ = 0.65) and between LDH and total cfDNA (R = 0.67, ρ = 0.76). These data suggest that cfDNA tissues of origin can be applied to resolve the specific tissues contributing to non-specific detection of LDH in blood. To evaluate whether cfDNA offers diagnostic value beyond conventional protein biomarkers (ALT, AST, C-reactive protein [CRP], total bilirubin, LDH, RBC distribution width- variation coefficient [RDW-CV], hematocrit, Hgb, creatinine), we developed a composite machine learning ensemble method (Extra Trees method35) incorporating all biochemical markers; the proportions of erythroblast, kidney, liver, lung and neutrophil cfDNA; and total cfDNA concentration (Method Details). Feature importance analysis identified erythroblast and kidney-derived cfDNA as the most informative for COVID-19 mortality prediction by this model (Figures S3A and S3B). ROC analysis with cross-validation indicated that the composite model using cfDNA outperforms a model that only used conventional biomarkers (AUC 0.91 versus 0.73, respectively; Figures S3A and S3B).
We found no differences between lung, liver, kidney, or erythroblast-derived cfDNA for patients receiving standard of care, or the experimental lopinavir/ritonavir treatment (Figure S3C). These data are in line with the results of recent clinical trials that treatment with lopinavir/ritonavir is not significantly different from standard of care treatment for COVID-19.36 , 37 Finally, we tested the prognostic value of biophysical properties of cfDNA and the burden of mitochondrial cfDNA since these factors have previously been shown to have diagnostic value in other disease settings.24 , 38, 39, 40, 41 We investigated the distribution of cfDNA fragment lengths represented in the sample (mean, median), fragmentation patterns (short:long ratio, dinucleosome fraction), and the burden of mitochondrial cfDNA. We found no significant differences for these metrics (deceased versus discharged; Figures S3D and S3E).
Discussion
We find significant support for the utility of cfDNA profiling as a prognostic tool for the early detection and monitoring of cell and tissue injury associated with COVID-19. An easy-to-obtain molecular blood test that can inform cell-, tissue-, and organ-specific injury due to COVID-19 has the potential to alleviate the impact of the COVID crisis by (1) providing quantifiable prognostic parameters and a more granular assessment of clinical severity at the time of presentation; and (2) providing a surrogate biomarker that can be included in clinical trials of candidate COVID-19 treatments.
In line with the diverse clinical manifestations of COVID-19, we find evidence for lung, liver, and kidney injury in hospitalized patients with COVID-19. While lung-derived cfDNA was elevated in COVID-19 patients, we did not find it to be a major contributor to plasma cfDNA. The level of lung-specific cfDNA in plasma was similar to the levels observed in lung transplant patients who suffer acute lung transplant rejection20 and lung cancer patients.42 , 43 We observed a striking correlation between the total abundance of circulating cfDNA in plasma and the WHO ordinal scale for disease progression. We propose that the total abundance of cfDNA, which can be measured within 1 h at low cost, can be used in the context of clinical trials and patient management in the near term.
In addition to the practical application of cfDNA profiling to patient monitoring and COVID-19 risk stratification, the cfDNA methylation assay and data reported may help elucidate aspects of COVID-19 pathogenesis. The most significant cfDNA signature observed in the two cohorts relative to controls was an increase in cfDNA derived from erythroid or red blood progenitor cells. Given that cfDNA is estimated to have a half-life of ~1 h44 and that the proportion of the erythroid lineage was relatively stable over several days, the elevated erythroid cfDNA is likely due to an increased erythroid turnover. In support of elevated erythroid turnover and production, two recent studies have identified RDW, a measure of the variation in size of RBCs, as an important prognostic predictor for severe COVID-19.15 , 16 The increased RDW is possibly associated with the increased turnover of RBCs since increased reticulocytes or newly formed RBCs have a wider diameter.16 However, our analysis demonstrated that there was no association with RDW and patient outcomes (mean 15.4 versus 14.0 between deceased and discharged, p = 0.2, Wilcoxon) and that erythroblast cfDNA was not strongly correlated with RDW (R = 0.26, ρ = 0.13, with data from UCSF and McGill University Health Center [MUHC]).
Increased erythroid turnover may be due to erythroid destruction as the primary driver, followed by compensatory production, and is supported by anemia (Hgb <13.5 g/dL for men and Hgb <12 for women) found in 26 of 33 COVID-19 patients across both studies. Possible mechanisms include (1) excessive inflammation and cytokine storm,45 , 46 (2) hemophagocytosis in relation to inflammation,47 and (3) consumption in microthrombi.6, 7, 8 , 10 We note that in 18 of 33 patients in all studies, CRP was elevated (>10 mg/L). It is notable, however, that megakaryocytes proportions were not increased in either cohort and would not support microthrombi as the predominant reason for increased erythroid turnover. Alternatively, past work has shown that angiotensin II regulates normal erythropoiesis and stimulates early erythroid proliferation through unclear downstream mechanisms.48, 49, 50 The binding of SARS-CoV-2 to the host ACE-2 may dysregulate erythropoiesis through the downstream angiotensin II pathway. The significant increase in cfDNA derived from red blood progenitor cells may alternatively be due to injury to red cell precursors51 through direct or indirect processes. These hypotheses are further testable through various routes, including comprehensive evaluation of erythrocytosis in patients with COVID-19, for example, through the evaluation of circulating reticulocytes and evaluation of the bone marrow; these measures were not systematically in place during the initial rapid wave of the pandemic and were not implemented in this study.
In summary, we report the application of cfDNA profiling to quantify cellular and tissue specific injury due to COVID-19.
Limitations of study
This study has several limitations. First, we assayed samples from only hospitalized patients, and we have not evaluated cfDNA profiles for mild COVID-19 cases. Second, while this study spans two independent cohorts, with patient groups that are genetically and geographically unrelated, the overall sample size and patient numbers may not be sufficient to generalize our findings to the entire spectrum of COVID-19 cases. Nonetheless, our analysis of cfDNA tissues of origin can provide immediate insights into the dynamics and pathogenesis of COVID-19. Last, the resolution of our measurements is limited by the availability of isolated cells and tissue methylation patterns. Our current reference dataset does not include all known human cell types and tissue types. Therefore, we are not sensitive to those rarer tissues that may play a role in the pathogenesis of COVID-19. More comprehensive investigations are therefore needed to confirm and further refine the observations reported here.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Plasma from COVID-19 patients (UCSF samples) | University of California, San Francisco | N/A |
| Plasma from patients with viral infections (excluding SARS-CoV-2) | University of California, San Francisco | N/A |
| Plasma from COVID-19 patients (MUHC samples) | McGill University Health Center | N/A |
| Plasma from healthy individuals | Cornell University | N/A |
| Critical Commercial Assays | ||
| QIAGEN MinElute Circulating Nucleic Acid Kit | QIAGEN | 55204 |
| QIAGEN EZ1 Virus Mini Kit v2.0 | QIAGEN | 955134 |
| QIAGEN Circulating Nucleic Acid Kit | QIAGEN | 55114 |
| Zymo EZ Methylation-Gold kit (for bisulfite conversion) | Zymo | D5005 |
| Swift Biosciences Accel-NGS Methyl-Seq DNA Library Kit | Swift | 30024 |
| Deposited Data | ||
| Genomic data | This manuscript | PRJNA687910 (NCBI Sequence Read Archive) |
| Methylation data | This manuscript | https://www.github.com/alexpcheng/cfDNAme |
| Oligonucleotides | ||
| cfDNA concentration control 5′- TTTAACGCATAAACATGCGTTT TGGGTAGTGTTTTTTGGAAACACAG ATCCGTGCGCACACCTGGTGGAG-3′ |
Integrated DNA Technologies | https://www.idtdna.com/pages |
| cfDNA concentration control 2 5′- ATAAACATGCGTTTTGGG TAGTGTTTTTTGGAAA CACAGATCCGTGCGCACACCT-3′ |
Integrated DNA Technologies | https://www.idtdna.com/pages |
| cfDNA concentration control 3 5′- GCGTTTTGGGTAGTGTTTTTT GGAAACACAGATCCGTGCG-3′ |
Integrated DNA Technologies | http://www.idtdna.com/pages |
| cfDNA concentration control 4 5′-GGTAGTGTTTTTTGG AAACACAGAT-3′ |
Integrated DNA Technologies | http://www.idtdna.com/pages |
| Software and Algorithms | ||
| Custom scripts | This manuscript | https://www.github.com/alexpcheng/cfDNAme |
| BBTools | 52 | https://jgi.doe.gov/data-and-tools/bbtools/ |
| Bismark | 53 | https://github.com/FelixKrueger/Bismark |
| SAMtools | 54 | http://www.htslib.org/ |
| Metilene | 55 | https://www.bioinf.uni-leipzig.de/Software/metilene/ |
| MethPipe | 56 | https://github.com/smithlabcode/methpipe |
| Other | ||
| DELFI pipeline | 41 | https://github.com/Cancer-Genomics/delfi_scripts |
| Illumina NextSeq 500 | Illumina | https://www.illumina.com |
| Illumina NovaSeq | Illumina | https://www.illumina.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Iwijn De Vlaminck (vlaminck@cornell.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Genomic data is hosted on the Sequence Read Archive under project number PRJNA687910. The binned methylation data, and the code used to process the data is available at https://www.github.com/alexpcheng/cfDNAme.
Experimental model and subject details
High frequency sampling
Clinical samples from UCSF were processed through protocols approved by the UCSF Institutional Review Board (protocol number 10-00476, 18-25287). Patient characteristics can be found in Table S1.
Randomized clinical trial
Individuals diagnosed with COVID-19 were recruited to a randomized, controlled clinical trial at the McGill University Health Center, where they received either Lopinavir/ritonavir, or standard-of-care (https://clinicaltrials.gov/ct2/show/NCT04330690). Blood samples were collected under MUHC Research Ethics Board protocol 10-256. Patient characteristics can be found in Table S2.
Healthy controls
Volunteers were recruited for blood donations through a protocol approved by the Cornell Institutional Review Board (protocol number 1910009101).
Method details
High frequency sampling
Clinical samples from UCSF were processed through informed (n = 1) and waived consent (n = 4) protocols approved by the UCSF Institutional Review Board (protocol number 10-00476, 18-25287) and followed guidelines established by the Helsinki Declaration.57 Residual plasma was collected as part of routine clinical testing and stored at 4°C for up to 5 days and subsequently stored at −80°C until batched extraction. Plasma was initially isolated from blood by the clinical laboratory after centrifugation at approximately 800 g for 10 minutes. Blood samples were collected in heparin tubes and processed within one hour of collection.58 After storage, the plasma was centrifuged at 16,000 g for 10 minutes. cfDNA extraction was performed according to manufacturer recommendations (QIAGEN MinElute Circulating Nucleic Acid Kit, reference #55204 or QIAGEN EZ1 Virus Mini Kit v2.0 955134) at 0.4-1 mL plasma input.
Randomized clinical trial
Individuals diagnosed with COVID-19 were recruited to a randomized, controlled clinical trial at the McGill University Health Center, where they received either Lopinavir/ritonavir (400 mg/100 mg for 14 days, or until discharge from hospital, whichever occurs first), or standard-of-care (https://clinicaltrials.gov/ct2/show/NCT04330690). Informed consent was obtained from all study participants. Blood samples were collected under MUHC Research Ethics Board protocol 10-256 through standard venipuncture in standard blood collection tubes and immediately centrifuged at 850 g for 10 minutes. The supernatant is then transferred to new tubes, and centrifuged at 16,000 g for 10 minutes. Plasma-containing supernatant is collected and stored in DNA cryostorage vials (Eppendorf, reference #0030079400) at −80°C. Plasma was shipped overnight on dry ice from the McGill University Health Center (Montreal, Canada) to Cornell University (Ithaca, United-States). Plasma was stored at −80°C until used for cfDNA extraction. cfDNA extraction was performed according to manufacturer recommendations (QIAGEN Circulating Nucleic Acid Kit, reference #55114).
Healthy controls
Volunteers were recruited for blood donations through an informed consent protocol approved by the Cornell Institutional Review Board (protocol number 1910009101). Blood was collected in K2 EDTA tubes (BD, reference #366643) and immediately centrifuged at 1600 g for 10 minutes. The supernatant was transferred to new tubes, and centrifuged at 16,000 g for 10 minutes. Supernatant is then stored in DNA cryostorage vials (Thermo Scientific #363401) at −80°C until cfDNA extraction. cfDNA extraction was performed according to manufacturer recommendations (QIAGEN Circulating Nucleic Acid Kit, reference #55114).
Whole genome bisulfite sequencing
Bisulfite treatment of DNA converts cytosine residues to uracil but leaves methylated cytosines unaffected.59 DNA sequencing of bisulfite-treated cfDNA can be used to reveal methylation patterns with single nucleotide resolution. Because these patterns are cell, tissue, and organ types specific, they can inform the origins of cfDNA. Following treatment with bisulfite (Zymo EZ Methylation-Gold kit, #D5005), whole-genome sequencing (WGS) libraries were prepared according to manufacturer’s protocols (Swift Biosciences Accel-NGS Methyl-Seq DNA Library Kit #30024) using a dual indexing barcode strategy (Swift biosciences #38096, NEBNext Multiplex Oligos for Illumina E7500L, or custom primers). Paired-end DNA sequencing was performed on the Illumina NextSeq 500 (2x75bp) at Cornell University or the Illumina NovaSeq (2x150bp) at University of California San Francisco. Resulting paired-end fastq files were trimmed to 75bp for downstream analysis.
Human genome alignment
Adaptor sequences were trimmed using BBDUK (BBTools software suite52). Resulting sequences were aligned to the human genome (version hg19) and deduplicated using Bismark.53 Alignment files were filtered with a minimum mapping quality of 10 using SAMtools.54
Reference methylomes and tissues of origin
Reference methylation profiles were obtained from publicly available datasets and international epigenetic consortium projects (Data S1) and processed as previously described.25 Briefly, files were downloaded and normalized to a standard 4 column BED format at single nucleotide resolution using hg19 coordinates. Differentially methylated regions (DMRs) were found using Metilene.55 Methylation densities within these DMRs were averaged. Tissues with methylation profiles highly dissimilar from the same tissues were removed. cfDNA methylation densities were extracted using Bismark53 and averaged over the DMRs. Tissues of origin were deconvoluted using a non-negative least-squares approach.
cfDNA concentration measurement - MUHC patients
Plasma samples were processed in batches of 4 to 10 alongside a control containing 8 μL of approximately 150 ng/μL of synthetic oligos. DNA concentration measurements were performed after cfDNA extraction (Qubit Fluorometer 3.0) and the normalized concentration was calculated by multiplying the sample’s concentration by the input/output ratio of the control.
Depth of coverage
The depth of DNA sequencing coverage was calculated by dividing the number of mapped nucleotides to the autosomal chromosomes to the size of the non-N hg19 autosomal genome.
Bisulfite conversion efficiency
The bisulfite conversion efficiency achieved in experiments was estimated using MethPipe56 by calculating the reported methylation density of cytosines present at C[A/T/G] dinucleotides, which are rarely methylated in mammalian genomes.
Quality control filtering
Samples from the high frequency sampling cohort were selected for analysis if 10 or more spike-in molecules were identified after sequencing and were also filtered for sufficient depth of sequencing (> 0.2x human genome). Samples from the randomized control trial cohort were sequenced to a minimum depth of 0.7x human genome coverage. All samples had a minimum bisulfite conversion efficiency of 96%.
Lengths of cfDNA fragments and cfDNA fragmentation patterns to discriminate deceased and recovered COVID-19 patients (MUHC cohort)
The lengths of cfDNA molecules were quantified after end-to-end alignment using Bismark.53 In addition to the mean and median fragment lengths, we investigated i) the ratio of short-to-long molecules described in Cristiano et al.41 and ii) the fraction of reads attributed to dinucleosome-bound cfDNA fragments (approximately 320 bp). For i), we calculated the number of short and long fragments found in consecutive, non-overlapping 100kb bins, after correction for PCR-induced GC bias using locally weighted scatterplot smoothing. In each bin, we calculated a fragmentation score defined as the ratio of short to long fragments. The mean for each sample was used to compare recovered COVID-19 patients to the deceased using a non-parametric Wilcoxon test. For ii), we calculated the fraction of reads of lengths greater or equal to 300 bp and shorter or equal to 340 bp and compared the recovered COVID-19 patients to deceased using a non-parametric Wilcoxon test.
Mitochondrial cfDNA burden
The proportion of reads mapped to the mitochondrial genome was multiplied by the cfDNA concentration to obtain the mitochondrial cfDNA burden.
Multivariate analysis
An Extra Trees Classifier was created in Python using the sklearn package (v0.23.2).35 , 60 We used this classifier to predict mortality or discharge of patients using two different models: a composite model containing cfDNA features (erythroblast, kidney, liver, lung and neutrophil cfDNA fraction, and total cfDNA concentration) and protein biomarkers (ALT, AST, CRP, total bilirubin, LDH, RDWCV, hematocrit, hemoglobin, creatinine) and a model based on only the protein biomarkers. Feature importance was extracted after training the classifier on the entire 52-sample datasets of either model. An AUC for predicting eventual mortality was calculated by averaging AUCs obtained by resampling training and testing groups. When clinical biomarker information was not available for a sample, imputation was performed using the mean value.
Quantification and statistical analysis
All statistical analyses were performed in R, version 3.5.0 or Python, version 3.6.1. Groups were compared using the two-sided, nonparametric Wilcoxon test. If the data distributions were zero-skewed, a two-sided, 2-sample proportions test without continuity correction was performed. Odds ratio p values were obtained using Fisher’s exact test. Boxplots span from the 25th and 75th percentiles. The band in the box indicates the median, lower and higher whiskers extend to the smallest and largest values at most 1.5 × IQR of the hinge, respectively. Significance was defined by a p value below 0.05.
Acknowledgments
We thank Dr. Peter Schweitzer and colleagues at the Cornell Genomics Center and the UCSF Center for Advanced Technology for help with sequencing assays. We thank Dr. Lynn Johnson and the Cornell Statistical Consulting Unit for help with the statistical analysis. We thank Dr. Lucie Roussel, Dr. Marianna Orlova, and Pauline Cassart for technical assistance. This work was supported by NIH grants 1DP2AI138242 (to I.D.V.), R01AI146165 (to I.D.V., M.P.C., F.M.M., and J.R.), 1R01AI151059 (to I.D.V.), K08-CA230156 (to W.G.), and R33-AI129455 to C.Y.C., a Synergy award from the Rainin Foundation (to I.D.V.), a SARS-CoV-2 seed grant at Cornell (to I.D.V.), a National Sciences and Engineering Research Council of Canada fellowship PGS-D3 (to A.P.C.), and a Burroughs-Wellcome CAMS Award (to W.G.). D.C.V. is supported by a Fonds de la Recherche en Sante du Quebec Clinical Research Scholar Junior 2 award. C.Y.C. is supported by the California Initiative to Advance Precision Medicine, and the Charles and Helen Schwab Foundation. The graphical abstract was created using BioRender.com.
Author contributions
A.P.C., M.P.C., W.G., C.Y.C., D.C.V., and I.D.V. designed the study. M.P.C., W.G., C.Y.C., and D.C.V. consented patients and obtained clinical data. J.S.L., E.H., and W.G. performed the experiments. A.P.C., M.P.C., W.G., and I.D.V. analyzed the data. A.P.C., W.G., M.P.C., D.C.V., and I.D.V. wrote manuscript. All of the authors provided input and comments.
Declaration of interests
A.P.C., M.P.C., W.G., C.Y.C., D.C.V., and I.D.V. are inventors on a patent application submitted by Cornell University Center for Technology Licensing.
Published: January 16, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.medj.2021.01.001.
Supplemental information
Tab 1: patient and sample-specific data. Tab 2: reference methylome data. Tab 3: ordinal scale definitions.
References
- 1.Fox S.E., Akmatbekov A., Harbert J.L., Li G., Quincy Brown J., Vander Heide R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir. Med. 2020;8:681–686. doi: 10.1016/S2213-2600(20)30243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puelles V.G., Lütgehetmann M., Lindenmeyer M.T., Sperhake J.P., Wong M.N., Allweiss L., Chilla S., Heinemann A., Wanner N., Liu S., et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020;383:590–592. doi: 10.1056/NEJMc2011400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta A., Madhavan M.V., Sehgal K., Nair N., Mahajan S., Sehrawat T.S., Bikdeli B., Ahluwalia N., Ausiello J.C., Wan E.Y., et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020;26:1017–1032. doi: 10.1038/s41591-020-0968-3. [DOI] [PubMed] [Google Scholar]
- 4.Varga Z., Flammer A.J., Steiger P., Haberecker M., Andermatt R., Zinkernagel A.S., Mehra M.R., Schuepbach R.A., Ruschitzka F., Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Julien P., Julien G., Morgan C., et al. Pulmonary Embolism in COVID-19 Patients: Awareness of an Increased Prevalence. Circulation. 2020;142:184–186. doi: 10.1161/CIRCULATIONAHA.120.047430. [DOI] [PubMed] [Google Scholar]
- 6.Goshua G., Pine A.B., Meizlish M.L., Chang C.H., Zhang H., Bahel P., Baluha A., Bar N., Bona R.D., Burns A.J., et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020;7:e575–e582. doi: 10.1016/S2352-3026(20)30216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klok F.A., Kruip M.J.H.A., van der Meer N.J.M., Arbous M.S., Gommers D.A.M.P.J., Kant K.M., Kaptein F.H.J., van Paassen J., Stals M.A.M., Huisman M.V., Endeman H. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kashi M., Jacquin A., Dakhil B., Zaimi R., Mahé E., Tella E., Bagan P. Severe arterial thrombosis associated with Covid-19 infection. Thromb. Res. 2020;192:75–77. doi: 10.1016/j.thromres.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sungnak W., Huang N., Bécavin C., Berg M., Queen R., Litvinukova M., Talavera-López C., Maatz H., Reichart D., Sampaziotis F., et al. HCA Lung Biological Network SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26:681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., Zeng X., Jiao Y., Li Z., Liu Q., Ye J., Yang M. Mechanisms involved in the development of thrombocytopenia in patients with COVID-19. Thromb. Res. 2020;193:110–115. doi: 10.1016/j.thromres.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitra A., Dwyre D.M., Schivo M., Thompson G.R., III, Cohen S.H., Ku N., Graft J.P. Leukoerythroblastic reaction in a patient with COVID-19 infection. Am. J. Hematol. 2020 doi: 10.1002/ajh.25793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foy B.H., Carlson J.C.T., Reinertsen E., Padros Valls R., Pallares Lopez R., Palanques-Tost E., Mow C., Westover M.B., Aguirre A.D., Higgins J.M. Elevated RDW is Associated with Increased Mortality Risk in COVID-19. medRxiv. 2020 doi: 10.1101/2020.05.05.20091702. [DOI] [Google Scholar]
- 13.Gong J., Ou J., Qiu X., Jie Y., Chen Y., Yuan L., Cao J., Tan M., Xu W., Zheng F., et al. A Tool to Early Predict Severe Corona Virus Disease 2019 (COVID-19) : A Multicenter Study using the Risk Nomogram in Wuhan and Guangdong, China. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pirofski L., Casadevall A. Pathogenesis of COVID-19 from the Perspective of the Damage-Response Framework. MBio. 2020;11 doi: 10.1128/mBio.01175-20. e01175-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye Q., Wang B., Mao J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020;80:607–613. doi: 10.1016/j.jinf.2020.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dufort E.M., Koumans E.H., Chow E.J., Rosenthal E.M., Muse A., Rowlands J., Barranco M.A., Maxted A.M., Rosenberg E.S., Easton D., et al. New York State and Centers for Disease Control and Prevention Multisystem Inflammatory Syndrome in Children Investigation Team Multisystem Inflammatory Syndrome in Children in New York State. N. Engl. J. Med. 2020;383:347–358. doi: 10.1056/NEJMoa2021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burnham P., Khush K., De Vlaminck I. Myriad applications of circulating cell-free DNA in precision organ transplant monitoring. Ann. Am. Thorac. Soc. 2017;14(Suppl 3):S237–S241. doi: 10.1513/AnnalsATS.201608-634MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan H.C., Blumenfeld Y.J., Chitkara U., Hudgins L., Quake S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA. 2008;105:16266–16271. doi: 10.1073/pnas.0808319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu R.W., Chan K.C., Gao Y., Lau V.Y., Zheng W., Leung T.Y., Foo C.H., Xie B., Tsui N.B., Lun F.M., et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc. Natl. Acad. Sci. USA. 2008;105:20458–20463. doi: 10.1073/pnas.0810641105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Vlaminck I., Martin L., Kertesz M., Patel K., Kowarsky M., Strehl C., Cohen G., Luikart H., Neff N.F., Okamoto J., et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proc. Natl. Acad. Sci. USA. 2015;112:13336–13341. doi: 10.1073/pnas.1517494112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Vlaminck I., Valantine H.A., Snyder T.M., Strehl C., Cohen G., Luikart H., Neff N.F., Okamoto J., Bernstein D., Weisshaar D., et al. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci. Transl. Med. 2014;6:241ra77. doi: 10.1126/scitranslmed.3007803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snyder T.M., Khush K.K., Valantine H.A., Quake S.R. Universal noninvasive detection of solid organ transplant rejection. Proc. Natl. Acad. Sci. USA. 2011;108:6229–6234. doi: 10.1073/pnas.1013924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun K., Jiang P., Chan K.C.A., Wong J., Cheng Y.K., Liang R.H., Chan W.K., Ma E.S., Chan S.L., Cheng S.H., et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA. 2015;112:E5503–E5512. doi: 10.1073/pnas.1508736112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder M.W., Kircher M., Hill A.J., Daza R.M., Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164:57–68. doi: 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng A.P., Burnham P., Lee J.R., Cheng M.P., Suthanthiran M., Dadhania D., De Vlaminck I. A cell-free DNA metagenomic sequencing assay that integrates the host injury response to infection. Proc. Natl. Acad. Sci. USA. 2019;116:18738–18744. doi: 10.1073/pnas.1906320116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann-Werman R., Neiman D., Zemmour H., Moss J., Magenheim J., Vaknin-Dembinsky A., Rubertsson S., Nellgård B., Blennow K., Zetterberg H., et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA. 2016;113:E1826–E1834. doi: 10.1073/pnas.1519286113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng A.P., Cheng M.P., Sesing Lenz J., Chen K., Burnham P., Timblin K.M., Orejas J.L., Silverman E., Marty F.M., Ritz J., et al. Cell-free DNA Tissues-of-Origin Profiling to Predict Graft versus Host Disease and Detect Infection after Hematopoietic Cell Transplantation. bioRxiv. 2020 doi: 10.1101/2020.04.25.061580. [DOI] [Google Scholar]
- 28.Lam W.K.J., Gai W., Sun K., Wong R.S.M., Chan R.W.Y., Jiang P., Chan N.P.H., Hui W.W.I., Chan A.W.H., Szeto C.C., et al. DNA of erythroid origin is present in human plasma and informs the types of anemia. Clin. Chem. 2017;63:1614–1623. doi: 10.1373/clinchem.2017.272401. [DOI] [PubMed] [Google Scholar]
- 29.WHO Working Group on the Clinical Characterisation and Management of COVID-19 Infection A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect. Dis. 2020;20:E192–E197. doi: 10.1016/S1473-3099(20)30483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan L., Zhang H.-T., Goncalves J., Xiao Y., Wang M., Guo Y., Sun C., Tang X., Jing L., Zhang M., et al. An interpretable mortality prediction model for COVID-19 patients. Nat. Mach. Intell. 2020;2:283–288. [Google Scholar]
- 32.Kato G.J., McGowan V., Machado R.F., Little J.A., Taylor J., 6th, Morris C.R., Nichols J.S., Wang X., Poljakovic M., Morris S.M., Jr., Gladwin M.T. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood. 2006;107:2279–2285. doi: 10.1182/blood-2005-06-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henderson R.F., Damon E.G., Henderson T.R. Early damage indicators in the the lung I. Lactate dehydrogenase activity in the airways. Toxicol. Appl. Pharmacol. 1978;44:291–297. doi: 10.1016/0041-008x(78)90191-6. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez-Cruz E., Escartin P., Bootello A., Kreisler M., Segovia de Arana J.M. Hepatocyte damage induced by lymphocytes from patients with chronic liver diseases, as detected by LDH release. Clin. Exp. Immunol. 1978;31:436–442. [PMC free article] [PubMed] [Google Scholar]
- 35.Geurts P., Ernst D., Wehenkel L. Extremely randomized trees. Mach. Learn. 2006;63:3–42. [Google Scholar]
- 36.World Health Organization WHO discontinues hydroxychloroquine and lopinavir/ritonavir treatment arms for COVID-19. 2020. http://www.who.int/news-room/detail/04-07-2020-who-discontinues-hydroxychloroquine-and-lopinavir-ritonavir-treatment-arms-for-covid-19
- 37.Chief Investigators of the Randomised Evaluation of COVid-19 thERapY (RECOVERY) Trial on Lopinavir-Ritonavir No clinical benefit from use of lopinavir-ritonavir in hospitalised COVID-19 patients studied in RECOVERY — RECOVERY Trial. 2020. https://www.recoverytrial.net/news/no-clinical-benefit-from-use-of-lopinavir-ritonavir-in-hospitalised-covid-19-patients-studied-in-recovery
- 38.Fan H.C., Blumenfeld Y.J., Chitkara U., Hudgins L., Quake S.R. Analysis of the size distributions of fetal and maternal cell-free DNA by paired-end sequencing. Clin. Chem. 2010;56:1279–1286. doi: 10.1373/clinchem.2010.144188. [DOI] [PubMed] [Google Scholar]
- 39.Jiang P., Chan C.W.M., Chan K.C.A., Cheng S.H., Wong J., Wong V.W., Wong G.L., Chan S.L., Mok T.S., Chan H.L., et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA. 2015;112:E1317–E1325. doi: 10.1073/pnas.1500076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mouliere F., Robert B., Arnau Peyrotte E., Del Rio M., Ychou M., Molina F., Gongora C., Thierry A.R. High fragmentation characterizes tumour-derived circulating DNA. PLoS ONE. 2011;6:e23418. doi: 10.1371/journal.pone.0023418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cristiano S., Leal A., Phallen J., Fiksel J., Adleff V., Bruhm D.C., Jensen S.Ø., Medina J.E., Hruban C., White J.R., et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–389. doi: 10.1038/s41586-019-1272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moss J., Magenheim J., Neiman D., Zemmour H., Loyfer N., Korach A., Samet Y., Maoz M., Druid H., Arner P., et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018;9:5068. doi: 10.1038/s41467-018-07466-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun K., Jiang P., Cheng S.H., Cheng T.H.T., Wong J., Wong V.W.S., Ng S.S.M., Ma B.B.Y., Leung T.Y., Chan S.L., et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 2019;29:418–427. doi: 10.1101/gr.242719.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu S.C.Y., Lee S.W.Y., Jiang P., Leung T.Y., Chan K.C., Chiu R.W., Lo Y.M. High-resolution profiling of fetal DNA clearance from maternal plasma by massively parallel sequencing. Clin. Chem. 2013;59:1228–1237. doi: 10.1373/clinchem.2013.203679. [DOI] [PubMed] [Google Scholar]
- 45.Roy N.B.A., Babbs C. The pathogenesis, diagnosis and management of congenital dyserythropoietic anaemia type I. Br. J. Haematol. 2019;185:436–449. doi: 10.1111/bjh.15817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodero M.P., Tesser A., Bartok E., Rice G.I., Della Mina E., Depp M., Beitz B., Bondet V., Cagnard N., Duffy D., et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat. Commun. 2017;8:2176. doi: 10.1038/s41467-017-01932-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dorward D.A., Russell C.D., Hwa Um I., Elshani M., Armstrong S.D., Penrice-Randal R., Millar T., Lerpiniere C.E., Tagliavini G., Hartley C.S., et al. Tissue-specific tolerance in fatal COVID-19. Am. J. Respir. Crit. Care Med. 2020:.. doi: 10.1164/rccm.202008-3265OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mrug M., Stopka T., Julian B.A., Prchal J.F., Prchal J.T. Angiotensin II stimulates proliferation of normal early erythroid progenitors. J. Clin. Invest. 1997;100:2310–2314. doi: 10.1172/JCI119769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim Y.C., Mungunsukh O., McCart E.A., Roehrich P.J., Yee D.K., Day R.M. Mechanism of erythropoietin regulation by angiotensin II. Mol. Pharmacol. 2014;85:898–908. doi: 10.1124/mol.113.091157. [DOI] [PubMed] [Google Scholar]
- 50.Vlahakos D.V., Marathias K.P., Madias N.E. The role of the renin-angiotensin system in the regulation of erythropoiesis. Am. J. Kidney Dis. 2010;56:558–565. doi: 10.1053/j.ajkd.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 51.Cavezzi A., Troiani E., Corrao S. COVID-19: hemoglobin, iron, and hypoxia beyond inflammation. A narrative review. Clin. Pract. 2020;10:1271. doi: 10.4081/cp.2020.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bushnell B., Rood J., Singer E. BBMerge – accurate paired shotgun read merging via overlap. PLoS ONE. 2017;12:e0185056. doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krueger F., Andrews S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jühling F., Kretzmer H., Bernhart S.H., Otto C., Stadler P.F., Hoffmann S. metilene: fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 2016;26:256–262. doi: 10.1101/gr.196394.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song Q., Decato B., Hong E.E., Zhou M., Fang F., Qu J., Garvin T., Kessler M., Zhou J., Smith A.D. A Reference Methylome Database and Analysis Pipeline to Facilitate Integrative and Comparative Epigenomics. PLoS ONE. 2013;8:e81148. doi: 10.1371/journal.pone.0081148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.World Medical Association World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 58.Lam N.Y.L., Rainer T.H., Chiu R.W.K., Lo Y.M.D. EDTA is a better anticoagulant than heparin or citrate for delayed blood processing for plasma DNA analysis. Clin. Chem. 2004;50:256–257. doi: 10.1373/clinchem.2003.026013. [DOI] [PubMed] [Google Scholar]
- 59.Frommer M., McDonald L.E., Millar D.S., Collis C.M., Watt F., Grigg G.W., Molloy P.L., Paul C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buitinck L., Louppe G., Blondel M., Pedregosa F., Mueller A., Grisel O., Niculae V., Prettenhofer P., Gramfort A., Grobler J., et al. API design for machine learning software: experiences from the scikit-learn project. arXiv. 2013 https://arxiv.org/abs/1309.0238 1309.0238. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tab 1: patient and sample-specific data. Tab 2: reference methylome data. Tab 3: ordinal scale definitions.
Data Availability Statement
Genomic data is hosted on the Sequence Read Archive under project number PRJNA687910. The binned methylation data, and the code used to process the data is available at https://www.github.com/alexpcheng/cfDNAme.