

Abstract
Sustained proliferative potential of cancer cells creates heightened energetic and biosynthetic demands. The resulting overt dependence of cancer cells on unperturbed nutrient supply, has prompted a wide-spread interest in amino acid restriction strategies as potential cancer therapeutics. However, owing to rapid signaling and metabolic reprogramming in cancer cells, the prospects for success of amino acid restriction approaches remain unclear. We thus recognize that the identification of co-vulnerabilities of amino acid-restricted cancers may inform actionable targets for effective combined interventions. In this perspective, we outline the current state of key cellular mechanisms underlying adaptation to amino acid restriction and discuss the role of signal transduction pathways governing cancer cell resistance to amino acid restriction, with potential ramifications for the design of future therapeutic efforts.
Keywords: ATF4, c-MYC, NRF2, mTORC1, MAPK, amino acids, adaptation, cancer, resistance, metabolism
Amino acid restriction is a promising strategy for cancer treatment; however, adaptive signaling and metabolic reprogramming confer resistance to the approach. In this Perspective, Pathria and Ronai discuss how the identification and co-targeting of signaling pathways underlying the resistance promises to offer durable therapeutic responses to amino acid restriction.
Introduction
One of the hallmarks of tumors is the abundance of rapidly proliferating cells. This entails a constant and ample supply of nutrients to support anabolism that drives biosynthesis of macromolecules such as proteins, fatty acids, cholesterol and nucleotides, the cellular building blocks. Consistently, “starving cancer to death”, a rather facile concept, has not only prompted major research initiatives by the cancer research community, but has also captured the imagination of the general public. However, targeting glucose metabolism, a logical and potentially effective nutrient restriction strategy in cancer, results in extensive toxicities and has fallen short of translating into clinically viable therapeutics (Ganapathy-Kanniappan and Geschwind, 2013); in addition to providing biosynthetic carbons, glucose is also the chief fuel for cellular energy production, a crucial liability, which underlies the general toxicity associated with anti-glycolytic approaches (Granchi and Minutolo, 2012).
The recognition of less optimal glucose metabolism targeting approaches brought about a number of alternative amino acid restriction approaches (Kang, 2020). Unlike glucose, amino acids primarily serve a biosynthetic role –in protein, nucleotide, and fatty acid biosynthesis– in cells with sufficient nutrient access, with glycine, cysteine, and glutamate also being critical for the biosynthesis of the anti-oxidative molecule glutathione (GSH). Therefore, limiting amino acid metabolism may enable selective targeting of highly proliferative cancer cells. As our cells lack the enzymes required to synthesize a subset of amino acids, referred to as essential amino acids (EAA), restriction of EAA for therapeutic purposes may resemble the challenges associated with the inhibition of glucose metabolism. On the other hand, with the underlying premise that the de novo biosynthesis of non-essential amino acids (NEAA) alone is insufficient to meet the exorbitant needs of rapidly dividing cancer cells, approaches focusing on curtailing vascular NEAA availability have shown promise (Egler et al., 2016; Kang, 2020; Knott et al., 2018; Maddocks et al., 2017). However, like other anti-cancer approaches, amino acid restriction strategies are not immune to cellular resistance mechanisms (Lowman et al., 2019; Maddocks et al., 2013; Tajan et al., 2018; Tsai et al., 2009; Tsai et al., 2012). Responding to low intracellular amino acid levels, cells activate an elaborate transcriptional program, referred to as Amino Acid Response (AAR) signaling (Kilberg et al., 2009). AAR signaling in turn reprograms metabolic pathways to regain cellular amino acid homeostasis, thus presenting a major challenge towards an effective clinical translation of amino acid restriction approaches.
In this perspective, while highlighting the key mechanisms of cancer cell adaptation to amino acid restriction, we discuss co-targetable signaling vulnerabilities of amino acid-restricted cancer cells.
Amino Acid Restriction in Cancer
Restriction of glutamine, the most abundant amino acid in the circulation; serine , a precursor of one-carbon metabolism and glycine biosynthesis; arginine, an amino acid which is also a metabolic intermediate in the urea cycle ; cysteine, a key component of cellular anti-oxidative program; and asparagine (Combs and DeNicola, 2019; Covini et al., 2012; Kang, 2020; Knott et al., 2018; Maddocks et al., 2017; Riess et al., 2018), have all been evaluated as potential therapeutic approaches across various cancer types. However, dietary restriction of specific amino acids for therapeutic purposes is yet to be developed. Therefore, for the foreseeable future, chemical inhibition of amino acid biosynthetic enzymes and transporters, frequently exhibiting elevated expression in cancer and associated with poor prognosis, and use of amino acid hydrolyzing enzymes, will likely be the prevailing amino acid restriction approaches (Abou-Alfa et al., 2018; Cluntun et al., 2017; Kang, 2020; Lieu et al., 2020; Pieters et al., 2011; Scalise et al., 2018; Zhao et al., 2020). Argininosuccinate synthetase (ASS) and asparagine synthetase (ASNS), the key biosynthetic enzymes for arginine and asparagine biosynthesis respectively, exhibit either very low or no expression in multiple malignancies (Bachet et al., 2015; Dillon et al., 2004; Lorenzi et al., 2006), rendering these tumors auxotrophic for these amino acids. Accordingly, depleting vascular supplies of these amino acids offered selective targeting of cancer cell proliferation. This led to the clinical evaluation of arginine and asparagine restriction in solid tumors, utilizing the corresponding hydrolyzing enzymes arginine deiminase and L-asparaginase, respectively (Abou-Alfa et al., 2018; Hammel et al., 2019; Phillips et al., 2013). Notably, L-asparaginase, initially discovered as the active agent in guinea pig serum that conferred anti-lymphoma activity (Broome, 1963; Kidd, 1953), received clinical approval for acute lymphoblastic leukemia (Pieters et al., 2011). In contrast to a direct depletion of asparagine and arginine available to cancer cells through vascular depletion, limiting cellular glutamine stores has overwhelmingly relied on blocking its assimilation. This has been achieved through chemical inhibition of glutamine hydrolyzing enzyme glutaminase (GLS), a c-MYC target gene, which is frequently overexpressed in multiple cancer types and catalyzes a key step (glutamine to glutamate conversion) in cellular glutamine assimilation (Cluntun et al., 2017; Dang, 2013). Chemical inhibition of phoshoglycerate dehydrogenase (PHGDH), the enzyme catalyzing the conversion of the glycolytic intermediate 3-phosphoglycerate to 3-hydroxypyruvate, the first step in serine/glycine biosynthesis pathway, and associated with poor prognosis in cancer, has emerged as an approach for depleting cellular serine and glycine levels (Pacold et al., 2016; Zhao et al., 2020). However, as the inhibitor lacks specificity and efficacy, dietary restriction of serine and glycine remains the method of choice for an effective depletion of these amino acids (Maddocks et al., 2017). In addition to the inhibition of amino acid biosynthesis and less-developed dietary restriction approaches, chemical inhibition of amino acid transporters such as solute carrier family (SLC) 1 member A5 (SLC1A5) and SLC7A5, which facilitate the cellular uptake of glutamine and EAA respectively is being evaluated, albeit with limited success due to limited specificity of these inhibitors (Lieu et al., 2020; Napolitano et al., 2017; Schulte et al., 2018).
Cancer Cell Adaptation to Amino Acid Restriction: A Way of Life
As rapid tumor growth outpaces blood vessel formation (angiogenesis), spatiotemporal fluctuations in nutrient access are inherent to tumor evolution. Cancer cells are therefore adept at enduring poorly perfused environments (Tameire et al., 2019; Wang and Kaufman, 2014). Along these lines, the propensity of tumors to reprogram intrinsic metabolic pathways allows them to cope with nutrient shortage and avoid a lasting perturbation of tumor homeostasis that may be caused by therapeutic amino acid restriction. Given that most amino acid restriction approaches focus on NEAA, it is conceivable that cancer cells will attempt to compensate for limited amino acid availability by upregulating the respective de novo amino acid biosynthesis. Here we discuss key mediators of cancer cell adaptation to amino acid restriction.
ATF4
Decrease in cellular amino acid levels engages an adaptive response, known as AAR signaling, which is a component of the more expansive Integrated Stress Response (ISR) pathway. Activation of AAR signaling is set to maintain steady-state amino acid levels, by upregulating the expression of amino acid biosynthetic enzymes and amino acid transport proteins (Guan et al., 2017; Harding et al., 2003; Kilberg et al., 2009). With a decrease in cellular amino acid concentration, there is a concomitant decrease in the levels of tRNAs that are bound to amino acids. These uncharged tRNAs in turn activate general control non-derepressible 2 (GCN2) kinase, which initiates AAR signaling through phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) (Dever et al., 1992; Harding et al., 2000). eIF2α phosphorylation depresses the levels of the ternary complex (f-Met-tRNA-eIF2α-GTP), attenuating the translation initiation for cap (m7G)-containing mRNAs. Conversely, eIF2α phosphorylation promotes translation of transcripts such as ATF4, harboring unique upstream open reading frames (uORFs), otherwise not translated under unstressed conditions (unphosphorylated eIF2α) (Dever et al., 1992; Vattem and Wek, 2004) (Figure 1). ATF4 directly promotes transcription of a large number of genes that harbor unique promoter sequences, known as amino acid responsive elements (AARE; (Kilberg et al., 2009)). A vast majority of these ATF4 transcriptional targets encode proteins that are either critical for the resolution of stress or facilitation of programmed cell death, a determination influenced by a host of poorly defined factors (Wortel et al., 2017). It is notable that although ATF4 induction in response to amino acid restriction in some cancer cell lines in culture conditions has been shown to promote apoptosis, in vivo ATF4 ablation invariably leads to effective attenuation of tumor growth (Tameire et al., 2019; Ye et al., 2010). Among the genes whose expression is induced by ATF4 are the enzymes of serine, alanine, aspartate, glutamate and asparagine biosynthesis, and membrane transporters that facilitate amino acid uptake (Harding et al., 2003) (Figure 2). Therefore, restriction of NEAA, due to an associated upregulation of ATF4, inadvertently tips the amino acid biosynthesis and uptake machinery for reinforcement, precluding sufficient suppression of cellular amino acid levels to limit tumor growth (Gwinn et al., 2018; Pathria et al., 2019). Indeed, effective suppression of asparagine levels in tumors entails suppression of both vascular asparagine by L-asparaginase and de novo asparagine biosynthesis, a synthetic vulnerability of asparagine-restricted tumors, by genetic depletion of asparagine biosynthetic enzyme asparagine synthetase (ASNS) or concomitant suppression of AAR signaling by GCN2 inhibition (Knott et al., 2018; Nakamura et al., 2018).
Figure 1. Cellular Adaptive Machinery to Amino Acid Restriction.
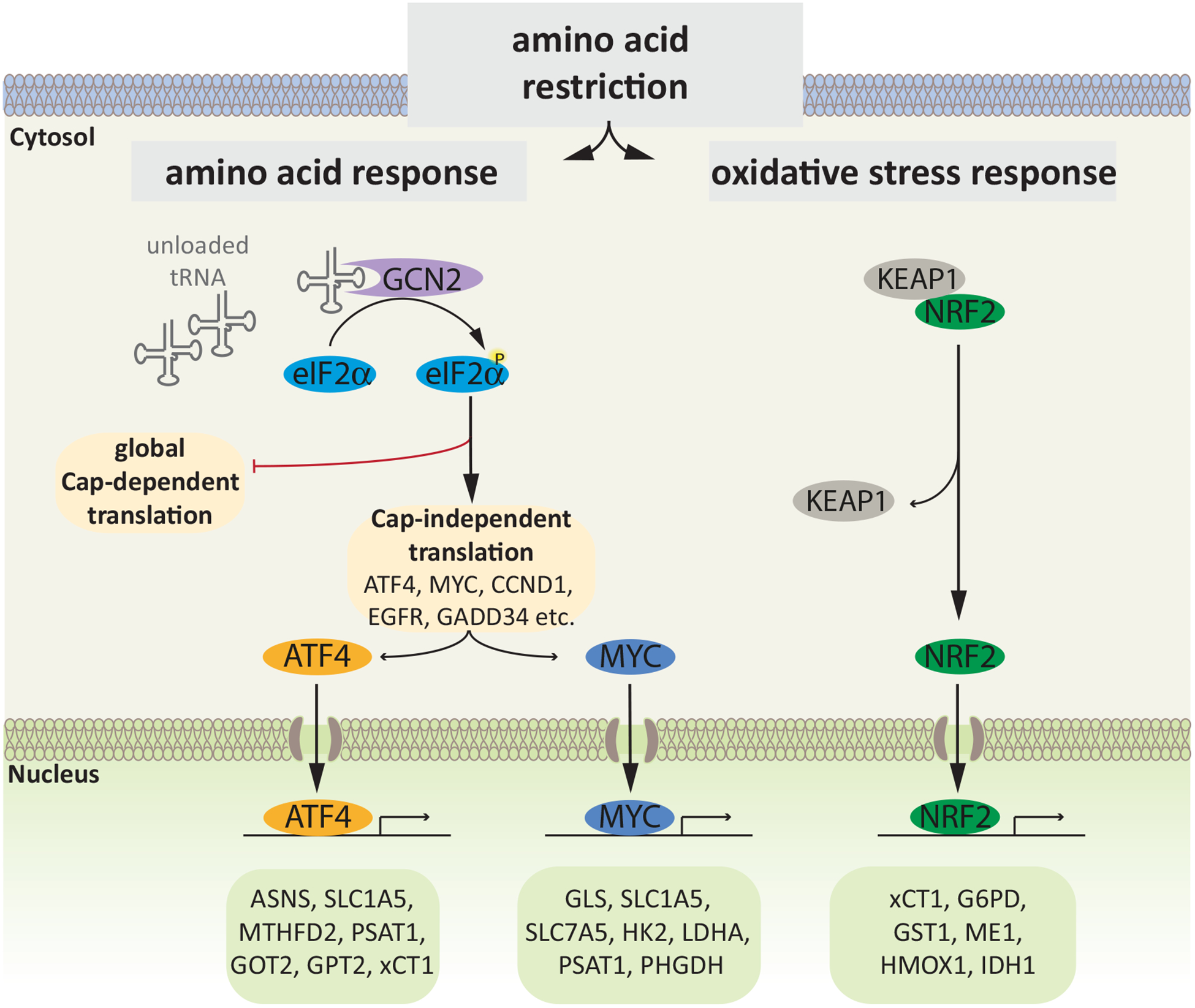
Decrease in intracellular amino acid levels leads to accumulation of unloaded tRNAs, which activate GCN2 kinase. GCN2 phosphorylates the translation initiation factor eIF2α, and the resulting suppression of the ternary complex formation impairs cap-dependent translation. Conversely, low levels of the ternary complex promote cap-independent translation of transcripts that harbor uORFs (ATF4, GADD34) or IRES (c-MYC, EGFR, CCND1). Additional stress pathways, e.g. the oxidative stress response, are activated independent of GCN2 in response to amino acid restriction., leading to activation of NRF2. Concerted activity of the transcription factors ATF4, MYC and NRF2 initiates a transcriptional program that orchestrates the cellular response to amino acid restriction. Representative candidates involved in amino acid biosynthesis/metabolism (PSAT1, PHGDH, GPT2, GOT2, GLS) and transport (SLC1A5, SLC7A5, xCT1), resolution of oxidative stress (HMOX1, xCT1, GST1, G6PD), glycolysis (ME1, HK2, LDHA), the TCA cycle (IDH1), and nucleotide biosynthesis (MTHFD2) are shown. ATF4, activating transcription factor 4; NRF2, nuclear factor E2-related factor 2; PSAT1, phosphoserine amino transferase 1; PHGDH, phosphoglycerate dehydrogenase; GPT2, glutamic-pyruvate transaminase 2; GOT2, glutamic-oxaloacetic transaminase 2; GLS, glutaminase; xCT1, cystine/glutamate transporter; HMOX1, heme oxygenase 1; GST1, glutathione S-transferase; G6PD, glucose 6-phosphate dehydrogenase; ME1, Malic Enzyme 1; HK2, hexokinase 2; LDHA, lactate dehydrogenase A; IDH1, isocitrate dehydrogenase 1; MTHFD2, methylene tetrahydrofolate dehydrogenase 2.
Figure 2. Regulation of Amino Acid Response Signaling by Growth Factor Signaling Pathways.
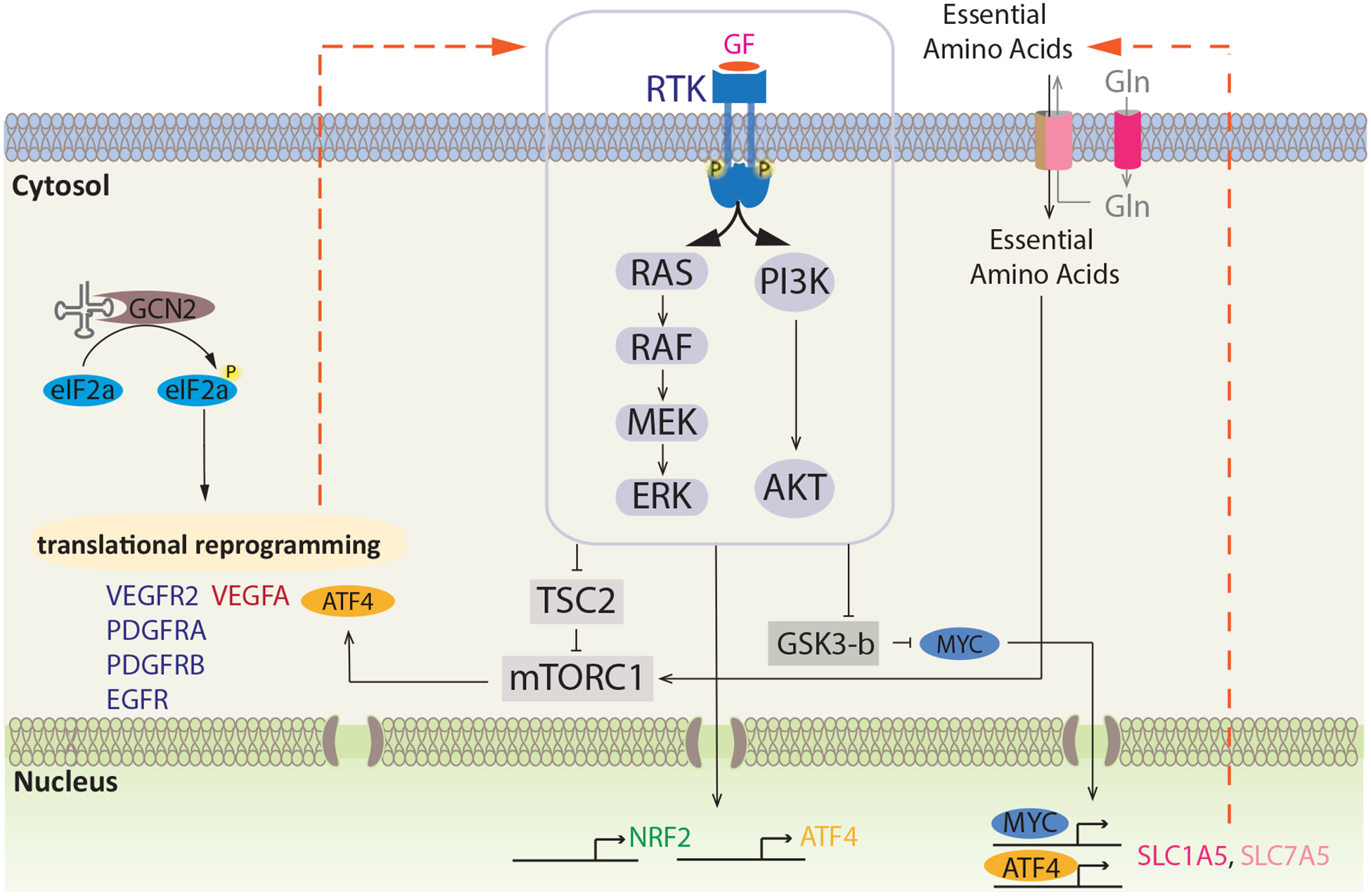
Both MAPK and PI3K-Akt signaling pathways employ multiple downstream mechanisms to support expression/activity of ATF4, NRF2, c-MYC, and mTORC1. Translational reprogramming elevates the expression of receptor tyrosine kinases (RTK) and growth factors (GF) leading to sustained RTK downstream signaling during stress. PI3K-Akt and MAPK signaling in turn results in transcriptional upregulation of stress-related transcription factors NRF2 and ATF4. ATF4 translation is further elevated by sustained mTORC1 signaling, mediated by the inhibitory effect of MAPK and PI3K-Akt signaling on TSC2. Additionally, MAPK and PI3K-Akt pathways elevate c-MYC via inhibition of GSK3-β. MYC works in concert with ATF4 to elevate the expression of amino acid transporters, which stimulate the uptake of essential amino acids (EAA) into the cell, ultimately supporting sustained mTORC1 signaling. Concerted activity of these mechanisms is essential for stress remediation and cancer cell survival, providing targetable nodes for therapeutic intervention.
c-MYC
c-MYC is another key regulator of cellular resistance to amino acid restriction, which is upregulated in cancer cells experiencing amino acid limitation (Cheng et al., 2017; Sun et al., 2015; Tsai et al., 2012). While transcriptional and post-translational control of c-MYC upregulation in amino acid-restricted cells has been reported, possible contribution of its translational induction –owing to c-MYC mRNA harboring an Internal Ribosome Entry Site (IRES)– under such stress conditions remains to be evaluated (Shi et al., 2005; Sun et al., 2015; Tsai et al., 2012). c-MYC-regulated mechanisms of resistance against amino acid restriction include the induction of (i) serine glycine one-carbon (SGOC) pathway enzymes (Sun et al., 2015), (ii) glutamine biosynthetic enzyme glutamine synthetase (GS) and glutamine transporter SLC1A5 (Bott et al., 2015; Chen et al., 2014), (iii) ASS, a rate-limiting enzyme in arginine biosynthesis (Tsai et al., 2009), and (iv) the mediation of epigenetically-regulated enhanced translational capacity in amino acid-restricted cells (Cheng et al., 2017). While not as widely studied in the context of amino acid restriction as ATF4, c-MYC has emerged as an important regulator of glucose and glutamine metabolism in cancer cells. c-MYC supports non-oxidative glucose metabolism through transcriptional upregulation of glucose transporter GLUT1, multiple glycolytic enzymes, and lactate-generating enzyme lactate dehydrogenase (LDH), responsible for the Warburg effect (Stine et al., 2015). Both glutamine cellular uptake and the first step in its cellular assimilation are catalyzed by c-MYC target genes GLS and SLC1A5 respectively (Sun et al., 2015). Some of the c-MYC target genes upregulated in response to amino acid restriction are depicted in Figure 1.
Notably, tumors initiated by c-MYC overexpression have been shown to induce the expression of ATF4 and crucially depend on it. This ATF4 engagement relies on enhanced protein synthesis, which is c-MYC-dependent and results in the suppression of free amino acid levels and charged tRNAs, culminating in a concomitant activation of GCN2 kinase (Tameire et al., 2019).
NRF2
Because amino acid restriction can activate unfolded protein response, a known trigger for oxidative stress , anti-oxidative mechanisms are also an inalienable component of cellular adaptive response to amino acid limitation (Chong et al., 2017; Gwinn et al., 2018; Wanders et al., 2016). Not surprisingly, therefore, Cap N’ Collar transcription factor nuclear factor erythroid 2-related factor 2 (NRF2)-regulated anti-oxidative gene expression program is induced not only in cells deprived of sulfur-containing (methionine, cysteine, and cystine) but also of other amino acids (Gwinn et al., 2018; Rojo de la Vega et al., 2018; Wanders et al., 2016). NRF2 strives to alleviate cellular oxidative stress through regulation of an expansive transcriptional program, including the expression of system X cystine transporter (xCT1) and cystathionine gamma lyase (CGL). Responsible for the uptake of cysteine precursor cystine and cysteine biosynthesis respectively, xCT1 and CGL are critical for maintaining cellular levels of cysteine, a precursor for the anti-oxidative molecule glutathione (Hassan et al., 2012; Koppula et al., 2018). Moreover, NRF2 is a key regulator of ATF4 transcription under both basal (DeNicola et al., 2015) and amino acid restriction states (Gwinn et al., 2018). This ability of NRF2 to induce ATF4 transcription points to its role in the control of AAR signaling. Inasmuch, NRF2 not only regulates cellular response to cysteine or methionine restriction but amino acid restriction at large. Figure 1 captures some of the key NRF2 target genes upregulated in response to amino acid restriction.
p53: An Emerging Regulator of Cancer Cell Adaptation to Amino Acid Restriction
One of the most extensively studied gene in cancer is the tumor suppressor p53 (TP53). Historically linked to the regulation of cell cycle and programmed cell death (apoptosis), the role of p53 in reprogramming metabolism in cancer cells (Simabuco et al., 2018) and cancer cell adaptation to amino acid restriction have been emerging. Among those, the role of p53 in the adaptation of colorectal carcinoma cells to serine/glycine deprivation was shown (Maddocks et al., 2013). In this work, p53 transcriptional target p21 (CDKN1A), along with suppressing cell cycle progression, was shown to promote the synthesis of GSH over nucleotides to support cancer cell survival in the absence of serine/glycine availability. As part of HCT116 cell adaptation to glutamine restriction, p53 was shown to be essential for the upregulation of aspartate transporter SLC1A3 (Tajan et al., 2018). p53 also contributes to cancer cell adaptation to glutamine restriction through induction of the arginine transporter SLC7A3. Arginine uptake supports mTORC1 activity and sustains cell viability in glutamine-restricted RAS-transformed mouse embryonic fibroblasts (Lowman et al., 2019). Furthermore, p53 transactivates ASS1, the urea cycle enzyme responsible for arginine biosynthesis (Miyamoto et al., 2017), suggesting a potential role for the upregulated ASS1 in cancer cell adaptation to arginine-depleting arginine deiminase. Notably, mouse double minute 2 (MDM2), a known negative regulator of p53, supports p53 independent and ATF3/4-dependent de novo serine metabolism in serine starved non-small cell lung cancer (NSCLC) cells (Riscal et al., 2016).
Growth Factor Signaling Mechanisms Regulating AAR
The mechanistic underpinnings of the translational induction of ATF4 in response to a multitude of stress stimuli, including amino acid restriction, have been extensively studied (Vattem and Wek, 2004). Curiously, the role of growth factor signaling pathways, which are frequently altered in cancer cells and closely tied to cancer pathophysiology, has gone largely unexplored with regards to AAR signaling. However, recent studies have started to shed light on the role of growth factor signaling in amino acid sensing (Gwinn et al., 2018; Pathria et al., 2019; Shin et al., 2015; Thiaville et al., 2008). Crucially, appreciating the challenges associated with targeting the master regulators (ATF4, c-MYC and NRF2) of cellular adaptation to stress stimuli, mapping their upstream signaling regulators could help identify actionable vulnerabilities of amino acid-deprived cancers. We discuss the emerging role of growth factor signaling in cancer cell adaptation to amino acid restriction, along with approaches to co-target these mechanisms.
Growth Factor Signaling Converges on NRF2 for ATF4 Regulation
Linking the growth factor and AAR signaling pathways, NRF2 expression induced by phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-Akt signaling pathway was shown to regulate ATF4 expression in mutant KRAS NSCLC cells (Gwinn et al., 2018). While attenuated asparagine biosynthesis underlies the loss of viability in glutamine-restricted cells, asparagine supplementation largely curtails apoptotic cell death in these cells (Pavlova et al., 2018; Zhang et al., 2014). NRF2-mediated induction of ATF4 promotes ASNS expression and increases cellular asparagine levels, offering respite to glutamine-deprived NSCLC cells (Gwinn et al., 2018; Zhang et al., 2014). Both mitogen-activated protein kinase kinase-extracellular signal-regulated kinase (MEK-ERK) (hereafter referred to as MAPK) and PI3K-Akt signaling pathways positively regulate NRF2 expression and nuclear activity (DeNicola et al., 2011; Koundouros and Poulogiannis, 2018; Zipper and Mulcahy, 2003), highlighting the role of growth factor receptor signaling in this regulatory pathway. Furthermore, ATF4 can dimerize with NRF2 to promote the expression of heme oxygenase 1 (HMOX-1), a key suppressor of oxidative stress (He et al., 2001). Since direct targeting of NRF2 remains a pharmacological challenge, elucidation of the upstream modulators of this key regulator of oxidative stress response and AAR signaling offers new opportunities for sensitizing cancer cells to amino acid restriction approaches (Figure 2). As a proof-of-concept, Akt inhibition, which suppresses NRF2 expression, together with asparagine restriction, effectively attenuates the growth of NSCLC tumors (Gwinn et al., 2018). Notably, a series of pharmacological inhibitors of PI3K-Akt signaling, a pathway frequently activated in various cancers and directly linked to disease pathophysiology, have been developed, albeit with limited clinical success (Yang et al., 2019). Hopefully, future efforts combining amino acid restriction with inhibitors targeting components along the PI3K-Akt signaling axis will prove effective and offer a way forward for eventual clinical successes.
MAPK Signaling Upregulates ATF4 via Translational Enhancement
The MAPK signaling pathway activity is enhanced and is critical for ATF4 induction in response to glucose (Shin et al., 2015) and histidine (Thiaville et al., 2008) restriction. Likewise, MAPK dependent induction of ATF4 was shown to be important for enhanced amino acid transport by L-type amino acid transporters (Franchi-Gazzola et al., 1999). Notably, several questions remain to be addressed for understanding the regulation of MAPK-ATF4 axis in cellular adaptive response to amino acid restriction. These include (i) how do suppressed amino acid levels induce MAPK signaling activity? and (ii) what is the mechanism underlying MAPK signaling-dependent upregulation of ATF4? Initial insight into these questions comes from our recent study, which identified the MAPK signaling pathway as a synthetic vulnerability of asparagine-restricted tumors (Pathria et al., 2019). Mechanistically, eIF2α phosphorylation in response to asparagine restriction induced translational reprogramming, enhancing the expression of receptor tyrosine kinases (RTKs). These include vascular endothelial growth factor receptor 2 (VEGFR2), platelet-derived growth factor receptor B (PDGFRB) and VEGFR2 ligand vascular endothelial growth factor A (VEGFA), which activated the MAPK signaling pathway. Activated MAPK signaling in turn proved critical for ATF4 translational upregulation through suppression of tuberous sclerosis 2 (TSC2) function and resulting mTORC1 activation (Ma et al., 2005; Pathria et al., 2019) (Figure 2). In demonstrating the importance of growth factor signaling for cancer cell adaptation to asparagine restriction, these findings formed the mechanistic basis for clinical evaluation of L-asparaginase with MAPK pathway inhibitors (Pathria et al., 2019). Moreover, as noted above, since ATF4 is also essential for p53 and MDM2-mediated resistance to amino acid restriction (Lowman et al., 2019; Riscal et al., 2016), inhibition of MAPK signaling with concomitant suppression of ATF4 may attenuate adaptation conferred by these emerging mechanisms. It is plausible that the regulation of ATF4 and the associated transcriptional changes are part of a greater adaptive program elicited by activated MAPK signaling – aspects discussed below.
c-MYC Regulation by Growth Factor Signaling as Part of AAR Signaling
The important role c-MYC plays in the regulation of AAR signaling argues for the possible targeting of c-MYC. However, because pharmacological inhibition of c-MYC remains an unmet need, it is critical to define alternative pathways for overcoming c-MYC function in amino acid-restricted cells. Although reports of c-MYC upregulation in amino acid-depleted cells date back ~30 years (Pohjanpelto and Holtta, 1990), the mechanistic basis for this upregulation emerged only in the past decade. Tsai and colleagues have demonstrated the role of MAPK and PI3K-Akt signaling in promoting c-MYC levels in arginine-restricted (arginine deiminase-treated) melanoma cells (Tsai et al., 2012). The authors demonstrated the requirement of both PI3K/Akt and MAPK signaling pathways in enhancing the inhibitory phosphorylation of glycogen synthase kinase 3-β (GSK3-β), alleviating c-MYC phosphorylation-dependent ubiquitination and degradation (Tsai et al., 2012), a regulatory axis that had previously been reported (Sears et al., 2000). Further, c-MYC transcriptionally induced ASS expression, promoting de novo arginine biosynthesis, a mechanism of resistance to arginine restriction. Consistently, combined arginine depletion and PI3K-Akt signaling inhibition achieved robust suppression of tumor growth (Tsai et al., 2012). Of note, enhanced c-MYC translation from a non-AUG initiation codon in methionine-deprived cells (Hann et al., 1992) should be considered when evaluating the potential translational value of methionine restriction approaches in cancer (Gao et al., 2019). With an established role of MAPK signaling as a regulator of c-MYC expression (Kerkhoff et al., 1998; Sears et al., 2000), the MAPK-c-MYC signaling emerges as an essential axis in amino acid sensing (Figure 2).
Concerted Growth Factor and AAR Signaling Sustain mTORC1 Activity - A Pre-Requisite to Stress Remediation
Ability of cells to successfully endure amino acid restriction depends on their ability to carry out new protein synthesis. This is crucial even when stress is severe and unresolvable, given that the facilitation of programmed cell death requires new protein synthesis (Sano and Reed, 2013). In this context, alternative mechanisms for translating gene transcripts appear essential to help cells resolve stress (Falletta et al., 2017; Kilberg et al., 2009). Along these lines, the indispensability of mTORC1 activity for the translational induction of ATF4, with the concomitant enhancement of ATF4-associated transcriptional program, in cells experiencing amino acid limitation has been shown (Chen et al., 2014; Nofal et al., 2017; Pathria et al., 2019) (Figure 2).
Critical for the regulation of mTORC1 activity are growth factor signaling pathways, including the MAPK and PI3K-Akt signaling axes (Dibble and Cantley, 2015; Inoki et al., 2002; Ma et al., 2005) (Figure 2). However, sole input from growth factor signaling pathways is insufficient to activate mTORC1, which requires additional activating signals from amino acids (Bar-Peled and Sabatini, 2014; Casas-Terradellas et al., 2008). Restriction of all 20 amino acids or all EAA, expectedly, compromises mTORC1 activity, and chemical inhibition of mTORC1 activity was shown to increase lysosomal catabolism, conferring survival benefit in leucine-restricted, albumin-supplemented KRASG12D-expressing mouse embryonic fibroblasts (Palm et al., 2015). A combination of increased protein scavenging and protein synthesis suppression was proposed, using the same experimental model, as mechanisms underlying survival benefit conferred by mTORC1 inhibition in amino acid-restricted cells (Nofal et al., 2017). Notably, studies in cancer cells have reported either maintenance or in some cases induction of mTORC1 activity in response to the restriction of select amino acids (Chen et al., 2014; Lowman et al., 2019; Pathria et al., 2019; Pathria et al., 2018). Moreover, no impairment of mTORC1 activity was seen in breast cancer xenograft of mice fed with leucine-free diet (Singh et al., 2011). Among mTORC1 regulators is Sestrin2 (SESN2), an ATF4 transcriptional target shown to sense low leucine levels and suppress mTORC1 activity (Kimball et al., 2016; Ye et al., 2015). However, since SESN2 is also regulated by the frequently mutated tumor suppressor gene, TP53 (Budanov et al., 2002; Velasco-Miguel et al., 1999), the possible changes in mTORC1 function in cancer due to deregulation of the ATF4-SESN2 and/or leucine-SESN2 axes remains to be established. A growing body of literature addresses the mechanistic underpinnings of seemingly paradoxical maintenance, and in some instances induction, of mTORC1 activity in response to select amino acid restriction. Combined ATF4 and c-MYC induction upregulates the expression of L-type amino acid transporters SLC7A5 and SLC1A5. These in concert with SLC3A2, facilitating the membrane localization and transporter channel activity of SLC1A5 and SLC7A5 (Nicklin et al., 2009), increase the cellular uptake of amino acids, including leucine and arginine, the bona-fide activators of mTORC1 (Bar-Peled and Sabatini, 2014; Chen et al., 2014; Nicklin et al., 2009; Pathria et al., 2018) (Figure 2). Also among the genes upregulated by ATF4 are the mediators of autophagy (B’Chir et al., 2013), which regulates cellular homeostasis and confers protection against starvation, while autophagy-deficient cells experience reduction in intracellular amino acid pools (Onodera and Ohsumi, 2005; Thomas et al., 2018). Growth factor signaling pathways (Inoki et al., 2002; Ma et al., 2005), cellular energy state (via AMPK-mediated TSC2 regulation (Inoki et al., 2003)), and intracellular EAA levels (Bar-Peled and Sabatini, 2014), are among known modulators of mTORC1 activity, a key negative regulator of autophagy (Dossou and Basu, 2019). Therefore, understanding how the interplay between autophagy-regulatory mechanisms in amino acid-restricted cells impacts mTORC1 function needs to be further explored. Along with autophagy, macropinocytosis, a process of internalization of extracellular proteins that are degraded in a lysosome-dependent fashion (Commisso et al., 2013; Davidson et al., 2017), has emerged as another key mechanism contributing to the maintenance of intracellular amino acid pools in amino acid-restricted cells. Coupled with suppressed mTORC1 activity prompting enhanced lysosomal degradation in amino acid-depleted cells, EGFR-Pak signaling has been show to enhance macropinocytosis in response to glutamine depletion (Lee et al., 2019; Nofal et al., 2017; Palm et al., 2015).
In aggregate, these studies present a myriad of mechanisms utilized by amino acid-restricted cells to retain growth factor signaling activity and amino acid cues, both of which are critical to maintain mTORC1 activity. A number of mTOR inhibitors, including rapalogs and ATP competitive inhibitors are being evaluated in pre-clinical studies and clinical trials for multiple indications (Xie et al., 2016) and may present opportunities for combination with amino acid restriction approaches. While in this perspective we primarily focus on cancer cell-specific mechanisms of adaptation to experimental amino acid restriction, it should be noted that during their normal evolutionary course tumors constantly experience nutrient-limiting environments, entailing intimate crosstalk between cancer cells and the cells of tumor microenvironment. Some notable examples are pancreatic stellate cells supplying alanine as a carbon source to poorly vascularized pancreatic ductal adenocarcinoma (PDAC) cells (Sousa et al., 2016); cancer-associated fibroblasts synthesizing and supplying glutamine as an energy source and biosynthetic precursor to ovarian carcinoma cells (Yang et al., 2016); and p62-deficient prostate cancer stromal fibroblasts upregulating ATF4 levels with consequent expression of pyruvate carboxylase and ASNS and biosynthesis of asparagine as a source of nitrogen both for stromal and tumor epithelial cell proliferation (Linares et al., 2017). Moving forward, it will be important to evaluate whether experimentally defined signaling co-vulnerabilities of amino acid-restricted tumors are potential drug targets in poorly perfused tumors such as PDAC. Moreover, future efforts should also be directed towards understanding how cells in the tumor microenvironment, are impacted by systemic amino acid restriction, and affect the adaptation/resistance of amino acid-restricted tumor cells.
MAPK-Interacting Kinase 1 (MNK1) Controls ATF4 Translation
MAPK signaling inhibition has been extensively used as part of therapeutic regimens for BRAF mutant melanomas. However, common to these and other targeted therapies are acquired and intrinsic resistance (Kakadia et al., 2018). Given these limitations, it is important to explore additional vulnerabilities of amino acid-restricted tumors. Our recent studies identified MAP kinase-interacting serine/threonine-protein kinase 1 (MKNK1/MNK1), a kinase substrate of ERK and p38 MAPK (Bramham et al., 2016), as a synthetic lethal partner of ASNS (Pathria et al., 2019). Analysis of TCGA data across multiple cancer types showed that concomitant downregulation of MNK1 and ASNS correlates with better patient survival. Mechanistically, MNK1 was found to be required for ATF4 induction/AAR signaling in asparagine-restricted cancer cells (Figure 3). Consistent with its essentiality for ATF4-mediated adaptation to asparagine restriction, combined asparagine restriction and MNK1 ablation effectively suppressed cancer cell proliferation (Pathria et al., 2019). Notably, in contrast to the inhibition of PI3K-Akt or MAPK signaling pathways, MNK1 targeting on its own didn’t impair cancer cell proliferation, pointing to a context-dependent (asparagine restriction-associated) role for MNK1. While MNK1-dependent ATF4 induction in asparagine-restricted cells depends on enhanced ATF4 mRNA translation, the underlying mechanism of this regulation remains to be explored. The availability of MNK inhibitors, such as eFT-508 (Webster et al., 2015), provides the opportunity to assess their in vivo efficacy in asparagine-deprived tumors, and in combination with the restriction of other amino acids.
Figure 3. MNK1 is Required for ATF4 Translation in Asparagine-Restricted Cancer Cells.
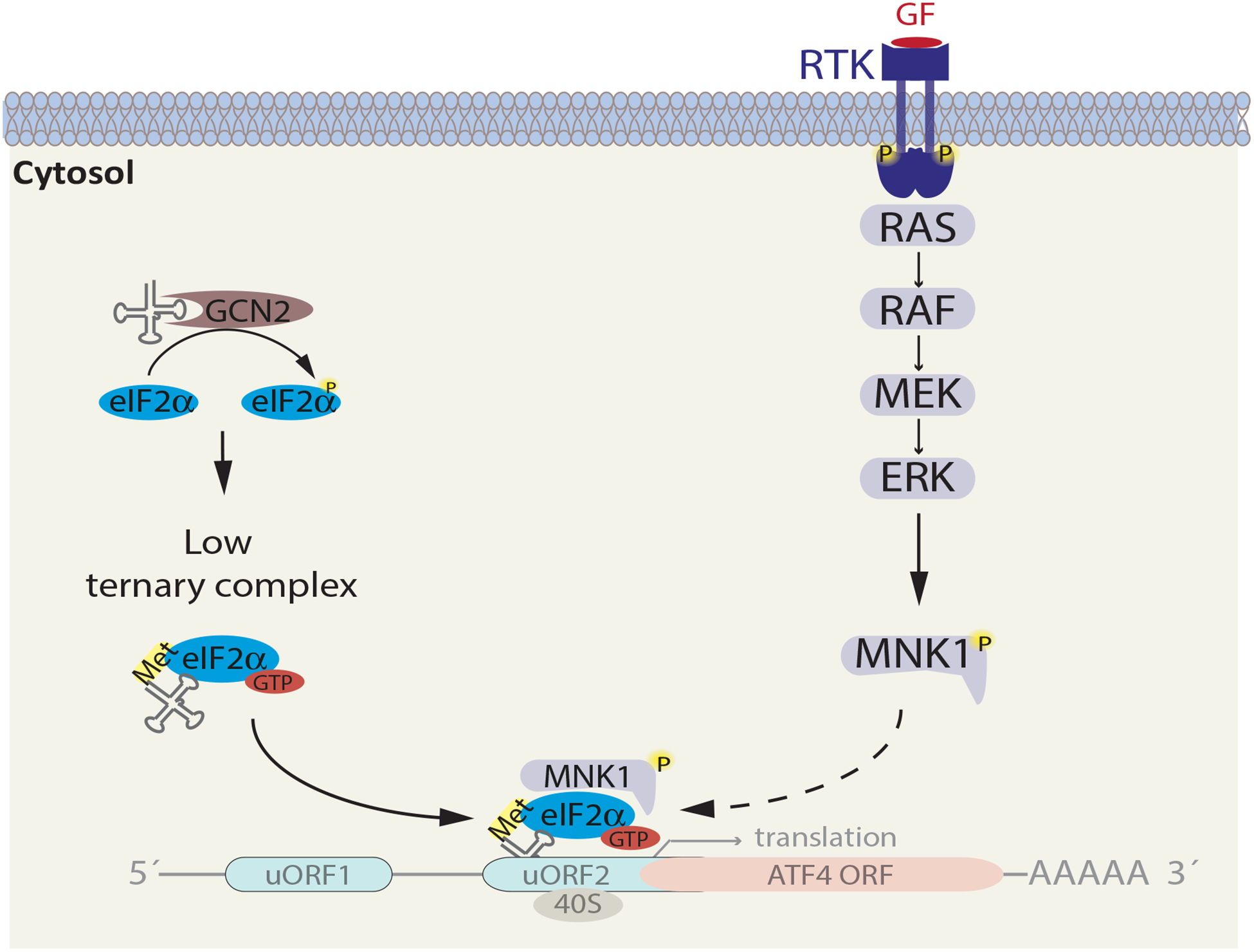
Asparagine restriction activates GCN2 and MAPK-MNK1 signaling. As a result of eIF2α phosphorylation, cellular levels of the ternary complex (eIF2α -GTP-methionine tRNA) are low, leading to selective translation of ATF4 mRNA. In asparagine-restricted cancer cells, MNK1 ablation suppresses ATF4 translation, indicating a crucial role of MNK1 in this process. Dotted arrow represents a potential yet unproven mechanism, whereby binding of MNK1 to the eIF2α complex is required for ATF4 translation.
Engagement of Growth Factor Signaling Following Amino Acid Restriction
It is noteworthy that not only are the MAPK and PI3K-Akt signaling pathways critical for ATF4 induction, in amino acid-restricted cells, there is a notable increase in the activity of these growth factor signaling cascades (Gwinn et al., 2018; Pathria et al., 2019; Thiaville et al., 2008). Initial clue for the mechanism underlying growth factor signaling activation in response to amino acid restriction came from the observation that GCN2 kinase is required for promoting the MAPK signaling activity in histidine-deprived hepatoma cells (Thiaville et al., 2008), although the underlying mechanism remained unclear. In studying asparagine restriction, our work identified enhanced translation of multiple RTKs (VEGFR2, PDGFRA, PDGFRB, and EGFR), as part of a feed-forward mechanism which enhances MAPK signaling (Pathria et al., 2019). While pointing to the role of VEGFR2 and PDGFRB as the upstream modulators of MAPK signaling in asparagine-restricted melanoma and pancreatic cancer cells, different set of RTKs may be involved in other cancer cell types or upon restriction of other amino acids. Mechanistically, a select set of transcripts that harbor specific uORF elements, such as the ones present in VEGFR2 and PDGFRA mRNA, could be differentially translated in response to the activation of ISR (Guan et al., 2017). However, since only a small fraction of the ~45% of transcripts whose 5’untranslated regions carry at least one uORF are selectively translated during ISR (Calvo et al., 2009; Sidrauski et al., 2015), additional, hitherto unknown factors likely determine which mRNAs should undergo enhanced translation under distinct stress conditions.
Notably, the induction of NRF2 in amino acid-restricted cancer cells is consistent with the activation of oxidative stress response (Jonsson et al., 2019). Oxidative stress/reactive oxygen species (ROS) suppresses the expression of dual specificity phosphatase (DUSPs), a family of serine/threonine and tyrosine phosphatases that dephosphorylate and inactivate the MAP kinase ERK (Cheung et al., 2020; Son et al., 2011). Future investigations will define whether limitation of distinct amino acids induces DUSPs and consequently cellular signaling pathways.
The role of ROS in metabolic signaling brings to light the possible importance of NRF2, a key regulator in the cellular response to oxidative stress. NRF2 has been implicated in the activation of PI3K-Akt signaling, via transcriptional upregulation of multiple RTK ligands, transforming growth factor A, amphiregulin, platelet-derived growth factor C, and NAD(P)H quinone dehydrogenase 1 (He et al., 2020). Notably, a recent report has shown activation of Wnt signaling components, albeit downstream of the membrane receptor, in glutamine-restricted colorectal cancer cells (Tran et al., 2020). This work adds another layer of complexity to the regulation of growth factor signaling in response to amino acid restriction, showing how suppression of α-ketoglutarate levels in response to low glutamine results in hypomethylation of DNA and histone H3K4me3, leading to a downregulation of Wnt target genes (Tran et al., 2020). Given altered histone methylation, one could expect wide-spread pleitropic changes that may encompass response to amino acid restriction.
Among other signaling mechanisms, Rac signaling pathway was induced upon deprivation of distinct EAA in mouse embryonic fibroblasts, with concomitant activation of jun N-terminal kinase 2 (JNK2) and its substrate transcription factor ATF2 (Chaveroux et al., 2009). ATF2 transcriptionally enhances the expression of ATF3, a key regulator of cellular response to amino acid restriction (Pan et al., 2007), pointing to a possible link between stress-activated kinases and AAR signaling. Among the stress activated kinases is p38 MAPK, another MNK1 kinase, which is activated following amino acid restriction (Li et al., 2017). Future studies will help establish the importance of p38 MAPK signaling in amino acid-restricted cancer cells.
Predicting Targetable Co-Vulnerabilities of Amino Acid-Restricted Cancers using Identification of Clinically Relevant Synthetic LEthality (ISLE)
Loss-of-function chemical and genetic screens along with genome-wide computational approaches predict synthetic lethal interactions from both cancer cell lines and patient samples. These tools are expected to guide the identification of potential synthetic vulnerabilities of amino acid-restricted cancer cells (Thompson et al., 2015). Often coupled with exhaustive functional and mechanistic studies, these approaches allow one to ascertain physiologically relevant synthetic lethal relationships. ISLE, a recently developed data-driven approach (Lee et al., 2018), which defined focal adhesion kinase as a synthetic lethal partner of GNAQ activation in uveal melanoma (Feng et al., 2019), can also serve as a tool for –in silico– prediction of synthetic lethal partners of tumors that may experience amino acid paucity. This computational approach effectively integrates patient-derived gene expression datasets, the corresponding clinical information, and the results of published loss-of-function genetic and chemical screens to predict putative synthetic lethal (SL) partner pairs of a gene of interest. As a proof-of-concept, ISLE predicted MAPK signaling as a synthetic lethal partner of asparagine-restricted cancers, a relationship confirmed in in vitro and in vivo studies (Pathria et al., 2019). Therefore, ISLE could be successfully put to practice to identify actionable vulnerabilities of tumors deprived of non-essential amino acids. Amino acid biosynthetic enzymes that may serve as proxies for employing ISLE to predict synthetic vulnerabilities of tumors deprived of other amino acids include- PHGDH, phosphoserine amino transferase 1 (PSAT1), or phosphoserine phosphatase for serine and glycine; GS for glutamine; glutamic-oxaloacetic transaminase 2 (GOT2) for aspartate; glutamic-pyruvate transaminase 2 (GPT2) for alanine; CGL for cysteine; GLS for glutamate; and phenylalanine hydroxylase for phenylalanine. Due to a lack of the corresponding cellular biosynthetic enzymes, employing ISLE for EAA is a challenge.
Nutrient Sensing- Not Just About Survival
The MAPK and PI3K-Akt signaling pathways, c-MYC, ATF4, and NRF2, are all well-documented pro-tumorigenic factors (Braicu et al., 2019; Dang, 2013; Janku et al., 2018; Rojo de la Vega et al., 2018; Wortel et al., 2017). Therefore, the activation of these pathways and molecules in cancer cells experiencing amino acid restriction may bestow upon them capabilities that render them more aggressive– “What doesn’t kill you makes you stronger”. For example, MAPK signaling invariably activated in response to amino acid restriction regulates every aspect of cancer cell biology, including enhancement of migratory, invasive and metastatic abilities (Braicu et al., 2019). Furthermore, translational reprogramming was shown to play a direct role in promoting the metastatic potential of glutamine-deprived melanoma cells (Falletta et al., 2017). Glutamine restriction in pancreatic ductal adenocarcinoma cells (PDAC) was also shown to increase macropinocytosis through EGFR-PAK activation (Lee et al., 2019). Moreover, p38 MAPK signaling, a stress-activated pathway, was shown to promote the expression of cyclooxygenase 2 (COX-2) in glioblastoma multiforme (GBM) cells deprived of a combination of glutamine, arginine, and lysine or methionine and cysteine (Li et al., 2017). COX-2 and its major product in brain, prostaglandin E2 have been proposed to facilitate the initiation and progression of GBM (Qiu et al., 2017).
With an expansive target gene profile, the fallout from rampant ATF4 and c-MYC induction in amino acid-restricted cells isn’t only limited to reestablishing the amino acid homeostasis. For one, the gene expression programs elicited by these master transcription regulators are non-discriminatory (independent of the actual trigger for AAR signaling activation), enhancing the biosynthesis of all NEAA and the uptake of all amino acids (Kilberg et al., 2009). Moreover, combined ATF4 and c-MYC-mediated transcriptional programs go well beyond just impacting amino acid metabolism, enhancing expression of genes involved in nucleotide biosynthesis (Ben-Sahra et al., 2016; Liu et al., 2008); autophagy (B’Chir et al., 2013; Chen et al., 2014); fatty acid and cholesterol metabolism (Chen et al., 2016; Dang, 2013); glycolysis (Lee et al., 2015; Stine et al., 2015); and angiogenesis, via induction of vascular endothelial growth factor A (VEGFA) (Baudino et al., 2002; Longchamp et al., 2018; Pereira et al., 2014).
As tumors are in a state of constant stress (hypoxia and limited nutrient supply), it is conceivable that the plethora of mechanisms discussed above –in the context of forced amino acid restriction– are at play even under steady-state conditions. Hypoxia-mediated induction of hypoxia inducible factor-1α and a consequent upregulation of VEGFA provides some support to such a hypothesis (Semenza, 2001), which, future investigations will likely be testing.
A vast majority of cancer cells comprising the tumor mass, owing to their high proliferative capacity, are expected to be adversely impacted by amino acid restriction. However, low frequency, metabolically less active dormant/quiescent cancer cells, which are reversibly arrested in the G0/G1 phase of cell cycle (Gomis and Gawrzak, 2017; Recasens and Munoz, 2019), may be able to successfully bypass amino acid restriction challenge and even be further selected. Therefore, moving forward, it will be quintessential to determine the impact of amino acid restriction on this important cancer cell subset, and develop combination strategies that may also target them.
Concluding Remarks
Amino acid restriction strategies are logical therapeutic interventions for nutrient addicted cancer cells. However, there is an increasing appreciation of the challenges associated with their further development as effective cancer treatments. Therefore, in addition to devising more efficacious and clinically translatable approaches for amino acid restriction, there is an urgent need to further delineate the mechanisms that underlie cancer cell adaptation to amino acid restriction. Along this process, hopefully, druggable adaptive signal transduction mechanisms will be defined and concurrently targeted for further clinical development. In recognizing the importance of tumor dormancy and slow-cycling cancer stem-like cells, which exhibit distinct and rather low metabolic needs (Hoang-Minh et al., 2018), targeting metabolic cascades will likely be combined with the inhibition of select signaling pathways that are unique to these small yet important tumor sub-populations. Lastly, computer-aided predictions for synthetic lethal interaction between metabolic and signal transduction pathways are expected to define effective treatment modalities which are rather personalized, based on individual’s tumor makeup.
Acknowledgements
We thank Daniela Senft for comments and figure preparation, Support by NCI grants R35CA197465, R01CA202021 and DOD grant CA1810216 to Z. A. Ronai is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abou-Alfa GK, Qin S, Ryoo BY, Lu SN, Yen CJ, Feng YH, Lim HY, Izzo F, Colombo M, Sarker D, et al. (2018). Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann Oncol 29, 1402–1408. [DOI] [PubMed] [Google Scholar]
- B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, and Bruhat A (2013). The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41, 7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachet JB, Gay F, Marechal R, Galais MP, Adenis A, Ms CD, Cros J, Demetter P, Svrcek M, Bardier-Dupas A, et al. (2015). Asparagine Synthetase Expression and Phase I Study With L-Asparaginase Encapsulated in Red Blood Cells in Patients With Pancreatic Adenocarcinoma. Pancreas 44, 1141–1147. [DOI] [PubMed] [Google Scholar]
- Bar-Peled L, and Sabatini DM (2014). Regulation of mTORC1 by amino acids. Trends Cell Biol 24, 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, and Cleveland JL (2002). c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 16, 2530–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, and Manning BD (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, Neidler S, Sun Y, Jaber N, Krokowski D, et al. (2015). Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab 22, 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L, Rusu A, Irimie A, Atanasov AG, Slaby O, et al. (2019). A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Jensen KB, and Proud CG (2016). Tuning Specific Translation in Cancer Metastasis and Synaptic Memory: Control at the MNK-eIF4E Axis. Trends Biochem Sci 41, 847–858. [DOI] [PubMed] [Google Scholar]
- Broome JD (1963). Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. II. Lymphoma 6C3HED cells cultured in a medium devoid of L-asparagine lose their susceptibility to the effects of guinea pig serum in vivo. J Exp Med 118, 121–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et al. (2002). Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 21, 6017–6031. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Pagliarini DJ, and Mootha VK (2009). Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci U S A 106, 7507–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas-Terradellas E, Tato I, Bartrons R, Ventura F, and Rosa JL (2008). ERK and p38 pathways regulate amino acid signalling. Biochim Biophys Acta 1783, 2241–2254. [DOI] [PubMed] [Google Scholar]
- Chaveroux C, Jousse C, Cherasse Y, Maurin AC, Parry L, Carraro V, Derijard B, Bruhat A, and Fafournoux P (2009). Identification of a novel amino acid response pathway triggering ATF2 phosphorylation in mammals. Mol Cell Biol 29, 6515–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Yuan R, Zhang Y, Zhang X, Chen L, Zhou X, Yuan Z, Nie Y, Li M, Mo D, et al. (2016). ATF4 regulates SREBP1c expression to control fatty acids synthesis in 3T3-L1 adipocytes differentiation. Biochim Biophys Acta 1859, 1459–1469. [DOI] [PubMed] [Google Scholar]
- Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, Liu X, Zhu C, Yang M, Ye W, et al. (2014). The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol 206, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Su T, Morselli M, and Kurdistani SK (2017). Histone demethylation and c-MYC activation enhance translational capacity in response to amino acid restriction. bioRxiv, 154419. [Google Scholar]
- Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, and Vousden KH (2020). Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 37, 168–182 e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong WC, Shastri MD, and Eri R (2017). Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluntun AA, Lukey MJ, Cerione RA, and Locasale JW (2017). Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 3, 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs JA, and DeNicola GM (2019). The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covini D, Tardito S, Bussolati O, Chiarelli LR, Pasquetto MV, Digilio R, Valentini G, and Scotti C (2012). Expanding targets for a metabolic therapy of cancer: L-asparaginase. Recent Pat Anticancer Drug Discov 7, 4–13. [DOI] [PubMed] [Google Scholar]
- Dang CV (2013). MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR, et al. (2017). Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med 23, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. (2015). NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, and Hinnebusch AG (1992). Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68, 585–596. [DOI] [PubMed] [Google Scholar]
- Dibble CC, and Cantley LC (2015). Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 25, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, and Clark MA (2004). Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer 100, 826–833. [DOI] [PubMed] [Google Scholar]
- Dossou AS, and Basu A (2019). The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egler RA, Ahuja SP, and Matloub Y (2016). L-asparaginase in the treatment of patients with acute lymphoblastic leukemia. J Pharmacol Pharmacother 7, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falletta P, Sanchez-Del-Campo L, Chauhan J, Effern M, Kenyon A, Kershaw CJ, Siddaway R, Lisle R, Freter R, Daniels MJ, et al. (2017). Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev 31, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Arang N, Rigiracciolo DC, Lee JS, Yeerna H, Wang Z, Lubrano S, Kishore A, Pachter JA, Konig GM, et al. (2019). A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 35, 457–472 e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi-Gazzola R, Visigalli R, Bussolati O, Dall’Asta V, and Gazzola GC (1999). Adaptive increase of amino acid transport system A requires ERK1/2 activation. J Biol Chem 274, 28922–28928. [DOI] [PubMed] [Google Scholar]
- Ganapathy-Kanniappan S, and Geschwind JF (2013). Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 12, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Sanderson SM, Dai Z, Reid MA, Cooper DE, Lu M, Richie JP Jr., Ciccarella A, Calcagnotto A, Mikhael PG, et al. (2019). Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis RR, and Gawrzak S (2017). Tumor cell dormancy. Mol Oncol 11, 62–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granchi C, and Minutolo F (2012). Anticancer agents that counteract tumor glycolysis. ChemMedChem 7, 1318–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan BJ, van Hoef V, Jobava R, Elroy-Stein O, Valasek LS, Cargnello M, Gao XH, Krokowski D, Merrick WC, Kimball SR, et al. (2017). A Unique ISR Program Determines Cellular Responses to Chronic Stress. Mol Cell 68, 885–900 e886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D, and Sweet-Cordero EA (2018). Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 33, 91–107 e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel P, Berardi R, Cutsem EV, Feliu J, Greil R, Wasan HS, Metges J-P, Nygren P, Osterlund PJ, Parner V, et al. (2019). Trybeca-1: A randomized, phase 3 study of eryaspase in combination with chemotherapy versus chemotherapy alone as second-line treatment in patients with pancreatic adenocarcinoma (NCT03665441). Journal of Clinical Oncology 37, TPS471–TPS471. [Google Scholar]
- Hann SR, Sloan-Brown K, and Spotts GD (1992). Translational activation of the non-AUG-initiated c-myc 1 protein at high cell densities due to methionine deprivation. Genes Dev 6, 1229–1240. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, and Ron D (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Hassan MI, Boosen M, Schaefer L, Kozlowska J, Eisel F, von Knethen A, Beck M, Hemeida RA, El-Moselhy MA, Hamada FM, et al. (2012). Platelet-derived growth factor-BB induces cystathionine gamma-lyase expression in rat mesangial cells via a redox-dependent mechanism. Br J Pharmacol 166, 2231–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, and Alam J (2001). Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem 276, 20858–20865. [DOI] [PubMed] [Google Scholar]
- He F, Antonucci L, Yamachika S, Zhang Z, Taniguchi K, Umemura A, Hatzivassiliou G, Roose-Girma M, Reina-Campos M, Duran A, et al. (2020). NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J Hepatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang-Minh LB, Siebzehnrubl FA, Yang C, Suzuki-Hatano S, Dajac K, Loche T, Andrews N, Schmoll Massari M, Patel J, Amin K, et al. (2018). Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, and Guan KL (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4, 648–657. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, and Guan KL (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Janku F, Yap TA, and Meric-Bernstam F (2018). Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol 15, 273–291. [DOI] [PubMed] [Google Scholar]
- Jonsson WO, Margolies NS, and Anthony TG (2019). Dietary Sulfur Amino Acid Restriction and the Integrated Stress Response: Mechanistic Insights. Nutrients 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B, Mina L, Dragovich T, Gimbel M, and Mahmoud F (2018). Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther 11, 7095–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS (2020). Dietary restriction of amino acids for Cancer therapy. Nutr Metab (Lond) 17, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkhoff E, Houben R, Loffler S, Troppmair J, Lee JE, and Rapp UR (1998). Regulation of c-myc expression by Ras/Raf signalling. Oncogene 16, 211–216. [DOI] [PubMed] [Google Scholar]
- Kidd JG (1953). Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. II. Studies on the nature of the active serum constituent: histological mechanism of the regression: tests for effects of guinea pig serum on lymphoma cells in vitro: discussion. J Exp Med 98, 583–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilberg MS, Shan J, and Su N (2009). ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab 20, 436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball SR, Gordon BS, Moyer JE, Dennis MD, and Jefferson LS (2016). Leucine induced dephosphorylation of Sestrin2 promotes mTORC1 activation. Cell Signal 28, 896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, Turgeon MO, Fish L, Erard N, Gable AL, et al. (2018). Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppula P, Zhang Y, Zhuang L, and Gan B (2018). Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond) 38, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koundouros N, and Poulogiannis G (2018). Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front Oncol 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Oney M, Frizzell K, Phadnis N, and Hollien J (2015). Drosophila melanogaster activating transcription factor 4 regulates glycolysis during endoplasmic reticulum stress. G3 (Bethesda) 5, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Das A, Jerby-Arnon L, Arafeh R, Auslander N, Davidson M, McGarry L, James D, Amzallag A, Park SG, et al. (2018). Harnessing synthetic lethality to predict the response to cancer treatment. Nat Commun 9, 2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Zhang Y, Jung M, Cruz N, Alas B, and Commisso C (2019). EGFR-Pak Signaling Selectively Regulates Glutamine Deprivation-Induced Macropinocytosis. Dev Cell 50, 381–392 e385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chang CM, Wang L, Zhang P, and Shu HG (2017). Cyclooxygenase-2 Induction by Amino Acid Deprivation Requires p38 Mitogen-Activated Protein Kinase in Human Glioma Cells. Cancer Invest 35, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu EL, Nguyen T, Rhyne S, and Kim J (2020). Amino acids in cancer. Exp Mol Med 52, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares JF, Cordes T, Duran A, Reina-Campos M, Valencia T, Ahn CS, Castilla EA, Moscat J, Metallo CM, and Diaz-Meco MT (2017). ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab 26, 817–829 e816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI, and Dang CV (2008). Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 3, e2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longchamp A, Mirabella T, Arduini A, MacArthur MR, Das A, Trevino-Villarreal JH, Hine C, Ben-Sahra I, Knudsen NH, Brace LE, et al. (2018). Amino Acid Restriction Triggers Angiogenesis via GCN2/ATF4 Regulation of VEGF and H2S Production. Cell 173, 117–129 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi PL, Reinhold WC, Rudelius M, Gunsior M, Shankavaram U, Bussey KJ, Scherf U, Eichler GS, Martin SE, Chin K, et al. (2006). Asparagine synthetase as a causal, predictive biomarker for L-asparaginase activity in ovarian cancer cells. Mol Cancer Ther 5, 2613–2623. [DOI] [PubMed] [Google Scholar]
- Lowman XH, Hanse EA, Yang Y, Ishak Gabra MB, Tran TQ, Li H, and Kong M (2019). p53 Promotes Cancer Cell Adaptation to Glutamine Deprivation by Upregulating Slc7a3 to Increase Arginine Uptake. Cell Rep 26, 3051–3060 e3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, and Pandolfi PP (2005). Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193. [DOI] [PubMed] [Google Scholar]
- Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, and Vousden KH (2013). Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F, et al. (2017). Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Lo PHY, Saichi N, Ueda K, Hirata M, Tanikawa C, and Matsuda K (2017). Argininosuccinate synthase 1 is an intrinsic Akt repressor transactivated by p53. Sci Adv 3, e1603204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Nambu T, Ebara S, Hasegawa Y, Toyoshima K, Tsuchiya Y, Tomita D, Fujimoto J, Kurasawa O, Takahara C, et al. (2018). Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc Natl Acad Sci U S A 115, E7776–E7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano L, Scalise M, Koyioni M, Koutentis P, Catto M, Eberini I, Parravicini C, Palazzolo L, Pisani L, Galluccio M, et al. (2017). Potent inhibitors of human LAT1 (SLC7A5) transporter based on dithiazole and dithiazine compounds for development of anticancer drugs. Biochem Pharmacol 143, 39–52. [DOI] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nofal M, Zhang K, Han S, and Rabinowitz JD (2017). mTOR Inhibition Restores Amino Acid Balance in Cells Dependent on Catabolism of Extracellular Protein. Mol Cell 67, 936–946 e935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera J, and Ohsumi Y (2005). Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J Biol Chem 280, 31582–31586. [DOI] [PubMed] [Google Scholar]
- Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ, Possemato R, Chen WW, Sullivan LB, Fiske BP, et al. (2016). A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol 12, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, and Thompson CB (2015). The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 162, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YX, Chen H, Thiaville MM, and Kilberg MS (2007). Activation of the ATF3 gene through a co-ordinated amino acid-sensing response programme that controls transcriptional regulation of responsive genes following amino acid limitation. Biochem J 401, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathria G, Lee JS, Hasnis E, Tandoc K, Scott DA, Verma S, Feng Y, Larue L, Sahu AD, Topisirovic I, et al. (2019). Translational reprogramming marks adaptation to asparagine restriction in cancer. Nat Cell Biol 21, 1590–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathria G, Scott DA, Feng Y, Sang Lee J, Fujita Y, Zhang G, Sahu AD, Ruppin E, Herlyn M, Osterman AL, et al. (2018). Targeting the Warburg effect via LDHA inhibition engages ATF4 signaling for cancer cell survival. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, White RM, Rabinowitz JD, Thompson CB, and Zhang J (2018). As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab 27, 428–438 e425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira ER, Frudd K, Awad W, and Hendershot LM (2014). Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF). J Biol Chem 289, 3352–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MM, Sheaff MT, and Szlosarek PW (2013). Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res Treat 45, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieters R, Hunger SP, Boos J, Rizzari C, Silverman L, Baruchel A, Goekbuget N, Schrappe M, and Pui CH (2011). L-asparaginase treatment in acute lymphoblastic leukemia: a focus on Erwinia asparaginase. Cancer 117, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohjanpelto P, and Holtta E (1990). Deprivation of a single amino acid induces protein synthesis-dependent increases in c-jun, c-myc, and ornithine decarboxylase mRNAs in Chinese hamster ovary cells. Mol Cell Biol 10, 5814–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Shi Z, and Jiang J (2017). Cyclooxygenase-2 in glioblastoma multiforme. Drug Discov Today 22, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens A, and Munoz L (2019). Targeting Cancer Cell Dormancy. Trends Pharmacol Sci 40, 128–141. [DOI] [PubMed] [Google Scholar]
- Riess C, Shokraie F, Classen CF, Kreikemeyer B, Fiedler T, Junghanss C, and Maletzki C (2018). Arginine-Depleting Enzymes - An Increasingly Recognized Treatment Strategy for Therapy-Refractory Malignancies. Cell Physiol Biochem 51, 854–870. [DOI] [PubMed] [Google Scholar]
- Riscal R, Schrepfer E, Arena G, Cisse MY, Bellvert F, Heuillet M, Rambow F, Bonneil E, Sabourdy F, Vincent C, et al. (2016). Chromatin-Bound MDM2 Regulates Serine Metabolism and Redox Homeostasis Independently of p53. Mol Cell 62, 890–902. [DOI] [PubMed] [Google Scholar]
- Rojo de la Vega M, Chapman E, and Zhang DD (2018). NRF2 and the Hallmarks of Cancer. Cancer Cell 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano R, and Reed JC (2013). ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833, 3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalise M, Pochini L, Console L, Losso MA, and Indiveri C (2018). The Human SLC1A5 (ASCT2) Amino Acid Transporter: From Function to Structure and Role in Cell Biology. Front Cell Dev Biol 6, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, Kondo J, Coffey RJ, Johnson MO, Rathmell JC, et al. (2018). Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med 24, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, and Nevins JR (2000). Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14, 2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2001). HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 107, 1–3. [DOI] [PubMed] [Google Scholar]
- Shi Y, Sharma A, Wu H, Lichtenstein A, and Gera J (2005). Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J Biol Chem 280, 10964–10973. [DOI] [PubMed] [Google Scholar]
- Shin S, Buel GR, Wolgamott L, Plas DR, Asara JM, Blenis J, and Yoon SO (2015). ERK2 Mediates Metabolic Stress Response to Regulate Cell Fate. Mol Cell 59, 382–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, McGeachy AM, Ingolia NT, and Walter P (2015). The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simabuco FM, Morale MG, Pavan ICB, Morelli AP, Silva FR, and Tamura RE (2018). p53 and metabolism: from mechanism to therapeutics. Oncotarget 9, 23780–23823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Akcakanat A, Sharma C, Luyimbazi D, Naff KA, and Meric-Bernstam F (2011). The effect of leucine restriction on Akt/mTOR signaling in breast cancer cell lines in vitro and in vivo. Nutr Cancer 63, 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, and Pae HO (2011). Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J Signal Transduct 2011, 792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, and Dang CV (2015). MYC, Metabolism, and Cancer. Cancer Discov 5, 1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Song L, Wan Q, Wu G, Li X, Wang Y, Wang J, Liu Z, Zhong X, He X, et al. (2015). cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res 25, 429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajan M, Hock AK, Blagih J, Robertson NA, Labuschagne CF, Kruiswijk F, Humpton TJ, Adams PD, and Vousden KH (2018). A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab 28, 721–736 e726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tameire F, Verginadis II, Leli NM, Polte C, Conn CS, Ojha R, Salas Salinas C, Chinga F, Monroy AM, Fu W, et al. (2019). ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol 21, 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiaville MM, Pan YX, Gjymishka A, Zhong C, Kaufman RJ, and Kilberg MS (2008). MEK signaling is required for phosphorylation of eIF2alpha following amino acid limitation of HepG2 human hepatoma cells. J Biol Chem 283, 10848–10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Davis T, Loos B, Sishi B, Huisamen B, Strijdom H, and Engelbrecht AM (2018). Autophagy is essential for the maintenance of amino acids and ATP levels during acute amino acid starvation in MDAMB231 cells. Cell Biochem Funct 36, 65–79. [DOI] [PubMed] [Google Scholar]
- Thompson JM, Nguyen QH, Singh M, and Razorenova OV (2015). Approaches to identifying synthetic lethal interactions in cancer. Yale J Biol Med 88, 145–155. [PMC free article] [PubMed] [Google Scholar]
- Tran TQ, Hanse EA, Habowski AN, Li H, Ishak Gabra MB, Yang Y, Lowman XH, Ooi AM, Liao SY, Edwards RA, et al. (2020). α-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nature Cancer 1, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WB, Aiba I, Lee SY, Feun L, Savaraj N, and Kuo MT (2009). Resistance to arginine deiminase treatment in melanoma cells is associated with induced argininosuccinate synthetase expression involving c-Myc/HIF-1alpha/Sp4. Mol Cancer Ther 8, 3223–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, and Kuo MT (2012). Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res 72, 2622–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem KM, and Wek RC (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 101, 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Miguel S, Buckbinder L, Jean P, Gelbert L, Talbott R, Laidlaw J, Seizinger B, and Kley N (1999). PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene 18, 127–137. [DOI] [PubMed] [Google Scholar]
- Wanders D, Stone KP, Forney LA, Cortez CC, Dille KN, Simon J, Xu M, Hotard EC, Nikonorova IA, Pettit AP, et al. (2016). Role of GCN2-Independent Signaling Through a Noncanonical PERK/NRF2 Pathway in the Physiological Responses to Dietary Methionine Restriction. Diabetes 65, 1499–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, and Kaufman RJ (2014). The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer 14, 581–597. [DOI] [PubMed] [Google Scholar]
- Webster KR, Goel VK, Hung IN, Parker GS, Staunton J, Neal M, Molter J, Chiang GG, Jessen KA, Wegerski CJ, et al. (2015). eFT508, a Potent and Selective Mitogen-Activated Protein Kinase Interacting Kinase (MNK) 1 and 2 Inhibitor, Is Efficacious in Preclinical Models of Diffuse Large B-Cell Lymphoma (DLBCL). Blood 126, 1554–1554. [Google Scholar]
- Wortel IMN, van der Meer LT, Kilberg MS, and van Leeuwen FN (2017). Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol Metab 28, 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Wang X, and Proud CG (2016). mTOR inhibitors in cancer therapy. F1000Res 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Nie J, Ma X, Wei Y, Peng Y, and Wei X (2019). Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer 18, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Achreja A, Yeung TL, Mangala LS, Jiang D, Han C, Baddour J, Marini JC, Ni J, Nakahara R, et al. (2016). Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab 24, 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, and Koumenis C (2010). The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 29, 2082–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Palm W, Peng M, King B, Lindsten T, Li MO, Koumenis C, and Thompson CB (2015). GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev 29, 2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fan J, Venneti S, Cross JR, Takagi T, Bhinder B, Djaballah H, Kanai M, Cheng EH, Judkins AR, et al. (2014). Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell 56, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Fu J, Du J, and Xu W (2020). The Role of D-3-Phosphoglycerate Dehydrogenase in Cancer. Int J Biol Sci 16, 1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipper LM, and Mulcahy RT (2003). Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol Sci 73, 124–134. [DOI] [PubMed] [Google Scholar]