

Abstract
On January 22, 2020, China National Center for Bioinformation (CNCB) released the 2019 Novel Coronavirus Resource (2019nCoVR), an open-access information resource for the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). 2019nCoVR features a comprehensive integration of sequence and clinical information for all publicly available SARS-CoV-2 isolates, which are manually curated with value-added annotations and quality evaluated by an automated in-house pipeline. Of particular note, 2019nCoVR offers systematic analyses to generate a dynamic landscape of SARS-CoV-2 genomic variations at a global scale. It provides all identified variants and their detailed statistics for each virus isolate, and congregates the quality score, functional annotation, and population frequency for each variant. Spatiotemporal change for each variant can be visualized and historical viral haplotype network maps for the course of the outbreak are also generated based on all complete and high-quality genomes available. Moreover, 2019nCoVR provides a full collection of SARS-CoV-2 relevant literature on the coronavirus disease 2019 (COVID-19), including published papers from PubMed as well as preprints from services such as bioRxiv and medRxiv through Europe PMC. Furthermore, by linking with relevant databases in CNCB, 2019nCoVR offers data submission services for raw sequence reads and assembled genomes, and data sharing with NCBI. Collectively, SARS-CoV-2 is updated daily to collect the latest information on genome sequences, variants, haplotypes, and literature for a timely reflection, making 2019nCoVR a valuable resource for the global research community. 2019nCoVR is accessible at https://bigd.big.ac.cn/ncov/.
Keywords: 2019nCoVR, SARS-CoV-2, Database, Genomic variation, Haplotype
Introduction
Coronavirus disease 2019 (COVID-19) is a severe respiratory disease that is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1]. It has rapidly spread as a pandemic after its outbreak in late December 2019. As of July 14, 2020, 12,964,809 confirmed cases have been reported in 216 countries/territories/areas (WHO Situation Report Number 176; https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/). SARS-CoV-2 samples have been extensively isolated and sequenced by different laboratories across many countries [2], resulting in a considerable number of viral genome sequences worldwide. Therefore, public sharing and free access to a comprehensive collection of SARS-CoV-2 genome sequences is of great significance, which would help to accelerate scientific research and knowledge discovery and also help develop medical countermeasures and sensible decision-making [3].
To date, unfortunately, SARS-CoV-2 genome sequences generated worldwide were scattered around different database resources, primarily including the Global Initiative on Sharing All Influenza Data (GISIAD) [4] repository and NCBI GenBank [5]. Many sequences are available in multiple repositories but their updates are not synchronized. This makes it extremely challenging for worldwide users to effectively retrieve a non-redundant and most updated set of sequence data, and to collaboratively and rapidly deal with this global pandemic. Toward this end, we constructed the 2019 Novel Coronavirus Resource (2019nCoVR, https://bigd.big.ac.cn/ncov/) [6]. Through comprehensive integration and value-added annotation and analysis, we provide public, free, and rapid access to a complete collection of non-redundant global SARS-CoV-2 genomes. Since its inception on January 22, 2020, 2019nCoVR is updated on a daily basis, leading to unprecedentedly dramatic data expansion from 86 genomes in its first release to 64,789 genomes in its current version (as of July 14, 2020). Moreover, it has been substantially upgraded by implementing enhanced data curation and analysis pipelines and online functionalities. Specifically, we enrich 2019nCoVR by including data quality evaluation, variant calling, variant spatiotemporal dynamic tracking, viral haplotype construction, and interactive visualization with more user-friendly web interfaces (Table 1). Here we report these significant updates of 2019nCoVR and present the global landscape of SARS-CoV-2 genomes, variants, and haplotypes.
Table 1.
Comparison of functional modules between two versions of 2019nCoVR
 |
Data collection and processing
Data collection and integration
All genome sequences as well as their related metadata were integrated from SARS-CoV-2 resources worldwide, including GISAID [4], NCBI [5], National Genomics Data Center (NGDC) [7], National Microbiology Data Center (NMDC) [8], and China National GeneBank (CNGB) [9]. To provide a non-redundant dataset, duplicated records from different databases were identified and merged.
Quality control and curation
To ensure the integrity of genome sequences, a sequence is defined as ‘complete’ if it is longer than 29,000 bp and covers all protein-coding regions of SARS-CoV-2 (nt 266–29674 of GenBank: MN908947.3); otherwise, it is defined as “partial”. Furthermore, to examine the quality of genome sequences, unknown bases (Ns) and degenerate bases (Ds, more than one possible base at a particular position and sometimes referred as “mixed bases”) were counted for each sequence. According to our definition, a sequence is considered “high-quality” if it contains ≤ 15 Ns and ≤ 50 Ds, and “low-quality” otherwise. In addition, a sequence is clearly labeled if the number of variants is ≥ 15 or the total number of deletions is ≥ 2, or the distribution of sequence variations is more aggregated (the ratio of the number of variants divided by the total number of bases in a window is ≥ 0.25).
Variant identification and haplotype network construction
Only complete and high-quality genome sequences were used for downstream analyses, including sequence comparison, variant identification, functional annotation, and haplotype network construction. Genome sequence alignment was performed with MUSCLE (3.8.31) [10] by comparing against the earliest released SARS-CoV-2 genome (GenBank: MN908947.3). Sequence variation was identified directly using an in-house Perl program. The effect of variants was determined using Ensembl Variant Effect Predictor (VEP) [11].
SARS-CoV-2 haplotypes were constructed based on short pseudo sequences that consist of all variants (filtering out variations located in UTR regions). Then, all these pseudo sequences were clustered into groups, and each group (a haplotype) represents a unique sequence pattern. The haplotype network was inferred from all identified haplotypes, where the reference sequence haplotype was set as the starting node, and its relationship with other haplotypes was determined according to the inheritance of mutations. As a result, nine major haplotype network clades (denoted as C01–C09) were obtained according to the phylogenetic tree-and-branch structure and the shared landmark mutations (Table 1). Specifically, mutations with population mutation frequency (PMF) ≥ 0.05 (except for ATG deletion at position 1605, PMF ≈ 0.03) were selected, and the co-occurring mutations were determined by LD linkage analysis. A clade refers to sequences with the co-occurring landmark mutations.
Implementation
2019nCoVR was built based on a browser/server (B/S) architecture. Web interfaces was developed by the Java Server Pages (JSP), HTML, Cascading Style Sheet (CSS), Asynchronous JavaScript and XML (AJAX), JQuery (a cross-platform and feature-rich JavaScript library; http://jquery.com), as well as Semantic-UI (an open source web development framework; https://semantic-ui.com). The database server was implemented by using the Spring Boot (a rapid application development framework based on Spring; https://spring.io). MySQL (https://mysql.com) was used for data storage. For interactive visualization, we implemented HighCharts (a modern SVG-based multi-platform charting library; https://highcharts.com), D3.js (a JavaScript library for manipulating documents based on data; https://d3js.org), and 3Dmol.js (a JavaScript library for visualizing protein structure associated with mutated amino acid residues) [12] in 2019nCoVR. The haplotype network was visualized using D3.js, Leaflet (http://leafletjs.com), and Echarts (http://echarts.baidu.com/).
Database content and features
Statistics of SARS-CoV-2 genome assemblies
Since the outbreak of COVID-19, the number of SARS-CoV-2 genome sequences released globally has been increasing at an unprecedented rate. To facilitate free public access to all genome assemblies and help worldwide researchers better understand the variation and transmission of SARS-CoV-2, we perform daily updates for 2019nCoVR by integrating all available genomes throughout the world and conducting value-added curation and analysis. As of July 14, 2020, 2019nCoVR hosted a total of 64,789 non-redundant genome sequences and provided a global distribution of SARS-CoV-2 genome sequences in 97 countries/regions across 6 continents. Duplicated sequences from different databases are merged with all IDs cross-referenced. Sequences are contributed primarily by United Kingdom (28,823, 44.5%), United States (13,556, 20.9%), Australia (2351, 3.6%), Spain (1852, 2.9%), Netherlands (1605, 2.5%), India (1581, 2.4%), and China (1431, 2.2%). According to our statistics, SARS-CoV-2 genome sequences started to grow rapidly from mid-March (https://bigd.big.ac.cn/ncov/release_genome), concordant with the outset of global pandemic of COVID-19. A full list of our sequence datasets, including strain name, accession number, and source, is provided in Table S1.
To provide high-quality genome sequences that are critically essential for downstream analyses (ranging from variant calling to haplotype construction), we perform sequence integrity and quality assessment for all newly-collected sequences. Among all the human-derived genome sequences released (64,700), 60,970 (94%) are complete, and 31,689 (49%) are high-quality (Figure 1A). Most of the low-quality sequences (29,281, 99.7%) contain different numbers of unknown bases (Ns). Among these sequences, 60% have 16–500 Ns (median 258), and 40% have more than 500 Ns (Figure 1B). Further investigation of the genomic locations reveals that some genomic regions with high frequency of Ns (Figure 1C). Sequence integrity and quality assessment analytic data are available for all genome sequences, and can be used as filters for sequence browse and search.
Figure 1.

Statistics and distribution of all released SARS-CoV-2 genomes in 2019nCoVR as of July 14, 2020
A. Number and percentage of complete and high-quality genomes. B. Distribution of sequence number across different ranges of Ns for low-quality genomes. C. Frequency distribution of Ns across the whole genome. A sequence is defined as “complete” if it is longer than 29,000 bp and covers all protein-coding regions of SARS-CoV-2 (nt 266–29674 of GenBank: MN908947.3); otherwise, it is defined as “partial”. A sequence is considered “high-quality” if it contains ≤ 15 Ns and ≤ 50 Ds, and “low-quality” otherwise. N, unknown base; D, degenerate base.
Landscape of genomic variants
Bases on the 31,689 human-derived high-quality complete genome sequences obtained globally (only high-quality complete genome sequences are used for downstream analysis if not indicated otherwise), we investigate the landscape of SARS-CoV-2 genomic variants in comparison with the reference genome (GenBank: MN908947.3) (Figure 2). By July 14, 2020, a total of 13,428 variants had been identified, including 12,828 (95.5%) single-nucleotide polymorphisms (SNPs), 437 deletions, 116 insertions, and 47 indels (a combination of an insertion and a deletion, affecting 2 or more nucleotides) (Figure 2A). More than half of these SNPs (6770, 50.4%) are nonsynonymous, causing amino acid changes. To evaluate the impact of missense variants of S protein on the interaction with its receptor human angiotensin-converting enzyme 2 (ACE2) (e.g., in the key binding region), mutated amino acids are projected onto protein 3D structures, which can be viewed by 360 degree rotation (Figure 2B). We further explore distribution of variants across different genes. Noticeably, three genes ORF1ab, S, and N accumulate more variants (Figure 2C). In addition, SNP densities (i.e., number of mutations per nucleotide in the genic region) are higher in several genic regions, including ORF7a, ORF3a, ORF6, and N (https://bigd.big.ac.cn/ncov/variation/annotation).
Figure 2.

Landscape of genomic variants
A. Number of mutations in different mutation types. The orange bar indicates the number of all mutations, and the blue bar indicates the number of mutations with PMF > 0.001. B. 3D structure display for nonsynonymous mutations in S protein (PDB: 6VSB, http://www.rcsb.org/structure/6VSB). The structure is shown in sphere (left panel) and stick (right panel). The three chains (A, B, and C) of S protein are displayed in blue, yellow, and green, respectively. The binding region (amino acid residues: 336–516) of the S protein with its receptor human ACE2 is shown in cyan for all three chains. C. Pie chart showing variant annotation for each gene of SARS-CoV-2. D. Distribution of PMF for all variants. Coordinate information for representative variants (including positions 241, 1059, 3037, 8782, 14408, and 23403) is provided. PMF, population mutation frequency; ACE2, angiotensin-converting enzyme 2.
For each variant, we investigate its PMF (the ratio of the number of mutated genomes to the total number of complete high-quality genomes) (Figure 2D). Clearly, there are 62 variants with PMF > 0.01 and 18 variants with PMF > 0.05. In particular, there are 4 variants with PMF > 0.75, including positions 241 in 5′UTR, 3037 and 14408 in ORF1ab, as well as position 23403 in S. These may potentially represent the main prevalent virus genotypes across the globe. All identified variants and their functional annotations are publicly available in the database. In addition, an online pipeline for variant identification and functional annotation is also provided for free access at https://bigd.big.ac.cn/ncov/analysis [13].
Spatiotemporal dynamics of genomic variants
To track the dynamics of SARS-CoV-2 genomic variants, particularly de novo mutations, we explore the spatiotemporal change of PMF for each variant according to sampling dates and locations (Figure 3). Among the 18 sites with PMF > 0.05, a few mutations occurred simultaneously in multiple sequences and in a linkage manner (Figure 3A), such as mutations at positions 8782 and 28144 as reported previously [14]. It is of note that mutations at these two sites appeared in the early stage of the outbreak on December 30, 2019. Their mutation frequencies reach ~ 0.33 around January 22, 2020, and then gradually declined to 0.10 on July 14, 2020. In contrast, some variants appear only at the middle stage around March 3, 2020. For instance, mutation at position 23403 (resulting in an amino acid change D614G in the S protein) is accompanied by three other mutations, namely, a C-to-U mutation at position 241 in the 5′UTR of SARS-CoV-2 genome, a silent C-to-U mutation in the gene nsp3 at position 3037, and a missense C-to-U mutation in the gene RdRp at position 14408 (P4715L). To make it easier for users to investigate any variant of interest, we provide an interactive heatmap in 2019nCoVR (https://bigd.big.ac.cn/ncov/variation/heatmap) to dynamically display and cluster the mutation patterns over all sampling dates, with customized options available that allow users to select specific variant frequency, annotated gene/region, variant effect type, and transcription regulation sequence (TRS).
Figure 3.

Spatiotemporal dynamics of genomic variants
A. Heatmap of variant PMF (PMF > 0.01) over sampling date. B. Distribution of PMF and cumulative growth curve of the sequence with mutation at position 23403 (D614G). C. Cumulative growth of the sequence with mutation at position 23403 (D614G) in top 10 countries. Data were downloaded from 2019nCoVR on July 14, 2020.
Moreover, we investigate dynamic patterns of SARS-CoV-2 genomic variants across different sampling locations over time. Taking the variant at position 23403 (D614G) as an example, its PMF has dramatically increased from 0 at the end of February to 0.76 in the middle of July, and the mutant form G614 became dominant gradually along with the development of pandemic (Figure 3B), presumably indicating that the mutated genotypes may have higher transmissibility [15]. In terms of the absolute number of mutations across different countries/regions, G614 form was dominantly reported in Europe and North America (Figure 3C). (https://bigd.big.ac.cn/ncov/variation/annotation/variant/23403). When investigating the mutation pattern for each country (Figure 4), we find that sequences from some Asian countries (such as South Korea, Malaysia, and Nepal) have no or very few G614 mutant form, whereas countries from Europe and America (e.g., Argentina, Czech Republic, and Serbia) have the G614 form that is dominant among the available samples. In some countries, both the D614 and G614 forms co-existed early in the epidemic, but the mutant form quickly became dominant, such as in Australia, Belgium, Canada, Chile, France, Israel, United States, and United Kingdom. The accumulation of this mutation varies in different parts of the world, possibly due to the prevention and control measures implemented in some countries/regions. Taken together, 2019nCoVR features spatiotemporal dynamics tracking of SARS-CoV-2 genomic variants, and thus bears great potential to help decipher viral transmission and adaptation to the host.
Figure 4.

PMF of variant 23,403 for each country across different sampling dates
Number of accumulated sequences as of July 14, 2020 is provided in parenthesis after country name.
Haplotype network construction and characterization
To better characterize the diversity of virus sequences, we built SARS-CoV-2 haplotypes based on all identified variants of non-UTR regions. As a result, 17,624 haplotypes were identified from 31,689 complete high-quality genome sequences as of July 14, 2020. We construct a haplotype network for SARS-CoV-2 (Figure 5), a graphical representation of relationships between individual genotypes inferred from genomic variations. The haplotype network is built based on the principle of the shortest set of connections that link all nodes (genotypes), where the length of each connection represents the genetic distance [16]. The SARS-CoV-2 haplotype network can be visualized according to sample collection date and across different countries/regions, thus providing an overview of the pandemic transmission in a spatiotemporal manner. It not only allows users to easily obtain a landscape of SARS-CoV-2 haplotypes and their relationships, but also helps users to navigate a set of haplotypes for a specific country/region. In addition to the haplotype network, the associated information could also be accessed, such as the number of genomes, as well as sampling time and location (Figure 5A).
Figure 5.

Haplotype network and clade identification and distribution
A. Snapshot of haplotype network dashboard, dynamically showing the development of haplotype (I) across countries (II) and over time (III). Each node in the network represents a haplotype and the node size is proportional to the number of viral genome sequences. The edge between any two nodes represents the genetic distance between two haplotypes. Number of newly-released genome sequences each day is dynamically displayed on the respective date. B. Schematic diagram of haplotype clades (C01–C09). C. Schematic diagram of three lineages and nine clades, and the common mutation sites for each clade. D. Percentage of sequences in clades C01–C09 across different continents. E. Sequence number distribution of different lineages (S, L, and G) and clades (C01–C09) throughout the globe and in three representative countries (United States, United Kingdom, and China).
According to the haplotype network, we classified all genome sequences into nine major clades (labeled as C01–C09; see Methods for details) (Figure 5B and C; Table 2). As the pandemic spread of SARS-CoV-2 is still ongoing, new branches that evolve and spread faster are constantly emerging, such as clades C04, C06, C08, and C09 (Table 2). The dominant clades are C06 (8681, 27.4%), C08 (7889, 24.9%), and C09 (6940, 21.9%) (Figure 5D), which are characterized by the signature mutations of C-to-U mutation at positions 241, 3037, and 14408, and A-to-G mutation at position 23403. These clades are defined as the G lineage (as the mutation at position 23403 leads to an amino acid change D614G of S protein). The G lineage sequences have been reported in 82 countries across the globe, and become the main epidemic virus type in most countries in Europe, North America, South America, Africa, and West Asia, etc. For example, there are 6827 (71.5%), 8305 (83.4%), and 970 (18.5%) sequences from the G lineage reported in the United States, United Kingdom, and China, respectively (Figure 5E). The widespread and prevalence of the G lineage in different countries suggest the adaptability of this lineage to humans [15].
Table 2.
Signature mutations of haplotype clades
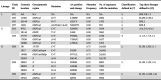 |
Note: AA, amino acid; NA, not applicable.
Discussion
Genome sequencing is vital to understand the epidemiology of SARS-CoV-2, which is not only useful for deciphering genomic composition of the virus and investigating its evolution and transmission, but also highly effective at determining whether individuals belong to the same transmission chain [17]. According to 2019nCoVR, the ratio of the number of sequenced samples to the number of confirmed cases is very low in some countries/regions (Figure S1), and genome sequences are even unavailable in some affected countries/regions. The SARS-CoV-2 sampling bias and limited sequencing depth may lead to inaccurate transmission patterns and phylogenetic relationships [18]. Sequencing all infected cases in a single region reveals that the transmission of Clostridium difficile from symptomatic patients accounts for only one third of all infected cases [19]. Given our current understanding of SARS-CoV-2 is still limited, we call for more efforts and collaborations in sequencing more SARS-CoV-2 genomes from both symptomatic and asymptomatic cases.
The SARS-CoV-2 genome sequences currently released were generated by multiple different laboratories on different sequencing platforms. This raises concerns on the quality of genome sequences, such as the Ns of genome, which may affect variant calling and lead to biased population frequency estimation. As mentioned above, the frequency of Ns in some genomic regions is high, possibly due to the low sequencing coverage, low sequence complexity, low efficiency of PCR primers used in sequencing library construction, presence of RNA secondary structure, etc. However, sequencing coverage information is largely unavailable, making it challenging to evaluate whether the Ns are due to low sequencing coverage. We further investigated the genomic regions with high frequency of Ns and had the following findings. (1) GC and AG contents of these regions are close to the average GC and AG contents of the whole genome, excluding the possibility of low sequence complexity. (2) The length of these regions ranges from 210 bp to 320 bp (similar to the length of PCR product) and more than 60% of the related sequences are generated on Illumina platform (based on PCR amplification), suggesting that these Ns may result from low efficiency of PCR primers during sequencing library construction. (3) Minimum free energy of RNA secondary structure of these regions is lower than that of randomly extracted regions, indicating that the secondary structure of these regions is more stable and may affect the determination of genome sequences (Figure S2). In future, we plan to construct a golden benchmark dataset with for quality assessment and data filtration.
Compared to the early overly simplified L-S classification [14] and the comprehensive lineages defined by Rambaut et al. [20], our classification scheme with nine clades provides a moderate system that can be correlated with the two classifications mentioned above (Table 2). The nine clades could also be grouped into three lineages defined previously [14], namely, S (C02 and C04), G (C06, C08, and C09), and L (the remaining clades). Although haplotype network cannot provide a precise evolutionary position as phylogenetic trees do, it can be used to quickly inform the clustering of viruses according to signature mutations in each haplotype. Definitely, new clades will be introduced as the virus is continuing to evolve.
A data-driven response to SARS-CoV-2 requires a public, free, and open-access data resource that contains complete high-quality genome sequence data, and equips with automated online pipelines to rapidly analyze genome sequences. Thus, 2019nCoVR (together with other resources in CNCB) provides a wide range of data services, including raw sequencing data archive, genome sequence and meta information management with quality control and curation, variation analysis, as well as data presentation and visualization. Additionally, compared to GISAID and NCBI Virus, 2019nCoVR features spatiotemporal dynamic tracking for all identified variants. This makes it easier for users worldwide to monitor any variant that may be associated with rapid transmission and high virulence. To better understand the epidemiology of SARS-CoV-2, future efforts are needed to collect ever more genome sequences worldwide, to include other types of omics data (such as transcriptome and epitranscriptome, if available) [21], and also to provide more friendly interfaces and online tools in support of research activities worldwide.
Data availability
SARS-CoV-2 genomes, variants (in vcf format), and their annotations are publicly available at https://bigd.big.ac.cn/ncov/.
CRediT author statement
Shuhui Song: Conceptualization, Methodology, Writing - original draft. Lina Ma: Data curation, Methodology, Writing - original draft. Dong Zou: Resources, Visualization, Writing - original draft. Dongmei Tian: Methodology. Cuiping Li: Methodology. Junwei Zhu: Software. Meili Chen: Data curation. Anke Wang: Software. Yingke Ma: Resources. Mengwei Li: Methodology. Xufei Teng: Visualization. Ying Cui: Data curation. Guangya Duan: Data curation. Mochen Zhang: Data curation. Tong Jin: Data curation. Chengmin Shi: Methodology. Zhenglin Du: Methodology. Yadong Zhang: Methodology. Chuandong Liu: Methodology. Rujiao Li: Data curation. Jingyao Zeng: Data curation. Lili Hao: Data curation. Shuai Jiang: Methodology. Hua Chen: Supervision. Dali Han: Supervision. Jingfa Xiao: Supervision, Methodology. Zhang Zhang: Conceptualization, Supervision, Writing - review & editing. Wenming Zhao: Conceptualization, Supervision, Methodology. Yongbiao Xue: Conceptualization, Supervision. Yiming Bao: Conceptualization, Supervision, Writing - review & editing. All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by grants from the Strategic Priority Research Program of Chinese Academy of Sciences (Grant Nos. XDA19090116, XDA19050302, and XDB38030400) awarded to SS, ZZ, and ML; the National Key R&D Program of China (Grant Nos. 2020YFC0848900, 2020YFC0847000, 2016YFE0206600, and 2017YFC0907502); the 13th Five-year Informatization Plan of Chinese Academy of Sciences (Grant No. XXH13505-05); Genomics Data Center Construction of Chinese Academy of Sciences (Grant No. XXH-13514-0202); the Open Biodiversity and Health Big Data Programme of International Union of Biological Sciences, International Partnership Program of Chinese Academy of Sciences (Grant No. 153F11KYSB20160008); the Professional Association of the Alliance of International Science Organizations (Grant No. ANSO-PA-2020-07). This work was also supported by KC Wong Education Foundation to ZZ, as well as the Youth Innovation Promotion Association of Chinese Academy of Sciences (Grant Nos. 2017141 and 2019104) awarded to SS and ML. We thank our colleagues and students for their hard work on the 2019nCoVR (https://bigd.big.ac.cn/ncov). We also thank a number of users and CNCB members for reporting bugs and sending comments. Complete genome sequences used for analyses were obtained from the Genome Warehouse of CNCB, CNGBdb, GenBank, GISAID, and NMDC resources. We acknowledge the sample providers and data submitters listed in Table S1.
Handled by Feng Gao
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2020.09.001.
Contributor Information
Zhang Zhang, Email: zhangzhang@big.ac.cn.
Wenming Zhao, Email: zhaowm@big.ac.cn.
Yongbiao Xue, Email: ybxue@big.ac.cn.
Yiming Bao, Email: baoym@big.ac.cn.
Supplementary material
The following are the Supplementary data to this article:
Distribution of genome sequence count divided by the number of confirmed cases for each country as of July 14, 2020.
Genome compositional dynamics of SARS-CoV-2 and two representative genomic regions with high frequency of Ns. A. GC and AG compositional variability of SARS-CoV-2. B. Two representative genomic regions (top: ORF1ab at positions19,276–19,458; bottom: S at positions 22325–22545) with high frequency of Ns and the number (percentage) of sequences obtained using different sequencing platforms. C. Examples of secondary structure. Representative regions enriched with Ns (positions 19276–19458) and a region without Ns (positions 782–1004) are shown on the left and right, respectively. MFE, minimum free energy.
References
- 1.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Z., Song S., Yu J., Zhao W., Xiao J., Bao Y. The elements of data sharing. Genomics Proteomics Bioinformatics. 2020;18:1–4. doi: 10.1016/j.gpb.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shu Y., McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao W.M., Song S.H., Chen M.L., Zou D., Ma L.N., Ma Y.K. The 2019 novel coronavirus resource. Yi Chuan. 2020;42:212–221. doi: 10.16288/j.yczz.20-030. (in Chinese with an English abstract) [DOI] [PubMed] [Google Scholar]
- 7.National Genomics Data Center Members and Partners Database resources of the National Genomics Data Center in 2020. Nucleic Acids Res. 2020;48:D24–D33. doi: 10.1093/nar/gkz913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi W., Qi H., Sun Q., Fan G., Liu S., Wang J. gcMeta: a global catalogue of metagenomics platform to support the archiving, standardization and analysis of microbiome data. Nucleic Acids Res. 2019;47:D637–D648. doi: 10.1093/nar/gky1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao S.Z., Armit C., Edmunds S., Goodman L., Li P., Tuli M.A. Increased interactivity and improvements to the GigaScience database GigaDB. Database (Oxford) 2019;2019:1–9. doi: 10.1093/database/baz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rego N., Koes D. 3Dmol.js: molecular visualization with WebGL. Bioinformatics. 2015;31:1322–1324. doi: 10.1093/bioinformatics/btu829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong Z., Zhu J.W., Li C.P., Jiang S., Ma L.N., Tang B.X. An online coronavirus analysis platform from the National Genomics Data Center. Zool Res. 2020;41:705–708. doi: 10.24272/j.issn.2095-8137.2020.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang X., Wu C., Li X., Song Y., Yao X., Wu X. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. 2020;7:1012–1023. doi: 10.1093/nsr/nwaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korber B, Fischer W, Gnanakaran S, Yoon H, Theiler J, Abfalterer W. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827. doi: 10.1016/j.cell.2020.06.043. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bandelt H.J., Forster P., Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 17.Croucher N.J., Didelot X. The application of genomics to tracing bacterial pathogen transmission. Curr Opin Microbiol. 2015;23:62–67. doi: 10.1016/j.mib.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Mavian C., Pond S.K., Marini S., Magalis B.R., Vandamme A.M., Dellicour S. Bias and incorrect rooting make phylogenetic network tracing of SARS-CoV-2 infections unreliable. Proc Natl Acad Sci U S A. 2020;117:12522–12523. doi: 10.1073/pnas.2007295117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eyre D.W., Cule M.L., Wilson D.J., Griffiths D., Vaughan A., O'Connor L. Diverse sources of C. difficile infection identified on whole-genome sequencing. N Engl J Med. 2013;369:1195–1205. doi: 10.1056/NEJMoa1216064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rambaut A., Holmes E.C., O'Toole A., Hill V., McCrone J.T., Ruis C. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim D., Lee J.Y., Yang J.S., Kim J.W., Kim V.N., Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–921. doi: 10.1016/j.cell.2020.04.011. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of genome sequence count divided by the number of confirmed cases for each country as of July 14, 2020.
Genome compositional dynamics of SARS-CoV-2 and two representative genomic regions with high frequency of Ns. A. GC and AG compositional variability of SARS-CoV-2. B. Two representative genomic regions (top: ORF1ab at positions19,276–19,458; bottom: S at positions 22325–22545) with high frequency of Ns and the number (percentage) of sequences obtained using different sequencing platforms. C. Examples of secondary structure. Representative regions enriched with Ns (positions 19276–19458) and a region without Ns (positions 782–1004) are shown on the left and right, respectively. MFE, minimum free energy.
Data Availability Statement
SARS-CoV-2 genomes, variants (in vcf format), and their annotations are publicly available at https://bigd.big.ac.cn/ncov/.