

Abstract
Cancer immunotherapies that train or stimulate the inherent immunological systems to recognize, attack, and eradicate tumor cells with minimal damage to healthy cells have demonstrated promising clinical responses in recent years. However, most of these immunotherapeutic strategies only benefit a small subset of patients and cause systemic autoimmune side effects in some patients. Immunogenic cell death (ICD)-inducing modalities not only directly kill cancer cells but also induce antitumor immune responses against a broad spectrum of solid tumors. Such strategies for generating vaccine-like functions could be used to stimulate a “cold” tumor microenvironment to become an immunogenic, “hot” tumor microenvironment, working in synergy with immunotherapies to increase patient response rates and lead to successful treatment outcomes. This Minireview will focus on nanoparticle-based treatment modalities that can induce and enhance ICD to potentiate cancer immunotherapy.
Keywords: cancer, immunogenic cell death, immunotherapy, nanoparticles
Graphical Abstract
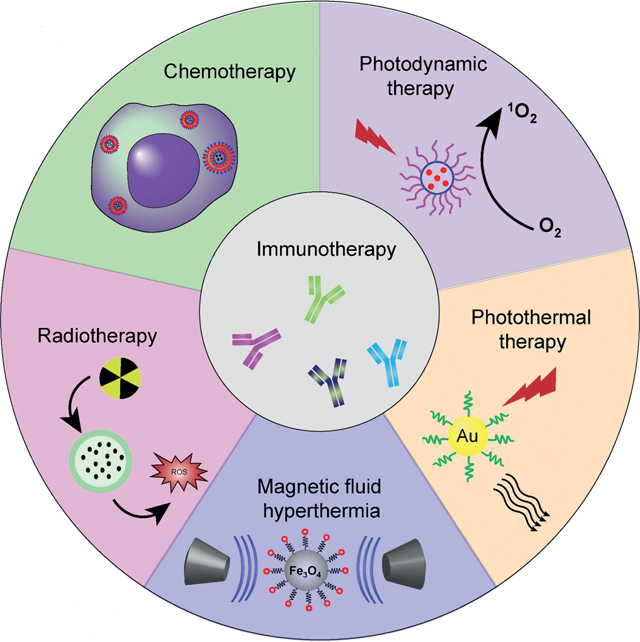
1. Introduction
Cancer immunotherapies have enjoyed rapid clinical progress over the past few years, particularly in the areas of chimeric antigen receptor (CAR)-modified T cells and immune modulation by blocking suppressive immune checkpoints.[1] The generation of an anti-cancer immune response consists of several key steps (Figure 1):[2] Antigens released from cancer cells are captured by antigen-presenting cells (APCs). Then, danger associated molecular patterns (DAMPs), such as pro-inflammatory cytokines and factors, released by the dying tumor cells signal APC maturation. Activated APCs travel to the lymph nodes to present the tumor antigens on major histocompatibility complex (MHC) I and MHC II molecules to T cells, resulting in the priming and activation of effector T cell responses against cancer-specific antigens. Finally, activated effector T cells traffic to and infiltrate the tumor bed where they specifically recognize cancer cells through interactions between T cell receptor (TCR) and MHC I-bound cognate antigen and kill cancer cells. Killing the cancer cell releases additional tumor-associated antigens (TAAs) to increase the breadth and depth of the immune response in subsequent revolutions of the cycle.
Figure 1.
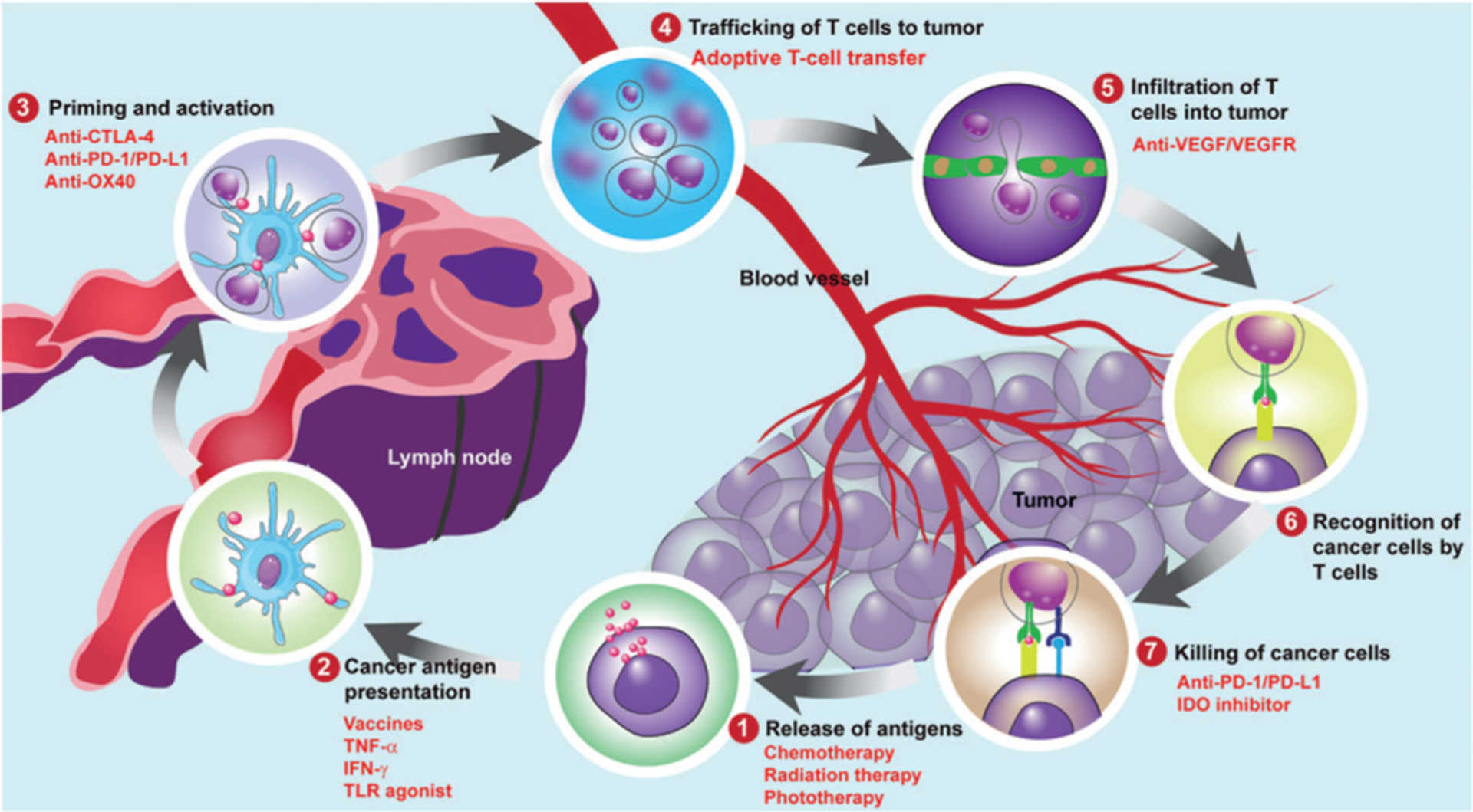
The cancer-immunity cycle and therapies that might affect the cycle.
However, cancer cells are able to evade the host immune system by blocking one or more of these steps.[3] This can occur by down-regulating surface antigen and MHC I expression to inhibit recognition and attack by the immune system, inducing expression of immunosuppressive molecules, and infiltrating inhibitory immune cells into the tumor microenvironment (TME) to inhibit effector T cell homing and activity.[4] Current cancer immunotherapies usually focus on one of two strategies: 1) To stimulate key players of the immune system, such as cancer vaccines,[5] cytokine therapy,[6] and adoptive T-cell transfer[7] and 2) to eliminate or inhibit immunosuppressive factors, such as immune checkpoint-blockade (ICB) therapy.[8] Although cancer immunotherapies have demonstrated some exciting clinical responses, they are limited by high costs, efficacy restricted to certain subsets of patients, resistance to treatment, and dose-limiting autoimmune effects like cytokine release syndrome.[9]
It is now established that ablative cancer treatments, such as radiotherapy (RT), photodynamic therapy (PDT), hyperthermia (HT) and photothermal therapy (PTT), and certain chemotherapeutics can cause tumor cell death in an immunogenic way.[10] This immunogenic cell death (ICD) is characterized by release of TAAs, DAMPs, and pro-inflammatory cytokines, which facilitates the presentation of TAAs to adaptive immune cells, eliciting an antigen-specific immune response against a broad spectrum of solid tumors.[11] Ultimately, ICD can enhance immune stimulatory or subvert immune suppressive effects for the activation, proliferation, and tumor infiltration of T cells to synergize with current immunotherapies. Such in situ “tumor vaccines” provide a new way to broaden and enhance immunotherapy by combining it with ICD-inducing modalities, such as radio-, photo-, and chemotherapy. In this Minireview, we will first discuss the characteristics and design parameters of nanoparticles (NPs) that allow for maximal immunotherapeutic efficacy, then provide an overview of nanoparticle platforms used to combine ICD-inducing modalities and immunotherapy in the emerging and rapidly growing field of NP-mediated cancer immunotherapy.
2. Immunogenic Cell Death (In Situ Cancer Vaccination)
During ICD, DAMPs and TAAs are released, captured by dendritic cells (DCs) and macrophages, then processed and presented to adaptive immune cells, leading to an antigen-specific immune response. Many immunogenic factors throughout apoptotic cell death have been identified as DAMPs, such as pre-apoptotic calreticulin (CRT) exposure on the cell surface,[12] ATP release during the blebbing phase of apoptosis,[13] and post-apoptotic exodus of high mobility group box 1 (HMGB-1)[14] and heat shock proteins (HSPs).[15] ATP serves as a chemoattractant to recruit APCs; CRTacts as an “eat-me” signal to facilitate the engulfment of dying tumor cells and their debris by APCs.[16] Finally, HMGB-1 and HSPs stimulate optimal antigen presentation to T cells (Figure 2).[14a,17] ICD may also induce a strong inflammatory response by releasing pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β.[11a,18] Such strategies to generate vaccine-like effects in situ do not require sophisticated procedures, have no ethical concerns, and are able to efficiently elicit selective and strong protections by local in situ cell death and systemic cell-mediated immune responses. Leveraging the ICD effects of traditional treatment modalities can increase infiltration of immune effector cells to convert an immunosuppressive TME to an immunogenic TME, and ultimately increase patient response rates to immune adjuvants and checkpoint blockade immunotherapy.
Figure 2.
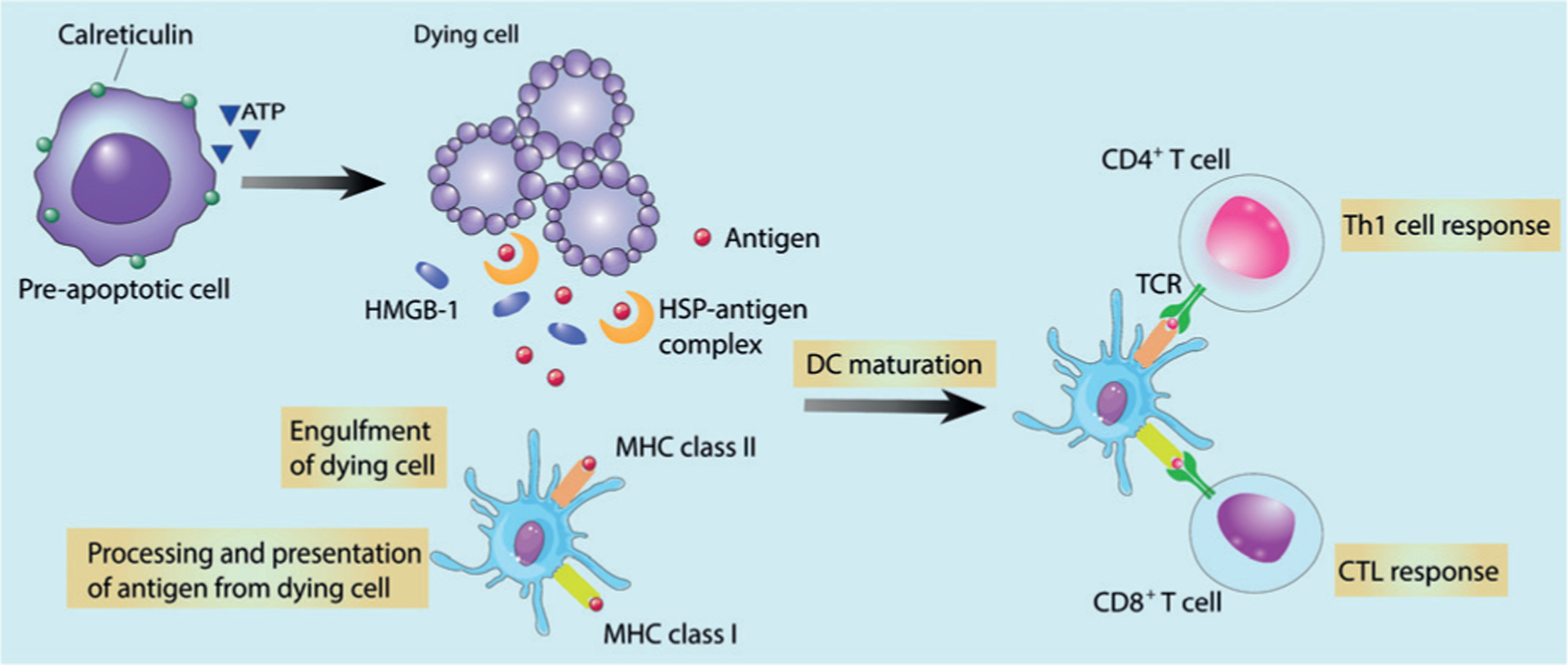
In immunogenic death, dying tumor cells expose CRT, secrete ATP, and release HMGB-1, HSPs, TSAs, and HSP-TSA complexes, all of which favor the engulfment of cell corpses and debris by antigen-presenting cells (mainly DCs) and promote DC maturation. Activated DCs can then prime CD4+ and CD8+ T cells and thereby trigger immunogenic T helper 1 (Th1) cell and cytotoxic T lymphocyte (CTL) responses, respectively. Adapted from Ref. [19] with permission. Copyright 2009, Nature Publishing Group.
3. Design of Nanoparticles for ICD
For in situ cancer vaccines, the ICD inducer needs to specifically target tumors. Compared to conventional approaches, nanotechnology offers an opportunity for the efficient delivery of an optimal dose of ICD inducers to specific tissues or cell types, enhancing their potency while reducing their side effects.[20] NPs can co-load multi-components for simultaneous delivery, protect the payloads from degradation and premature release, and passively or actively target tumors by the enhanced permeability and retention (EPR) effect or surface modification with ligands, respectively.[21] Typically, polymeric NPs are designed with selective shapes and sizes, for intracellular release of loaded small molecules, and surface functionalization for delivery beyond biological barriers.[22] Furthermore, inorganic NPs can be used as a localized source of ICD-inducing treatment or accentuate treatment by external energy fields to minimize damage to healthy tissues. Metals in inorganic NPs are chosen for biocompatibility and intrinsic behavior such as magnetic susceptibility and colloidal stability.[23]
NPs with imaging functions can monitor the localization and treatment effect in real-time, while NPs with an immunomodulatory effect can act as adjuvants or immune potentiators. Furthermore, NPs can be tailor-made in terms of size, shape, structure, payload, and surface properties for transporting themselves and their cargoes through biological barriers and enhancing accumulation in the solid tumor.[24]
4. Application of Nanoparticles in Enhancing Cancer Immunotherapy
4.1. Hyperthermia
Hyperthermia (HT), the heating of tissue to 39–45°C, was shown to activate an antitumor immune response by inducing a cell stress known as the unfolded protein response, which leads to up-regulation of HSPs on the cell membrane and TAAs within the tumor cells. The released TAAs and TAA–HSP complexes facilitate the uptake by APCs and aid in the presentation of antigens through MHC class I, activating antigen-specific killer T cells that undergo clonal expansion and traffic to all tumors (primary and metastatic) to kill tumor cells directly.[25] In addition, the fever-like temperature created by HT has been shown to enhance antigen presentation, improve tumor blood flow, and accelerate leukocyte trafficking to the tumor.[26] Both treatment efficacy and elicited immune response were found to be dependent on the thermal dose, which requires careful optimization to balance treatment of the primary tumors and the induction of immunological effects against distant tumors. For example, coagulative ablation strategies, in which high temperatures “melt” the tumor tissue and cut off intratumoral (i.t.) blood flow, showed the most destructive effects on the primary tumor but likely also formed a barrier for immune cell infiltration and immune induction.[27] Another important consideration is that normal tissues may also be severely damaged under conventional hyperthermia treatment, thus, specifically heating the tumor region to the desired temperature without damaging surrounding normal tissues is a major technical barrier. Fortunately, NP-assisted hyperthermia offers a means for controlling the thermal dose and localizing the treatment.[28]
4.1.1. Magnetic Fluid Hyperthermia
Magnetic fluid hyperthermia (MFH) involves the combination of magnetic NPs with an alternating magnetic field (AMF), which induces local hyperthermia in tumors in a controlled and uniform manner without the limitation of penetration depth. Toraya-Brown et al. stimulated 100 nm Fe3O4 BNF-Starch magnetic NPs with an external magnetic field to heat tumors to 43°C for 30 min,[29] while the Kobayashi group used cationic liposomes containing magnetic NPs (MNHT) to heat the local tumor tissue to above 43°C without damaging surrounding normal tissues in the presence of AMF.[30] In both instances, MFH triggered an antitumor immune response mediated by both CD8+ and CD4+ T cells, leading to elimination of both heat-treated primary tumors and unheated distant tumors and subsequent rechallenge rejection.[29,31] Interestingly, the differences in temperature did not show obvious differences in immune response. To enhance MNHT, magnetic NPs were conjugated with a melanogenesis substrate, N-propionyl cysteaminylphenol (NPrCAP), to be specifically taken up by melanoma cells and react with tyrosinase to produce highly cytotoxic free radicals, resulting in chemotherapeutic and immunotherapeutic cell death.[32] In parallel, they extended their localized hyperthermia approach in combination with immunostimulatory factors; i.t. injection of IL-2 or GM-CSF 24 h after intracellular HT resulted in complete tumor regression in 75% and 40% of mice, respectively.[33] Recombinant mouse HSP70 gene[34] and protein[35] were also able to enhance the therapeutic efficacy of MNHT.
4.1.2. Photothermal Therapy
Light-induced hyperthermia, or photothermal therapy (PTT), involves irradiation of light-absorbing agents accumulated in the tumor with NIR light to convert optical energy into heat for thermal ablation of cancer cells. Theoretically, photothermal agents are non-toxic in the dark and the light is locally applied on the tumor area, leading to high treatment efficacy with few side effects.[36] Recently, PTT has shown the ability to generate antitumor immunological effects by producing TSAs from ablated tumor cell residues.
A number of inorganic nano-agents (e.g. AuNPs and nanoshells,[37] CuS NPs,[38] GO,[39] MoS2 nanosheets,[40] or carbon nanotubes[41]) with intrinsic capacity for NIR light absorption have been developed to deliver thermal energy and immunoadjuvants. Yata et al. modified AuNPs with CpG oligodeoxynuleotides and then mixed them with a hexapod-like structured DNA containing CpG sequences, obtaining an immunostimulatory Au–DNA hydrogel. Intratumoral injection of the Au–DNA hydrogel followed by laser irradiation increased the local temperature and HSP70 mRNA levels in the tumor, improved TAA-specific IgG levels in the serum, and induced TAA-specific IFN-γ production in splenocytes, significantly retarding tumor growth and extending the survival time of tumor-bearing mice.[37] Guo et al. designed chitosan-coated hollow CuS NPs containing CpG, which could break down after laser excitation, reassemble, and transform into polymer complexes, improving CpG tumor retention, effectively eliminating the primary tumor and simultaneously inhibiting the growth of distant untreated tumors.[38]
Small molecule photothermal agents can also be co-loaded with immunostimulators into liposomes or polymeric NPs for effective combination of PTT and immunotherapy. Li et al. coated a hyaluronic acid (HA)-CpG conjugate onto fluorophore (IR-7)-loaded liposomes to activate and increase tumor infiltration of DCs and CD8+ T cells, thereby eradicating tumors in mice and inhibiting tumor metastasis.[42] Owing to its immunostimulatory effect, glycated chitosan (GC) can be used as both an immunoadjuvant and a carrier to enhance the uptake and presentation of tumor antigens, increase tumor immunogenicity, and synergize with PTT. Kumar and Srivastava developed biocompatible and biodegradable monodisperse polycaprolactone/GC/Poloxamer blend NPs encapsulating photothermal agent IR 820 for imaging and photo-immunotherapy. Though the NPs enhanced toxicity on MCF-7 cells upon laser treatment, the immune response and efficacy in vivo were not investigated.[43]
As a potent stimulator of the immune system, PTT has also shown synergy with ICB to reverse immunosuppression. Wang et al. demonstrated the first combination of anti-CTLA-4 therapy with PTT using single-wall carbon nanotubes. This stimulated DC maturation in tumor-draining lymph nodes (TDLNs), induced infiltration of effector T cells, and greatly abrogated Treg cells in non-heated distant tumors, thereby reducing tumor burden in both subcutaneous and lung metastasis models and leading to prolonged survival.[44] Similar long-term survival was later observed using Prussian blue NP-based PTT complemented with anti-CTLA-4 in a preclinical neuroblastoma model.[45] In addition, all of the mice that survived past 100 days also rejected a tumor rechallenge, illustrating immune memory. Chen et al. also demonstrated the long-term survival by combining anti-CTLA-4 with a PLGA NP co-loaded with a photothermal agent indocyanine green (ICG) and a TLR-7 agonist imiquimod (R837, Figure 3a). PLGA-ICG-R837 plus anti-CTLA-4 therapy additionally demonstrated long-term survival (70% at 70 days) in a 4T1 lung metastasis rechallenge model compared to no survival for the anti-CTLA-4 only treated mice (Figure 3b).[46] More recently, Liu et al. demonstrated that plasmonic gold nanostar-mediated PTT in combination with anti-PD-L1 immunotherapy dramatically enhanced the efficacy of immunotherapy, achieving complete eradication of primary treated tumors and distant untreated tumors in some mice implanted with MB49 bladder cancer cells. Furthermore, the treatment induced effective long-lasting immunity, rejecting the formation of challenged tumor in a period of 60 days.[47]
Figure 3.
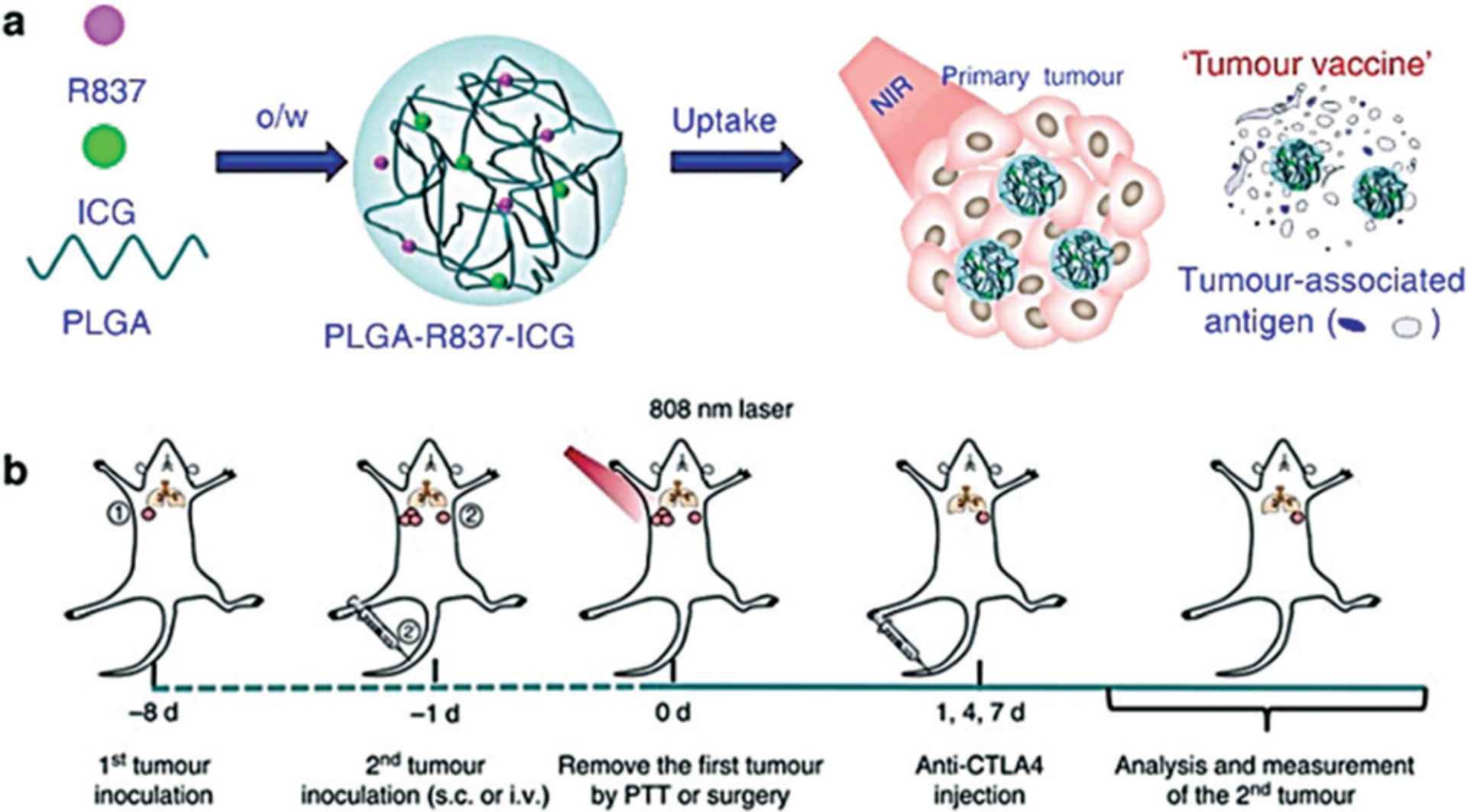
Formulation of PLGA-ICG-R837 and their immune-stimulation abilities. a) Structure of PLGA-ICG-R837 and the mechanism of antitumor immune responses induced by PTT in combination with anti-CTLA-4. b) Treatment schedule to induce a tumour vaccine and reject secondary and rechallenge tumors. Adapted from Ref. [46] with permission. Copyright 2016, Nature Publishing Group.
4.2. Photodynamic Therapy
Photodynamic therapy (PDT) is a clinically used, minimally invasive therapeutic procedure with a two-step modus operandi involving the administration of a photosensitizer (PS) followed by irradiation with a specific wavelength light. Upon irradiation, PS in the presence of oxygen generates highly cytotoxic ROS to cause oxidative stress-based cell death and disrupts tumor vasculature. In addition to local tumor ablation, PDT can increase tumor immunogenicity by inducing CRT exposure and release of tumor cell debris, which improves tumor antigen presentation and activation of T lymphocytes, resulting in the destruction of residual tumor cells and reduction in the risk of distant metastasis.[48]
Typically PSs are hydrophobic and aggregate in aqueous media, which deleteriously affects their photophysical (decreased 1O2 formation), chemical (decreased solubility), and biological (insufficient tumor localization) properties.[49] NPs can overcome these limitations and selectively deliver PSs to tumors, minimize damage to normal tissues, and reduce systemic toxicity when accompanied by spatially controlled light irradiation.[50] Yu et al. functionalized graphene oxide (GO) with an integrin αvβ6-specific HK peptide and loaded the PS 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbidea (HPPH) onto the surface of GO through hydrophobic interactions and π–π stacking. The nanophotosensitizer (nPS) exhibited high tumor uptake after intravenous (i.v.) injection and increased infiltration of cytotoxic CD8+ T lymphocytes within tumors, suppressing tumor growth upon irradiation in both subcutaneous and lung metastatic mouse models.[51]
Though PDT alone can activate the immune system, combination with immune adjuvants can further potentiate the immune response. Marrach et al. encapsulated a long-wavelength-absorbing PS, zinc phthalocyanine (ZnPc), within a polymeric NP core composed of PLGA-b-PEG, and then coated the polymeric core with CpG-modified gold NPs. Such NP-treated 4T1 cell lysate primed murine bone marrow-derived dendritic cells (BMDCs) to recognize and phagocytose PDT-killed tumor cells. This phagocytosis led to pro-inflammatory cytokines (IL-2, IL-6, IL-12, and TNF-α) release, evidence of DC maturation and activation.[52]
Combination with ICB further enhanced the effects of PDT. Duan et al. synthesized a nontoxic core–shell NP (ZnP@pyro) with a coordination polymer of Zn2+ and pyrophosphate in the core and the PS pyrolipid in the shell. In vivo, the NPs improved tumor immunogenicity upon light irradiation, particularly upon combination with PD-L1. ZnP@pyro with anti-PD-L1 eradicated the irradiated tumors, suppressed non-irradiated tumors, and prevented lung meta-stases in a 4T1 metastatic triple-negative breast cancer murine model (Figure 4). In addition, the combination therapy produced an efficient abscopal effect on two bilateral syngeneic mouse models, leading to complete inhibition of pre-existing non-irradiated tumors by generating a systemic tumor-specific cytotoxic T cell response.[53]
Figure 4.
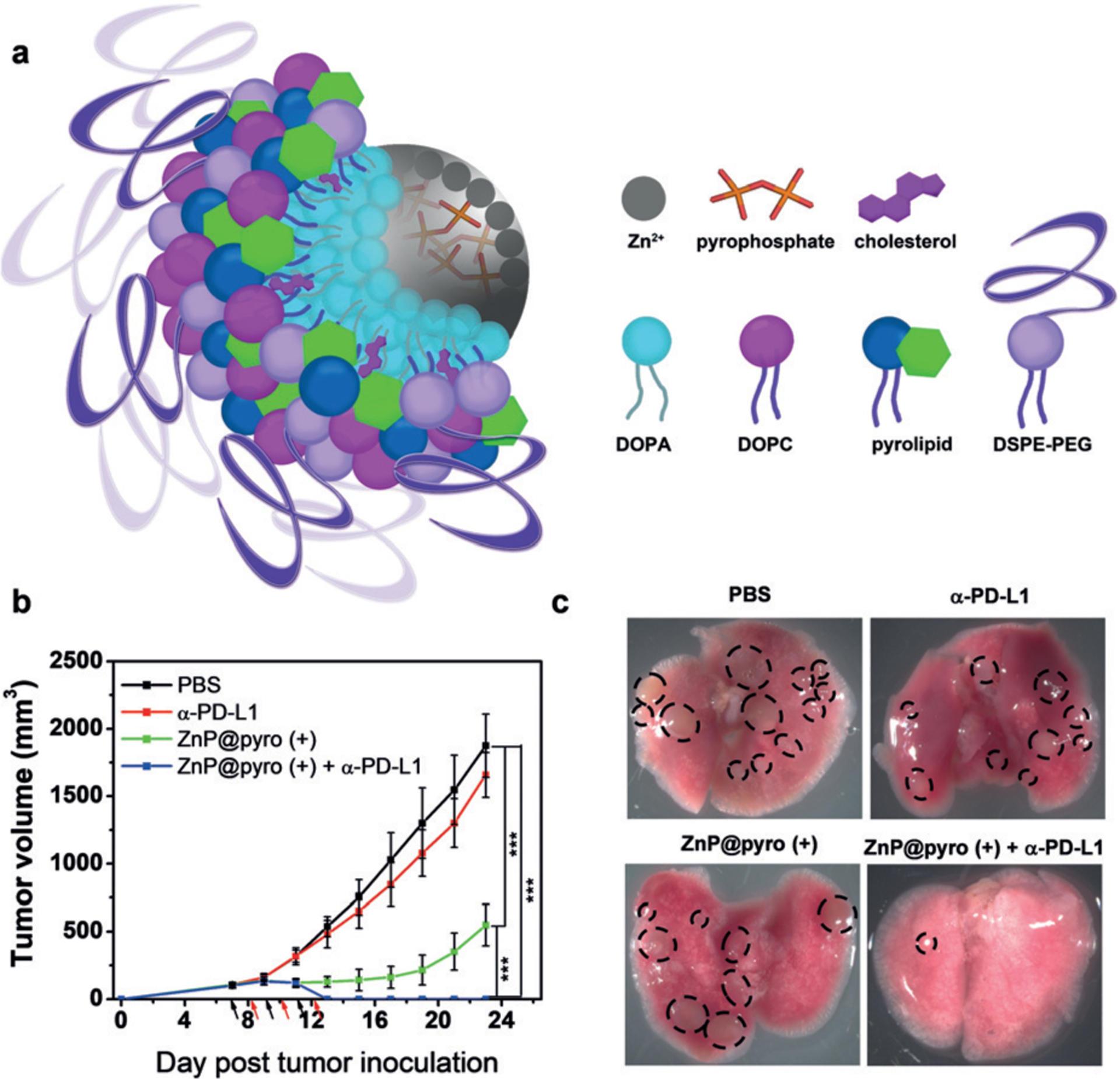
ZnP@pyro PDT enhances PD-L1 blockade immunotherapy. a) The core–shell structure of ZnP@pyro. b,c) The treatment with PDT plus anti-PD-L1 eradicated primary 4T1 cancer and reduced the metastases in the lung. Adapted from Ref. [53] with permission. Copyright 2016, American Chemical Society.
Nanoparticles are ideal to overcome tumor conditions that limit PDT, such as the requirement for oxygen to generate ROS. To overcome hypoxia in tumor, Lan et al. reported a novel nanoscale metal–organic framework (nMOF), Fe-TBP (TBP = 5,10,15,20-tetra(p-benzoato)porphyrin), consisting of Fe3O clusters and TBP ligands. When irradiated under hypoxic conditions, Fe3O clusters decomposed intracellular H2O2 to produce O2 through a Fenton-like reaction, and the generated O2 was then converted to cytotoxic 1O2 by photo-excited porphyrins. Fe-TBP mediated PDT induced significant tumor infiltration of cytotoxic T cells, sensitizing anti-PD-L1 treatment and eliciting the abscopal effect in a mouse model of colorectal cancer with greater than 90% regression of tumors (Figure 5).[54]
Figure 5.
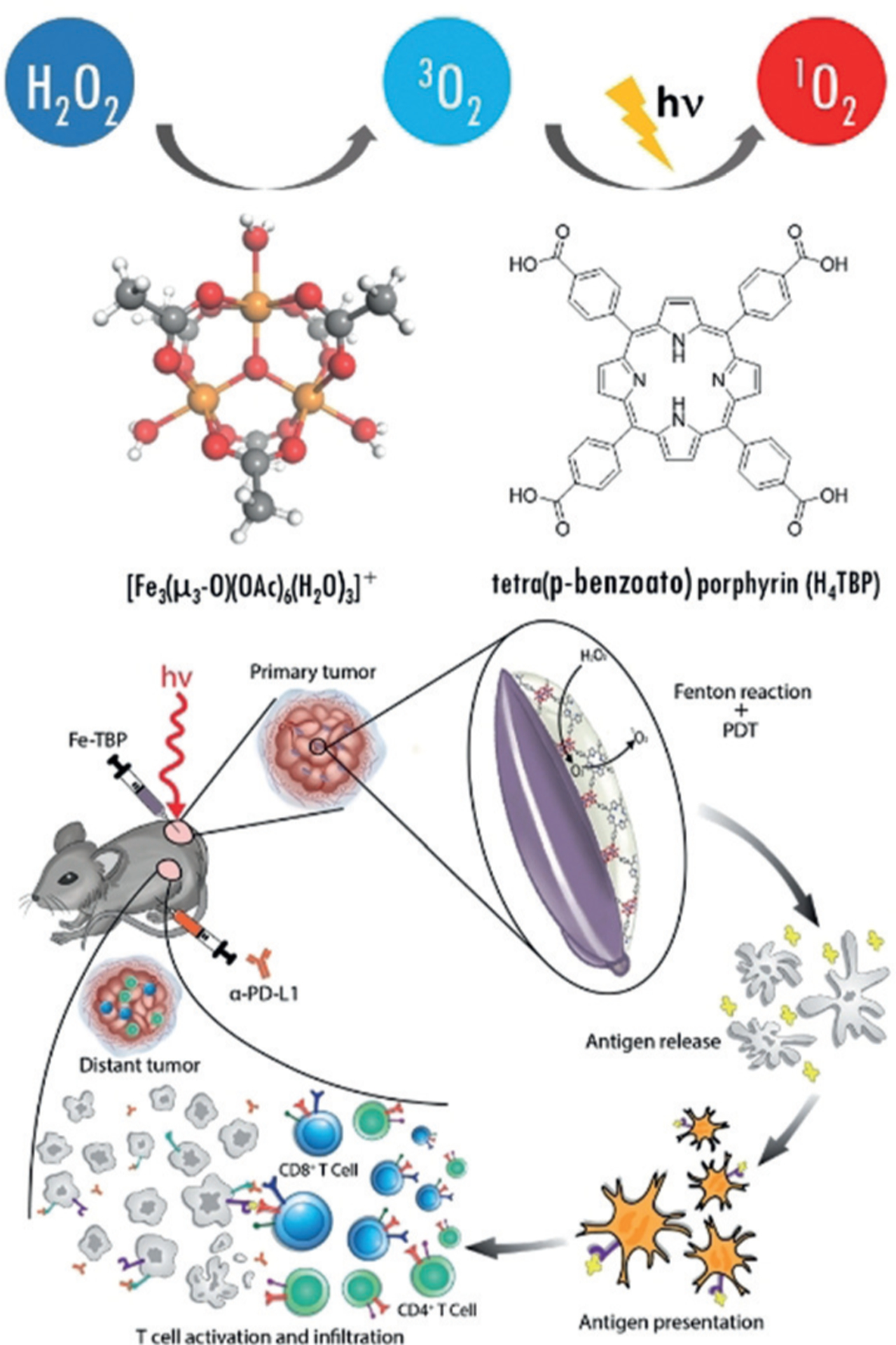
Fe-TBP decomposes H2O2 to O2 through a Fenton-like reaction, then converts O2 to cytotoxic 1O2 upon light irradiation (top). Fe-TBP overcomes tumor hypoxia for PDT-primed cancer immunotherapy. The Fe-TBP catalyzed Fenton-like reaction leads to significant antitumor response and sensitizing anti-PD-L1 treatment to induce abscopal effect (bottom). Reprinted from Ref. [54] with permission. Copyright 2018, American Chemical Society.
Wang et al. combined PDT and ICB onto a single particle with a versatile micelleplex, integrating an acid-activatable cationic micelle, a PS pheophorbide A (PPa) and a siRNA that targeted PD-L1 (Figure 6a). The micelleplex was inert at physiological pH conditions and activated only upon internalization in the acidic endocytic vesicles of tumor cells for fluorescence imaging and PDT (Figure 6b). Compared to PDT alone, the combination of PDT and PD-L1 knockdown showed significantly enhanced efficacy for inhibiting tumor growth and distant metastasis in a B16F10 melanoma xenograft tumor model (Figure 6c–e).[55]
Figure 6.
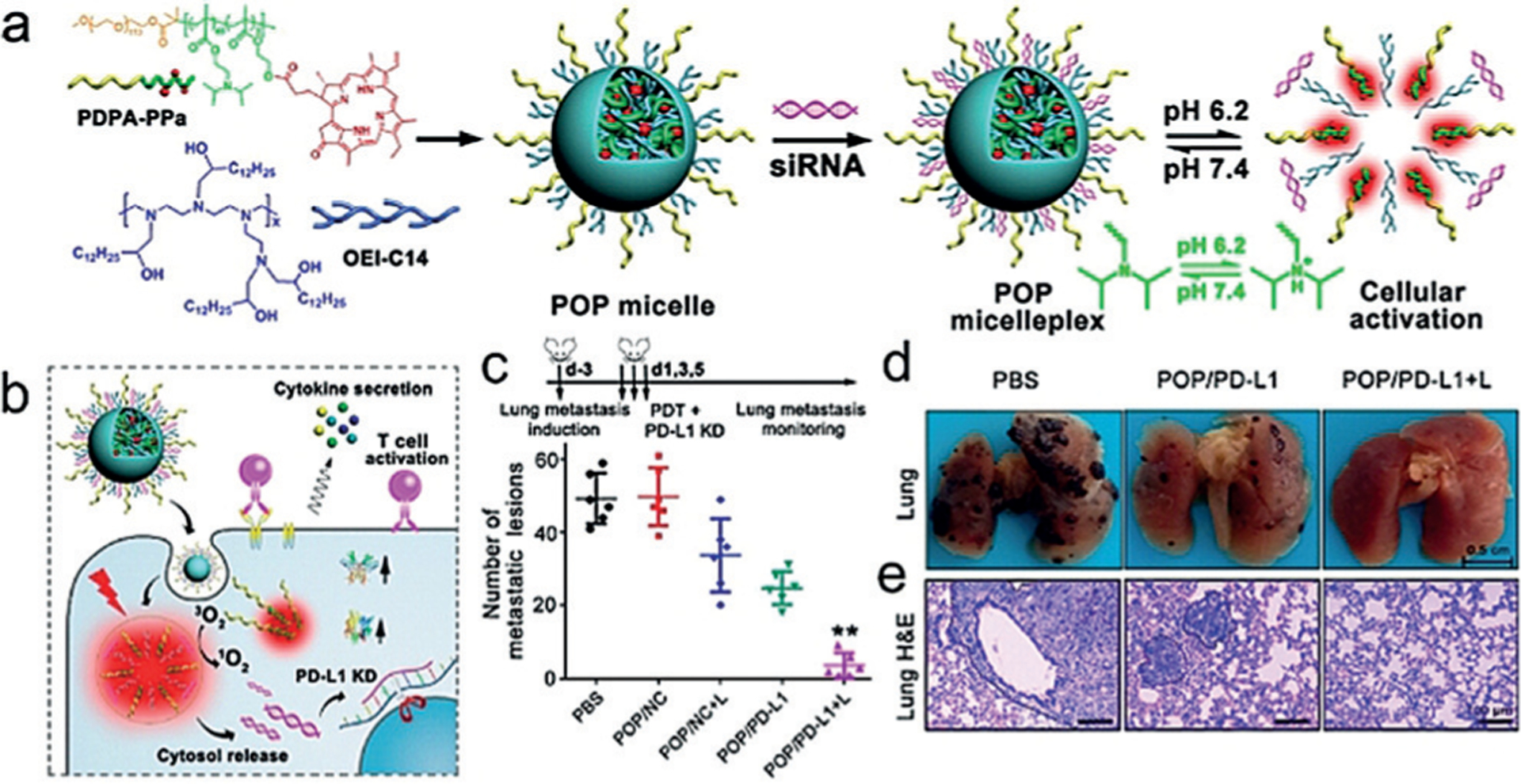
Acid-activatable micelleplexes for the combination of PDT with PD-L1 knockdown. a) Chemical structure of the acid-activatable POP micelleplexes co-loaded with PPa and siRNA. The micelleplexes dissociate in an acidic microenvironment owing to the protonation of the tertiary amino groups of PDPA. b) POP-PD-L1 micelleplex induction of ROS and release of RNAi upon cell uptake. c,d,e) Metastatic inhibition in metastatic tumor-bearing mice. Reprinted from Ref. [55] with permission. Copyright 2016, American Chemical Society.
Indoleamine 2,3-dioxygenase (IDO) inhibition has also shown significant synergy with PDT. Recently, Lu et al. showed that an IDO inhibitor (IDOi) could be loaded into the channels of a chlorin-based nMOF, TBC-Hf (TBC is 5,10,15,20-tetra(p-benzoato)chlorin), for anticancer efficacy and immune activation. In addition to increased T cell infiltration and consistent abscopal responses in mouse models of colorectal cancers, the i.t. infiltration of neutrophils and B cells was also observed, which may play compensatory roles in presenting TAAs to T cells.[56]
Song et al. recently synthesized a chimeric peptide, PpIX-1MT, by integrating the PS PpIX with a small-molecule IDOi 1MT (1-methyltryptophan). Light irradiation of PpIX-1MT NPs induced apoptosis and facilitated the expression of caspase-3 and the production of TAAs, triggering an intense immune response. Upon caspase-3 cleavage, the subsequently released 1MT inhibited the IDO pathway to help activate CD8+ T cells and synergistically inhibiting both primary and lung metastasis (Figure 7).[57]
Figure 7.
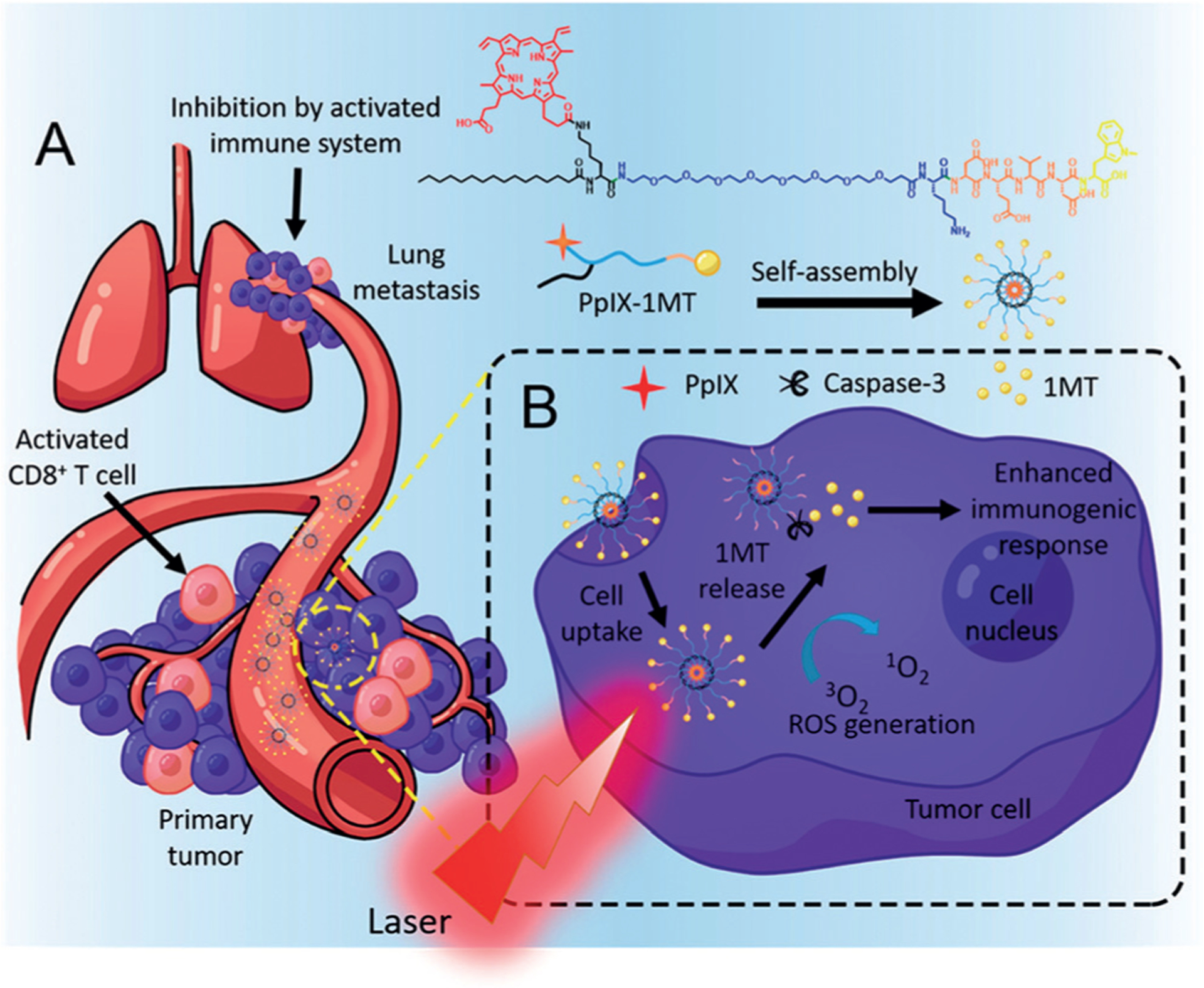
a) Structure of the chimeric peptide PpIX-1MT. The PpIX-1MT NPs accumulated in the tumor area through the EPR effect, activated the CD8+ T cells by a series of cascade activations, and inhibited both the primary tumor and lung metastasis effectively. b) In situ PDT in the primary tumor caused apoptosis of tumor cells, production of caspase-3, and release of 1MT from PpIX-1MT NPs. Reprinted from Ref. [57] with permission. Copyright 2018, American Chemical Society.
Subsequently, Xu et al. used upconversion NPs (UCNPs) to simultaneously deliver a PS chlorin e6 (Ce6) and R837. Upon irradiation of i.t. injected UCNP-Ce6-R837, the effective photodynamic destruction of tumors generated a pool of TAAs, which were able to trigger DC maturation, promote cytokines secretion, and resulted in strong antitumor immune responses. PDT with UCNP-Ce6-R837 in combination with anti-CTLA-4 not only showed excellent efficacy in eliminating tumors exposed to the NIR laser but also resulted in strong in situ vaccination, inhibiting the growth of distant tumors and protecting treated mice from tumor rechallenge.[58] Though the combination of PDT immunostimulatory and/or ICB agents consistently led to impressive anticancer efficacy and immune cell infiltration, PDT is reliant on external light stimulation. While the technology is promising, there is limited clinical potential owing to the shallow penetration depth of light.
4.3. Radiotherapy
Radiotherapy (RT) is a powerful therapeutic modality for cancer, commonly used for its ability to kill cancer cells by causing DNA double strand breaks, which is not limited by tissue penetration. Surface exposure of CRT[16] and release of HSP70[59] and HMGB-1[60] suggest that RT induces ICD in situ,[61] stimulates DC maturation, and induces IFN γ-producing T cells in vitro and in vivo.[62] Therefore, RT combined with i.t. injection of CpG adjuvant has shown preclinical success and has been tested in humans in whom it induced rejection of the irradiated tumor as well as tumors outside the radiation field (abscopal effect).[63] RT has also been reported to synergistically promote antitumor immunity with ICB.[64] However, not all RT-induced modifications of the tumor and its microenvironment favor immune activation. There has been new evidence of pro-tumorigenic M2 macrophages accumulating in hypoxic areas of irradiated tumors[65] and increased immunosuppressive Treg cells post-RT.[66] Intriguingly, the dose and fractionation of RT may play a role in modulating the expansion of effector versus Treg cells.[67]
Min et al. engineered antigen-capturing NPs (AC-NPs) with different surface chemistry to capture TAAs released after radiation and transport them to APCs to promote anticancer immunity (Figure 8). Mechanistic studies revealed that AC-NPs induced an expansion of CD8+ cytotoxic T cells and increased both CD4+ T/Treg and CD8+ T/Treg ratios, thus significantly improving the efficacy of anti-PD-1 treatment on the B16F10 melanoma model with up to a 20% cure rate compared to 0% without AC-NPs.[68]
Figure 8.
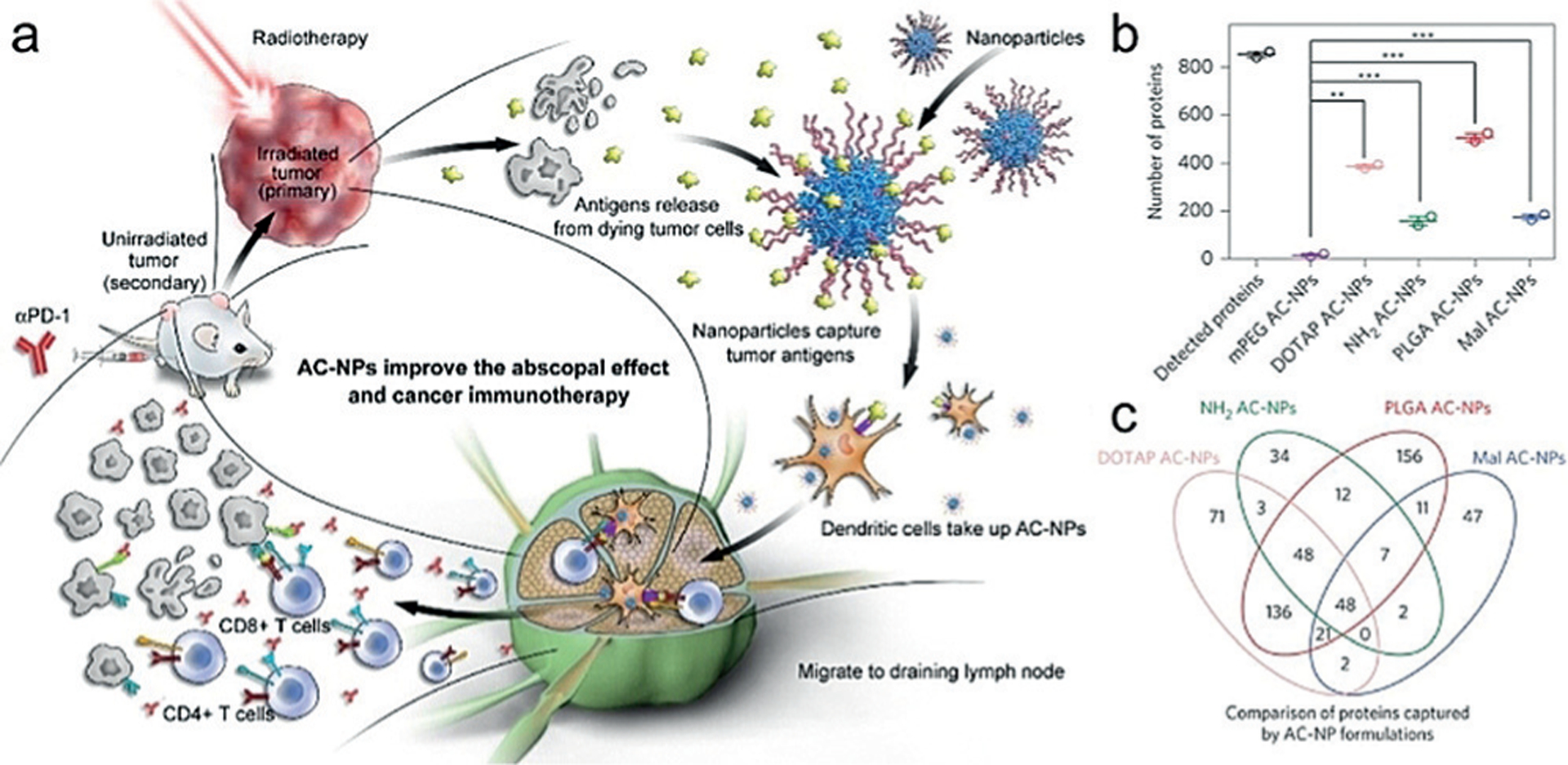
a) Schematic of utilizing AC-NPs to improve cancer immunotherapy. Radiation of the primary tumor induces the release of antigens, which were captured by AC-NPs and presented to DCs. The improved immune activation combined with anti-PD-1 treatment eradicates the unirradiated secondary tumor. b,c) Binding of unique TAAs to AC-NPs with different surface chemistry. Reprinted from Ref. [68] with permission. Copyright (2017), Nature Publishing Group.
Nanotechnology also allows for nanovectorized ionizing radiation that could boost the quality and magnitude of an immune response in a predictable and designable fashion.[69] Hindré’s group used lipid nanocapsules loaded with a lipophilic complex of Rhenium-188 (LNC188Re-SSS) for fractionated internal radiation in glioblastoma and hepatocellular carcinoma models.[35] Intratumoral infusion of LNCs by convection-enhanced delivery led to their complete distribution throughout the tumor and peritumoral space without leaking into the contralateral hemisphere except when large volumes were used. 70% of the 188Re-SSS activity was present in the tumor region 24 h after injection, and no toxicity was observed in the healthy brain. Double fractionated internal radiotherapy with LNC188Re-SSS resulted in a cure rate of 50% in a human glioblastoma model on T-cell deficient nude mice,[70a,b] whereas a higher cure rate of 83% was observed in the immunocompetent 9L rat glioma model.[70c] Increased cytokine (IL-2 and IFN-γ) production in peripheral blood after internal radiation, recruitment of immune and inflammatory cells (DCs, CD4, CD8, NK, macrophages, micro-glia) into the tumor site, and increased expression of MHC class I and class II were all observed, indicating the immune system plays a role in the treatment efficacy.[71]
4.4. Chemotherapeutic Drugs
Though apoptosis has historically been regarded as a non-inflammatory, immunologically silent or even tolerogenic, recent evidence has shown that a subset of chemotherapeutics can be pro-inflammatory and induce ICD.[72] As a systemic agent, chemotherapy has the potential to initiate an immune response in multiple sites. Furthermore, NP-mediated chemotherapy has been reported to enhance ICD and consequently improve antitumor effects of the free ICD inducer. For example, Zhao et al. found that immunogenic oxaliplatin encapsulated in PLGA-mPEG NPs released more DAMPs and induced more dendritic cell and T lymphocyte activation and infiltration than free oxaliplatin, improving anticancer efficacy in immunocompetent mice.[73]
Doxorubicin (DOX) is a bonafide ICD inducer that has already been widely used in NP formulations. Zheng et al. developed a pH- and GSH-dual-sensitive delivery system for systemic treatment of highly metastatic triple-negative breast cancer by loading DOX into highly integrated mesoporous silica NPs, which induced DC maturation and antitumor cytokine release.[74] The antitumor efficacy and immunity induced by DOX can be enhanced by combination with immunotherapy. Su et al. found that pre-treatment with TNF-α pDNA polyplexes 48 h prior to liposomal DOX (Doxil) promoted its accumulation in tumors, likely owing to TNF-α-mediated opening of the tumor endothelial tight junctions. Three treatment cycles with TNF-α gene vectors and Doxil significantly delayed tumor growth in subcutaneous murine Neuro2A neuroblastoma, and prevented liver metastasis in systemic Neuro2A metastasis or human LS174T colon carcinoma metastasis models.[75] Strong intercalation between DOX and DNA has been exploited for the development of DOX/CpG DNA hydrogels, which elevated the levels of cytokines (IL-12, IL-6, and IFN-γ) in serum as well as in tumor tissue, thus inhibiting tumor growth in various tumor models.[76] More recently, Liu et al. designed a dual pH-responsive multifunctional NP system by coating TLR-7/8 agonist (R848)-loaded poly(l-histidine) (PHIS) nanocore with acid-cleavable HA-DOX conjugates to treat breast cancer. The components separated in the TME, leading to internalization of HA-DOX through CD44-mediated endocytosis by tumor cells. Intracellular HA–DOX released DOX by hydrolysis of the hydrazone bond at pH≈5.5 to induce ICD. Extracellular PHIS/R848 released R848 by ionization of PHIS at pH≈6.5 to potentiate the immune response. The combined chemoimmunotherapy led to remarkable tumor growth inhibition.[77] Kuai et al. reported that high-density lipoprotein-mimicking nanodiscs loaded with DOX induced antitumor T cell responses and enhanced therapeutic efficacy of anti-PD-1, leading to eradication of CT26 and MC38 tumors in 80 to 88% of mice and long-term immunity against rechallenge.[78]
Another chemotherapeutic with a clinically approved nanoformulation, paclitaxel (PTX), is also known to induce ICD. Roy et al. combined chemo- and immunotherapy using PLGA NPs loaded with PTX and a TLR-4 agonist (SP-LPS). The NPs have both direct cytotoxicity and immunostimulatory activity in vitro, but the in vivo antitumor activity was not significantly different from PTX alone because of suboptimal encapsulation of SP-LPS.[79] Instead, they conjugated PTX with SP-LPS, which subsequently self-assembled into a NP. These NPs showed higher in vivo antitumor activity and a higher percentage of activated immune cells in the TME than the Taxol-treated group.[80] They also increased the loading of TLR-4 agonist from 20% to 65% by replacing SP-LPS with the similar P-LPS. The higher P-LPS loading showed improved synergy with PTX when co-encapsulated into PLGA NPs, leading to significant reduction in tumor growth compared with either stand-alone modality. Flow cytometric analysis of tumor-infiltrating immune cells indicated high infiltration and activation of APCs and T cells (CD4+ and CD8+), correlating to the enhanced survival of mice.[81] Similarly, Seth et al. demonstrated the synergy between PTX and a TLR-7 agonist for treatment of B16F10 melanoma by using poly(γ-glutamic acid) to co-deliver PTX and imiquimod. The co-delivery system enhanced the proliferation (250%) of DCs and secretion of pro-inflammatory and Th1 cytokines, exemplifying drastic inhibition of tumor growth after i.t. injection and leading to 70% survival as compared to individual components with 0% survival at day 41. The antitumor response generated was also found to have systemic memory response since the vaccinated mice significantly delayed secondary tumor development at a distant site six weeks after treatment.[82] Heo et al. also demonstrated that primary injection with the HA/PTX complex generated TSAs and enhanced their uptake by tumor-recruited BMDCs. With a secondary injection of separate CpG- or IL-10 siRNA-loaded PLGA NPs, the BMDCs became activated and migrated to TDLNs. As a result, the combination not only efficiently inhibited tumor growth but also increased the animal survival rate.[83]
4.5. Multipronged Approaches
Immunogenic chemotherapy has also been combined with other ablative therapies, such as PTT and PDT. Tao et al. designed a multifunctional platform for combining chemo-, photothermal-, and immunotherapy, constituted of DOX intercalated into CpG sequences, which in turn were conjugated to gold nanorods. The nanosystem localized gold rods into the tumor site for PTT, whereas the CpG induced immune response thereby enhancing the cytotoxic effects of DOX. The combination resulted in significant antitumor efficacy and also led to a long-term tumor-specific immunity.[84]
He et al. first demonstrated the synergistic effect of chemotherapy with PDT to elicit antitumor immunity, which could be used to augment the antitumor efficacy of ICB. NCPs carrying oxaliplatin (OxPt) in the core and the PS pyrolipid in the shell were prepared for effective chemotherapy and PDT, which provoked a systemic tumor-specific T-cell response. When combined with anti-PD-L1, the NCP led to not only the regression of the irradiated primary tumors but also regression of the non-irradiated tumors by inducing CD8+ and CD4+ T cell infiltration in bilateral mouse models of syngeneic colorectal cancer (Figure 9).[85] Subsequently, Yang et al. co-loaded a PDT agent Ce6 and DOX into hollow H-MnO2 nanoshells modified with PEG. The obtained H-MnO2-PEG/C&D dissociated under reduced pH within the TME to release the payloads, while simultaneously decomposing tumor H2O2 to relieve tumor hypoxia. As a result, a remarkable in vivo synergistic therapeutic effect was achieved through the combined chemo-photodynamic therapy and the subsequent antitumor immune response. Further combination with ICB led to inhibition of tumors at distant sites, showing promise for the treatment of tumor metastases.[86]
Figure 9.
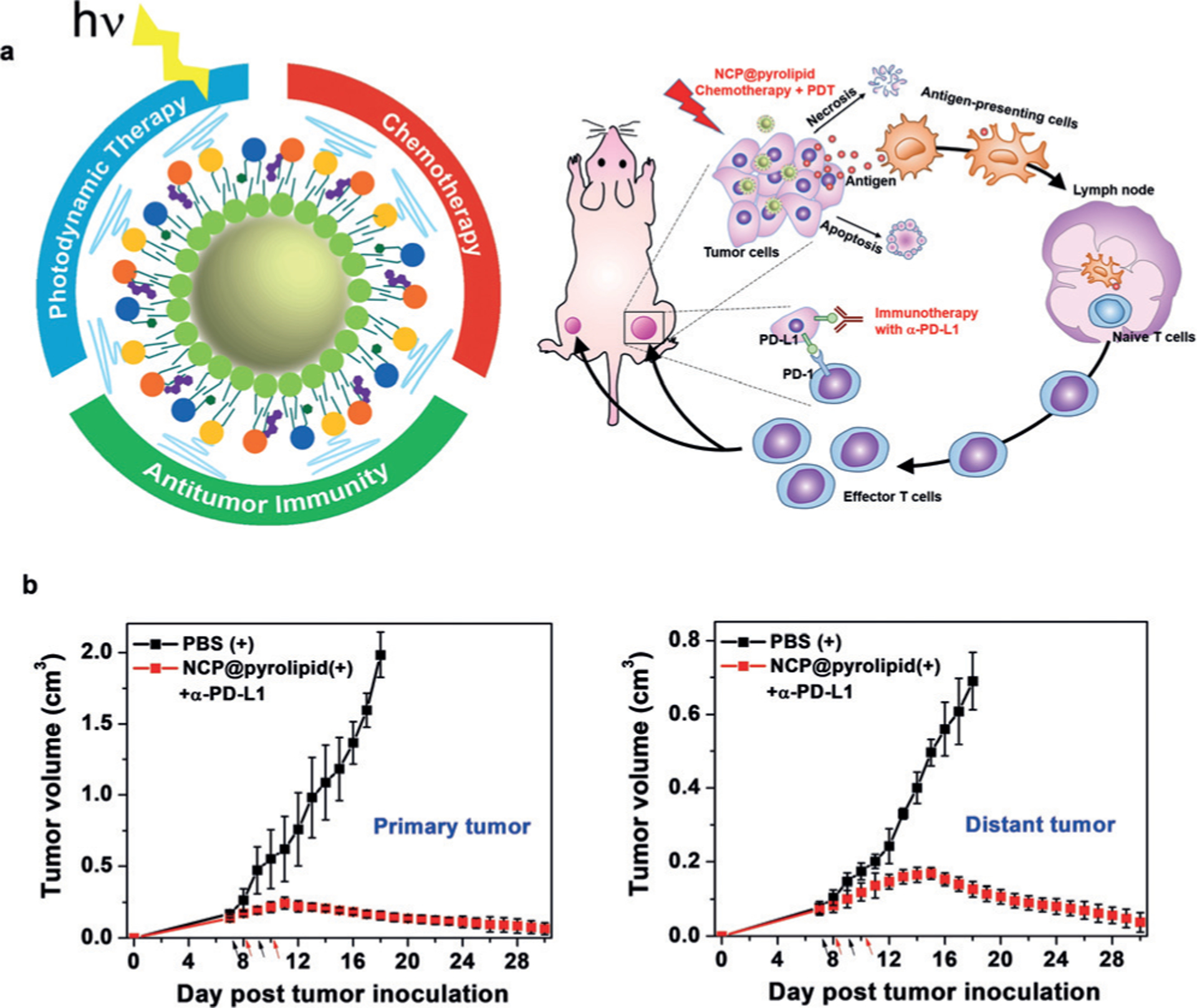
Chemotherapy and PDT of NCP@pyrolipid potentiate PD-L1 blockade to induce systemic antitumor immunity. a) Chemotherapy and PDT of NCP@pyrolipid induce ICD and an inflammatory environment, leading to the release of TAAs, which are processed and presented by infiltrated APCs, to elicit the proliferation of tumor-specific effector T cells in lymphoid organs. b) Combined with PD-L1 blockade, the NCP@pyrolipid chemotherapy/PDT significantly resulted in tumor eradication in the primary sites and also a systemic anti-tumor immune response to reject distant tumors. Adapted from Ref. [84] with permission. Copyright 2016, Nature Publishing Group.
5. Future perspectives
Cancer immunotherapy has shown exciting clinical responses owing to its unique advantages, such as the induction of specific antitumor immunity and long term immunological memory response, but the low response rate and the potential side effects are still significant hurdles for the broad application of cancer immunotherapy in the clinic. As shown in this review, a number of known and novel inorganic and polymeric nanoparticles such as AuNPs, MOFs, and micelles have been evaluated as ICD-inducing modalities, which can synergize with immunotherapies to improve the treatment outcomes while limiting systemic toxicities. However, the potency of different nanoparticle platforms or applications cannot be easily compared as each experiment was independently performed. Furthermore, more work is needed to monitor the dynamic immune response and understand the specific impact of each combinatorial approach on the TME, with the goal of providing rationales or guidance for selecting the best combinatorial approach for an individual patient. Specifically, it is important to elucidate whether a NP-mediated treatment is more suited towards combination with an immune adjuvant or ICB. In addition, the timing of the ICD-inducing modalities and immunotherapies might also have an impact on the therapeutic efficacy and should be investigated in more detail. So far, only a few stimuli have been shown to induce bonafide ICD; the identification of more compounds or modalities that render cell death immunogenic is clinically urgent. In order to preferentially deliver ICD-inducing agents to tumors, especially metastatic tumors, nanoparticles need to be injected systemically. However, most of the studies to date have utilized i.t. delivery, possibly owing to low stability, inefficient tumor accumulation or high toxicity; thus, the development of new nanoparticle platforms that can enhance tissue localization and response after systemic administration is critically needed.
Acknowledgements
We acknowledge the National Cancer Institute (U01-CA198989 and 1R01CA216436) and the University of Chicago Medicine Comprehensive Cancer Center (NIH CCSG: P30 CA014599) for funding support.
Biography

Xiaopin Duan obtained a B.S. degree in pharmacy from Hebei University (China) in 2008 and received a Ph.D. degree in Pharmaceutics jointly at Shenyang Pharmaceutical University and Shanghai Institute of Materia Medica, Chinese Academy of Sciences in 2013. She is currently a postdoctoral fellow with Prof. Wenbin Lin at the University of Chicago. Her research focuses on combination therapy and immunotherapy of metastatic cancers using nanomedicine.

Christina Chan received a B.A. degree in chemistry from the University of Chicago in 2013. She is a PhD candidate in the NIH Chemistry-Biology Interface Predoctoral Training Program at the University of Chicago. Her research addresses the combination and delivery of active chemical and biological molecules for the treatment of solid cancers.

Wenbin Lin studied chemical physics at University of Science and Technology of China, received a Ph.D. in chemistry at University of Illinois at Urbana-Champaign, and carried out NSF postdoctoral research with Prof. Tobin J. Marks at Northwestern University. He is currently the James Franck Professor of Chemistry, Radiation and Cellular Oncology, and Ludwig Center for Metastasis Research at the University of Chicago. His group has pioneered the application of metal–organic frameworks in cancer therapy, bioimaging, earth-abundant metal catalysis, artificial photosynthesis, asymmetric catalysis, and second-order nonlinear optics.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Xiaopin Duan, Department of Chemistry, The University of Chicago, Chicago, IL 60637 (USA).
Christina Chan, Department of Chemistry, The University of Chicago, Chicago, IL 60637 (USA).
References
- [1].Ribas A, Wolchok JD, Science 2018, 359, 1350 – 1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Chen DS, Mellman I, Immunity 2013, 39, 1 – 10; [DOI] [PubMed] [Google Scholar]; b) Chen DS, Mellman I, Nature 2017, 541, 321 – 330. [DOI] [PubMed] [Google Scholar]
- [3].a) Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD, Nat. Immunol 2002, 3, 991 – 998; [DOI] [PubMed] [Google Scholar]; b) Schreiber RD, Old LJ, Smyth MJ, Science 2011, 331, 1565 – 1570. [DOI] [PubMed] [Google Scholar]
- [4].a) Dewitte H, Verbeke R, Breckpot K, De Smedt SC, Lentacker I, Nano Today 2014, 9, 743 – 758; [Google Scholar]; b) Sau S, Alsaab HO, Bhise K, Alzhrani R, Nabil G, Iyer AK, J. Controlled Release 2018, 274, 24 – 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, Science 2015, 348, 803 – 808; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Palucka K, Banchereau J, Nat. Rev. Cancer 2012, 12, 265 – 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dranoff G, Nat. Rev. Cancer 2004, 4, 11 – 22. [DOI] [PubMed] [Google Scholar]
- [7].a) Restifo NP, Dudley ME, Rosenberg SA, Nat. Rev. Immunol 2012, 12, 269 – 281; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Amrolia PJ, Pule M, Lancet 2015, 385, 488 – 490. [DOI] [PubMed] [Google Scholar]
- [8].a) Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, Nature 2014, 515, 577 – 581; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pardoll DM, Nat. Rev. Cancer 2012, 12, 252 – 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ledford H, Nature 2015, 519, 17 – 18. [DOI] [PubMed] [Google Scholar]
- [10].a) Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G, Clin. Cancer Res 2010, 16, 3100 – 3104; [DOI] [PubMed] [Google Scholar]; b) Dudek AM, Garg AD, Krysko DV, Ruysscher D. De, Agostinis P, Cytokine Growth Factor Rev. 2013, 24, 319 – 333. [DOI] [PubMed] [Google Scholar]
- [11].a) Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P, Nat. Rev. Cancer 2012, 12, 860 – 875; [DOI] [PubMed] [Google Scholar]; b) Kroemer G, Galluzzi L, Kepp O, Zitvogel L, Annu. Rev. Immunol 2013, 31, 51 – 72. [DOI] [PubMed] [Google Scholar]
- [12].a) Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, Castedo M, Mignot G, Panaretakis T, Casares N, Nat. Med 2007, 13, 54 – 61; [DOI] [PubMed] [Google Scholar]; b) Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, Fimia G, Kepp O, Piacentini M, Froehlich K, Cell Death Differ. 2008, 15, 1499 – 1509. [DOI] [PubMed] [Google Scholar]
- [13].a) Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, EMBO J. 2012, 31, 1062 – 1079; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala A, Shen S, Kepp O, Metivier D, Galluzzi L, Perfettini J, Cell Death Differ. 2014, 21, 79 – 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, Andre F, Tursz T, Kroemer G, Zitvogel L, Immunol. Rev 2007, 220, 47 – 59; [DOI] [PubMed] [Google Scholar]; b) Scaffidi P, Misteli T, Bianchi ME, Nature 2002, 418, 191 – 195. [DOI] [PubMed] [Google Scholar]
- [15].a) Binder RJ, J. Immunol 2014, 193, 5765 – 5771; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G, Cell Death Differ. 2008, 15, 3 – 12. [DOI] [PubMed] [Google Scholar]
- [16].Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, Van Endert P, Zitvogel L, Kroemer G, Cell Death Differ. 2007, 14, 1848 – 1850. [DOI] [PubMed] [Google Scholar]
- [17].Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L, Nat. Med 2007, 13, 1050 – 1059. [DOI] [PubMed] [Google Scholar]
- [18].Kono H, Rock KL, Nat. Rev. Immunol 2008, 8, 279 – 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Green DR, Ferguson T, Zitvogel L, Kroemer G, Nat. Rev. Immunol 2009, 9, 353 – 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Irvine DJ, Hanson MC, Rakhra K, Tokatlian T, Chem. Rev 2015, 115, 11109 – 11146; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jo SD, Nam G-H, Kwak G, Yang Y, Kwon IC, Nano Today 2017, 17, 23 – 37; [Google Scholar]; c) Ng KK, Lovell JF, Zheng G, Acc. Chem. Res 2011, 44, 1105 – 1113; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang Z, Cao W, Jin H, Lovell JF, Yang M, Ding L, Chen J, Corbin I, Luo Q, Zheng G, Angew. Chem. Int. Ed 2009, 48, 9171 – 9175; Angew. Chem. 2009, 121, 9335 – 9339. [DOI] [PubMed] [Google Scholar]
- [21].a) Zheng G, Chen J, Li H, Glickson JD, Proc. Natl. Acad. Sci. USA 2005, 102, 17757 – 17762; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shao S, Geng J, Yi HA, Gogia S, Neelamegham S, Jacobs A, Lovell JF, Nat. Chem 2015, 7, 438; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang Y, Jeon M, Rich LJ, Hong H, Geng J, Zhang Y, Shi S, Barnhart TE, Alexandridis P, Huizinga JD, Nat. Nanotechnol 2014, 9, 631 – 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Elsabahy M, Wooley KL, Chem. Soc. Rev 2012, 41, 2545 – 2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Reddy LH, Arias JL, Nicolas J, Couvreur P, Chem. Rev 2012, 112, 5818 – 5878; [DOI] [PubMed] [Google Scholar]; b) Dykman L, Khlebtsov N, Chem. Soc. Rev 2012, 41, 2256 – 2282. [DOI] [PubMed] [Google Scholar]
- [24].a) Duan X, Li Y, Small 2013, 9, 1521 – 1532; [DOI] [PubMed] [Google Scholar]; b) Hong E, Dobrovolskaia MA, Adv. Drug Deliv. Rev 2018, 10.1016/j.addr.2018.01.005; [DOI] [PubMed] [Google Scholar]; c) Sykes EA, Chen J, Zheng G, Chan WC, ACS Nano 2014, 8, 5696 – 5706; [DOI] [PubMed] [Google Scholar]; d) Sun Y, Xia Y, Science 2002, 298, 2176 – 2179; [DOI] [PubMed] [Google Scholar]; e) Lu Y, Yin Y, Mayers BT, Xia Y, Nano Lett. 2002, 2, 183 – 186. [Google Scholar]
- [25].a) Homma T, Fujii J, Exp. Cell Res 2016, 349, 128 – 138; [DOI] [PubMed] [Google Scholar]; b) Chen T, Guo J, Han C, Yang M, Cao X, J. Immunol 2009, 182, 1449 – 1459; [DOI] [PubMed] [Google Scholar]; c) Slovak R, Ludwig JM, Gettinger SN, Herbst RS, Kim HS, J. Immunother. Cancer 2017, 5, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Evans SS, Repasky EA, Fisher DT, Nat. Rev. Immunol 2015, 15, 335 – 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nguyen HT, Tran KK, Sun B, Shen H, Biomaterials 2012, 33, 2197 – 2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Moy AJ, Tunnell JW, Adv. Drug Delivery Rev 2017, 114, 175 – 183. [DOI] [PubMed] [Google Scholar]
- [29].Toraya-Brown S, Sheen MR, Zhang P, Chen L, Baird JR, Demidenko E, Turk MJ, Hoopes PJ, Conejo-Garcia JR, Fiering S, Nanomed. Nanotechnol. Biol. Med 2014, 10, 1273 – 1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a) Kobayashi T, Biotechnol. J 2011, 6, 1342 – 1347; [DOI] [PubMed] [Google Scholar]; b) Kobayashi T, Kakimi K, Nakayama E, Jimbow K, Nanomedicine 2014, 9, 1715 – 1726. [DOI] [PubMed] [Google Scholar]
- [31].a) Yanase M, Shinkai M, Honda H, Wakabayashi T, Yoshida J, Kobayashi T, Cancer Sci. 1998, 89, 775 – 782; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kawai N, Ito A, Nakahara Y, Futakuchi M, Shirai T, Honda H, Kobayashi T, Kohri K, Prostate 2005, 64, 373 – 381. [DOI] [PubMed] [Google Scholar]
- [32].a) Jimbow K, Tamura Y, Yoneta A, Kamiya T, Ono I, Yamashita T, Ito A, Honda H, Wakamatsu K, Ito S, J. Biomater. Nanobiotechnol 2012, 3, 140 – 153; [Google Scholar]; b) Jimbow K, Ishii-Osai Y, Ito S, Tamura Y, Ito A, Yoneta A, Kamiya T, Yamashita T, Honda H, Wakamatsu K, J. Skin Cancer 2013, 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ito A, Yamaguchi M, Okamoto N, Sanematsu Y, Kawabe Y, Wakamatsu K, Ito S, Honda H, Kobayashi T, Nakayama E, Nanomedicine 2013, 8, 891 – 902. [DOI] [PubMed] [Google Scholar]
- [33].Ito A, Tanaka K, Kondo K, Shinkai M, Honda H, Matsumoto K, Saida T, Kobayashi T, Cancer Sci. 2003, 94, 308 – 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ito A, Matsuoka F, Honda H, Kobayashi T, Cancer Gene Ther. 2003, 10, 918 – 925. [DOI] [PubMed] [Google Scholar]
- [35].Ito A, Matsuoka F, Honda H, Kobayashi T, Cancer Immunol. Immunother 2004, 53, 26 – 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zou L, Wang H, He B, Zeng L, Tan T, Cao H, He X, Zhang Z, Guo S, Li Y, Theranostics 2016, 6, 762 – 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yata T, Takahashi Y, Tan M, Nakatsuji H, Ohtsuki S, Murakami T, Imahori H, Umeki Y, Shiomi T, Takakura Y, Biomaterials 2017, 146, 136 – 145. [DOI] [PubMed] [Google Scholar]
- [38].Guo L, Yan DD, Yang D, Li Y, Wang X, Zalewski O, Yan B, Lu W, ACS Nano 2014, 8, 5670 – 5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tao Y, Ju E, Ren J, Qu X, Biomaterials 2014, 35, 9963 – 9971. [DOI] [PubMed] [Google Scholar]
- [40].Han Q, Wang X, Jia X, Cai S, Liang W, Qin Y, Yang R, Wang C, Nanoscale 2017, 9, 5927 – 5934. [DOI] [PubMed] [Google Scholar]
- [41].Zhou F, Wu S, Song S, Chen WR, Resasco DE, Xing D, Biomaterials 2012, 33, 3235 – 3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li L, Yang S, Song L, Zeng Y, He T, Wang N, Yu C, Yin T, Liu L, Wei X, Theranostics 2018, 8, 860 – 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kumar P, Srivastava R, Mater. Sci. Eng. C 2015, 57, 321 – 327. [DOI] [PubMed] [Google Scholar]
- [44].Wang C, Xu L, Liang C, Xiang J, Peng R, Liu Z, Adv. Mater 2014, 26, 8154 – 8162. [DOI] [PubMed] [Google Scholar]
- [45].Cano-Mejia J, Burga RA, Sweeney EE, Fisher JP, Bollard CM, Sandler AD, Cruz CRY, Fernandes R, Nanomed. Nanotechnol. Biol. Med 2017, 13, 771 – 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen Q, Xu L, Liang C, Wang C, Peng R, Liu Z, Nat. Commun 2016, 7, 13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liu Y, Maccarini P, Palmer GM, Etienne W, Zhao Y, Lee C-T, Ma X, Inman BA, Vo-Dinh T, Sci. Rep 2017, 7, 8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].a) Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, CA: Cancer J. Clin 2011, 61, 250 – 281; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Thong PS-P, Ong K-W, Goh NS-G, Kho K-W, Manivasager V, Bhuvaneswari R, Olivo M, Soo K-C, Lancet Oncol. 2007, 8, 950 – 952; [DOI] [PubMed] [Google Scholar]; c) Castano AP, Mroz P, Hamblin MR, Nat. Rev. Cancer 2006, 6, 535 – 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S, Pogue BW, Hasan T, Chem. Rev 2010, 110, 2795 – 2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].a) Chatterjee DK, Fong LS, Zhang Y, Adv. Drug Delivery Rev 2008, 60, 1627 – 1637; [DOI] [PubMed] [Google Scholar]; b) Lucky SS, Soo KC, Zhang Y, Chem. Rev 2015, 115, 1990 – 2042. [DOI] [PubMed] [Google Scholar]
- [51].Yu X, Gao D, Gao L, Lai J, Zhang C, Zhao Y, Zhong L, Jia B, Wang F, Chen X, ACS Nano 2017, 11, 10147 – 10158. [DOI] [PubMed] [Google Scholar]
- [52].Marrache S, Choi JH, Tundup S, Zaver D, Harn DA, Dhar S, Integr. Biol 2013, 5, 215 – 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Duan X, Chan C, Guo N, Han W, Weichselbaum RR, Lin W, J. Am. Chem. Soc 2016, 138, 16686 – 16695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lan G, Ni K, Xu Z, Veroneau SS, Song Y, Lin W, J. Am. Chem. Soc 2018, 140, 5670 – 5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang D, Wang T, Liu J, Yu H, Jiao S, Feng B, Zhou F, Fu Y, Yin Q, Zhang P, Nano Lett. 2016, 16, 5503 – 5513. [DOI] [PubMed] [Google Scholar]
- [56].Lu K, He C, Guo N, Chan C, Ni K, Weichselbaum RR, Lin W, J. Am. Chem. Soc 2016, 138, 12502 – 12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Song W, Kuang J, Li C-X, Zhang M, Zheng D, Zeng X, Liu C, Zhang X-Z, ACS Nano 2018, 12, 1978 – 1989. [DOI] [PubMed] [Google Scholar]
- [58].Xu J, Xu L, Wang C, Yang R, Zhuang Q, Han X, Dong Z, Zhu W, Peng R, Liu Z, ACS Nano 2017, 11, 4463 – 4474. [DOI] [PubMed] [Google Scholar]
- [59].Stangl S, Themelis G, Friedrich L, Ntziachristos V, Sarantopoulos A, Molls M, Skerra A, Multhoff G, Radiother. Oncol 2011, 99, 313 – 316. [DOI] [PubMed] [Google Scholar]
- [60].Suzuki Y, Mimura K, Yoshimoto Y, Watanabe M, Ohkubo Y, Izawa S, Murata K, Fujii H, Nakano T, Kono K, Cancer Res. 2012, 72, 3967 – 3976. [DOI] [PubMed] [Google Scholar]
- [61].a) Golden E, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC, Front. Oncol 2012, 2, 88; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rubner Y, Wunderlich R, Rühle P-F, Kulzer L, Werthmçller N, Frey B, Weiss E-M, Keilholz L, Fietkau R, U. S. Gaipl, Front. Oncol 2012, 2, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].a) Prasad SJ, Farrand KJ, Matthews SA, Chang JH, McHugh RS, Ronchese F, J. Immunol 2005, 174, 90 – 98; [DOI] [PubMed] [Google Scholar]; b) Schaue D, Ratikan JA, Iwamoto KS, McBride WH, Int. J. Radiat. Oncol. Biol. Phys 2012, 83, 1306 – 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Mason KA, Hunter NR, Front. Oncol 2012, 2, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].a) Binder DC, Fu Y-X, Weichselbaum RR, Trends Mol. Med 2015, 21, 463 – 465; [DOI] [PubMed] [Google Scholar]; b) Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Nature 2015, 520, 373 – 377; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu Y-X, J. Clin. Invest 2014, 124, 687 – 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chiang C-S, Fu S-Y, Wang S-C, Yu C-F, Chen F-H, Lin C-M, Hong J-H, Front. Oncol 2012, 2, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Schaue D, Xie MW, Ratikan JA, McBride WH, Front. Oncol 2012, 2, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Demaria S, Formenti SC, Front. Oncol 2012, 2, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Min Y, Roche KC, Tian S, Eblan MJ, McKinnon KP, Caster JM, Chai S, Herring LE, Zhang L, Zhang T, Nat. Nanotechnol 2017, 12, 877 – 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vanpouille-Box C, Hindré F, Front. Oncol 2012, 2, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].a) Cikankowitz A, Clavreul A, Tétaud C, Lemaire L, Rousseau A, Lepareur N, Dabli D, Bouchet F, Garcion E, Menei P, J. Neuro-Oncol 2017, 131, 49 – 58; [DOI] [PubMed] [Google Scholar]; b) Jestin E, Mougin-Degraef M, Faivre-Chauvet A, Remaud-Le Saec P, Q. J. Nucl. Med. Mol. Imaging 2007, 51, 51 – 60; [PubMed] [Google Scholar]; c) Vanpouille-Box C, Lacoeuille F, Roux J, Aubé C, Garcion E, Lepareur N, Oberti F, Bouchet F, Noiret N, Garin E, PLoS One 2011, 6, e16926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Vanpouille-Box C, Lacoeuille F, Belloche C, Lepareur N, Lemaire L, LeJeune J-J, Benoit J-P, Menei P, Couturier OF, Garcion E, Biomaterials 2011, 32, 6781 – 6790. [DOI] [PubMed] [Google Scholar]
- [72].a) Albert ML, Sauter B, Bhardwaj N, Nature 1998, 392, 86 – 89; [DOI] [PubMed] [Google Scholar]; b) Inoue H, Tani K, Cell Death Differ. 2014, 21, 39 – 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhao X, Yang K, Zhao R, Ji T, Wang X, Yang X, Zhang Y, Cheng K, Liu S, Hao J, Biomaterials 2016, 102, 187 – 197. [DOI] [PubMed] [Google Scholar]
- [74].Zheng D-W, Chen J-L, Zhu J-Y, Rong L, Li B, Lei Q, Fan J-X, Zou M-Z, Li C, Cheng S-X, Nano Lett. 2016, 16, 4341 – 4347. [DOI] [PubMed] [Google Scholar]
- [75].Su B, Cengizeroglu A, Farkasova K, Viola JR, Anton M, Ellwart JW, Haase R, Wagner E, Ogris M, Mol. Ther 2013, 21, 300 – 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].a) Bagalkot V, Lee I-H, Yu MK, Lee E, Park S, Lee J-H, Jon S, Mol. Pharm 2009, 6, 1019 – 1028; [DOI] [PubMed] [Google Scholar]; b) Mizuno Y, Naoi T, Nishikawa M, Rattanakiat S, Hamaguchi N, Hashida M, Takakura Y, J. Controlled Release 2010, 141, 252 – 259; [DOI] [PubMed] [Google Scholar]; c) Nishikawa M, Mizuno Y, Mohri K, Matsuoka N, Rattanakiat S, Takahashi Y, Funabashi H, Luo D, Takakura Y, Biomaterials 2011, 32, 488 – 494. [DOI] [PubMed] [Google Scholar]
- [77].Liu Y, Qiao L, Zhang S, Wan G, Chen B, Zhou P, Zhang N, Wang Y, Acta Biomater. 2018, 66, 310 – 324. [DOI] [PubMed] [Google Scholar]
- [78].Kuai R, Yuan W, Son S, Nam J, Xu Y, Fan Y, Schwendeman A, Moon JJ, Sci. Adv 2018, 4, eaao1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Roy A, Singh MS, Upadhyay P, Bhaskar S, Mol. Pharm 2010, 7, 1778 – 1788. [DOI] [PubMed] [Google Scholar]
- [80].Roy A, Chandra S, Mamilapally S, Upadhyay P, Bhaskar S, Pharm. Res 2012, 29, 2294 – 2309. [DOI] [PubMed] [Google Scholar]
- [81].Roy A, Singh MS, Upadhyay P, Bhaskar S, Int. J. Pharm 2013, 445, 171 – 180. [DOI] [PubMed] [Google Scholar]
- [82].Seth A, Heo MB, Lim YT, Biomaterials 2014, 35, 7992 – 8001. [DOI] [PubMed] [Google Scholar]
- [83].Heo MB, Kim S-Y, Yun WS, Lim YT, Int. J. Nanomed 2015, 10, 5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Tao Y, Ju E, Liu Z, Dong K, Ren J, Qu X, Biomaterials 2014, 35, 6646 – 6656. [DOI] [PubMed] [Google Scholar]
- [85].He C, Duan X, Guo N, Chan C, Poon C, Weichselbaum RR, Lin W, Nat. Commun 2016, 7, 12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yang G, Xu L, Chao Y, Xu J, Sun X, Wu Y, Peng R, Liu Z, Nat. Commun 2017, 8, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]