

Abstract
Background
Obstructive sleep apnea (OSA) is associated with pulmonary fibrosis. Chronic intermittent hypoxia (CIH) is considered to be a surrogate of OSA. However, its exact role in pulmonary fibrosis remains uncertain. Therefore, we investigated the mechanism underlying CIH-induced pulmonary fibrosis and the role of the anti-fibrotic agent in bleomycin (BLE) induced lung injury.
Methods
Mice were divided into eight groups: the normoxia (NOR), CIH, NOR plus BLE, CIH plus BLE, NOR plus pirfenidone (PF), CIH plus PF, NOR plus BLE and PF, and CIH plus BLE and PF groups. BLE was administered intratracheally on day 14 following CIH or NOR exposure. Subsequently, the mice were exposed to CIH or NOR for an additional 4 weeks. PF was administered orally on day 5 after BLE instillation once daily for 3 weeks.
Results
In the BLE-treated groups, CIH-induced more collagen deposition in lung tissues than NOR, and significantly increased hydroxyproline and transforming growth factor-β expression. The CIH and BLE-treated groups showed increased lung inflammation compared to NOR or CIH groups. Following CIH with BLE treatment, nuclear factor-κB (NF-κB) protein expression was significantly increased, whereas nuclear factor-erythroid-related factor 2 (Nrf2) and heme oxygenase-1 protein levels were decreased. After PF treatment, NF-κB and Kelch-like ECH-associated protein 1 expression were suppressed, and Nrf2 expression was increased.
Conclusion
CIH accelerated lung fibrosis in BLE-induced lung injury in mice, potentially by regulating the NF-κB/Nrf2 signaling pathway. Our results implicate PF as a potential therapeutic agent for treating pulmonary fibrosis in individuals with OSA and idiopathic pulmonary fibrosis.
Keywords: Sleep Apnea, Obstructive; Pulmonary Fibrosis; Bleomycin
Introduction
Obstructive sleep apnea (OSA) is characterized by complete or partial recurrent episodes of upper airway obstruction during sleep. It is usually associated with nocturnal intermittent hypoxia (IH)/reoxygenation, which is otherwise known as chronic intermittent hypoxia (CIH). OSA is a highly prevalent condition that has been associated with morbidities and mortalities associated with systemic hypertension, stroke, ischemic heart disease, and congestive heart failure.
OSA is common among patients with idiopathic pulmonary fibrosis (IPF). In a prospective study conducted by Gille et al. [1], the prevalences of moderate-to-severe and severe OSA were 62% and 40%, respectively, in patients with newly diagnosed IPF. Indeed, in a population of patients with IPF, severe OSA was strongly associated with the presence of cardiovascular disease. Importantly, OSA may adversely influence the progression of IPF and related mortality [2].
The mechanisms whereby OSA worsens IPF remains unknown. In patients with IPF, the potential influence of OSA on lung fibrosis has not fully evaluated. Several reports have shown that CIH is associated with oxidative stress and inflammation, which can lead to fibrosis in the systemic vasculature, as well as the myocardium [3,4], liver [5], and kidneys [6]. We hypothesized that, by increasing lung oxidative stress and inflammation, CIH can promote the development or progression of pulmonary fibrosis in a mouse mode of bleomycin (BLE)-induced lung injury, thereby mimicking IPF. To test this hypothesis, we established an experimental model wherein mice were first exposed to CIH for 2 weeks and then were treated with BLE to induce lung fibrosis. Additionally, the potential protective effect of the anti-fibrotic agent, pirfenidone (PF) was evaluated.
Materials and Methods
1. Experimental design and animal model
The experimental design of this study is illustrated in Figure 1. Male C57BL/6J mice (7–8 weeks old) were purchased from DBL (Eumseong, Korea). After 1 week of acclimation, the mice were divided into the (1) normoxia (NOR) control, (2) CIH control, (3) NOR plus BLE, (4) CIH plus BLE, (5) NOR plus PF, (6) CIH plus PF, (7) NOR plus BLE and PF, and (8) CIH plus BLE and PF groups. Mice were administered by intratracheal instillation of 0.01 U BLE once on day 14 following CIH (fraction of inspired oxygen of approximately 7%±0.5% within 30 seconds, followed by reoxygenation to NOR levels within the next 30 seconds; 20 episodes/hr, 8 hours during the daytime) or NOR exposure. After BLE instillation, the mice were exposed to CIH or NOR for an additional 4 weeks. PF was administered on day 5 after BLE challenge by oral gavage (400 mg/kg), once daily, until the mice were euthanized. Food and drinking water were provided ad libitum. The body weight (BW) of each mouse was monitored once per week. Oxygen levels were measured with a ProOx 110 analyzer (BioSpherix, Redfield, NY, USA). Gases were exposed to coincide with mouse sleep cycles, and a gas control delivery system was designed to regulate air, nitrogen, and oxygen flow.
Figure 1.
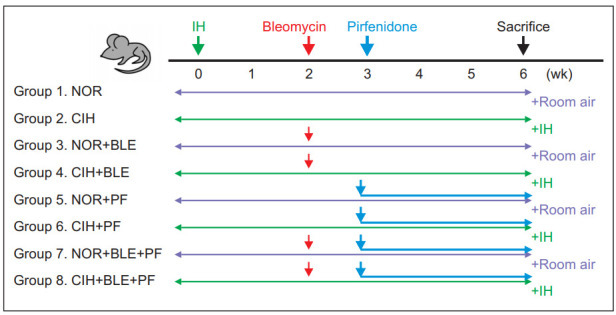
Schematic representation of the animal experimental protocol. Mice were exposed to normoxia (NOR) or chronic intermittent hypoxia (CIH) for 2 weeks, and then administered an intratracheal instillation of bleomycin (BLE) or saline on day 14. After BLE instillation, the mice were exposed to CIH or NOR for an additional 4 weeks. Pirfenidone (PF) was administered on day 5 after the BLE challenge by oral gavage (400 mg/kg), once daily, until the mice were sacrificed. IH: intermittent hypoxia.
BLE was purchased from Sigma (St. Louis, MO, USA) and dissolved in sterile normal saline. PF was provided from ILDong Pharmaceutical Co. (Shionogi & Co., Ltd., Osaka, Japan) and dissolved in 0.5% carboxymethylcellulose (CMC). Control mice received an equal volume of normal saline or 0.5% CMC. This study was approved by the Ethical Committee on Animal Experiments of the Catholic University of Korea (approval number SPH-20170530–01) and was compliant with their animal welfare guidelines.
2. Histopathology
After sacrifice, the trachea was exposed and the left lung tissues of each group were fixed with 10% formalin for 24 hours and then processed in paraffin wax for histological examination. Four-micrometer-thick sections were prepared using a microtome. The deparaffinized tissue sections were stained with hematoxylin and eosin (H&E) for histological examination and with Masson’s trichrome stain and Sirius red to detect fibrosis. Images were scanned with a Panoramic MIDI slide scanner (3DHISTECH, Ltd., Budapest, Hungary).
3. Bronchoalveolar lavage fluid
Bronchoalveolar lavage (BAL) fluid was obtained by cannulating mice with silicone tubing attached to a 23-G needle on a tuberculin syringe. BAL fluid was collected via instillation of 0.8 mL phosphate-buffered saline and centrifuged at 1,500 rpm for 5 minutes at 4°C. Total cell counts were performed using a hemocytometer. Differential cell counts were determined by performing cytospins (5 minutes at 750 ×g) onto microscope slides, followed by staining with Diff-Quick (Sysmex, Tokyo, Japan). The percentages of BAL fluid macrophages, neutrophils, lymphocytes, and eosinophils were obtained by counting 500 leukocytes on randomly selected areas of the slides. Supernatants were stored at –70°C for subsequent measurement of total albumin and cytokine concentrations.
4. Enzyme-linked immunosorbent assay analysis
Commercial mouse enzyme-linked immunosorbent assay (ELISA) kits were used to measure albumin and the proinflammatory and/or fibrosis-related cytokines interleukin (IL)- 6, IL-1β, and tumor necrosis factor α (TNF-α; Thermo Fisher Scientific, Inc., Bartlesville, OK, USA) in BAL fluid, as well as myeloperoxidase (MPO; BioVision, Inc., Milpitas, CA, USA), the lipid peroxidation enzyme, malondialdehyde (MDA; Abcam, Burlingame, CA, USA), and the oxidative stress-related enzyme, superoxide dismutase (SOD; Bioassay Systems, Hayward, CA, USA) in lung tissues. The ELISAs were performed according to the manufacturers’ instructions.
5. Hydroxyproline content and transforming growth factor β
The collagen concentrations in lung tissues were measured using a Hydroxyproline Colorimetric Assay Kit (BioVision, Inc.), according to the manufacturer’s instructions. Briefly, the same volume of each lung homogenate was incubated with 12 N HCl for 16 hours at 110–120°C. Following centrifugation at 13,000 rpm for 3 minutes, the supernatants were mixed with chloramine T solution and DMAB reagent (provided in the kit) and incubated for 90 minutes at 60°C. Serum transforming growth factor β (TGF-β) levels were determined using a Mouse TGF-β ELISA Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA), according to the manufacturer’s instructions. The absorbances of the samples were read at 560 nm and 450 nm, respectively, using an automatic microplate reader (Infinite 200 PRO, Life Science, Horsham, PA, USA), and the concentrations were calculated from a standard curve. All samples and standards were run in duplicate.
6. Western blotting
Lung homogenate proteins were isolated using a Nuclear/Cytosol Fractionation Kit (Thermo Fisher Scientific, Inc., Bartlesville, OK, USA). Protein concentrations were determined by performing bicinchoninic acid assays. Equal amounts of proteins were separated by 10%–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). After blocking with 5% skim milk, the membrane was incubated overnight at 4°C with primary antibodies against nuclear factor-κB (NF-κB), the inhibitor of NF-κB, Kelch-like ECH-associated protein 1 (Keap1), nuclear factorerythroid-related factor 2 (Nrf2), heme oxygenase-1 (HO-1), β-actin, and histone H3 (Cell Signaling Technology, Inc., Danvers, MA, USA). After incubation with a secondary antibody for 2 hours, target proteins were detected using the enhanced chemiluminescence method (Bio-Rad, Hercules, CA, USA). Signals were detected using the ImageQuant LAS 500 Imaging System (GE Healthcare Life Sciences, Seoul, Korea).
7. Data analysis
The data were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test or two-way ANOVA (for studying BAL cell differentiation). The analyses were conducted using GraphPad Prism software, version 7.00 (GraphPad Software Inc., San Diego, CA, USA). The data are presented as the mean±standard deviation and a value of p<0.05 was considered to reflect a statistically significant difference.
Results
1. BW changes
The animals were weighed once per week. After BLE inhalation or treatment with PF, the weights of the mice decreased, and the mice in groups that received both BLE and PF showed significantly lower weights than those in the NOR group (p<0.05, p<0.01, vs. the NOR group) (Figure 2).
Figure 2.

The effects of bleomycin (BLE), chronic intermittent hypoxia (CIH), and pirfenidone (PF) on body weights (BWs) and weight gain. (A) The BWs of mice exposed to normoxia (NOR) or CIH, with or without PF administration. BWs were recorded weekly. (B) Comparison of weight gains in mice (ΔBW=BW at 6 weeks–BW at 1 week). The data shown are expressed as the mean±SD. *p<0.05, **p<0.01 vs. the NOR group.
2. Lung injury and inflammation
After H&E staining, alveolar septal thickening (Figure 3, upper) and peribronchial inflammatory cell infiltration (Figure 3, bottom) in lung sections were observed by light microscopy. Lung tissues in the NOR group showed normal alveolar septa and clear pulmonary alveoli, whereas alveolar damage and inflammation were observed in CIH, BLE, and CIH+BLE groups. These inflammatory changes or injuries in the lungs were attenuated in the groups treated with PF when compared with each corresponding control group that was not treated with PF.
Figure 3.
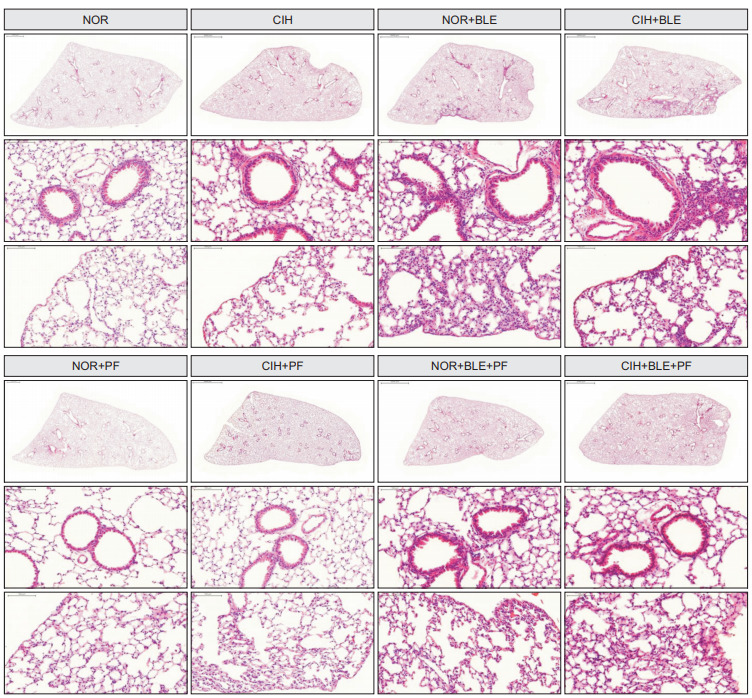
The effects of bleomycin (BLE), chronic intermittent hypoxia (CIH), and pirfenidone (PF) on lung morphology. NOR: normoxia. Representative photomicrographs of lung tissues represent stained tissues with hematoxylin and eosin. Vascular morphology (upper panels) or peribronchial morphology (bottom panels) were observed as a measure of lobular inflammation (×200).
Treatment with both CIH and BLE significantly increased the total numbers of cells and macrophages in BAL fluid specimens, whereas PF treatment significantly reduced the numbers of these cells. No significant differences were found in terms of other inflammatory cells such as neutrophils, lymphocytes, and eosinophils (Figure 4A). The MPO levels tended to be slightly higher in the groups treated with BLE and/or CIH, compared to the NOR group; however, these differences were not significant (Figure 4B). Albumin levels increased in BAL fluids after both CIH and BLE treatment; however, PF significantly decreased the albumin levels (Figure 4C). The concentrations of IL-6, IL-1β, and TNF-α in BAL fluids were significantly higher after treatment with BLE and/or CIH, and PF treatment significantly attenuated IL-6 levels when compared with each corresponding control group that was not treated with PF (Figure 4D–F). These results suggest that BLE and/or CIH-induced lung injury and inflammation, and that PF treatment attenuated these effects.
Figure 4.
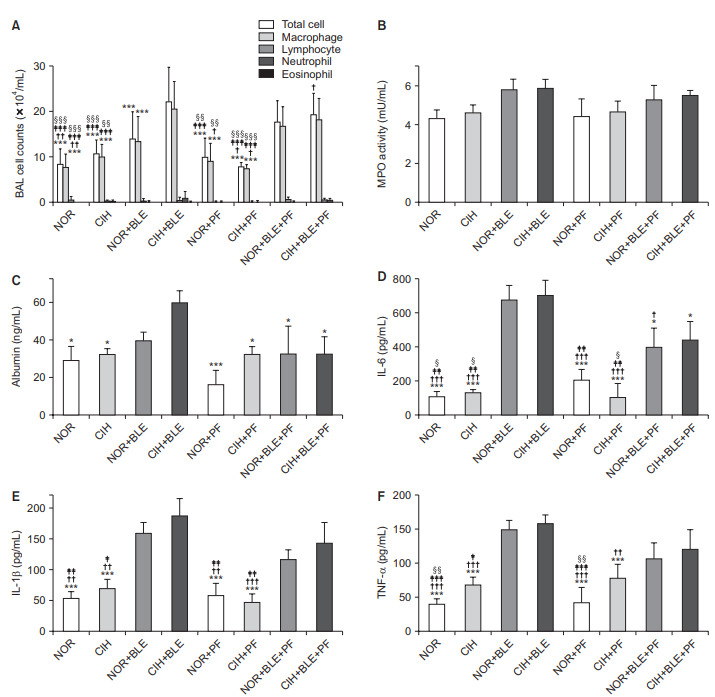
The effect of bleomycin (BLE), chronic intermittent hypoxia (CIH), and pirfenidone (PF) on lung inflammation. (A) Bronchoalveolar lavage (BAL) cells were isolated, after which total cell numbers and the numbers of different inflammatory cell types were counted. (B) Activity of the inflammatory enzyme myeloperoxidase (MPO) was measured in total lung homogenates from mice. (C–F) The levels of albumin (C) and the proinflammatory cytokines interleukin (IL)-6 (D), IL-1β (E), and tumor necrosis factor α (TNF-α) (F) in BAL fluid were measured by enzyme-linked immunosorbent assay. The data shown are expressed as the mean±SD. *p<0.05, **p<0.01, ***p<0.001 vs. the CIH+BLE group. †p<0.05, ††p<0.01, †††p<0.001 vs. the normoxia (NOR)+BLE group. ‡p<0.05, ‡‡p<0.01, ‡‡‡p<0.001 vs. the CIH+BLE+PF group. §p<0.05, §§p<0.01, §§§p<0.001 vs. the NOR+BLE+PF group.
3. Lung fibrosis
To demonstrate the lung fibrosis, tissue sections were stained with Masson’s trichrome stain and Sirius red, and the hydroxyproline and TGF-β contents were measured in lung tissues and serum, respectively. In the interalveolar space and alveoli, collagen deposition and scattered fibrotic changes were reflected by blue or red staining, which appeared higher in the NOR+BLE and CIH+BLE groups. Interstitial thickening with collagen deposition was more prominent in the CIH and BLE-treated group compared to NOR+BLE groups. Whereas PF treatment attenuated collagen deposition in the pleura and collagen fibers in the interstitium and perivascular spaces (Figure 5A, B).
Figure 5.
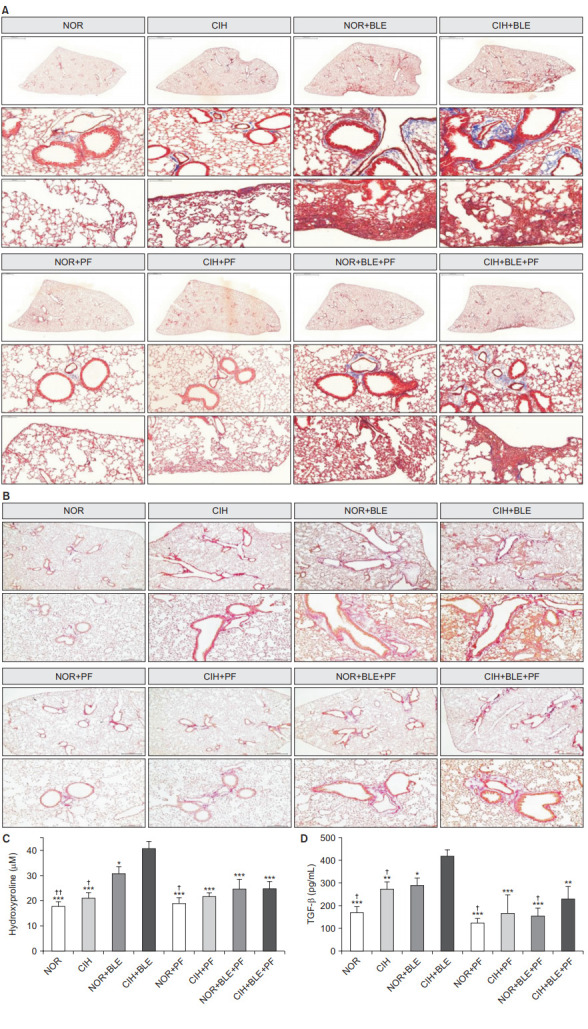
The effects of bleomycin (BLE), chronic intermittent hypoxia (CIH), and pirfenidone (PF) on lung fibrosis. (A) Representative photomicrographs of lung tissues were stained with Masson’s trichrome stain as a measure of collagen accumulation. The upper panels show images of whole lung sections, and the bottom panels represent the enlarged images. Scale bars=2,000 μm, middle panels (×10); 100 μm, bottom panels (×200). (B) Sirius red staining (×200). (C) Hydroxyproline content was measured in lung tissues. (D) Serum transforming growth factor β (TGF-β) levels were measured by performing enzyme-linked immunosorbent assays. The data shown are expressed as the mean±SD. *p<0.05, **p<0.01, ***p<0.001 vs. the CIH+BLE group. †p<0.05, ††p<0.01 vs. the normoxia (NOR)+BLE group.
Hydroxyproline levels significantly increased in the NOR+BLE and CIH+BLE groups (Figure 5C) and a similar trend was observed with the TGF-β levels (Figure 5D). In addition, the hydroxyproline and TGF-β levels were significantly higher in the CIH+BLE group than in the NOR+BLE group (p<0.05 vs. CIH+BLE). After treatment with PF, hydroxyproline and TGF-β levels were markedly decreased in each PF-treatment group than in the CIH+BLE group. In addition, TGF-β production was significantly decreased in the NOR+BLE+PF group than in the NOR+BLE group. These results suggest that CIH exacerbated BLE-induced pulmonary fibrosis, but that PF treatment attenuated the effects of BLE and/or CIH.
4. Oxidative stress
We investigated whether BLE or CIH affected oxidative damage (MDA) or the balance of a biochemical redox marker (SOD). The MDA levels in lung homogenates was significantly higher in the CIH+BLE group than in the NOR group, and the MDA levels significantly decreased in each PF-treatment group, except for the CIH+BLE+PF group (p<0.05, p<0.01 vs. CIH+BLE) (Figure 6A). Although SOD levels did not significantly change in each group, they tended to be slightly lower in the groups treated with BLE and/or CIH than in the NOR group, but not in the groups treated with PF (Figure 6B). These results suggest that CIH and BLE treatment affected the levels of pulmonary enzymes that are usually involved in oxidative stress and antioxidant defense mechanisms.
Figure 6.

The effects of bleomycin (BLE), chronic intermittent hypoxia (CIH), and pirfenidone (PF) on oxidative stress. (A, B) The levels of malondialdehyde (MDA; as a lipid peroxidation marker) (A) and superoxide dismutase (SOD; as an indicator of oxidative stress) (B) were measured in the total lung homogenates from the mice in each group. NOR: normoxia. The data shown are expressed as the mean±SD. *p<0.05, **p<0.01 vs. the CIH+BLE group.
5. NF-κB/Nrf2 signaling pathway
To investigate the intracellular molecular mechanism of lung fibrosis associated with CIH and BLE, and the therapeutic effects of PF, the expression levels of several genes related to the TGF-β–signaling pathway were measured. Our results revealed that NF-κB could increase inflammation and oxidative stress via the TGF-β–signaling pathway. NF-κB expression was significantly higher in the CIH+BLE group than in the other groups, whereas Nrf2 and HO-1 levels decreased in mice treated with BLE and CIH. In contrast, treatment with PF suppressed NF-κB and Keap1 levels and increased Nrf2 levels (Figure 7). Our findings suggested that CIH augmented BLEinduced protein expression of NF-κB, whereas treatment with PF suppressed NF-κB expression by upregulating the antioxidant factor, Nrf2. Representative results are shown in Figure 8.
Figure 7.
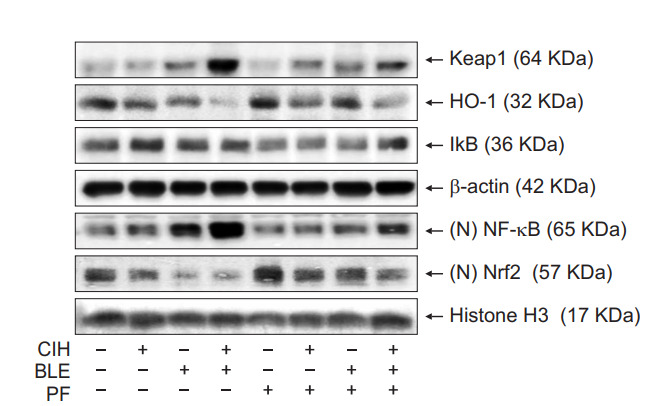
The effects of bleomycin (BLE), chronic intermittent hypoxia (CIH), and pirfenidone (PF) on the nuclear factor-κB (NFκB)/nuclear factor-erythroid-related factor 2 (Nrf2) signaling pathway. Protein-expression levels in the total and nuclear fractions of lung tissues were analyzed by Western blotting. β-Actin or histone H3 were detected as loading controls for the total and nuclear fractions, respectively. Keap1: Kelch-like ECH-associated protein 1; HO1: heme oxygenase-1.
Figure 8.
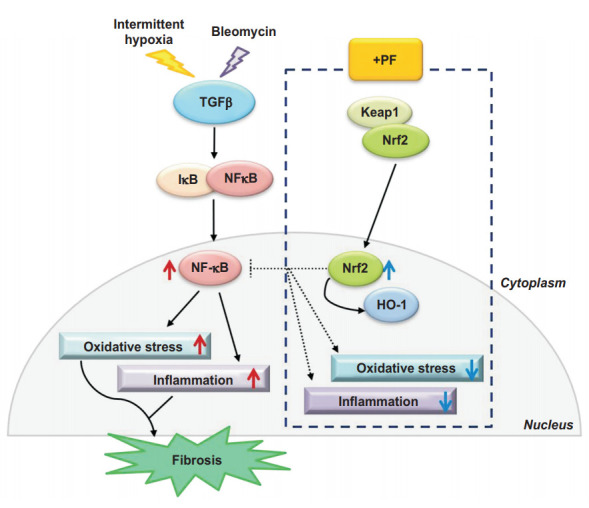
Summary of the lung fibrosisrelated intracellular signaling pathways induced by bleomycin, chronic intermittent hypoxia, and anti-fibrotic drug mechanisms. PF: pirfenidone; Keap1: Kelch-like ECH-associated protein 1; Nrf2: nuclear factor-erythroid-related factor 2; HO-1: heme oxygenase-1.
Discussion
In this study, we demonstrated that CIH treatment for 6 weeks induced significant alveolar septal thickening and parenchymal fibrosis in a mouse model of BLE-induced lung injury and fibrosis. After BLE treatment, CIH promoted increased oxidative stress markers and the release of proinflammatory mediators, which contributed to pulmonary fibrosis via the NF-κB/Nrf2 signaling pathway. PF treatment attenuated CIH-associated pulmonary fibrosis in BLE-treated mice.
We evaluated the effect of CIH on BLE-induced pulmonary fibrosis progression in mice. CIH led to an increase in the total collagen content. Additionally, we observed that CIH exposure, independently of BLE, caused focal pathological changes in the parenchyma. These changes included a tendency for increase hydroxyproline and TGF-β levels in uninjured, CIHtreated mice, suggestive of a potential profibrogenic effect of OSA. The mouse model of BLE-induced lung injury used in this study is the most extensively used model for studying the pathogenesis of pulmonary fibrosis. The commonly employed model of single exposure to BLE only resulted in temporary fibrosis. A single intratracheal BLE instillation can upregulate collagen gene expression within a few days, and most fibrosis appears between days 7–28 and then revolve within 6 weeks [7]. Considering this time course of fibrosis in BLE-induced lung injury, CIH treatment delayed recovery in accordance with increasing lipid peroxidation and collagen deposition in the BLE-treated groups. These results suggest that CIH exposure promoted lung fibrosis following BLE treatment.
In previous animal studies, CIH worsened lung fibrosis in BLE-treated groups. However, in those studies, rodents received BLE instillation and were then exposed to CIH. Sprague-Dawley rats exposed to CIH for 35 days showed structural changes in their lung tissues, and only an increasing trend in hydroxyproline levels was detected [8]. Gill et al. [9] evaluated the effects of CIH on BLE-induced pulmonary fibrosis in mice using similar CIH regimens for 21 days. The authors observed significant increases in mortality, lung inflammation, and lung fibrosis in mice with BLE-induced lung injury, but they did not specifically evaluate the molecular mechanisms involved [9]. Several reports showed that CIH may lead to oxidative stress, inflammation, angiogenesis, and apoptosis in lung tissues. TNF-α levels, extracellular matrix deposition, and intra-alveolar septum thicknesses increased significantly during IH [10,11]. In addition, elevation of Kerbs von den Lungen 6 has been detected in patients with OSA [12], as a marker of subclinical lung injury.
Recent studies showed that CIH exerted positive effects on the lung by increasing the synthesis of antioxidant enzymes and enlarging the alveolar surface area [13,14]. With our model, we observed lung inflammation in mice treated with BLE, but not in mice treated with saline and exposed to either NOR or CIH. Lung inflammation was significantly higher in mice treated with CIH and BLE than in those treated with NOR or CIH, as indicated by histologically more intense inflammatory cell infiltration, an increasing trend in MPO expression in lung homogenates, and a significant increase in albumin, IL-6, IL-1β, and TNF-α in BAL fluid. In addition, pulmonary macrophages were markedly more abundant in BAL fluids from mice in the CIH+BLE group versus mice in the NOR+BLE group. IH can induce the accumulation of circulating monocytes in the lungs and promote their differentiation into proinflammatory pulmonary macrophages capable of expressing inducible nitric oxide synthase [15]. Consistent with previous studies [16], our present results suggest that increased pulmonary macrophages may accentuate inflammation and generate high levels of reactive oxygen species (ROS), and thereby aggravating oxidative stress.
Oxidative stress is considered a major pathogenic feature of CIH, but it is also induced by BLE. In this study, we observed increased MDA production and a trend towards decreased SOD production in lung homogenates from mice treated with BLE and exposed to CIH. No significant effects were observed due to CIH exposure per se, in that no difference was found between mice exposed to NOR and mice exposed to CIH. However, a significant increase in MDA expression was only observed in the CIH+BLE group, and antioxidant enzyme levels in lung homogenates showed some differences between the NOR+BLE and CIH+BLE groups. SOD-expression levels showed a decreasing trend in the CIH+BLE group when compared with those in other groups. These results suggest that antioxidant defense mechanisms were partially compromised in CIH+BLE group. Indeed, we observed that lung levels of the prooxidant MPO enzyme showed an increasing trend in mice subjected to CIH and treated with BLE. Elevated MPO expression was associated with inflammatory cell infiltration in parenchyma and abundant macrophages in BAL fluid, indicating that an imbalance occurred between prooxidant enzymes and antioxidant defense enzymes during treatment with CIH and BLE.
IPF is a chronic and almost irreversible pulmonary parenchymal disease, yet the precise molecular mechanisms involved are still not fully understood. However, many findings have demonstrated that oxidative stress is one of the major pathogenic disease mechanisms [17,18]. TGF-β, which is the most potent profibrogenic cytokine, plays a vital role in extracellular matrix deposition by regulating crucial cellular activities such as apoptosis and the epithelial–mesenchymal transition [19]. Oxidative stress can activate latent TGF-β by establishing a vicious profibrogenic cycle, including the activation of fibroblast proliferation and collagen production. Conversely, TGF-β stimulates ROS production, thereby leading to oxidative stress [20]. In this study, in addition to observing increased MDA expression, we found that TGF-β expression was significantly higher in mice subjected to CIH and BLE. These results suggest that interactive effects between oxidative stress and TGF-β are important for promoting fibrosis in CIH exposure following BLE treatment.
Nrf2 is involved in the expression of antioxidant factors, enabling it to further regulate the oxidation/antioxidation capacity [21]. Nrf2 binds to Keap1 in the cytoplasm and has low activity under normal physiological conditions. However, when cells are exposed to oxidative stress, Nrf2 uncouples with Keap1, translocates into the nucleus, and binds to the Nrf2 antioxidant response element (ARE), thus initiating the transcription and expression of a series of downstream antioxidases such as HO-1, SOD, and thioredoxin. NF-κB also regulates Nrf2 expression by promoting Keap1 and subsequent Nrf2 ubiquitination or interfering with interactions between Nrf2 and ARE sequences [22]. Nrf2 is believed to be an anti-inflammatory modulator involved in regulating NF-κB activity by decreasing NF-κB inhibitor-α phosphorylation, thereby reducing the nuclear accumulation of NF-κB [23]. Dysfunction of the Nrf2–AREsignaling pathway plays an important role in the development of pulmonary fibrosis. Nrf2 activation increases antioxidasedependent defenses and myofibroblastic dedifferentiation in pulmonary fibrosis, suggesting that Nrf2 can potentially serve as a novel therapeutic target for pulmonary fibrosis and other oxidative stress-induced fibrogenic diseases. The Nrf2–AREsignaling pathway was previously found to be dysfunctional in a mouse model of IH. In that study, decreased Nrf2 and HO-1 expression were observed in the kidneys of mice exposed to IH for 8 weeks [24]. The levels of Nrf2 mRNA and protein have also been shown to be reduced in the genioglossus muscles of IH mice [25]. When the IH mice received a HO-1 activator, IH-induced cellular oxidative stress and apoptosis were alleviated [26]. However, the role of the Nrf2-signaling pathway in OSA in patients with IPF has not been reported. In this study, we found significant decreases in nuclear Nrf2 and HO-1 expression, increased nuclear NF-κB expression, and markedly increased cytoplasmic Keap1 protein expression in mice treated with BLE and exposed to CIH. These results suggest that CIH helps regulate both the NF-κB and Nrf2 pathways by modulating the balance between inflammatory and antioxidant factors.
PF is known to inhibit the proliferation, differentiation, and the collagen synthesis of fibroblasts. It also inhibits the generation and activities of the fibrosis-related cytokines TGF-β1, fibronectin, and connective tissue growth factor, thus exerting anti-inflammatory and anti-fibrotic effects that have been regarded to treat IPF [27]. Data from a recent in vivo study indicated that PF suppressed chemotaxis and accumulation of fibroblasts in the lungs of mouse with BLE-induced pulmonary fibrosis [28]. A previous report showed that PF alleviated oxidation by suppressing NADPH-dependent lipid peroxidation in liver microsomes [29]. This finding suggested that the anti-fibrotic activity of PF was linked to oxidative stress. Data from other studies also showed that PF exerted an anti-fibrotic effect by alleviating lipid peroxidation and clearing the active intracellular oxygen [30], further demonstrating that the antifibrotic effect of PF was associated with oxidative stress.
However, the effect of PF in CIH-associated pulmonary fibrosis has not been evaluated. Our present data showed that administering PF attenuated inflammatory cell infiltration and histologically fibrotic changes in the CIH+BLE+PF group. Our results also demonstrated that after PF treatment, expression of fibrosis-related cytokine IL-6 decreased, and the levels of TGF-β and hydroxyproline significantly decreased in mice treated with CIH and BLE, whereas no significant difference occurred in the NOR+BLE group. These results suggest that PF inhibited production of a pro-fibrotic factor and the development of lung fibrosis, and that the influence of PF in BLEtreated mice intensified during CIH. To further explore the antioxidant and anti-fibrotic effects of PF, we investigated the Nrf2-signaling pathway. Following PF administration, the Nrf2 protein expression significantly increased in the BLE-treated groups, whereas NF-κB protein expression significantly decreased when compared with the groups exposed to CIH and BLE. Our results suggest that in BLE-induced lung fibrosis, PF promotes the expression of an antioxidant factor, such as Nrf2, by reversing the inhibitory effect of antioxidant activity induced by the NF-κB– and TGF-β–signaling pathways during CIH.
In conclusion, our results indicate that CIH-induced lung inflammation and lung fibrosis in BLE-treated mice, which was modulated by the NF-κB/Nrf2 signaling pathway. We also demonstrated a protective role of PF against CIH-induced lung injury in mice. These findings provide insights into the mechanisms whereby OSA affects patients with IPF. Further investigations are needed to understand the consequences of OSA and IPF, as well as the effects of PF in such patients.
Footnotes
Authors’ Contributions
Conceptualization: Kang HH, Lee SH. Methodology: Kang HH, Kim IK. Formal analysis: Kang HH, Kim IK, Yeo CD, Kim SW, Lee HY, Im JH, Kwon HY, Lee SH. Data curation: Kim IK, Im JH, Kwon HY. Validation: Kang HH, Lee SH, Kim IK. Investigation: Lee SH, Kim IK, Im JH, Kwon HY. Writing – original draft preparation: Kang HH, Lee SH, Kim IK. Writing – review and editing: Yeo CD, Kim SW, Lee HY. Approval of final manuscript: all authors.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Funding
This research was funded by the Korean Academy of Tuberculosis and Respiratory Diseases (2018).
REFERENCES
- 1.Gille T, Didier M, Boubaya M, Moya L, Sutton A, Carton Z, et al. Obstructive sleep apnoea and related comorbidities in incident idiopathic pulmonary fibrosis. Eur Respir J. 2017;49:1601934. doi: 10.1183/13993003.01934-2016. [DOI] [PubMed] [Google Scholar]
- 2.Mermigkis C, Bouloukaki I, Antoniou K, Papadogiannis G, Giannarakis I, Varouchakis G, et al. Obstructive sleep apnea should be treated in patients with idiopathic pulmonary fibrosis. Sleep Breath. 2015;19:385–91. doi: 10.1007/s11325-014-1033-6. [DOI] [PubMed] [Google Scholar]
- 3.Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep. 2014;16:411. doi: 10.1007/s11906-013-0411-y. [DOI] [PubMed] [Google Scholar]
- 4.Ramos P, Rubies C, Torres M, Batlle M, Farre R, Brugada J, et al. Atrial fibrosis in a chronic murine model of obstructive sleep apnea: mechanisms and prevention by mesenchymal stem cells. Respir Res. 2014;15:54. doi: 10.1186/1465-9921-15-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cannito S, Paternostro C, Busletta C, Bocca C, Colombatto S, Miglietta A, et al. Hypoxia, hypoxia-inducible factors and fibrogenesis in chronic liver diseases. Histol Histopathol. 2014;29:33–44. doi: 10.14670/HH-29.33. [DOI] [PubMed] [Google Scholar]
- 6.Pan SY, Chang YT, Lin SL. The role of hypoxia-inducible factors in renal fibrosis. J Formos Med Assoc. 2013;112:587–8. doi: 10.1016/j.jfma.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Chung MP, Monick MM, Hamzeh NY, Butler NS, Powers LS, Hunninghake GW. Role of repeated lung injury and genetic background in bleomycin-induced fibrosis. Am J Respir Cell Mol Biol. 2003;29:375–80. doi: 10.1165/rcmb.2003-0029OC. [DOI] [PubMed] [Google Scholar]
- 8.Braun RK, Broytman O, Braun FM, Brinkman JA, Clithero A, Modi D, et al. Chronic intermittent hypoxia worsens bleomycin-induced lung fibrosis in rats. Respir Physiol Neurobiol. 2018;256:97–108. doi: 10.1016/j.resp.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gille T, Didier M, Rotenberg C, Delbrel E, Marchant D, Sutton A, et al. Intermittent hypoxia increases the severity of bleomycin-induced lung injury in mice. Oxid Med Cell Longev. 2018;2018:1240192. doi: 10.1155/2018/1240192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.da Rosa DP, Forgiarini LF, Baronio D, Feijo CA, Martinez D, Marroni NP. Simulating sleep apnea by exposure to intermittent hypoxia induces inflammation in the lung and liver. Mediators Inflamm. 2012;2012:879419. doi: 10.1155/2012/879419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Xie M, Gao Y, Wei HH, Zheng JQ. Study on the effect and mechanism of hypoxia on the histological structure of rat’s lung. Sichuan Da Xue Xue Bao Yi Xue Ban. 2012;43:1–5. [PubMed] [Google Scholar]
- 12.Aihara K, Oga T, Harada Y, Chihara Y, Handa T, Tanizawa K, et al. Comparison of biomarkers of subclinical lung injury in obstructive sleep apnea. Respir Med. 2011;105:939–45. doi: 10.1016/j.rmed.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 13.Gonchar O, Mankovska I. Moderate hypoxia/hyperoxia attenuates acute hypoxia-induced oxidative damage and improves antioxidant defense in lung mitochondria. Acta Physiol Hung. 2012;99:436–46. doi: 10.1556/APhysiol.99.2012.4.8. [DOI] [PubMed] [Google Scholar]
- 14.Reinke C, Bevans-Fonti S, Grigoryev DN, Drager LF, Myers AC, Wise RA, et al. Chronic intermittent hypoxia induces lung growth in adult mice. Am J Physiol Lung Cell Mol Physiol. 2011;300:L266–73. doi: 10.1152/ajplung.00239.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagai H, Kuwahira I, Schwenke DO, Tsuchimochi H, Nara A, Ogura S, et al. Pulmonary macrophages attenuate hypoxic pulmonary vasoconstriction via beta3AR/iNOS pathway in rats exposed to chronic intermittent hypoxia. PLoS One. 2015;10:e0131923. doi: 10.1371/journal.pone.0131923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He C, Ryan AJ, Murthy S, Carter AB. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutaseinduced alternative activation of macrophages. J Biol Chem. 2013;288:20745–57. doi: 10.1074/jbc.M112.410720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schunemann HJ, Muti P, Trevisan M. Serum indicators of free radical activity in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:769. doi: 10.1164/ajrccm.155.2.9032227. [DOI] [PubMed] [Google Scholar]
- 18.Matsuzawa Y, Kawashima T, Kuwabara R, Hayakawa S, Irie T, Yoshida T, et al. Change in serum marker of oxidative stress in the progression of idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. 2015;32:1–6. doi: 10.1016/j.pupt.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Dong Z, Tai W, Lei W, Wang Y, Li Z, Zhang T. IL-27 inhibits the TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells. BMC Cell Biol. 2016;17:7. doi: 10.1186/s12860-016-0084-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med. 2010;48:1–15. doi: 10.1016/j.freeradbiomed.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med. 2011;32:234–46. doi: 10.1016/j.mam.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Yu M, Li H, Liu Q, Liu F, Tang L, Li C, et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell Signal. 2011;23:883–92. doi: 10.1016/j.cellsig.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Wang B, Du F, Su X, Sun G, Zhou G, et al. Epigallocatechin-3-gallate attenuates oxidative stress and inflammation in obstructive nephropathy via NF-kappaB and Nrf2/HO-1 signalling pathway regulation. Basic Clin Pharmacol Toxicol. 2015;117:164–72. doi: 10.1111/bcpt.12383. [DOI] [PubMed] [Google Scholar]
- 24.Sun W, Yin X, Wang Y, Tan Y, Cai L, Wang B, et al. Intermittent hypoxia-induced renal antioxidants and oxidative damage in male mice: hormetic dose response. Dose Response. 2012;11:385–400. doi: 10.2203/dose-response.12-027.Cai. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding WH, Li W, Chen XY, Shi JJ. The study of genistein attenuating genioglossus muscle fatigue under chronic intermittent hypoxia. Zhonghua Kou Qiang Yi Xue Za Zhi. 2016;51:46–50. doi: 10.3760/cma.j.issn.1002-0098.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 26.Han Q, Yeung SC, Ip MS, Mak JC. Intermittent hypoxia-induced NF-kappaB and HO-1 regulation in human endothelial EA.hy926 cells. Cell Biochem Biophys. 2013;66:431–41. doi: 10.1007/s12013-012-9491-6. [DOI] [PubMed] [Google Scholar]
- 27.Iyer SN, Hyde DM, Giri SN. Anti-inflammatory effect of pirfenidone in the bleomycin-hamster model of lung inflammation. Inflammation. 2000;24:477–91. doi: 10.1023/a:1007068313370. [DOI] [PubMed] [Google Scholar]
- 28.Inomata M, Kamio K, Azuma A, Matsuda K, Kokuho N, Miura Y, et al. Pirfenidone inhibits fibrocyte accumulation in the lungs in bleomycin-induced murine pulmonary fibrosis. Respir Res. 2014;15:16. doi: 10.1186/1465-9921-15-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misra HP, Rabideau C. Pirfenidone inhibits NADPH-dependent microsomal lipid peroxidation and scavenges hydroxyl radicals. Mol Cell Biochem. 2000;204:119–26. doi: 10.1023/a:1007023532508. [DOI] [PubMed] [Google Scholar]
- 30.Giri SN, Leonard S, Shi X, Margolin SB, Vallyathan V. Effects of pirfenidone on the generation of reactive oxygen species in vitro. J Environ Pathol Toxicol Oncol. 1999;18:169–77. [PubMed] [Google Scholar]