

Abstract
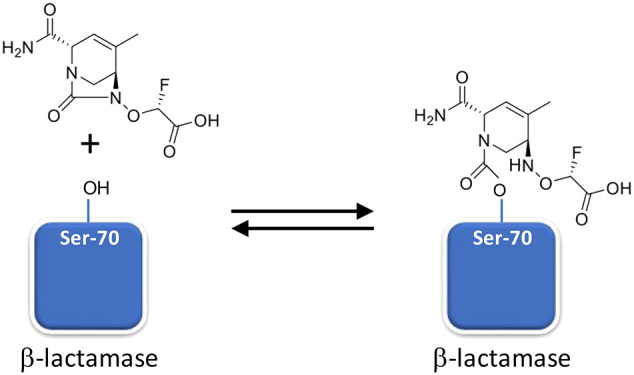
ETX0282 is an orally bioavailable prodrug of the diazabicyclooctane serine β-lactamase inhibitor ETX1317. The combination of ETX0282 with cefpodoxime proxetil is in clinical trials as an oral therapy for complicated urinary tract infections caused by Enterobacterales. Earlier diazabicyclooctane β-lactamase inhibitors, such as avibactam and durlobactam, contain a sulfate moiety as the essential anionic group and are administered intravenously. In contrast, ETX1317 contains a fluoroacetate moiety, which is esterified with an isopropyl group in ETX0282 to provide high oral bioavailability. Previous studies of avibactam and durlobactam showed that covalent inhibition of certain β-lactamases is reversible due to the ability of the ring-opened inhibitors to recyclize and dissociate in their original form. We investigated the interaction of ETX1317 with several β-lactamases commonly found in relevant bacterial pathogens, including CTX-M-15, KPC-2, SHV-5, and TEM-1 from Ambler Class A; Pseudomonas aeruginosa AmpC and Enterobacter cloacae P99 from Class C, and OXA-48 from Class D. The second-order rate constants for inhibition (kinact/Ki) of these enzymes show that ETX1317 is intermediate in potency between durlobactam and avibactam. The partition ratios were all approximately 1, indicating that the inhibitor is not also a substrate of these enzymes. The rate constants for dissociation of the covalent complex (koff) were similar to those for durlobactam and avibactam. Acylation exchange experiments demonstrated that ETX1317 dissociated in its original form. No loss of mass from the inhibitor was observed in the covalent inhibitor-enzyme complexes.
Keywords: β-lactamase, β-lactamase inhibitor, diazabicyclooctane
The diazabicyclooctane serine β-lactamase inhibitors have recently been introduced to help fight the increasingly severe threat of antibacterial drug resistance by Gram negative bacterial pathogens.1,2 Avibactam (Figure 1a) was the first member of the class to obtain regulatory approval. It is used in combination with ceftazidime for intravenous therapy of complicated urinary tract infections (cUTI), hospital-acquired bacterial pneumonia, and ventilator-associated bacterial pneumonia; and in combination with ceftazidime and metronidazole for intravenous therapy of complicated intraabdominal infections (cIAI).3,4 Another diazabicyclooctane, relebactam, in combination with imipenem/cilastatin, was recently approved for treatment of cUTI and cIAI.5 Durlobactam (ETX2514)6 (Figure 1d) is in Phase 3 testing in combination with sulbactam for treatment of carbapenem-resistant Acinetobacter baumannii pneumonia.
Figure 1.

Chemical structures of avibactam (a), ETX0282 (b), ETX1317 (c), and durlobactam (d).
ETX0282 (Figure 1b) was rationally designed to be orally bioavailable.7 It is currently in early clinical trials in combination with the β-lactam cefpodoxime proxetil, also an orally bioavailable antibacterial prodrug. The target pathogens for the combination therapy are the Enterobacterales causing urinary tract infections, including Escherichia coli and Klebsiella pneumoniae. The β-lactamase inhibitor protects the β-lactam from degradation by a range of serine β-lactamases commonly found in these bacteria. These include Ambler class A enzymes such as members of the TEM, CTX-M, and KPC families; class C cephalosporinases; and the class D oxacillinase OXA-48.8
ETX1317 (Figure 1c) is the active product of ETX0282 isopropyl ester hydrolysis, which occurs in the liver after absorption from the intestine.7 We investigated the interaction of ETX1317 with several serine β-lactamases representing those that are prevalent in relevant bacterial pathogens. We measured the second-order rate constants kinact/Ki for inactivation of the enzymes, or the dissociation constant Ki, where appropriate (previously reported in ref (7)). We also measured the partition ratios, i.e. the ratio of inhibitor molecules to enzyme molecules required to achieve complete inhibition. We investigated the reversibility of inhibition of the enzymes by ETX1317 by measuring the rate constants for dissociation of the inhibitor. Finally, we examined the ability of ETX1317 to dissociate from β-lactamases in its original form by recyclizing, a phenomenon that has been observed for avibactam9 and durlobactam.10
Results and Discussion
β-Lactamase Inhibition Kinetics
Table 1 shows the values of kinact/Ki or Ki for ETX1317, durlobactam, and avibactam measured for several Ambler class A, C, and D β-lactamases. The inhibitory potency of ETX1317 was comparable to, or somewhat lower than, that of durlobactam and higher than that of avibactam. The potency of ETX1317 was lowest for the class D enzymes OXA-10, −23, and −24. These enzymes are common in A. baumannii, the target pathogen for the sulbactam–durlobactam combination, but not in Enterobacterales, the target pathogens for the cefpodoxime proxetil–ETX0282 combination. For TEM-1, an equilibrium dissociation constant (Ki) was determined for ETX1317 instead of a second order rate constant (kinact/Ki) because the inhibition was not time-dependent on the time scale of the measurement (30 s between time points).
Table 1. Inhibition of β-Lactamases by ETX1317a.
| β-lactamase | ETX1317 kinact/Ki (M–1 s–1) or Ki (μM) | durlobactam kinact/Ki (M–1 s–1) | avibactam kinact/Ki (M–1 s–1) |
|---|---|---|---|
| class A | |||
| CTX-M-15 | 4.8 (±0.4) × 106 | 7 (±2) × 106 | 8 × 105 |
| KPC-2 | 4.9 (±0.2) × 104 | 9.3 (±0.6) × 105 | 6 × 103 |
| SHV-5 | 3.2 ± (0.1) × 106 | 6.4 (±0.5) × 106 | 1 × 105 |
| TEM-1 | Ki = 3.4 (±0.4) × 10–4 | 1.4 (±0.6) × 107 | 4 × 105 |
| class C | |||
| P. aeruginosa AmpC | 1.20 (±0.04) × 104 | 9 (±5) × 105 | 3 × 103 |
| E. cloacae P99 | 3.9 (±0.2) × 104 | 2.3 (±0.4) × 106 | 8 × 103 |
| class D | |||
| OXA-10 | 6.8 ± (0.3) × 102 | 9 (±2) × 103 | 70 |
| OXA-23 | 1.54 ± (0.06) × 103 | 5.1 (±0.2) × 103 | 100 |
| OXA-24 | 4.6 (±0.2) × 103 | 9 (±2) × 103 | 80 |
| OXA-48 | 5.3 (±0.2) × 104 | 8 (±2) × 105 | 5 × 103 |
Partition Ratios
The partition ratio can be defined as the molar ratio of the inhibitor to the target enzyme that is required, on average, to achieve 100% inhibition given sufficient time. It takes into account the possibility that the inhibitor may also be degraded by the enzyme, as is the case with β-lactamase inhibitors that are themselves β-lactams (see, for example, ref (11) concerning sulbactam). Partition ratios measured for ETX1317 with five β-lactamases were all approximately 1 (Figure 2), indicating that the enzymes do not degrade ETX1317 to a measurable extent, despite the fact that the enzyme concentrations during the incubation were very high, in the 1–10 μM range. Since it was previously observed that durlobactam is slowly degraded by KPC-2 such that the partition ratio increases with time,10 the ETX1317 + KPC-2 incubation time was extended from 1 h, as with the other enzymes, to 3 h. Nevertheless, the partition ratio was 1.1, the same as after a 15 min incubation (not shown), indicating that ETX1317 was not detectably hydrolyzed by KPC-2 on this time scale.
Figure 2.

Partition ratio measurements for inhibition by ETX1317 of several β-lactamases. The partition ratios were 1.1 for KPC-2, 0.7 for CTX-M-15, 1.1 for OXA-48, 1.0 for AmpC, and 0.9 for SHV-5. Incubations of enzyme and inhibitor were for 3 h with KPC-2 and 1 h with the other enzymes.
Dissociation Rate Constants
Jump dilution experiments were performed with ETX1317 and 7 β-lactamases to measure dissociation rate constants (koff). Enzyme activity recovery progress curves are shown in Figure 3. The dissociation rate constant measurements taken from the curves are shown in Table 2 and compared with published values for durlobactam and avibactam. Overall, the dissociation rate constants were similar between the three compounds for each enzyme but varied widely between enzymes. For TEM-1, koff was so high that dissociation was nearly instantaneous on the time scale of the measurement. This is consistent with the observation above that inhibition of TEM-1 was not time-dependent on a 30 s time scale. The lowest koff was measured for OXA-48, with almost no recovery of activity after 2 h. The value is reported as ∼0 in Table 2. By raising the temperature from ambient (about 22 °C) to 37 °C, a value of koff for durlobactam with OXA-48 was measured as 2.5 × 10–5 s–1 using a discontinuous jump dilution method covering 6 h.10 Thus, it is likely that the ambient temperature koff for ETX1317 from OXA-48 is lower than that value.
Figure 3.

Dissociation kinetics of ETX1317 from several β-lactamases. Progress curves are shown for enzymes preincubated without (blue) and with (red) ETX1317 before jump dilution. Dissociation rate constants are reported in Table 2.
Table 2. Dissociation Rate Constants (koff) for ETX1317, Durlobactam, and Avibactam from Several β-Lactamasesa.
| β-lactamase | ETX1317 koff (s–1) | durlobactam koff (s–1)10 | avibactam koff (s–1)11 |
|---|---|---|---|
| CTX-M-15 | 1.3 (±0.1) × 10–4 | 2.2 (±0.5) × 10–4 | 3 (±1) × 10–4 |
| KPC-2 | 5.2 (±0.6) × 10–4 | 1.0 (±0.1) × 10–3 | 1.4 (±0.1) × 10–4 |
| SHV-5 | 2.0 (±0.7) × 10–3 | 5.5 (±0.3) × 10–4 | ND |
| TEM-1 | 8 (±5) × 10–3 | 1.4 (±0.2) × 10–3 | 8 (±4) × 10–4 |
| P. aeruginosa AmpC | 6 (±1) × 10–4 | 4 (±1) × 10–3 | 1.9 (±0.6) × 10–3 |
| E. cloacae P99 | 9.2 (±0.8) × 10–5 | 3.4 (±0.5) × 10–4 | 3.8 (±0.2) × 10–5 |
| OXA-48 | ∼0 | 2.5 (±0.3) × 10–5 | 1.2 (±0.4) × 10–5 |
Dissociation of ETX1317 from OXA-48 was too slow to measure. ND, not done.
Reversible Covalent Inhibition and Recyclization
The formation of covalent complexes between ETX1317 with seven β-lactamases (AmpC, KPC-2, OXA-48, CTX-M-15, P99, SHV-5, and TEM-1) was observed by intact protein mass spectrometry, which is performed under protein denaturing conditions such that noncovalent complexes are not observed (Figures 4–6 and S1–S4). In every case, the only adduct observed was +273 Da, corresponding to the mass of ETX1317. This result contrasts with the results observed for avibactam12 and durlobactam,10 for which adducts with masses less than that of entire inhibitor were observed with some enzymes due, it is thought, to loss of an SO3 or SO4 moiety. Even after 17–19 h, no change in the mass of the ETX1317 adduct or loss of the entire adduct was observed for any of the enzymes (Figure 6), (Figure 3).
Figure 4.
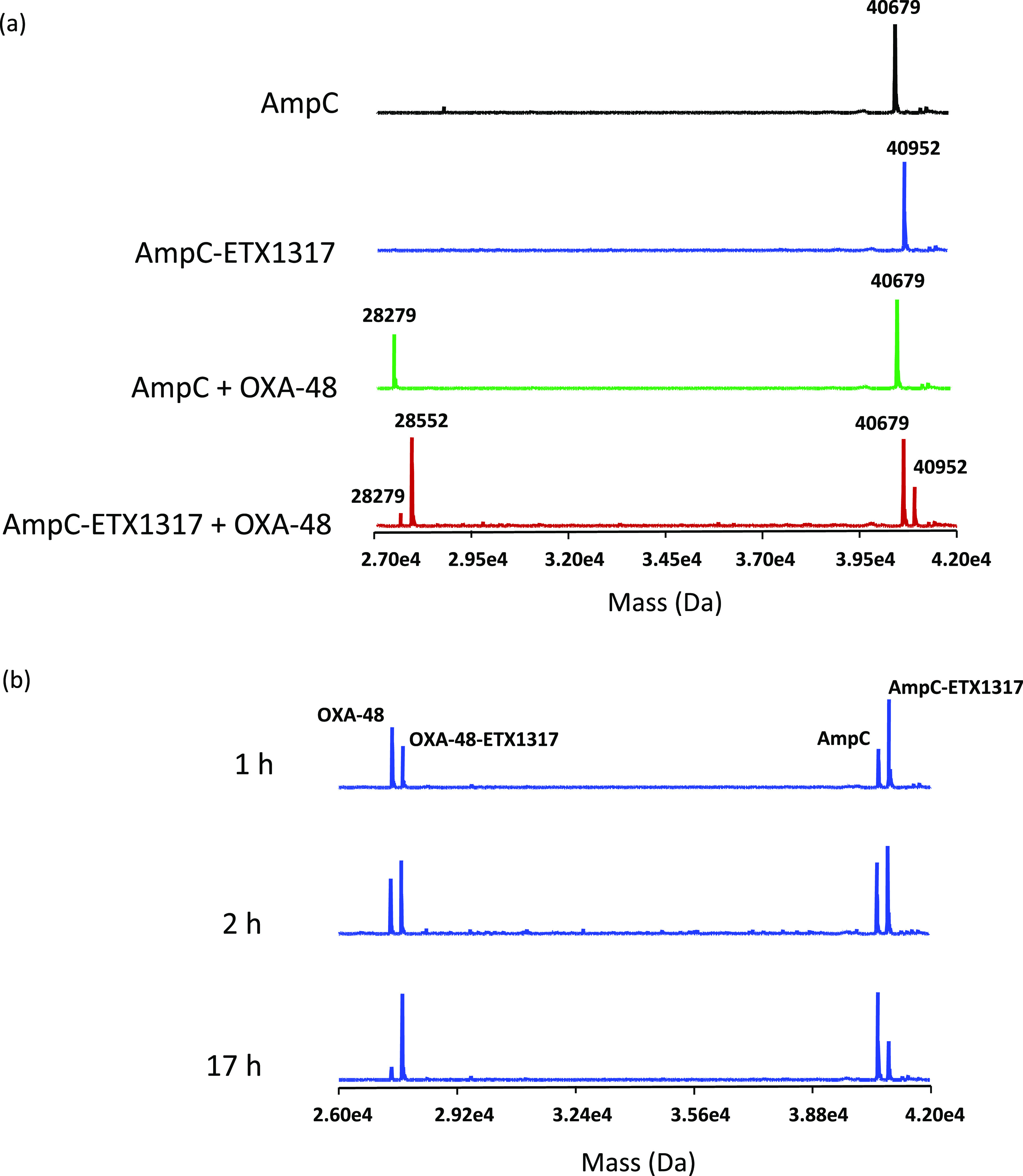
Intact protein mass spectrometry for exchange of covalently bound ETX1317 (273 Da) from P. aeruginosa AmpC (40 679 Da) to OXA-48 (28 279 Da) after 17 h. (a) Mass spectra of untreated AmpC (black), the AmpC–ETX1317 covalent complex (blue; 40 952 Da), the mixture of untreated AmpC and OXA-48 (green), and the mixture of the AmpC–ETX1317 covalent complex with OXA-48 (red). The mass of the OXA-48–ETX1317 covalent complex is 28 552 Da. (b) Time-dependence of exchange of ETX1317 from AmpC to OXA-48 at 1, 2, and 17 h of incubation.
Figure 6.

Intact protein mass spectrometry showing lack of exchange of covalently bound ETX1317 (273 Da) from OXA-48–ETX1317 covalent complex (28 553 Da) to CTX-M-15 (28 110 Da) after 19 h of incubation. Shown are the mass spectra of untreated OXA-48 (black; 28 280 Da), the OXA-48–ETX1317 covalent complex (blue), the mixture of untreated OXA-48 and CTX-M-15 (green), and the mixture of OXA-48–ETX1317 covalent complex with CTX-M-15 (red). The mass of a CTX-M-15–ETX1317 covalent complex would be 28 383 Da.
This result is consistent with the observation of measurable rates of dissociation (see above), because the protein concentrations in the jump dilution experiment are several orders of magnitude lower than in the mass spectrometry experiment. In the jump dilution experiment, if the inhibitor recyclizes and dissociates from the enzyme, the enzyme and inhibitor concentrations are so low that reformation of the covalent complex is very slow. In contrast, in the mass spectrometry experiment, the enzyme and inhibitor concentrations are so high that reformation of the covalent complex is essentially instantaneous.
This explanation presupposes that ETX1317 is capable of recyclizing and dissociating from the enzyme in its original state. To further test this concept, we employed the acylation exchange experiment. The covalent complex of ETX1317 with a donor β-lactamase was diafiltered to remove unbound ETX1317 and then mixed with an acceptor β-lactamase. The loss of ETX1317 from the donor enzyme and appearance of a covalent ETX1317–acceptor enzyme complex demonstrates recyclization and dissociation of intact ETX1317. This phenomenon was observed for donor enzymes Pseudomonas aeruginosa AmpC (Figure 4a), KPC-2 (Figure 5), CTX-M-15 (Figure S1), Enterobacter cloacae P99 (Figure S2a), SHV-5 (Figure S3), and TEM-1 (Figure S4a), but not with OXA-48 (Figure 6). The lack of acylation exchange from OXA-48 is consistent with the immeasurably low koff for ETX1317 with OXA-48 (Table 2). An additional factor may have been the lack of added bicarbonate ion in the acylation exchange experiment, since OXA family β-lactamase are activated by bicarbonate. In contrast, the time-dependent exchanges of ETX1317 from other donor enzymes are shown for AmpC (Figure 4b), P99 (Figure S2b), and TEM-1 (Figure S4b). Since dissociated ETX1317 partitions between the donor and acceptor enzymes in the acylation exchange experiment, the rate of exchange to the acceptor enzyme is slower than the koff measured by jump dilution.
Figure 5.

Intact protein mass spectrometry for exchange of covalently bound ETX1317 (273 Da) from KPC-2 (28 720 Da) to OXA-48 (28 280 Da) after 19 h. Shown are the mass spectra of untreated KPC-2 (black), the KPC-2–ETX1317 covalent complex (blue; 28 993 Da), the mixture of untreated KPC-2 and OXA-48 (green), and the mixture of KPC-2–ETX1317 covalent complex with OXA-48 (red). The mass of the OXA-48-ETX1317 covalent complex is 28 553 Da.
To create a diazabicyclooctane β-lactamase inhibitor with high oral bioavailability, the sulfate moiety of all previous members of the class was replaced with a fluoroacetate moiety, allowing an ester prodrug to be prepared.7 Interestingly, a side effect of replacement of the sulfate moiety is that the covalent complex of the inhibitor with certain β-lactamases, especially KPC-2, is no longer subject to loss of the anionic group that was observed with both durlobactam and avibactam.10,12 In the case of KPC-2, desulfated durlobactam and avibactam were gradually lost from the enzyme. Since the desulfated compounds were no longer inhibitory, this resulted in gradual loss of inhibition, demonstrated by a time-dependent increase in the partition ratio of durlobactam.10 ETX1317 was impervious to this effect.
As with the related DBO-class β-lactamase inhibitors avibactam and durlobactam, the rate of recovery of activity of the class D enzyme OXA-48 due to recyclization and dissociation of the inhibitor was much slower than for class A and C enzymes. Based on X-ray crystallography, Lahiri et al.13 hypothesized that this results from the requirement for a carboxylated lysine residue in the catalytic mechanism of class D enzymes and that binding of the inhibitor (avibactam in this case) changes the charge distribution in the vicinity of the carboxy-Lys residue, causing its decarboxylation. Since this carboxy-Lys residue participates in the recyclization of the inhibitor, it was proposed that recyclization is inhibited. Lysine decarboxylation of avibactam-acylated OXA-48 at neutral pH was also observed by King et al.15 Lohans et al.16 found that lysine carboxylation of OXA-48 was disfavored but not ablated by avibactam in solution using 13C NMR. Using a novel 19F NMR technique that did not require the presence of bicarbonate, van Groessen et al.17 later observed that avibactam derivatization of OXA-48 more substantially disfavored lysine carboxylation. Their suggested explanation for this effect was that a hydrogen bond from the nucleophilic serine, which is acylated by avibactam, stabilizes the carbamoyl lysine.
Observations with avibactam and durlobactam, which have a sulfate moiety instead of the fluoroacetate moiety in ETX1317, showed a loss of mass from the acyl–enzyme complexes with some β-lactamases, such as KPC-2.10,12 This is thought to be due to loss of the sulfate moiety, with additional chemical changes possible subsequently. In contrast, we observed no loss of mass from any of the ETX1317-enzyme complexes we studied. The bond strength and electronics between the core of the inhibitor and the 2-fluoroacetic acid activating group are different from those between the core and the sulfate activating group in avibactam and durlobactam. The proposed mechanism for the desulfation of avibactam12 cannot occur with ETX1317 due to the different chemical nature of the side chain. The chemical differences between the sulfate-containing and fluoroacetate-containing inhibitors likely also explains the inability of KPC-2 to gradually degrade ETX1317, as shown by the lack of a time-dependent increase in the partition ratio.
Avibactam was the first β-lactamase inhibitor of the DBO class introduced to clinical practice. Its spectrum of β-lactamase inhibition is largely limited to class A and C enzymes, except that it is also able to inhibit certain class D enzymes, particularly OXA-48, which is prevalent among the bacterial species commonly responsible for urinary tract infections. To expand the spectrum of DBOs to include a broader range of class D enzymes, such as those prevalent in the pathogen Acinetobacter baumannii, durlobactam was developed. Two factors are responsible for the enhanced inhibitory potency and class D spectrum of inhibition of durlobactam relative to avibactam.6 First, the double bond in the ring system increases the ring strain, making the compound more reactive. Second, the methyl substitution on the ring provides a hydrophobic interaction with the hydrophobic bridge found in the active site of most class D enzymes, but not in OXA-48. Both of these features were retained in ETX1317. The result is that ETX1317 has intermediate potency between avibactam and durlobactam, despite having a less-than-optimal anionic substituent needed for preparation of the oral prodrug, and it retains the broad spectrum of class D β-lactamase inhibition of durlobactam.
ETX1317 has been cocrystallized with only one β-lactamase to date, CTX-M-14.7 The potency of CTX-M-14 inhibition by ETX1317 is the same as that of durlobactam and about 10-fold higher than that of avibactam.7 Comparing the CTX-M-14 crystal structures with covalently bound ETX1317 and avibactam ring-opened products showed slight differences that could account for the difference in potencies.7 Further elucidation of the structural correlates of inhibitory potency and dissociation rates will await solution of additional acyl-enzyme structures. It should be noted, however, that these structures are unable to reveal the structure of the encounter complex prior to the acylation reaction.
Conclusion
ETX1317, the active component of the orally bioavailable β-lactamase inhibitor ETX0282, broadly inhibited Ambler class A, C, and D serine β-lactamases. The partition ratio was approximately 1 in each case tested (CTX-M-15, KPC-2, SHV-5, P. aeruginosa AmpC and OXA-48), indicating that there was no detectable hydrolysis of the inhibitor by the enzymes. The covalent inhibitor–enzyme complex consisted of the full mass of ETX1317 in every case. In most cases, the covalent complexes were reversible due to recyclization of the inhibitor and dissociation. For OXA-48, dissociation of ETX1317 was too slow to measure. The qualities of the interactions of ETX1317 with these serine β-lactamases support the usefulness of ETX0282 in combination with cefpodoxime proxetil for treatment of complicated urinary tract infections.
Methods
Chemicals
ETX1317,7 avibactam,14 and durlobactam6 were prepared by Entasis Therapeutics according to established methods. Purity was measured by HPLC at 95%, 90%, and 97.4%, respectively.
β-Lactamase Inhibition kinact/Ki Measurements
β-Lactamases were prepared as described previously.6,12,13 Measurements of kinact/Ki, or Ki, where appropriate, were made as described previously.6 The assay buffer (Buffer A) consisted of 100 mM sodium phosphate (pH 7.0), 10 mM sodium bicarbonate, and 0.005% Triton X-100 (Thermo-Fisher Scientific, Waltham, MA). The assay volume was 45 μL. Assays were performed at ambient temperature in clear polystyrene 384-well plates (Greiner Bio-one, Monroe, NC). Inhibitors were freshly dissolved in assay buffer. Fourteen 3-fold serial dilutions of inhibitors were prepared in assay buffer, starting from 300 μM and ending at 188 pM, and 15 μL of each was added to the assay plate. Two additional wells received 15 μL of assay buffer. To all wells was then added 15 μL of 300 μM nitrocefin substrate (custom-synthesized by Syngene, Bangalore, India). Reactions were initiated by addition of 15 μL of 3× β-lactamase enzyme diluted in assay buffer, except that one well without inhibitor received buffer to act as a blank. The final enzyme concentrations were as follows: 15 pM AmpC, 10 pM CTX-M-15, 55 pM KPC-2, 20 pM OXA-10, 10 pM OXA-23, 10 pM OXA-24, 10 pM OXA-48, 8 pM P99, 64 pM SHV-5 and 4 pM TEM-1. Absorbance at 490 nm was monitored at 30 s intervals for 1 h with a Spectramax Plus 384 plate reader (Molecular Devices, San Jose, CA).
The progress curve of the blank was subtracted from the progress curves of the enzyme-containing wells. The resulting 15 progress curves were then fit globally to the kinetic model below, using Global Kinetic Explorer (Kintek, Snow Shoe, PA). The units used were μM and s.
where E, S, ES, P, I, and EI represent, respectively, the enzyme, the nitrocefin substrate, the enzyme–substrate complex, the product of nitrocefin hydrolysis, the inhibitor, and the enzyme–inhibitor complex. The premeasured KM(nitrocefin) values, where KM = (k–1 + k+2)/k+1, were entered as k+1 = k–1 = 1 and k+2 = KM – 1. The value of k–2 was allowed to float in the fitting. The values of KM(nitrocefin) for the enzymes were 130 μM for AmpC, 8 μM for CTX-M-15, 64 μM for KPC-2, 6 μM for OXA-10, 160 μM for OXA-23, 30 μM for OXA-24, 16 μM for OXA-48, 110 μM for P99, 3 μM for SHV-5, and 20 μM for TEM-1. The value of k+3 corresponds to kinact/Ki and the value of k–3 corresponds to koff. The measurement of koff does not distinguish between reversible binding and hydrolysis of the inhibitor.
Partition Ratio Measurements
Partition ratios were measured as described previously10 at ambient temperature in Buffer A. ETX1317 was freshly dissolved in assay buffer and an arithmetic dilution series was prepared from 0 to 12 μM in 1.2 μM steps. Each of these was mixed with an equal volume of 6 μM β-lactamase. At each time point, the mixtures were diluted in buffer to 2-fold the final reaction concentration of enzyme. Then, 22.5 μL of enzyme + inhibitor solutions were mixed with 22.5 μL of 200 μM nitrocefin substrate in a clear polystyrene 384-well assay plate. The absorbance at 490 nm was monitored at 10-s intervals for 10 min with a Spectramax Plus 384 plate reader. The slopes of the progress curves (ΔA490/s) were calculated. The percent of the control value for each inhibitor concentration was plotted versus the inhibitor:enzyme molar ratio. A best-fit line was calculated with a fixed point at 100% on the y-axis.
β-Lactamase Inhibition koff Measurements
Dissociation rate constants (koff) were measured by the jump-dilution method at ambient temperature as described previously.10 Enzymes at 500 000-fold the final β-lactamase assay concentration (see above) were incubated for 15–20 min with 3-fold molar excess of freshly dissolved ETX1317 at ambient temperature in Buffer A. The mixtures were then diluted 250 000-fold with Buffer A, and 22.5 μL of the dilutions was mixed with 22.5 μL of 200 μM nitrocefin in Buffer A. The absorbance at 490 nm was monitored at 30 s intervals for 2 h at ambient temperature with a Spectramax Plus 384 plate reader. Progress curves were fit by nonlinear regression using XLfit (ID Business Solutions, Boston, MA) to the following equation:
where P is A490, C is the initial A490, V0 is the initial rate of absorbance increase, Vs is the final rate of absorbance increase, t is time in seconds, and k is the first order rate constant for enzyme reactivation due to inhibitor dissociation, koff.
β-Lactamase Acylation Exchange
β-Lactamase acylation exchange experiments and intact protein mass spectrometry were performed as described previously.10 OXA-48 was used as the acceptor enzyme in most cases because of the stability of the ETX1317–OXA-48 complex. When OXA-48 was the donor enzyme, CTX-M-15 was used as the acceptor. Each β-lactamase (5 μM) was incubated with or without 25 μM ETX1317 in a 60 μL volume of 50 mM HEPES (pH 7.0) buffer for 1 h at ambient temperature. The samples were diluted to 400 μL with ice-cold buffer and then concentrated to ∼40 μL with Microcon YM-10 centrifugal ultrafilters (Millipore-Sigma, Burlington, MA) at 14 000g for 45 min at 4 °C. The retentates were rediluted to 400 μL with ice-cold buffer and reconcentrated as before. This was repeated a further time, leaving no significant amount of unbound ETX1317. The sample volumes were restored to 60 μL with ice-cold buffer and the samples were divided into two equal aliquots of 30 μL. Buffer (6 μL) was added to one aliquot, and 6 μL of acceptor enzyme was added to the other. The final acceptor enzyme concentrations were 5 μM. Samples were incubated at ambient temperature for the times indicated and then stored at −80 °C until analysis. LC-MS analysis was performed on a Triple TOF5600+ (AB Sciex, Redwood City, CA) equipped with a DuoSpray Ion Source and a Shimadzu LC 20-AD HPLC system (Shimadzu Scientific Instruments, Marlborough, MA). A 2 μL portion of each sample was injected onto a Poroshell 300SB-C8 75 × 2.1 mm, 5 μm column (Agilent) at 30 °C with a gradient of acetonitrile (5–95%) in 0.1% formic acid for 3 min followed by a 1 min hold at 95% before returning to 5% acetonitrile at a flow rate of 0.4 mL/min. The mass spectrometer was operated in positive ion and intact protein mode with source temperature of 450 °C. LC-MS data were acquired in the TOF MS mode for m/z+ from 600 to 2000. Nebulizer gas (GS1), heater gas (GS2), and curtain gas were set at 60, 70, and 30 psi, respectively. Ion spray voltage was 5500 V. Declustering voltage was 100 V, and collision energy was 10 V. Peak masses for protein species were determined following spectrum deconvolution using PeakView version 2.2 software. Mass accuracy of intact proteins is ±1 Da.
Acknowledgments
We thank Ruben Tommasi, Alita Miller, Thomas Durand-Réville and John O’Donnell for critical reading of the manuscript and helpful advice. This research program was partially supported by Cooperative Agreement Number IDSEP160030 from ASPR/BARDA and by awards from Wellcome Trust and Germany’s Federal Ministry of Education and Research (BMBF), as administrated by CARB-X. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Health and Human Services Office of the Assistant Secretary for Preparedness and Response, other funders, or CARB-X.
Glossary
Abbreviations
- cUTI
complicated urinary tract infection
- cIAI
complicated intraabdominal infection
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00656.
Intact protein mass spectrometry for exchange of covalently bound ETX1317 from CTX-M-15, P99, SHV-5 and TEM-1 to OXA-48 (PDF)
Author Contributions
A.B.S performed biochemical experiments and prepared the manuscript. N.G. performed mass spectrometry.
The authors declare the following competing financial interest(s): A.B.S. is an employee of Entasis Therapeutics and may possess stock and/or stock options.
Supplementary Material
References
- Bush K.; Bradford P. A. (2019) Interplay between β-lactamases and new β-lactamase inhibitors. Nat. Rev. Microbiol. 17, 295–306. 10.1038/s41579-019-0159-8. [DOI] [PubMed] [Google Scholar]
- Bush K. (2018) Game changers: new β-lactamase inhibitor combinations targeting antibiotic resistance in Gram-negative bacteria. ACS Infect. Dis. 4, 84–87. 10.1021/acsinfecdis.7b00243. [DOI] [PubMed] [Google Scholar]
- Barber K. R.; Ortwine J. K.; Akins R. L. (2016) Ceftazidime/avibactam: who says you can’t teach an old drug new tricks?. J. Pharm. Pharm. Sci. 19, 448–464. 10.18433/J3X31R. [DOI] [PubMed] [Google Scholar]
- Shirley M. (2018) Ceftazidime-avibactam: a review in the treatment of serious Gram-negative bacterial infections. Drugs 78, 675–692. 10.1007/s40265-018-0902-x. [DOI] [PubMed] [Google Scholar]
- Smith J. R.; Rybak J. M.; Claeys K. C. (2020) Imipenem-cilastatin-relebactam: A novel β-lactam-β-lactamase inhibitor combination for the treatment of multidrug-resistant Gram-negative infections. Pharmacotherapy 40, 343. 10.1002/phar.2378. [DOI] [PubMed] [Google Scholar]
- Durand-Réville T. F.; Guler S.; Comita-Prevoir J.; Chen B.; Bifulco N.; Huynh H.; Lahiri S.; Shapiro A. B.; McLeod S. M.; Carter N. M.; Moussa S. H.; Velez-Vega C.; Olivier N. B.; McLaughlin R.; Gao N.; Thresher J.; Palmer T.; Andrews B.; Giacobbe R. A.; Newman J. V.; Ehmann D. E.; deJonge B.; O’Donnell J.; Mueller J. P.; Tommasi R. A.; Miller A. A. (2017) ETX2514 is a broad spectrum β-lactamase inhibitor for the treatment of drug resistant Gram-negative infections, including those caused by Acinetobacter baumannii. Nat. Microbiol. 2, 17104. 10.1038/nmicrobiol.2017.104. [DOI] [PubMed] [Google Scholar]
- Durand-Réville T. F.; Comita-Prevoir J.; Zhang J.; Wu X.; May-Dracka T. L.; Romero J. A. C.; Wu F.; Chen A.; Shapiro A. B.; Carter N. M.; McLeod S. M.; Giacobbe R. A.; Verheijen J. C.; Lahiri S. D.; Sacco M. D.; Chen Y.; O’Donnell J. P.; Miller A. A.; Mueller J. P.; Tommasi R. A. (2020) Discovery of an orally available diazabicyclooctane inhibitor (ETX0282) of class A, C and D serine β-lactamases. J. Med. Chem. 63 (21), 12511–12525. 10.1021/acs.jmedchem.0c00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K.; Bradford P. A. (2020) Epidemiology of β-lactamase-producing pathogens. Clin. Microbiol. Rev. 33, e00047–19. 10.1128/CMR.00047-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmann D. E.; Jahic H.; Ross P. L.; Gu R.-F.; Hu J.; Kern G.; Walkup G. K.; Fisher S. L. (2012) Avibactam is a covalent, reversible, non−β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 109, 11663–11668. 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro A. B.; Gao N.; Jahić H.; Carter N. M.; Chen A.; Miller A. A. (2017) Reversibility of covalent, broad-spectrum serine β-lactamase inhibition by the diazabicyclooctenone ETX2514. ACS Infect. Dis. 3, 833–844. 10.1021/acsinfecdis.7b00113. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B. (2017) Kinetics of sulbactam hydrolysis by β-lactamases, and kinetics of β-lactamase inhibition by sulbactam. Antimicrob. Agents Chemother. 61, e01612–17. 10.1128/AAC.01612-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmann D. E.; Jahić H.; Ross P. L.; Gu R.-F.; Hu J.; Durand-Réville T. F.; Lahiri S.; Thresher J.; Livchak S.; Gao N.; Palmer T.; Walkup G. K.; Fisher S. L. (2013) Kinetics of avibactam inhibition against Class A, C, and D β-lactamases. J. Biol. Chem. 288, 27960–27971. 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S. D.; Mangani S.; Jahić H.; Benvenuti M.; Durand-Réville T. F.; De Luca F.; Ehmann D. E.; Rossolini G. M.; Alm R. A; Docquier J.-D. (2015) Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: a structure-guided study of OXA-24 and OXA-48. ACS Chem. Biol. 10, 591–600. 10.1021/cb500703p. [DOI] [PubMed] [Google Scholar]
- Boyd J. A. (2012) Process for preparing heterocyclic compounds including trans-7-oxo-6-(sulphooxy-1,6-diazabicyclo[3,2]octane-2-carboxamide and salts thereof. Patent application WO2012/172368A1.
- King D. T.; King A. M.; Lal S. M.; Wright G. D.; Strynadka N. C. J. (2015) Molecular mechanism of avibactam-mediated β-lactamase inhibition. ACS Infect. Dis. 1, 175–184. 10.1021/acsinfecdis.5b00007. [DOI] [PubMed] [Google Scholar]
- Lohans C. T.; Wang D. Y.; Jorgensen C.; Cahill S. T.; Clifton I. J.; McDonough M. A.; Oswin H. P.; Spencer J.; Domene C.; Claridge T. D. W.; Brem J.; Schofield C. J. (2017) 13C-Carbamylation as a mechanistic probe for the inhibition of class D β-lactamases by avibactam and halide ions. Org. Biomol. Chem. 15, 6024. 10.1039/C7OB01514C. [DOI] [PubMed] [Google Scholar]
- van Groesen E.; Lohans C. T.; Brem J.; Aertker K. M. J.; Claridge T. D. W.; Schofield C. J. (2019) 19F NMR monitoring of reversible protein post-translational modifications: class D β-lactamase carbamylation and inhibition. Chem. - Eur. J. 25, 11837–11841. 10.1002/chem.201902529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.