

Abstract
Background & Aims:
Pancreatic tumors undergo rapid growth and progression, become resistant to chemotherapy, and recur after surgery. We studied the functions of the solute carrier family 39 member 4 (SLC39A4, also called ZIP4), which regulates concentrations of intracellular zinc and is increased in pancreatic cancer cells, in cell lines and mice.
Methods:
We obtained 93 pancreatic cancer specimens (tumor and adjacent non-tumor tissues) from patients who underwent surgery and gemcitabine chemotherapy and analyzed them by immunohistochemistry. ZIP4 and/or ITGA3 or ITGB1 were overexpressed or knocked down with small hairpin RNAs in AsPC-1 and MIA PaCa-2 pancreatic cancer cells lines, and in pancreatic cells from KPC and KPC-ZEB1 knockout mice, and pancreatic spheroids were established; cells and spheroids were analyzed by immunoblots, reverse transcription PCR, and liquid chromatography tandem mass spectrometry. We studied transcriptional regulation of ZEB1, ITGA3, ITGB1, JNK, and ENT1 by ZIP4 using chromatin precipitation and luciferase reporter assays. Nude mice were given injections of genetically manipulated AsPC-1 and MIA PaCa-2 cells and growth of xenograft tumors and metastases was measured.
Results:
In pancreatic cancer specimens from patients, increased levels of ZIP4 associated with shorter survival times. MIA PaCa-2 cells that overexpressed ZIP4 had increased resistance to gemcitabine, 5-FU, and cisplatin, whereas AsPC-1 cells with ZIP4 knockdown had increased sensitivity to these drugs. In mice, xenograft tumors grown from AsPC-1 cells with ZIP4 knockdown were smaller and more sensitive to gemcitabine. ZIP4 overexpression significantly reduced accumulation of gemcitabine in pancreatic cancer cells, increased growth of xenograft tumors in mice, and increased expression of the integrin subunits ITGA3 and ITGB1; expression of ITGA3 and ITGB1 was reduced in cells with ZIP4 knockdown. Pancreatic cancer cells with ITGA3 or ITGB1 knockdown had reduced proliferation and formed smaller tumors in mice, despite overexpression of ZIP4; spheroids established from these cells had increased sensitivity to gemcitabine. We found ZIP4 to activate STAT3 to induce expression of ZEB1, which induced expression of ITGA3 and ITGB1 in KPC cells. Increased ITGA3 and ITGB1 expression and subsequent integrin α3β1 signaling, via JNK, inhibited expression of the gemcitabine transporter ENT1, which reduced gemcitabine uptake by pancreatic cancer cells. ZEB1-knockdown cells had increased sensitivity to gemcitabine.
Conclusions:
In studies of pancreatic cancer cell lines and mice, we found that ZIP4 increases expression of the transcription factor ZEB1, which activates expression of ITGA3 and ITGB1. The subsequent increase in integrin α3β1 signaling, via JNK, inhibits expression of the gemcitabine transporter ENT1, so that cells take up smaller amounts of the drug. Activation of this pathway might help mediate resistance of pancreatic tumors to chemotherapeutic agents.
Keywords: Pancreas, EMT, signal transduction, oncogene
Graphical Abstract
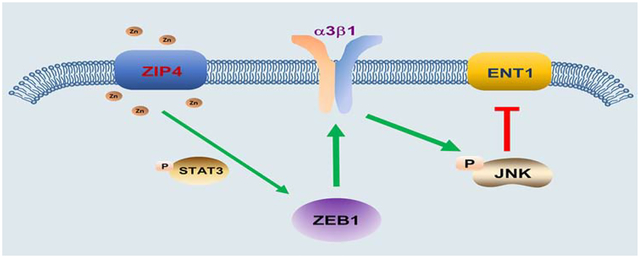
Lay summary
We identified a signaling pathway that is activated in pancreatic cancer cells that mediates their resistance to chemotherapeutic agents such as gemcitabine.
Introduction
Pancreatic cancer is the third leading cause of cancer deaths in the United States. Only 15–20% of patients are eligible for resection at the time of diagnosis, and most patients develop disease recurrence within a year1, 2. Due to the absence of effective screening and biomarkers for pancreatic cancer, the overall survival rate of patients remains poor3. Furthermore, current therapeutic options have had limited impact on the course of the disease, and pancreatic cancer patients frequently progress to metastasis and eventually succumb to this devastating disease4. Thus, it is paramount that new therapeutic options for pancreatic cancer be identified based on better understanding of the pathobiology of pancreatic cancer and its primary resistance to current treatment.
Gemcitabine, a pyrimidine nucleoside analogue, remains the first line treatment for pancreatic cancer, and often slows disease progression5. However, most patients demonstrate either primary resistance or will develop gemcitabine resistance over time and, ultimately, experience cancer recurrence6. Integrins are transmembrane receptors that regulate cell attachment to extracellular matrix (ECM)7 and promote tumor proliferation, adhesion, migration and invasion8. Most notably, integrin signaling plays a critical role in chemoresistance of multiple cancers including breast, colon, lung, and prostate cancer7. In pancreatic cancer, integrins have been shown to correlate with ECM-induced FAK, PI3K/AKT pathway activation, and promotes chemoresistance9, 10. Numerous reports have shown that a major event controlling gemcitabine resistance is the regulation of its cellular uptake through equilibrative or nucleoside transporters (ENTs or CNTs)11. ENT1 is highly expressed in pancreatic cancer comparing with other transporters12. Loss of ENT1 contributes to chemical drug resistance and high level of ENT1 predicts good response of pancreatic cancer to gemcitabine treatment13. Recent studies linked gemcitabine resistance to epithelial-to-mesenchymal transition (EMT)3, 14. It’s indicated that pancreatic cancer cells underwent gemcitabine resistance acquired a more motile and invasive phenotype suggesting a mechanistic relationship between chemoresistance and metastatic potential in pancreatic carcinoma. However, little is known about the mechanism of gemcitabine resistance and ENT1 regulation in pancreatic cancer. Thus, elucidation of the molecular pathway involved in ENT1 regulation and gemcitabine resistance will help improve responses to chemotherapeutic agents. New emerging technologies which detect the trace level of chemo-drugs in cancer cells on a single cell base also facilitated the development of new treatment in pancreatic cancer targeting resistance pathways. A rapid and sensitive liquid chromatography-mass spectrometry (LC-MS/MS) method was recently developed that accurately measures gemcitabine levels in human and rat plasma as well as in mouse tumor tissue15.
In the current study, we identify a novel function for ZIP4 in mediating pancreatic cancer tumorigenesis, EMT, metastasis and chemoresistance. ZIP4 plays a critical role in maintaining physiologic concentrations of intracellular zinc, an essential trace element and a co-factor in many proteins involved in diverse biological processes16, 17–19. In pancreatic cancer ZIP4 regulates cell proliferation, resistance to apoptosis, and promotes cachexia16, 20, 21. Here, we found that knock down of ZIP4 increased sensitivity of pancreatic cancer cells to gemcitabine. We further elucidate the regulatory cascade from ZIP4 overexpression to reduction of gemcitabine uptake and thus increased resistance: ZIP4 activates STAT3 which upregulates the zinc dependent EMT like transcription factor ZEB1. ZEB1-induced integrin α3β1 downregulates the gemcitabine uptake protein ENT1 to limit internalization of the drug through activating MAP kinase JNK. These findings not only provide evidence for a previously uncharacterized pathway controlling metastasis and chemoresistance but may have a significant impact on future treatment of pancreatic cancer.
Materials and methods
Cell lines, plasmids and chemical reagents.
Human pancreatic cancer cells AsPC-1 and MIA PaCa-2 were purchased from the American Type Culture Collection (ATCC, Rockville, MD) in 2016, and were cultured in RPMI 1640 medium or DMEM medium with 10% fetal bovine serum (FBS). KPC and KPC-ZEB1 knockout (KPCZ) cells were kindly provided by Dr. Thomas Brabletz from University of Erlangen-Nürnberg, Germany. All cell lines were verified to be mycoplasma free using MycoAlert™ kit (Lonza). The plasmids containing ZEB1, ITGA3 and ITGB1 (ORF vector) were purchased from Genecopoeia. Gemcitabine, Hygromycin B, WP1066 and SP600125 were purchased from Sigma.
Human tissue samples.
93 human pancreatic cancer specimens were obtained from The University of Oklahoma Health Sciences Center (OUHSC). All patients underwent surgery and gemcitabine chemotherapy. This study was approved by the Institutional Review Board (IRB) at OUHSC. Written consent from all subjects was obtained before they entered the study.
Stable cell line construction.
The stable cell lines MIA-V, MIA-ZIP4, AsPC-shV and AsPC-shZIP4 were constructed as previously described16, 17. Integrin α3 (ITGA3), β1 (ITGB1) knock down cells were selected in MIA-ZIP4 cells using a lentivirus vector (Genecopoeia), following the manufacturer’s instructions. Briefly, human ITGA3 (NM 002204.3) and ITGB1 (NM 002211.3) shRNAs were cloned into the LVRH1GH vector. Viral supernatants were collected and transduced to the target cells. Stable cell lines expressing ITGA3 and ITGB1 shRNAs (MIA-ZIP4-shITGA3 and MIA-ZIP4-shITGB1) or negative control vector (MIA-ZIP4 shV) were selected using 400μg/ml Hygromycin B.
Spheroid growth assay.
The spheroid growth assay was performed as previously described21. Cells were suspended in the culture medium containing 0.24% methylcellulose (Sigma, M0512), and then seeded onto 10 cm2 dishes’ inner lid with 20 μl/drop, and the lids were inverted over dishes containing 10 ml PBS. The solidified drops were kept on the dish lid for 2 to 24 h. Spheroids were imaged using a light microscope and the size of the spheroid was calculated by measuring the diameters using the line morphometry function. 10 spheroids were included and analyzed in each group.
Western blot analysis.
Cell lysate protein were collected and loaded on SDS polyacrylamide gels as previously described19. Membranes were incubated with appropriate antibodies against ZIP4 (Proteintech, 1:2000), ITGA3, ITGB1 (Millipore, 1: 1000), ENT1 (Proteintech, 1:1000), pSTAT3, STAT3 (Cell Signaling, 1:1000), pJNK, JNK (Cell signaling, 1:1000) or β-actin (Sigma, 1:10000) at 4 °C overnight. Following washes with 0.1% Tween 20-TBS membranes were incubated with a horseradish peroxidase-linked secondary antibody (1: 5000) for 2 h at room temperature. Immunoreactive bands were detected using an enhanced chemiluminescent (ECL) plus reagent kit.
Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS).
MIA PaCa-2 and AsPC-1 cells were harvested after the treatment with gemcitabine (200 μM for 18 h and 400 μM for 18 h), respectively. Cell supernatant was removed after centrifugation and 200 μL methanol was used to re-dissolve the dried cell lysate, and the internal standard (N15-gemcitabine) was added with 200 nM final concentration. A Dionex 3000 HPLC system (Thermo Scientific) was used in combination with an LTQ Orbitrap XL mass spectrometer (Thermo Scientific) for cell lysate analysis. The total quantity (mole) of gemcitabine in the lysate was obtained based on the relative peak areas of gemcitabine and N15-gemcitabine, and the average quantity of gemcitabine in each cell (i.e., mole/cell) was calculated using the total drug quantity in the lysate divided by the cell number used for lysate preparation.
Single cell Mass Spectrometry.
After treatment using 400 μM gemcitabine for 24 h, cells were washed with PBS to eliminate the drug residues prior to single cell mass spectrometry (SCMS) analysis. The SCMS analysis was achieved using the single-probe, a multifunctional ionization and sample device, coupled to a LTQ Orbitrap XL mass spectrometer as previously described. The mass spectra of gemcitabine treated cells were collected from 15 individual cells in each cell line. The normalized (total ion current (TIC) normalization)22 ion intensities of gemcitabine from individual cells were then utilized for the comparison of the relative intracellular drug abundances at the single-cell level.
Pancreatic cancer xenograft mouse model.
Stable pancreatic cancer cell lines (AsPC-shV, AsPC-shZIP4, MIA-V shV, MIA-ZIP4 shV, MIA-ZIP4 sh-ITGA3, MIA-ZIP4-sh-ITGB1) were used to establish the orthotopic xenograft tumor model as previously described16, 17. Cells were harvested by trypsinization and resuspended in RPMI or DMEM medium. 3 ×106 tumor cells in 50 μL were injected into the pancreases of 5-to-6-week-old male nude mice. All mice were cared for in accordance with the Office for Protection from Research Risks (OPRR) and Animal Welfare Act Guidelines under an animal protocol approved by the Animal Welfare Committee at OUHSC. Gemcitabine (25 mg/mL) was administered to mice by intraperitoneal injection twice a week, starting at 5 days after tumor implantation. The gemcitabine dosages were determined by mouse body weight: 80 μl for body weights <23.0 g, 100 μl for body weights of 23.0–27.0 g, and 120 μl for body weights >27.0 g. For AsPC-shV, AsPC-shZIP4 groups, survival rate of the mice was examined everyday up to 75 days. For MIA-V shV, MIA-ZIP4 shV, MIA-ZIP4 sh-ITGA3, MIA-ZIP4-sh-ITGB1 groups, after 6 weeks, all surviving mice were euthanized by CO2 asphyxiation.
Statistical analysis.
Quantitative results are shown as means ± SD. Overall difference among groups were assessed by ANOVA and subsequent Student’s t-tests were used to compare data from control and treated groups with multiple testing adjusted by Dunnett’s method. Two-group comparisons were analyzed by Student’s t-tests. Kaplan-Meier curves and the log-rank tests were used for the analyses of censored survival data. A P value of < .05 was considered statistically significant. All tests were two-sided.
Results
ZIP4 predicts poor survival, promotes tumor growth and confers gemcitabine resistance in pancreatic cancer cells.
To assess the impact of ZIP4 on pancreatic cancer growth and survival, we initially determined the expression of ZIP4 in human pancreatic cancer patients who had underwent gemcitabine treatment. In this cohort of 93 patients, 72 patients had low or negative ZIP4 expression and 21 had high ZIP4 expression, as determined by immunohistochemical staining (Fig. S1A). Kaplan-Meier analysis showed that patient survival was significantly reduced in the group with high ZIP4 (Fig. 1A). We further examined additional 117 patients from The Cancer Genome Atlas (TCGA) database; these analyses also showed that high ZIP4 levels predict poor overall survival for pancreatic cancer patients undergone gemcitabine treatment (Fig. S1B). To determine the biological impact of these clinical findings, next we examined the cell growth and gemcitabine sensitivity of pancreatic cancer cells with various levels of ZIP4. MIA PaCa-2 cells overexpressing ZIP4 showed increased resistance to gemcitabine, however, knock down of ZIP4 in AsPC-1 cells increased their sensitivity to this drug (Fig. 1B–1C, Supplementary table 1). We also examined the resistance of pancreatic cancer cells to 5-FU and cisplatin, and found that ZIP4 also contributed to 5-FU and cisplatin resistance both in MIA PaCa-2 and AsPC-1 cells (Fig. S1C–S1D, Supplementary table 2). To further investigate the effect of ZIP4 on pancreatic cancer growth and chemoresistance, we established a spheroid-based 3D tumor-culture model. Spheroids of the MIA-ZIP4 cells were less responsive to gemcitabine treatment than the vector control cells, i.e. Spheroids of MIA-ZIP4 cells were less responsive to gemcitabine treatment than control cells, while AsPC-shZIP4 cells with ZIP4 knocking down were more sensitive to gemcitabine (Fig. 1D). TUNEL staining showed significantly less apoptotic signal in MIA-ZIP4 spheroids comparing with MIA-V group under gemcitabine treatment (Fig. S1E), while in AsPC-shZIP4 spheroids there were more apoptotic cells (Fig. S1F). To further validate the function of ZIP4 in pancreatic cancer survival, metastasis, and chemoresistance, we examined the gemcitabine sensitivity of AsPC-1 cells with different levels of ZIP4, in an orthotopic xenograft mouse model. Compared to PBS control mice, the gemcitabine-treated mice had smaller tumors (Fig. S1G) and knockdown of ZIP4 further increased overall survival of gemcitabine treated mice (Fig. S1H). These results indicate that ZIP4 predicts poor survival, promotes tumor growth, and confers pancreatic cancer chemoresistance to gemcitabine both in vitro and in vivo.
Figure 1. ZIP4 predicts survival and promotes pancreatic cancer cell growth and chemoresistance to gemcitabine.

(A). Overall survival. All 93 patients were categorized into two groups based on ZIP4 level: negative/low and strong ZIP4 staining. The low ZIP4 group has significantly greater survival than the high ZIP4 group (P= .04). (B). Cell viability of MIA-V and MIA-ZIP4 cells, and (C). AsPC-shV and AsPC-shZIP4 cells following gemcitabine treatment. Data were collected on day 2. Cell viability under chemotherapeutic treatment normalized to regular media was calculated. (D). Spheroid growth assay. Spheroid growth was assessed at 48 h post gemcitabine treatment. The scale bar is 200 μm. (E). LC-MS/MS. The gemcitabine level in MIA-V and MIA-ZIP4 cells treated with 200 μM gemcitabine for 18 h was detected using LC-MS/MS. (F). LC-MS/MS. Gemcitabine level in AsPC-shV and AsPC-shZIP4 cells treated with 400 μM gemcitabine for 18 h was detected using LC-MS/MS. (G). Single cell MS. Gemcitabine level in MIA-V and MIA-ZIP4 cells treated with 400 μM gemcitabine for 24 h was detected using single cell MS.
ZIP4 inhibits gemcitabine accumulation in pancreatic cancer cells.
To further determine the impact of ZIP4 on gemcitabine accumulation in pancreatic cancer cell, we used a highly sensitive LC-MS/MS methodology6, 15 to measure gemcitabine concentration in pancreatic cancer cells after exposure to the drug for 18 h. Gemcitabine levels in MIA-V cells were 3 times higher than that in MIA-ZIP4 cells (Fig. 1E), and gemcitabine in AsPC-shZIP4 cells was 1.5 times higher than that in AsPC-shV cells (Fig. 1F). In order to alleviate the limitation of cancer cell heterogeneity for gemcitabine delivery, we used single cell mass spectrometry to quantify the drug concentration at the single cell level. We found the amount of gemcitabine in MIA-ZIP4 cells was significantly decreased comparing with that in MIA-V cells (Fig. 1G). These data demonstrated that ZIP4 overexpression significantly reduced the gemcitabine accumulation in pancreatic cancer cells. Combined with data from our in vitro and mouse model studies reported above, this finding strongly indicates that knock down of ZIP4 sensitizes pancreatic cancer cells to gemcitabine treatment while high ZIP4 levels endow pancreatic cancer with resistance to gemcitabine. Thus, ZIP4 may serve as a predictor for pancreatic cancer chemotherapy resistance.
ZIP4 is positively correlated with ITGA3 and ITGB1 in pancreatic cancer.
We next examined how ZIP4 affects pancreatic cancer survival, metastasis, and chemoresistance. Dysregulation of integrins has been shown to contribute to both disruption of tumor proliferation and chemoresistance7, 23. Among the various integrin subunits (18 α and 8 β subunits) we found only ITGA3 and ITGB1 were significantly upregulated by ZIP4 in MIA PaCa-2 and AsPC-1 cells. Overexpression of ZIP4 increased both ITGA3 and ITGB1 levels in MIA PaCa-2 cells whereas knockdown of ZIP4 significantly reduced ITGA3 and ITGB1 expression in AsPC-1 cells (Fig. 2A, Fig. S2A–S2F). In pancreatic cancer tissues, we found that ITGA3 and ITGB1 were highly expressed in tumor tissues compared to adjacent benign tissues (Fig. 2B), and ITGA3 and ITGB1 were closely correlated with ZIP4 level (Fig. S2G–S2H).
Figure 2. ZIP4 enhances pancreatic cancer growth and gemcitabine resistance through integrin α3β1.

(A). Expression of ITGA3 and ITGB1 in MIA PaCa-2 and AsPC-1 cells. (B). IHC staining of ITGA3 and ITGB1 in pancreatic cancer specimens and adjacent benign tissues. Quantified data are plotted on right. (C). Tumor weight. Representative xenograft pictures were shown (n=5). (D). Cell viability following gemcitabine treatment for 2 days. (E). Spheroid growth assay. Spheroid growth was assessed in MIA-ZIP4 siITGA3, MIA-ZIP4 siITGB1 cells at 48 h post gemcitabine treatment. The scale bar is 200 μm. (F). Stable cells were orthotopically implanted (n=5/group). All the mice were administrated with gemcitabine. Representative tumors were shown. Tumors in MIA-V shV group were circled in dotted white since the tumors were too small.
ITGA3 and ITGB1 contribute to pancreatic cancer growth, metastasis, and chemoresistance both in vitro and in vivo.
Having shown that ZIP4 enhances metastasis and chemoresistance and upregulates expression of ITGA3 and ITGB1 in multiple pancreatic cancer cells, we hypothesized that ZIP4 regulates the metastasis and chemoresistance phenotype via targeting the heterodimer integrin α3β1. We found that knockdown of integrin α3β1 using siRNA or shRNA inhibited pancreatic cancer cell proliferation by in vitro and IHC staining (Fig. S2I – S2M), and reduced mouse tumor weight compared to the vector control (Fig. 2C). These results demonstrate that integrin α3β1 plays a critical role in pancreatic cancer growth and metastasis.
Since ZIP4 contributes to pancreatic cancer resistance to gemcitabine, we also investigated whether integrin α3β1 was involved in chemoresistance. We found reduced cell viability from integrin α3β1 knockdown cell lines upon treatment with gemcitabine compared with the control cells (Fig. 2D). In the 3D tumor spheroid model, knock down of integrin α3β1 significantly increased the sensitivity of tumor spheroids to gemcitabine treatment (Fig. 2E). To confirm the role of integrin α3β1 in gemcitabine resistance in vivo, we orthotopically implanted the integrin α3β1 knock-down MIA-ZIP4 cells into the nude mice, and treated the mice with gemcitabine. The tumor weight in the MIA-ZIP4 shV group was significantly higher than that in the MIA-V shV group, however, when integrin α3β1 was knocked down, the tumor weight was significantly reduced (Fig. 2F). Ki67 staining confirmed that the tumor cell proliferation was increased with ZIP4 overexpression but decreased when integrin α3β1 was knocked down (Fig. S2N). These data indicate that knock down of integrin α3β1 attenuated the chemoresistance induced by ZIP4 overexpression in the orthotopic xenograft tumor model.
EMT like transcription factor ZEB1 regulates integrin α3β1 expression in pancreatic cancer cells.
We next investigated the underlying mechanism(s) by which ZIP4 regulates integrin α3β1 expression. A promoter analysis of integrin α3β1 indicates there are multiple binding sites of ZEB1, a zinc dependent EMT like transcription factor, at the promoter region of ITGA3 and ITGB1. Previously we have shown that ZEB1 is upregulated by ZIP4 in pancreatic cancer19, we hypothesized that ZIP4 may mediate ITGA3 and ITGB1 expression via ZEB1. We confirmed the binding of ZEB1 to the ITGA3 and ITGB1 promoter regions using ChIP assay in both MIA PaCa-2 and AsPC-1 cells (Fig. 3A). Luciferase reporter assay also confirmed that knock down of ZEB1 reduced ZIP4-mediated induction of the ITGA3 and ITGB1 promoter activity (Fig. 3B–3C). In a rescue experiment, when ZEB1 was overexpressed in the presence of shZIP4, the ITGA3 and ITGB1 promoter activity was increased (Fig. 3D–3E). Furthermore, we found that the ITGA3 and ITGB1 were increased when ZIP4 was overexpressed but were decreased when ZEB1 was knocked down in MIA PaCa-2 cells (Fig. 3F, Fig. S3A–S3C). On the contrary, ITGA3 and ITGB1 were decreased when ZIP4 was knocked down but were rescued when ZEB1 was overexpressed in AsPC-1 cells (Fig. 3G, Fig. S3D–S3F), indicating a close correlation between ZIP4, ZEB1 and ITGA3 and ITGB1 in pancreatic cancer cells.
Figure 3. ZIP4 upregulated integrin α3β1 expression is dependent on the EMT like transcription factor ZEB1 in pancreatic cancer cells.

(A). ChIP-PCR. ChIP binding assay with anti-ZEB1 confirms the binding of ZEB1 to the ITGA3 and ITGB1 promoter regions. (B-C). Relative reporter activity of ITGA3 and ITGB1 promoters in MIA-ZIP4 cells with ZEB1 blocked. (D-E). Relative reporter activity of ITGA3 and ITGB1 promoters in AsPC-shZIP4 cells with ZEB1 overexpressed. (F). The protein level of integrin α3β1 in MIA-ZIP4 with ZEB1 blocked and (G). AsPC-shZIP4 cells with ZEB1 overexpressed.
ZEB1 is indispensable for ITGA3 and ITGB1 upregulation in KPC mouse derived cell lines.
We further examined the regulation of ITGA3 and ITGB1 by ZEB1 in mouse pancreatic cancer cell lines, derived from a genetically engineered KPC mouse, and its ZEB1 knockout derivative KPCZ mouse24. Knockout of ZEB1 in KPCZ cells significantly decreased ITGA3 and ITGB1 expression (Fig. 4A); while exogenously overexpressed ZEB1 rescued ITGA3 and ITGB1 in KPCZ cells (Fig. 4B). Next, we examined whether ZIP4-regulated ITGA3 and ITGB1 is dependent on ZEB1 in KPC mouse cell lines. We overexpressed mouse ZIP4 (mZIP4) in both KPC and KPCZ cells, and found that overexpression of mZIP4 significantly increased ITGA3 and ITGB1 in KPC cells, but not in KPCZ cells where ZEB1 was totally abolished (Fig. S4A and S4B). Therefore, these results indicate that ZIP4-activated integrin upregulation is dependent on ZEB1 in KPC mouse derived cells. Considering this result in its entirety we have shown that ZIP4-dependent regulation of ITGA3 and ITGB1 promoter activity is mediated via direct binding of ZEB1 to the integrin promoter regions.
Figure 4. ZEB1 is indispensable for integrin α3β1 upregulation in KPC mouse derived cell lines.

(A). The expression of integrin α3β1 in two KPC and two KPCZ cells, and in (B). KPCZ-ZEB1 cells. (C). Cell viability of MIA-ZIP4 siZEB1 cells, and (D). KPCZ-V and KPCZ-ZEB1 cells following gemcitabine treatment. (E). Spheroid growth assay in MIA-ZIP4 cells with ZEB1 knockdown. The scale bar is 200 μm. (F). Spheroid growth assay in KPC-V, KPCZ-V and KPCZ-ZEB1 cells at 48 h post gemcitabine treatment. The scale bar is 100 μm.
We further investigated whether ZEB1 also contributed to chemoresistance in pancreatic cancer. When ZEB1 was knocked down in ZIP4 overexpressing cells, they became more sensitive to gemcitabine treatment (Fig. 4C). We also found that KPCZ vector control cells showed increased sensitivity to gemcitabine, while reintroducing ZEB1 back to the same cell line (KPCZ-ZEB1) rescued the gemcitabine resistance phenotype (Fig. 4D). We then used the tumor spheroid model to confirm the impact of ZEB1 on resistance to gemcitabine, and found that decreased ZEB1 expression resulted in increased sensitivity to gemcitabine in both human MIA-ZIP4 (Fig. 4E) and mouse KPC cells (Fig. 4F) derived spheroids.
ZIP4 upregulates ZEB1 through activating STAT3 in pancreatic cancer cells.
We previously identified ZIP4-mediated upregulation of ZEB1 expression both at the mRNA and protein levels, here we further investigated the mechanism of how ZIP4 regulates ZEB1 expression. Firstly, DNA sequence analysis of the ZEB1 promoter region analysis indicated putative STAT3 binding sites. Having previously shown that ZIP4 activates phosphorylation of STAT3 but does not affect total STAT3 levels in pancreatic cancer cells25, we hypothesized that ZIP4 upregulates ZEB1 via activating pSTAT3. ChIP assay confirmed the binding of STAT3 to the ZEB1 promoter region (Fig. 5A). We found that knockdown of STAT3 reduced ZEB1 promoter activity in both MIA PaCa-2 and AsPC-1 cells (Fig. 5B). Next, we knocked down STAT3 using siRNA in MIA PaCa-2 and AsPC-1 cells and found that ZEB1 expression was significantly decreased in both cells (Fig. 5C). Knock down of STAT3 also attenuated the upregulation of ZEB1 expression by ZIP4 (Fig. 5D). These results suggested that STAT3 was involved in ZEB1 regulation in pancreatic cancer cells. In order to further investigate whether phosphorylation of STAT3 contributes to upregulation of ZEB1, we treated pancreatic cancer cells with WP1066 which specifically inhibits phosphorylation of STAT326. This compound reduced pSTAT3 level and also decreased ZEB1 expression in parental MIA PaCa-2 and AsPC-1 cell lines (Fig. S5A). Inhibition of pSTAT3 with WP1066 also significantly suppressed promoter activity of ZEB1 in MIA PaCa-2 cells (Fig. S5B–S5C). Furthermore, WP1066 decreased both the expression and promoter activity of ZEB1 in MIA-ZIP4 cells (Fig. 5E–5F). All together these results reveal that STAT3 phosphorylation contributes to ZEB1 upregulation in pancreatic cancer cells.
Figure 5. ZIP4 upregulates ZEB1 through activating STAT3 in pancreatic cancer cells.

(A). ChIP binding assay with anti-STAT3 antibody confirms the binding of STAT3 to the ZEB1 promoter region. (B). Relative promoter activity of ZEB1 with STAT3 knocked down. (C). Protein level of ZEB1 in MIA PaCa-2 and AsPC-1 cells with STAT3 blocked and in (D). MIA-ZIP4 cells with STAT3 blocked. (E). Protein level of ZEB1 in MIA-ZIP4 cells treated with 10 μM WP1066 (STAT3 inhibitor) for 48 h. (F). Relative promoter activity of ZEB1 in MIA-ZIP4 cells treated with 10 μM WP1066 for 48 h. Cells were treated with DMSO as negative controls.
ZIP4, ZEB1 and integrins regulate pancreatic cancer metastasis and chemoresistance through gemcitabine transporter ENT1.
ENT1 is one of the major routes for gemcitabine uptake by pancreatic cancer cells27, 28. We sought to determine whether ZIP4 impacted ENT1 expression in cells treated with gemcitabine in vitro. ENT1 levels were reduced in cells overexpressing ZIP4 exposed to gemcitabine, but was increased when ZIP4 was knocked down (Fig. 6A). Having demonstrated that ZIP4 upregulates the transcription factor ZEB1, we hypothesized that ZIP4 might regulate ENT1 via its effect on ZEB1. Our data showed that ZEB1 mediated ZIP4-caused downregulation of ENT1 expression; knock down of ZEB1 restored the expression of ENT1 in MIA-ZIP4 cells, and overexpression of ZEB1 downregulated ENT1 in AsPC-shZIP4 cells (Fig. 6B and 6C). Furthermore, ENT1 expression was rescued by knockdown of integrin α3β1 in MIA-ZIP4 cells (Fig. 6D), and overexpression of integrin α3β1 decreased the ENT1 expression in ASPC-shZIP4 cells treated with gemcitabine (Fig. 6E). Those results suggest that ZIP4, ZEB1 and integrins regulate pancreatic cancer metastasis and chemoresistance through gemcitabine transporter ENT1.
Figure 6. ZIP4, ZEB1 and integrins regulate pancreatic cancer metastasis and chemoresistance through gemcitabine transporter ENT1.

After 48 h treatment with gemcitabine, ENT1 expression was determined in (A). AsPC-1 and MIA PaCa-2 cells. (B). MIA-ZIP4 siZEB1 cells. (C). AsPC-shZIP4-ZEB1 cells (transient transfection). (D). MIA-ZIP4 siITGA3 or siITGB1 cells. (E). AsPC-shZIP4 cells with ITGA3 or ITGB1 overexpressed (transient transfection).
Integrin α3β1 inhibits ENT1 expression through activation of JNK.
Next, we examined how integrin α3β1 regulates ENT1 expression. It has been shown that gemcitabine treatment induced MAP kinase activation such as JNK, which led to ENT1 inhibition and contributed to gemcitabine resistance29. We found that when integrin α3β1 was knocked down, pJNK was inhibited in MIA PaCa-2 and AsPC-1 cells under gemcitabine treatment (Fig. 7A–7B). Knocking down of integrin α3β1 led to upregulation of ENT1 both at mRNA and protein level (Fig. 7A–7B, Fig. S6A–S6B), and increased ENT1 promoter activity (Fig. 7C–7D). In order to investigate whether phosphorylation of JNK contribute to ENT1 downregulation, we did the luciferase reporter assay and transfected the cells with ENT1 promoter and treated the cells with JNK inhibitor SP600125 which specifically inhibits JNK phosphorylation30 and we found inhibition of pJNK increased ENT1 promoter activity (Fig. 7E–7F). We also found inhibition of JNK with the inhibitor SP600125 could increase ENT1 expression both in MIA PaCa-2 and AsPC-1 cells (Fig. 7G). These data indicate that ZIP4 inhibits ENT1 expression through the ZEB1-integrin-JNK signaling pathway.
Figure 7. Integrin α3β1 inhibits ENT1 expression through activation of JNK.

(A-B). Expression of ENT1 and JNK in MIA PaCa-2 and AsPC-1 cells with ITGA3 or ITGB1 blocked under 200 μM gemcitabine for 24 h. (C-D). Promoter activity of ENT1 with ITGA3 or ITGB1 blocked (siRNA). (E-F). Promoter activity of ENT1 with 20 μM JNK inhibitor SP600125 for 24h. (G). Protein levels of ENT1 and JNK in MIA PaCa-2 and AsPC-1 cells under 20 μM JNK inhibitor SP600125 treatment for 3 h and 200μM gemcitabine treatment for 24 h. (H). Schematic diagram of the ZIP4-STAT3-ZEB1-integrin-JNK-ENT1 signaling pathway.
Discussion
Our previous studies have shown that ZIP4 contributes to pancreatic cancer pathogenesis and progression17–19, 21. This finding raised the question of whether ZIP4 may play an additional and previously uncharacterized role in pancreatic cancer tumorigenesis, EMT, metastasis, and resistance to therapy. In the present study, we have identified the underlying mechanism contributing to pancreatic cancer metastasis and chemoresistance: the ZIP4-ZEB1-integrin signaling pathway (Fig. 7H). Low ZIP4 levels statistically correlated with better survival in a large cohort of pancreatic adenocarcinoma patients who had undergone gemcitabine treatment. Following this discovery, we investigated the association of ZIP4 and metastasis and gemcitabine resistance using gain- and loss-of-function approaches both in vitro and in vivo. We found that ZIP4 overexpression enhanced cell growth and chemoresistance in pancreatic cancer cells, while knock down of ZIP4 reduced metastasis and sensitized pancreatic cancer cells to chemotherapy, in cultured cell lines, tumor 3D spheroid model, xenograft and transgenic mouse models. ZIP4 rendered chemo-resistance is largely due to a zinc dependent EMT like transcription factor ZEB1, which was activated by phosphorylated STAT3 in pancreatic cancer cells. When taken together, these results suggest that targeted ZIP4 knock down in combination with gemcitabine may be a new and effective strategy for pancreatic cancer therapy. Additionally, we showed that knockdown of integrin α3β1, downstream target of ZIP4 and ZEB1, increased the sensitivity of pancreatic cancer cells to gemcitabine treatment through JNK mediated upregulation of ENT1.
In this study, we found ZIP4 promoted pancreatic cancer metastasis and chemoresistance to gemcitabine through activation of ZEB1, an important EMT like transcription factor which contributes to pancreatic cancer growth, metastasis and chemoresistance. Krebs et al found that ZEB1 acts as an EMT activator, which promotes pancreatic cancer progression from early precursor lesions formation to late stage metastasis in a ZEB1 knock out KPC mouse model24. They also found that ZEB1 conferred pancreatic cancer drug resistance through suppressing miR-20331. Wang et al showed that ZEB1 knockdown sensitized pancreatic cancer cells to chemotherapy32. However, previous studies have largely focused on the downstream target of ZEB1, little is known about how ZEB1 was activated and the role of ZEB1 in gemcitabine resistance of pancreatic cancer still remains unknown. Here we identified the mechanism underlying gemcitabine resistance in pancreatic cancer. This is the first time showing ZEB1 can be upregulated by a zinc transporter ZIP4 through phosphorylated STAT3 which directly binds to the promoter region of ZEB1 and activated its transcription. Activation of ZIP4-STAT3-ZEB1 signaling pathway leads to pancreatic cancer metastasis and chemoresistance.
Integrins have been shown to contribute to metastasis and resistance to chemotherapy. α2β1 integrin protects pancreatic cancer to 5-FU treatment by upregulating Bcl-27. Yang et al found ITGB1 promoted pancreatic cancer gemcitabine resistance through activating Cdc429. Collagen/ITGA1 signaling is required for gemcitabine resistance in pancreatic cancer33. However, little is known about how integrins are upregulated in pancreatic cancer. Although we found upregulation of integrin activated pJNK in pancreatic cancer, the mechanism that how integrin activates JNK and whether activation of JNK contributes to gemcitabine resistance still needs further investigation. It’s indicated that integrin mediated activation of JNK through FAK, Src or Rac. Tanaka et al found the complex of FAK/Src/p130CAS activates JNK by recruiting Crk to the plasma membrane34. Zhang at al indicated Rac is an important positive regulator of JNK activation dependent on integrin35. Upon activation, JNK phosphorylates and activates transcription factors c-Jun and AP-1 and contributes to tumor progress36. ENT1 is the most widely distributed nucleoside transporter that mediates cellular delivery of antineoplastic drugs such as gemcitabine. It was previously shown that HIF1α or inflammatory cytokines can downregulate ENT1 expression. Importantly JNK is a common response to chemotherapy which can be activated by gemcitabine and Leisewitz et al found chemical stress-induced JNK activation decreases ENT1 expression in a c-Jun-dependent manner29. In the current study we demonstrated that ZEB1 can regulate integrin α3β1 through binding to their promoter regions. Integrin α3β1 promoted gemcitabine resistance through activation of JNK which downregulated gemcitabine transporter ENT1. Together we identified the mechanism that activation of ZEB1-integrin-JNK-ENT1 led to metastasis and gemcitabine resistance in pancreatic cancer.
Until now, gemcitabine-based single or combination therapy has been the standard of care for advanced pancreatic cancer. However, no combined regimen has been shown to be significantly more effective than gemcitabine alone as first-line therapy. Membrane transporters and enzymes involved in the metabolism of gemcitabine have been reported to contribute to the drug resistance in pancreatic cancer. Several candidate genes and/or pathways that may serve as therapeutic targets to counter gemcitabine resistance in pancreatic cancer have been identified to date. For example, apoptosis-related genes Bcl-xL and Bcl-237 and an activated Notch signaling pathway38 have been linked to gemcitabine resistance. Pancreatic cancer cells became sensitized to gemcitabine following decreased HMGA1 gene expression, which itself has been reported to promote gemcitabine chemoresistance through an Akt-dependent mechanism39. In addition, several participants in pyrimidine nucleoside metabolism, such as DCK and RRM1 have been shown to be involved in gemcitabine resistance40, 41. However, the underlying mechanism of drug resistance in pancreatic cancer is still not clear. The current study described an important signaling cascade for pancreatic cancer chemoresistance, involving ZIP4 acting through a series of intermediate steps to modulate ENT1 (Fig. 6 and 7). ENT1 expression significantly correlates with the median survival of pancreatic cancer after gemcitabine treatment. Pancreatic cancer cells lacking ENT1 are highly resistant to gemcitabine. Following uptake gemcitabine is activated by deoxycytidine kinase (DCK)42, and the activated gemcitabine exerts its cytotoxicity by inhibiting ribonucleotide reductase (RRM1/RRM2), which drives DNA damage repair in the cells43. RRM1 gene upregulation is associated with gemcitabine resistance in non-small cell lung cancer44, and downregulation of DCK is reported in gemcitabine-resistant pancreatic cancer cell lines40. Our data indicate that ENT1 is the major downstream target in ZIP4-mediated chemoresistance in pancreatic cancer. Nanhan et al found that in multiple myeloma gemcitabine activity requires caspase activation45. Similarly, we previously reported that ZIP4 confers resistance against low zinc-induced apoptosis in pancreatic cancer cells by inhibiting caspases20. This suggests that ZIP4 may sensitize pancreatic cancer cells to gemcitabine by regulating caspase activity and via the apoptosis pathway. Next, we will determine the expression of DCK, RRM1 in regulation of pancreatic cancer gemcitabine resistance and investigate the correlations between ZIP4 and those markers. Further studies are warranted to understand the cross talk between the gemcitabine uptake and apoptosis in pancreatic cancer. New technologies allowing for detecting trace levels of chemotherapy drugs in cancer cells facilitated the development of new treatment in PC targeting resistance pathways. Recently, liquid chromatography-mass spectrometry (LC-MS) method was used to accurately measure gemcitabine levels in human pancreatic cancer tissue46, 47. In other studies, gemcitabine, dFdU, and their phosphorylated metabolites (e.g., dFdCTP) were measured in human and mouse plasma and tumor tissue. Importantly, LC-MS can simultaneously quantify gemcitabine, the active metabolite dFdCTP, and the inactive metabolite dFdU in both tumor tissue and plasma in pancreatic cancer studies6, 15.
Despite considerable efforts, little progress has been made towards improving the efficacy of gemcitabine-based chemotherapy in pancreatic cancer. Many other chemotherapeutic drugs also showed limited survival advantage in pancreatic cancer, and more specific targets with clinical importance in pancreatic cancer have not been identified. Thus, there is a great need to identify specific and potent targeted therapies to overcome drug resistance and prolong survival in pancreatic cancer. Our previous studies showed that ZIP4 downregulation can significantly increase the survival of mice with pancreatic cancer xenografts17. ZIP4 knock down and gemcitabine treatment significantly extended survival compared with gemcitabine alone in pancreatic cancer mouse models. Follow-up data from studies using human pancreatic cancer tissues further confirmed these findings. Our data support the hypothesis that ZIP4 plays a critical role in regulating gemcitabine resistance in pancreatic cancer, potentially through an ENT1-dependent mechanism. A combination of ZIP4 knockdown and gemcitabine could be a promising new therapy to conquer metastasis and drug resistance in pancreatic cancer.
In conclusion, we have shown that ZIP4 promotes pancreatic cancer tumorigenesis, metastasis, and resistance to therapy by upregulating its downstream target integrin α3β1, and that is mediated directly by an EMT like transcription factor ZEB1. Furthermore, ZIP4 inhibits gemcitabine uptake and accumulation into pancreatic cancer cells by downregulating ENT1. We have delineated a signaling pathway by which ZIP4 increases gemcitabine resistance by ultimately inhibiting ENT1-mediated gemcitabine uptake. ZIP4 acts on ENT1 through a newly identified ZEB1-integrin pathway. Our data suggest that ZIP4 is the crucial regulator of this ZEB1-integrin pathway and ultimately of metastasis and gemcitabine resistance and as a result may be an effective therapeutic strategy of pancreatic cancer.
Supplementary Material
What you need to know.
BACKGROUND AND CONTEXT: The solute carrier family 39 member 4 (SLC39A4, also called ZIP4) regulates concentrations of intracellular zinc and is increased in pancreatic cancer cells.
NEW FINDINGS: In studies of pancreatic cancer cell lines and mice, we found that ZIP4 increases expression of the transcription factor ZEB1, which activates integrin α3β1 signaling. This signaling inhibits expression of the gemcitabine transporter ENT1, so that the cells no longer take up this drug.
LIMITATIONS: This study was performed in cell lines and mice; further studies in humans are needed.
IMPACT: Activation of this pathway might help mediate resistance of pancreatic tumors to chemotherapeutic agents. Strategies to block this pathway might be developed to increase the sensitivity of pancreatic tumors to chemotherapy.
Grant support:
This work was supported in part by the National Institutes of Health (NIH) grants R01 CA186338-01A1, R01 CA203108, the William and Ella Owens Medical Research Foundation (Li M), and the NIH/NCI award P30CA225520.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Reymond N, d’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer 2013;13:858–70. [DOI] [PubMed] [Google Scholar]
- 3.Samulitis BK, Pond KW, Pond E, et al. Gemcitabine resistant pancreatic cancer cell lines acquire an invasive phenotype with collateral hypersensitivity to histone deacetylase inhibitors. Cancer Biol Ther 2015;16:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamisawa T, Wood LD, Itoi T, et al. Pancreatic cancer. Lancet 2016;388:73–85. [DOI] [PubMed] [Google Scholar]
- 5.da Rocha Lino A, Abrahao CM, Brandao RM, et al. Role of gemcitabine as second-line therapy after progression on FOLFIRINOX in advanced pancreatic cancer: a retrospective analysis. J Gastrointest Oncol 2015;6:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bapiro TE, Richards FM, Goldgraben MA, et al. A novel method for quantification of gemcitabine and its metabolites 2′,2′-difluorodeoxyuridine and gemcitabine triphosphate in tumour tissue by LC-MS/MS: comparison with (19)F NMR spectroscopy. Cancer Chemother Pharmacol 2011;68:1243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aoudjit F, Vuori K. Integrin signaling in cancer cell survival and chemoresistance. Chemother Res Pract 2012;2012:283181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grzesiak JJ, Ho JC, Moossa AR, et al. The integrin-extracellular matrix axis in pancreatic cancer. Pancreas 2007;35:293–301. [DOI] [PubMed] [Google Scholar]
- 9.Yang D, Tang Y, Fu H, et al. Integrin beta1 promotes gemcitabine resistance in pancreatic cancer through Cdc42 activation of PI3K p110beta signaling. Biochem Biophys Res Commun 2018;505:215–221. [DOI] [PubMed] [Google Scholar]
- 10.Cooper J, Giancotti FG. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019;35:347–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Visser F, King KM, et al. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev 2007;26:85–110. [DOI] [PubMed] [Google Scholar]
- 12.Hagmann W, Jesnowski R, Lohr JM. Interdependence of gemcitabine treatment, transporter expression, and resistance in human pancreatic carcinoma cells. Neoplasia 2010;12:740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farrell JJ, Elsaleh H, Garcia M, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology 2009;136:187–95. [DOI] [PubMed] [Google Scholar]
- 14.Shah AN, Summy JM, Zhang J, et al. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol 2007;14:3629–37. [DOI] [PubMed] [Google Scholar]
- 15.van Nuland M, Hillebrand MJX, Rosing H, et al. Ultra-sensitive LC-MS/MS method for the quantification of gemcitabine and its metabolite 2′,2′-difluorodeoxyuridine in human plasma for a microdose clinical trial. J Pharm Biomed Anal 2018;151:25–31. [DOI] [PubMed] [Google Scholar]
- 16.Li M, Zhang Y, Liu Z, et al. Aberrant expression of zinc transporter ZIP4 (SLC39A4) significantly contributes to human pancreatic cancer pathogenesis and progression. Proc Natl Acad Sci U S A 2007;104:18636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Zhang Y, Bharadwaj U, et al. Down-regulation of ZIP4 by RNA interference inhibits pancreatic cancer growth and increases the survival of nude mice with pancreatic cancer xenografts. Clin Cancer Res 2009;15:5993–6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Yang J, Cui X, et al. A novel epigenetic CREB-miR-373 axis mediates ZIP4-induced pancreatic cancer growth. EMBO Mol Med 2013;5:1322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu M, Yang J, Zhang Y, et al. ZIP4 Promotes Pancreatic Cancer Progression by Repressing ZO-1 and Claudin-1 through a ZEB1-Dependent Transcriptional Mechanism. Clin Cancer Res 2018;24:3186–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui X, Zhang Y, Yang J, et al. ZIP4 confers resistance to zinc deficiency-induced apoptosis in pancreatic cancer. Cell Cycle 2014;13:1180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Zhang Z, Zhang Y, et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice With Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 2019;156:722–734 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan N, Standke SJ, Kothapalli NR, et al. Quantification of Drug Molecules in Live Single Cells Using the Single-Probe Mass Spectrometry Technique. Anal Chem 2019;91:9018–9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naci D, Vuori K, Aoudjit F. Alpha2beta1 integrin in cancer development and chemoresistance. Semin Cancer Biol 2015;35:145–53. [DOI] [PubMed] [Google Scholar]
- 24.Krebs AM, Mitschke J, Lasierra Losada M, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol 2017;19:518–529. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Bharadwaj U, Logsdon CD, et al. ZIP4 regulates pancreatic cancer cell growth by activating IL-6/STAT3 pathway through zinc finger transcription factor CREB. Clin Cancer Res 2010;16:1423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin L, Deangelis S, Foust E, et al. A novel small molecule inhibits STAT3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells. Mol Cancer 2010;9:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spratlin J, Sangha R, Glubrecht D, et al. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res 2004;10:6956–61. [DOI] [PubMed] [Google Scholar]
- 28.Koay EJ, Truty MJ, Cristini V, et al. Transport properties of pancreatic cancer describe gemcitabine delivery and response. J Clin Invest 2014;124:1525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leisewitz AV, Zimmerman EI, Huang M, et al. Regulation of ENT1 expression and ENT1-dependent nucleoside transport by c-Jun N-terminal kinase. Biochem Biophys Res Commun 2011;404:370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li JY, Huang JY, Xing B, et al. SP600125, a JNK inhibitor, suppresses growth of JNK-inactive glioblastoma cells through cell-cycle G2/M phase arrest. Pharmazie 2012;67:942–6. [PubMed] [Google Scholar]
- 31.Meidhof S, Brabletz S, Lehmann W, et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med 2015;7:831–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, Chen Y, Lin Y, et al. Novel crosstalk between KLF4 and ZEB1 regulates gemcitabine resistance in pancreatic ductal adenocarcinoma. Int J Oncol 2017;51:1239–1248. [DOI] [PubMed] [Google Scholar]
- 33.Gharibi A, La Kim S, Molnar J, et al. ITGA1 is a pre-malignant biomarker that promotes therapy resistance and metastatic potential in pancreatic cancer. Sci Rep 2017;7:10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka S, Ouchi T, Hanafusa H. Downstream of Crk adaptor signaling pathway: activation of Jun kinase by v-Crk through the guanine nucleotide exchange protein C3G. Proc Natl Acad Sci U S A 1997;94:2356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Rivera Rosado LA, Moon SY, et al. Silencing of D4-GDI inhibits growth and invasive behavior in MDA-MB-231 cells by activation of Rac-dependent p38 and JNK signaling. J Biol Chem 2009;284:12956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brakebusch C, Bouvard D, Stanchi F, et al. Integrins in invasive growth. J Clin Invest 2002;109:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang M, Lu X, Dong X, et al. pERK1/2 silencing sensitizes pancreatic cancer BXPC-3 cell to gemcitabine-induced apoptosis via regulating Bax and Bcl-2 expression. World J Surg Oncol 2015;13:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Li Y, Ahmad A, et al. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim Biophys Acta 2010;1806:258–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liau SS, Whang E. HMGA1 is a molecular determinant of chemoresistance to gemcitabine in pancreatic adenocarcinoma. Clin Cancer Res 2008;14:1470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saiki Y, Yoshino Y, Fujimura H, et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem Biophys Res Commun 2012;421:98–104. [DOI] [PubMed] [Google Scholar]
- 41.Minami K, Shinsato Y, Yamamoto M, et al. Ribonucleotide reductase is an effective target to overcome gemcitabine resistance in gemcitabine-resistant pancreatic cancer cells with dual resistant factors. J Pharmacol Sci 2015;127:319–25. [DOI] [PubMed] [Google Scholar]
- 42.Giovannetti E, Del Tacca M, Mey V, et al. Transcription analysis of human equilibrative nucleoside transporter-1 predicts survival in pancreas cancer patients treated with gemcitabine. Cancer Res 2006;66:3928–35. [DOI] [PubMed] [Google Scholar]
- 43.Kim MP, Gallick GE. Gemcitabine resistance in pancreatic cancer: picking the key players. Clin Cancer Res 2008;14:1284–5. [DOI] [PubMed] [Google Scholar]
- 44.Lee JJ, Maeng CH, Baek SK, et al. The immunohistochemical overexpression of ribonucleotide reductase regulatory subunit M1 (RRM1) protein is a predictor of shorter survival to gemcitabine-based chemotherapy in advanced non-small cell lung cancer (NSCLC). Lung Cancer 2010;70:205–10. [DOI] [PubMed] [Google Scholar]
- 45.Nabhan C, Gajria D, Krett NL, et al. Caspase activation is required for gemcitabine activity in multiple myeloma cell lines. Mol Cancer Ther 2002;1:1221–7. [PubMed] [Google Scholar]
- 46.Duan Q, Zhao H, Zhang Z, et al. Mechanistic Evaluation and Translational Signature of Gemcitabine-induced Chemoresistance by Quantitative Phosphoproteomics Analysis with iTRAQ Labeling Mass Spectrometry. Sci Rep 2017;7:12891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coleman O, Henry M, O’Neill F, et al. A Comparative Quantitative LC-MS/MS Profiling Analysis of Human Pancreatic Adenocarcinoma, Adjacent-Normal Tissue, and Patient-Derived Tumour Xenografts. Proteomes 2018;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.