

Abstract
The SARS-CoV-2 virus is causing COVID-19, an ongoing pandemic, with extraordinary global health, social, and political implications. Currently, extensive research and development efforts are aimed at producing a safe and effective vaccine. In the interim, small molecules are being widely investigated for antiviral effects. With respect to viral replication, the papain-like (PLpro) and main proteases (Mpro), are critical for processing viral replicase polypeptides. Further, the PLpro possesses deubiquitinating activity affecting key signalling pathways, including inhibition of interferon and innate immune antagonism. Therefore, inhibition of PLpro activity with small molecules is an important research direction. Our aim was to focus on identification of potential inhibitors of the protease activity of SARS-CoV-2 PLpro. We investigated 300 small compounds derived predominantly from our OliveNet™ library (222 phenolics) and supplemented with synthetic and dietary compounds with reported antiviral activities. An initial docking screen, using the potent and selective noncovalent PLpro inhibitor, GRL-0617 as a control, enabled a selection of 30 compounds for further analyses. From further in silico analyses, including docking to scenes derived from a publicly available molecular dynamics simulation trajectory (100 μs PDB 6WX4; DESRES-ANTON-11441075), we identified lead compounds for further in vitro evaluation using an enzymatic inhibition assay measuring SARS-CoV-2 PLpro protease activity. Our findings indicate that hypericin possessed inhibition activity, and both rutin and cyanidin-3-O-glucoside resulted in a concentration-dependent inhibition of the PLpro, with activity in the micromolar range. Overall, hypericin, rutin, and cyanidin-3-O-glucoside can be considered lead compounds requiring further characterisation for potential antiviral effects in appropriate model systems.
Keywords: Coronavirus, COVID-19, SARS-CoV-2, Papain-like protease, GRL-0617, Hypericin, Rutin, Cyanidin-3-O-Glucoside, Molecular docking, Molecular dynamics simulations
Graphical abstract

1. Introduction
On December 31st, 2019, the World Health Organization China Country Office was informed of cases of atypical pneumonia identified in Wuhan City (Hubei Province, China) [1]. Within a week the causative pathogen was identified to be a novel coronavirus originally designated as 2019-nCoV [2]. The new coronavirus was shown to share genetic similarities with the severe acute respiratory syndrome coronavirus (SARS-CoV) identified in 2002 which caused an epidemic until 2004, and Middle East respiratory syndrome coronavirus (MERS-CoV) identified in 2012 which caused an outbreak that is still persisting, with the majority of cases in the Arabian Peninsula [[3], [4], [5], [6]]. The novel coronavirus shares approximately 50% sequence similarity with MERS-CoV and 80% with SARS-CoV, and was designated as SARS-CoV-2 [2]. Currently, SARS-CoV-2 is causing the ongoing COVID-19 pandemic with significant global health, social and political implications [7].
Like SARS-CoV, SARS-CoV-2 utilises the cell-surface angiotensin converting enzyme 2 (ACE2) via binding though the receptor binding domain of the viral surface spike (S) protein, to invade host cells [8,9]. Therefore, much research is devoted to modifying the interaction between the viral S protein and the ACE2 receptor. For example, soluble decoy ACE2 receptors have been widely investigated for their ability to prevent infection, and recombinant forms of the human ACE2 are undergoing evaluation in clinical trials [10,11]. The ultimate goal is the development and distribution of a safe and effective vaccine. Given the urgency, there are easily over 100 vaccines currently at different stages of development; there are numerous online vaccine trackers by reputable organizations that indicate between 120 and 170 vaccines currently in development [12]. There is conflicting information regarding the progress status of various vaccines however it appears that an mRNA-based vaccine (mRNA-1273), and two adenovirus vectored vaccines (ChAdOx1 nCoV-19 and Ad5-vectored COVID-19 vaccine), are currently the most advanced in clinical trials [[13], [14], [15], [16], [17], [18]].
Another potential therapeutic approach involves inhibiting viral replication. In this context, the adenine nucleotide analogue, remdesivir (Veklury®), initially identified from a broad antiviral screen against Ebola and emerging viruses, has received emergency use authorization for the treatment of hospitalized adult and paediatric patients with COVID-19 by the US Food and Drug Administration (FDA) [[19], [20], [21]]. By chain incorporation of the active form, remdesivir inhibits viral RNA-dependent RNA polymerase (RdRP), thereby terminating viral RNA synthesis [[22], [23], [24]]. Clinically, the first person (35 year old, male with no prior medical history), diagnosed with COVID-19 in the US was treated successfully with remdesivir [25]. Further case studies and clinical trials have highlighted modest clinical effects of remdesivir in people with severe COVID-19, with improvements in recovery times [[26], [27], [28], [29], [30]]. Numerous large-scale clinical trials are currently ongoing.
Along with the RdRP, the SARS-CoV-2 Mpro and PLpro are also critical antiviral targets [31]. These proteases are responsible for processing the open reading frame (ORF) 1a and 1 ab encoded viral polyproteins (pp) 1a and 1 ab to release the non-structural proteins (nsp) 1–16 [32]. Given their role in viral replication, potential inhibition of both of these proteases has been widely investigated. Development of antivirals against the Mpro has focused on covalent inhibitors of which the peptidomimetic α-ketoamides have emerged as an important class [33,34]. One of the lead compounds, α-ketoamide 13b, which was designed to possess a favourable pharmacokinetic profile, has been shown to inhibit Mpro activity with an IC50 of approximately 0.7 μM and to inhibit SARS-CoV-2 replication in vitro, with activity in the low micromolar range [34]. Other notable potential inhibitors of the SARS-CoV-2 Mpro include ebselen which emerged from a recent large-scale screening effort, and a covalent inhibitor denoted as compound 11a which has been shown to possess potent antiviral effects and favourable properties in vivo [34,35].
Our aim was to identify potential inhibitors of the SARS-CoV-2 PLpro. The PLpro is part of the larger multi-domain nsp3 protein [36,37]. Apart from protease activity critical for the viral life-cycle, the PLpro has deubiquitinating (DUB) activity and removes interferon-stimulated gene 15 (ISG15) from cellular proteins [38,39]. These DUB and de-ISG15 activities, result in the modulation of key cellular pathways, including NFκB inflammation and interferon (IFN)-1 which plays a role in host immunity [[40], [41], [42]]. A recent study has further clarified the role of the PLpro in attenuating viral spread and modulating innate immunity [43]. Therefore, inhibition of PLpro activity has been intensely investigated and promising covalent, and noncovalent, inhibitors have been identified [8]. We focused on noncovalent interactions and examined 300 small molecules as potential PLpro inhibitors, utilising our OliveNet™ library (222 phenolics and fatty acids) as well as a selection of dietary and synthetic compounds with reported antiviral activities [44]. The promising naphthalene-based noncovalent inhibitor, GRL-0617, was used as a control to enable selection of potential lead compounds [45].
2. Materials and methods
2.1. Protein structures and ligands
Several structures of the PLpro monomer were obtained from the RCSB Protein Data Bank [46]. This included 6XAA, 6W9C, 6WUU and 6WX4 [[47], [48], [49]]. Since PLpro crystallised as a complex for the 6WUU and 6W9C structures, a single chain was isolated. The ubiquitin-propargylamide chain and the peptide inhibitors VIR250 and VIR251 were removed from 6XAA, 6WUU and 6W9C, respectively. The crystal structure of PLpro with the naphthalene inhibitor GRL-0617 (PDB ID: 7JRN) was recently made available and a single chain was isolated for use [50]. The co-crystallised ligand was consequently removed. Zinc was retained in all of the crystal structures [51].
A total of 300 ligands were examined in this study and this included both pharmacological and dietary compounds. In addition to GRL-0617, which was used as a positive control, the antiviral drug α-ketoamide 13b was of interest [34,52]. A number of protease inhibitors (n = 8), antibiotics (n = 19) and sirtuin activators (n = 2) were examined, as well as compounds with anti-inflammatory, antiviral, antioxidant and anti-parasitic properties (n = 38) [[53], [54], [55], [56], [57], [58], [59], [60], [61], [62], [63], [64], [65], [66], [67], [68], [69], [70], [71], [72], [73], [74], [75], [76], [77]]. A selection of phenolic compounds (n = 220) and fatty acid esters (n = 13) from the OliveNet™ database were also utilised [44]. The structures of the ligands were sourced from the National Centre for Biotechnology Information (NCBI) PubChem database and if they were unavailable, they were drawn using Chem3D 19.0 (Perkin Elmer, Massachusetts, USA) [78]. The structure of hypericin was obtained from ChEMBL [79,80]. A full list of the compounds can be found in Table S1 of the Supplementary Information.
2.2. Initial screen for binding affinity and docking to the SARS-CoV-2 papain-like protease active pocket
The proteins were imported into the Schrödinger Suite and the Protein Preparation Wizard was used to preprocess, optimise and minimise the structures [81,82]. The protonation states of the amino acid residues were predicted using PROPKA with pH 7 and this was the default value. The ligands were also prepared using the LigPrep tool and this allowed for various conformers to be generated at a pH of 7.0 ± 2.0 [81]. The top ranking conformer was selected for docking. For the initial screen, 6WUU was used as the main crystal structure. A 20 × 20 × 20 Å receptor grid was centred around the residues that were surrounding the co-crystallised peptide inhibitor VIR250 [[83], [84], [85]]. The residues were M208, P247, T301, P248, D164, Y273, G163, Y112, L162, C111, N109, W106, H272, C270, Q269, Y268, Y264 and G271 [47]. These residues were also used to create the receptor grids for the 6W9C, 6XAA, 6WX4 and 7JRN structures. The optimized potential for liquid simulations 3e (OPLS3e) force field was used for the protein preparation, ligand preparation, receptor grid and ligand docking stages [[86], [87], [88], [89]].
The Glide ligand docking workflow was used to screen the library of 300 compounds using the standard precision (SP) mode [[83], [84], [85]]. Based on the Glide Energies (kcal/mol), 30 compounds and GRL-0617, were docked using the quantum-mechanics-polarised ligand docking (QPLD) protocol as previously described [[90], [91], [92], [93], [94], [95]]. The Glide Energies are shown in the Supplementary Information (Tables S1, S2 and S3). The lead compounds were docked to the 6W9C, 6XAA and 6WX4 crystal structures using the QPLD protocol [[90], [91], [92], [93], [94], [95]]. Based on the molecular docking results for 6WUU and the blind docking data, the receptor grid was adjusted for hypericin and its isomer. The residues that were used included K157, E167, D164, Y273, T301, Y264, P247, P248, Y268 and Q269 [52,96]. In regards to 7JRN, the positive control compound GRL-0617 was docked using the QPLD protocol [[90], [91], [92], [93], [94], [95]].
2.3. Blind docking to the SARS-CoV-2 papain-like protease and site mapping
The server PrankWeb was used to identify potential ligand binding sites within PLpro [97] (Table S4). Furthermore, blind docking of lead compounds was conducted on the crystal structures 6WUU, 6WX4, 6XAA and 6W9C. The structures of the protein and the ligands were processed in PyRx to generate the corresponding pdbqt files [98]. In this study, the protein was assumed to be rigid while all torsions of the ligands were activated. The receptor grid was generated around the entire protein. Docking was performed at an exhaustiveness of 2048 with AutoDock Vina using cloud computing services provided by Galileo (Hypernet Labs) [99,100]. Details can be found in the Supplementary Information (Tables S5 and S6).
2.4. Time-dependent docking using a publicly available molecular dynamics simulation trajectory
The 100 μs molecular dynamics (MD) simulation trajectory of the PLpro in its apo form was downloaded from D.E. Shaw Research [101]. Structures of the protein at six time points (1, 2, 10, 20, 50 and 100 μs) were extracted [102]. Hypericin, an isomer of hypericin, cyanidin-3-O-glucoside, rutin, (−)-epigallocatechin gallate, cefotaxime, α-ketoamide 13b and GRL-0617 were docked to the PLpro structure at each time point using the QPLD protocol of the Schrödinger Suite.
The protein structure alignment tool in Maestro was used to compare the protein structures obtained at these time points: 1 to 2, 2 to 10, 10 to 20, 20 to 50 and 50–100 μs [103]. The crystal structures 6XAA, 6W9C, 6WX4 and 7JRN were also aligned. The binding site was also separately aligned using the binding site alignment tool, using the receptor grid residues [103]. Moreover, the docked ligands were aligned using the superposition tool [103]. The root mean square deviation (RMSD) and alignment scores were recorded for each alignment process [103]. These details can be found in the Supplementary Information (Tables S7–S9).
2.5. Enzymatic inhibition assay
To investigate the inhibition of the SARS-CoV-2 PLpro in vitro, an enzymatic inhibition assay was performed. The proprietary papain-like protease (SARS-CoV-2) assay kit measuring protease activity was utilised (BPS Bioscience, San Diego, CA, USA). The assay was performed according to the manufacturer’s instructions, using a fluorogenic substrate with detection at an emission wavelength of 460 nm (excitation wavelength = 360 nm). The internal positive control was the nonconvalent GRL-0617 inhibitor which was used at a concentration of 100 μM in the assay (n = 3). The test small molecule inhibitors were hypericin (89%, HWI pharma services GmbH, Germany), cyanidin-3-O-glucoside (reference standard, PhytoLab, Germany), and rutin (>94%), (−)-epicatechin gallate (>98%), (−)-epigallocatechin gallate (>95%), and cefotaxime (European pharmacopoeia reference standard) from Sigma-Aldrich (St Luis, MO, USA). The inhibitors were prepared as 20 mM stock solutions and stored at −80 °C until use. For use in the assay, doubling dilutions to achieve a final concentration range of 3.1 μM–200 μM for cyanidin-3-O-glucoside, rutin, and (−)-epigallocatechin gallate, were performed in assay buffer. Hypericin, (−)-epicatechin gallate, and cefotaxime were tested at 50 and 100 μM. All test inhibitors at each concentration, GRL-0617 positive control, and background were assayed in triplicate; six determinations were made for the total PLpro protease activity. Absolute fluorescence intensity values were measured, and the % protease inhibition was calculated.
3. Results and discussion
3.1. Selection of compounds with relatively high affinity for SARS-CoV-2 papain-like protease active pocket
In order to narrow down the library of 300 compounds and identify potential lead compounds, molecular docking was used to screen the ligands. This included the positive control inhibitor GRL-0617. For the initial screen in standard precision mode, GRL-0617 was predicted to bind to PLpro with an affinity of −36.3 kcal/mol. In total, 295 compounds produced Glide Energies ranging from −13.8 to −69.1 kcal/mol (Table S1). In regards to the phenolic compounds from the OliveNet™ database, the molecular weights of the ligands were compared to the binding affinities and the correlation coefficient was found to be 0.7 (Figure S1). In comparison to other docking programs that are available, it has been demonstrated that Glide predicts the crystallographic orientation of compounds with greater accuracy [104]. The standard precision mode allows for libraries of ligands to be reliably docked to the specified binding site [83,85].
Based on the data generated, 30 compounds were selected for further analysis (Table S2). With the exception of the protease inhibitor oseltamivir and the anti-malarial medication hydroxychloroquine, the Glide Energies of these ligands (−37.0 to −62.4 kcal/mol) were stronger than the noncovalent inhibitor GRL-0617. They included α-ketoamide 13b, suramin, nüzhenide, ritonavir, remdesivir, acteoside, (−)-epigallocatechin gallate, rutin, hesperidin, lopinavir, ceftriaxone, saquinavir, amikacin, darunavir, indinavir, cyanidin-3-O-glucoside, ertapenem, SRT2104, cefuroxime, baricitinib, cefotaxime, salidroside, SRT1720, nelfinavir, doripenem, arbidol, hypericin and the hypericin isomer. Oseltamivir and hydroxychloroquine were predicted to bind with an affinity of −34.6 and −32.2 kcal/mol, respectively.
The PLpro crystal structure 6WUU was imported into PrankWeb and the protein was analysed for potential ligand binding sites. A binding pocket was identified just above the catalytic triad that consisted of residues G163, G164, R166, M208, P247, P248, Y264, Y268, Y273, T301 and D302 (Fig. 1 ). This was referred to as pocket 2 and it had a score of 3.6 (Table S4). This binding site has previously been characterised in the literature and a number of studies have showed that naphthalene-based inhibitors target this region [52,105,106]. In a recently published paper by Bosken et al. the naphthalene-based inhibitor 3k was investigated for its ability to target the SARS-CoV-2 PLpro and they demonstrated that the ligand binds to this site, adjacent to the catalytic triad [105]. The authors noted that 3k inhibited the enzymatic activity of PLpro, sterically interfering with the binding of ubiquitin and ISG15 [105].
Fig. 1.

Binding of the noncovalent inhibitor GRL-0617 to various forms of PLpro. The SARS-CoV-2 PLpro contains several domains highlighted in A), and catalytic triad residues C111, H272, and D286 are shown. PrankWeb analysis identified the naphthalene inhibitor binding pocket as a potential binding site (B). The known inhibitor GRL-0617 was docked to the binding pocket in various structures of the PLpro protein (C, D), as well as along time points of a molecular dynamics simulation trajectory in E). Residue interactions are shown as dashed lines, including hydrogen bonds (yellow), salt bridges (magenta), π−π interactions (cyan), and π−cation interactions (green). Amino acid residues are coloured according to their properties, namely hydrophobic residues (green), polar uncharged residues (cyan), negatively charged residues (red), and glycine residues (grey). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Blind docking was subsequently performed using GRL-0617 and the top 30 compounds. The aim was to determine whether the ligands would preferentially bind to the target site in PLpro. The ligands that were predicted to have poses within this naphthalene inhibitor binding pocket were amikacin, arbidol, baricitinib, cefotaxime, ceftriaxone, cyanidin-3-O-glucoside, cefuroxime, darunavir, doripenem, (−)-epigallocatechin gallate, ertapenem, hesperidin, hypericin, the hypericin isomer, nüzhenide, oseltamivir, rutin, salidroside, saquinavir, and SRT1720 (Table S5). In addition to GRL-0617, 11 compounds were selected for further examination. They included α-ketoamide 13b, amikacin, doripenem, saquinavir, SRT1720, (−)-epigallocatechin gallate, cefotaxime, rutin, cyanidin-3-O-glucoside, hypericin, and the hypericin isomer.
The top ligands were then re-docked to the binding site of the co-crystallised VIR250 peptide inhibitor using the QPLD protocol of the Schrödinger Suite (Table S3). The extra precision (XP) mode was selected for this step and as described by Freisner et al. this scoring function is important for eliminating false positives [84]. In a paper that was made available by Rut et al. details were provided about the crystal structures of PLpro with the peptide inhibitors VIR250 and VIR251 [47]. The catalytic triad residues of the SARS-CoV-2 PLpro are C111, H272 and D286. These inhibitors have been found to form a covalent thioether linkage with C111 through a Michael addition reaction [47]. GRL-0617 was predicted to bind with an affinity of −38.2 kcal/mol. Glide Energies and the inter-atomic contacts that were formed between protein residues, the positive control inhibitor and the top compounds are summarised in Table 1 .
Table 1.
The Glide Energies (kcal/mol) and structures of the top compounds are provided, as well as details about the inter-atomic contacts formed. Refined selection of potential lead compounds.
| Compounds and Glide Energies | Structures | Inter-atomic contacts |
|---|---|---|
| α-ketoamide 13b: −60.3 kcal/mol |
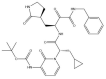 |
|
| GRL-0617: −38.2 kcal/mol |
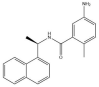 |
|
| (−)-Epigallocatechin gallate: −56.4 kcal/mol |
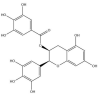 |
|
| Cyanidin-3-O-glucoside: −59.2 kcal/mol |
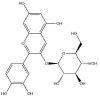 |
|
| Rutin: −59.9 kcal/mol |
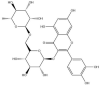 |
|
| Cefotaxime: −41.6 kcal/mol |
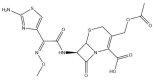 |
|
| Amikacin: −50.1 kcal/mol |
 |
|
| Doripenem: −44.9 kcal/mol |
 |
|
| Saquinavir: −47.2 kcal/mol |
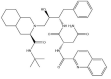 |
|
| SRT1720: −44.3 kcal/mol |
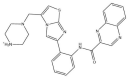 |
|
| Hypericin: −38.0 kcal/mol |
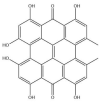 |
|
| Hypericin isomer: −38.0 kcal/mol |
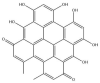 |
|
Most notably, these ligands were positioned in the naphthalene inhibitor binding pocket identified from blind docking and the PrankWeb analysis. GRL-0617 has previously been found to bind to this site in both SARS-CoV and SARS-CoV-2 [96,106]. It has been reported that this inhibitor interacts with several residues including K157, E167, D164, Y273, T301, Y264, P247, P248, Y268 and Q269 [52,96]. This is consistent with the results obtained from the present study as GRL-0617 was surrounded by M208, P247, P248, Y264, Y273, K157, R166, D164 and T301 (Fig. 1). The noncovalent inhibitor formed a hydrogen bond with Y264 (2.1 Å), a π-π interaction with Y264 (5.4 Å) and a hydrogen bond with Q269 (1.7 Å). GRL-0617 was also docked to the crystal structure 7JRN and its binding mode was compared to the experimentally determined structure. The co-crystallised ligand formed a single hydrogen bond with Q269 (2.1 Å) and this was also the case for the docked structure. The distance between residue Q269 and the oxygen atom of GRL-0617 was 2.0 Å. The docked ligand also formed three π-π interactions with Y268 and the Glide Energy was determined to be −43.2 kcal/mol.
Depending on the size and orientation of the compounds, some had side chains that were able to extend beyond the pocket and were positioned deeper in the active site. When examining ligands within 5 Å of cefotaxime, the hydrophobic residues Y112, C111, T273 and C270 were present. C111 and H272 are part of the catalytic triad, whereas Y112, Y273 and C270 are adjacent residues. Although cefotaxime formed inter-atomic contacts with residues in the naphthalene inhibitor binding site, it was positioned in proximity to the catalytic residues. Several of these amino acids also appeared in the ligand interaction diagrams of GRL-0617, α-ketoamide 13b, rutin, (−)-epigallocatechin gallate and cyanidin-3-O-glucoside.
GRL-0617, rutin, (−)-epigallocatechin gallate, cyanidin-3-O-glucoside, α-ketoamide 13b, cefotaxime, hypericin and the hypericin isomer were selected for further in silico analysis. Docking of these compounds to the naphthalene binding pocket is depicted in Fig. 2 . (−)-Epigallocatechin gallate is a polyphenol that is primarily found in green tea and is a type of catechin [107,108]. Cyanidin-3-O-glucoside is an anthocyanin and rutin is a plant pigment that can be found in a variety of fruits and vegetables [71,109]. These three compounds belong to the flavonoid subclass and their bioactivities have been greatly explored [76]. Conversely, cefotaxime is an antibiotic [110]. Hypericin, a naphthodianthrone constituent of St. John’s Wort, was also of interest [111]. Following adjustment of the grid for the 6WUU crystal structure, hypericin and its isomer were docked to the naphthalene inhibitor binding site within PLpro and both compounds had a Glide Energy of −38.0 kcal/mol. Hypericin formed hydrogen bonds with E167, Y264, G163 and L162, as well as a π−π cation with R166. The isomer of hypericin formed two hydrogen bonds with Y273 and G163, as well as a π-π interaction with Y268. The antidepressant effects of this compound have been of particular interest, and antiviral properties have also been reported [112,113]. Although hypericin has a binding affinity that is weaker than other compounds, it is important to consider its position, orientation and the interactions that are formed with the protein residues within the PLpro binding site. Hypericin has also been examined as a lead compound for the SARS-CoV-2 spike protein and Mpro [114,115].
Fig. 2.

Docking of lead compounds to the naphthalene inhibitor binding pocket and PLpro and blind docking. Lead compounds were docked to the naphthalene inhibitor binding pocket of PLpro using the QPLD protocol of Glide, and interactions with residues are depicted. Blind docking was performed using AutoDock Vina to produce 9 poses, which are shown for the respective compounds. Residue interactions are shown as dashed lines, including hydrogen bonds (yellow), salt bridges (magenta), π−π interactions (cyan), and π−cation interactions (green). Amino acid residues are coloured according to their properties, namely hydrophobic residues (green), polar uncharged residues (cyan), negatively charged residues (red), and glycine residues (grey). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
In order to validate the results and compare the binding properties of the ligands to various PLpro structures available on the RCSB PDB, the compounds were docked to the structures 6XAA (ubiquitin-bound), 6W9C (apo) and 6WX4 (peptide inhibitor-bound). These crystal structures were analysed using PrankWeb and in addition to the main catalytic triad residues, the pocket consisting of the amino acids G163, G164, R166, M208, P247, P248, Y264, Y268, Y273, T301 and D302 was identified (Table S4). The site ranked as pocket 2 in 6XAA, pocket 1 in 6WX4 and pocket 4 in 6W9C. Their ligandability scores were 3.5, 4.2 and 1.4, respectively [97]. Blind docking was conducted on these crystal structures of PLpro and the results demonstrated that GRL-0617 had poses within the naphthalene inhibitor binding site (Table S6).
The crystal structures were aligned, producing scores of 0.1 for all alignments (6WUU to 6W9C, 6WUU to 6XAA, 6WUU to 6WX4 and 6WUU to 7JRN) [103]. The RMSD values were 1.7 Å for 6WUU and 6W9C, 1.8 Å for 6WUU and 6WX4, 1.5 Å for 6WUU and 6XAA, and 1.7 Å for 6WUU and 7JRN. Bosken et al. and Báez-Santos et al. have discussed the dynamics of the SARS-CoV-2 PLpro and note that the fingers domain, particularly the zinc-binding region, blocking loop 2 (BL2) and the N-terminal Ub-like (UBL) domain are highly flexible [51,105].
When the lead compounds were docked to the naphthalene inhibitor binding pocket of 6XAA, 6WX4 and 6W9C, inter-atomic contacts with the residues Y264, Y268, G271, T301, D164, E167, G163 and Y273 were found in all three structures (Fig. 3, Fig. 4 ). K157 and R166 were also prominent residues. Interestingly, cyanidin-3-O-glucoside formed two π−π interactions with the catalytic triad residue H272 in the 6W9C crystal structure. This flavonoid compound had a strong binding affinity for all three PLpro structures and this was also true for rutin.
Fig. 3.

Docking of lead pharmacological compounds to different structures of PLpro. Lead pharmacological compounds were docked to the naphthalene inhibitor binding pocket of different structures of PLpro (PDB ID: 6XAA, 6W9C, and 6WX4) using the QPLD protocol of Glide. Residue interactions are shown as dashed lines, including hydrogen bonds (yellow), salt bridges (magenta), π−π interactions (cyan), and π−cation interactions (green). Amino acid residues are coloured according to their properties, namely hydrophobic residues (green), polar uncharged residues (cyan), negatively charged residues (red), and glycine residues (grey). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Fig. 4.

Docking of lead dietary compounds to different structures of PLpro. Lead dietary compounds were docked to the naphthalene inhibitor binding pocket of different structures of PLpro (PDB ID: 6XAA, 6W9C, and 6WX4) using the QPLD protocol of Glide. Residue interactions are shown as dashed lines, including hydrogen bonds (yellow), salt bridges (magenta), π−π interactions (cyan), and π−cation interactions (green). Amino acid residues are coloured according to their properties, namely hydrophobic residues (green), polar uncharged residues (cyan), negatively charged residues (red), and glycine residues (grey). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Molecular docking was also performed at various time points (1, 2, 10, 20, 50 and 100 μs) of a publicly available MD simulation trajectory (D.E. Shaw Research). As aforementioned, various conformations of the BL2 and fingers domain in the coronavirus PLpro crystal structures have previously been observed [51,105]. Through docking the compounds to the structures of PLpro obtained from D.E. Shaw’s 100 μs MD simulation trajectory, the aim was to determine whether there were any differences in the binding properties of the ligands over time. The crystal structures of PLpro at these time points were compared, producing alignment scores of 0.1 for 1–2 μs (RMSD = 1.4 Å), 0.1 for 2–10 μs (RMSD = 1.9 Å), 0.2 for 10–20 μs (RMSD = 2.0 Å), 0.3 for 20–50 μs (RMSD = 2.7 Å), and 0.3 for 50–100 μs (RMSD = 2.9 Å). When comparing the 1 μs structure of PLpro to 100 μs, the alignment score was also 0.1 and the RMSD was 1.7 Å.
The compounds were docked to each structure and the protein-ligand complexes were examined (Table 2 , Fig. 5, Fig. 6 ). Docked compounds were superimposed and the RMSD was calculated (Table S7). Proteins were also aligned based on the receptor grid residues and the RMSD values were recorded. The RMSD scores for this region were found to be 0.9 Å for 1–2 μs, 0.9 Å for 2–10 μs, 0.7 Å for 10–20 μs, 1.3 Å for 20–50 μs and 1.7 Å for 50–100 μs. While there were some differences in orientation of the ligands at each time point, this may be attributed to more flexible key residues.
Table 2.
The protein residues that formed hydrogen bonds with the lead ligands at each time point of the D.E. Shaw SARS-CoV-2 PLpro MD simulation trajectory are provided.
| 1 μs | 2 μs | 10 μs | 20 μs | 50 μs | 100 μs | |
|---|---|---|---|---|---|---|
| GRL-0617 | None | Y273, G163 | Y273, G271 | Y264 | D164, Y273, D302 | Y264 |
| Rutin | L162, G163, D164, R166, Y273, G266 | Y273, D164, E167, K157, C155, Y171 | Y264, Y268, Q269, T301, R166, D164 | Y273, D164, R166, G266 | N267, Y273, G163, D164, R166 | D164, G163, Q269, N267, G266, Y264 |
| Cyanidin-3-O-glucoside | G163, D164, G266 | N267, Y268, Q269, E161, E167 | G271, Y268, L162, D164 | Q269, G271, Y273, D164 | K157, R166 | Q269, N267, G266, Y264, R166, A246 |
| Hypericin | R166, Y273, G163, Y268 | K157, L162, N267, R166 | Y273, R166, Y264 | D164, R166, Y273 | K157, R166, E167 | Y268, R166 |
Fig. 5.

Docking of lead pharmacological compounds to snapshots of a 100 μs dynamic simulation of PLpro (100 μs PDB 6WX4; DESRES-ANTON-11441075). Molecular docking was performed to multiple snapshots of a dynamic simulation of PLpro, with comparisons between the different time points shown. Binding site residues were aligned, and RMSDs between time points are stated. Residues with an RMSD greater than 2 following alignment are labelled and highlighted in a ball and stick representation. Lead pharmacological compounds docked to the naphthalene inhibitor binding pocket are depicted, with Glide Energies for docking to each structure stated. Residue interactions are shown as dashed lines, including hydrogen bonds (yellow), salt bridges (magenta), π−π interactions (cyan), and π−cation interactions (green). Amino acid residues are coloured according to their properties, namely hydrophobic residues (green), polar uncharged residues (cyan), negatively charged residues (red), and glycine residues (grey). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Fig. 6.

Docking of lead dietary compounds to snapshots of a dynamic simulation of PLpro (100 μs PDB 6WX4; DESRES-ANTON-11441075). Molecular docking was performed to multiple snapshots of a dynamic simulation of PLpro, with comparisons between the different time points shown. Lead dietary compounds docked to the naphthalene inhibitor binding pocket are depicted with Glide Energies for docking to each structure stated. Residue interactions are shown as dashed lines, including hydrogen bonds (yellow), salt bridges (magenta), π−π interactions (cyan), and π−cation interactions (green). Amino acid residues are coloured according to their properties, namely hydrophobic residues (green), polar uncharged residues (cyan), negatively charged residues (red), and glycine residues (grey). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Several receptor grid residues produced a higher RMSD value (>2 Å) when comparing the proteins for 20–50 μs and 50–100 μs. They included L162, N109, W106, C270, P248, H272, G271, Q269 and Y268 (Table S8). Interestingly, the RMSD for Y268 was above 3 Å for all comparisons made. The residue Y268 is important for the binding mode of GRL-0617 and Shin et al. have recently demonstrated this through conducting MD simulations [43]. In saying this, Y268 also had a higher RMSD value in the protein alignment of 6XAA, 6W9C and 7JRN with 6WUU as the reference structure (Table S9). The RMSD scores for this residue were 3.8 Å, 4.4 Å and 4.6 Å in the respective structures. The flexibility that was identified in the binding site residues may explain the different binding orientations observed for the compounds in the current study.
Hypericin, rutin, and cyanidin-3-O-glucoside as potential lead compounds: inhibition of protease activity in vitro.
To confirm inhibition of the SARS-CoV-2 PLpro in vitro, we performed an enzymatic inhibition assay, which specifically measures protease activity. Overall, our findings indicate that, at least to some extent, all of our selected ligands inhibited PLpro activity (Fig. 7 ). The noncovalent inhibitor GRL-0617 was used as an internal positive control in our assay. The IC50 value for inhibition of the SARS-CoV-2 protease activity by GRL-0617 has been determined to be 1.6 μM in this assay system (BPS Bioscience), and our findings indicated that a 100 μM concentration of the compound inhibited protease activity by approximately 94% in our experiments (Fig. 7B). Interestingly, our findings indicate a similar potency for hypericin, with approximately 97% inhibition of protease activity at 100 μM (Fig. 7B). Our data may suggest a concentration dependence as we measured an approximately 87% inhibition in protease activity at a concentration of 50 μM hypericin. However, further experiments are required to clarify the concentration-dependence and precise IC50 value for hypericin with a broader range of ligand concentrations; autofluorescence and/or fluorescence quenching properties of the ligand may also require additional consideration given its well-known photoreactive qualities. Nevertheless, our results show that both rutin and cyanidin-3-O-glucoside result in a concentration-dependent inhibition of the SARS-CoV-2 PLpro activity (Fig. 7A). Although, the IC50 values were not clarified in our experiment, it is evident that both ligands are less potent than the GRL-0617 positive control (∼94% inhibition of protease activity at 100 μM), with rutin and cyanidin-3-O-glucoside inhibiting approximately 38% and 20% of the protease activity at a concentration of 100 μM. Indeed, at 100 μM the inhibitory activity observed for cyanidin-3-O-glucoside was more closely related to those measured for epigallocatechin gallate (13%), epicatechin gallate (20%), and cefotaxime (16%). In summary, our results are encouraging and indicate inhibition of the SARS-CoV-2 PLpro activity with an order of potency of GRL-0617 and hypericin > rutin > cyanidin-3-O-glucoside and epicatechin gallate > cefotaxime and epigallocatechin gallate.
Fig. 7.

Inhibition of the SARS-CoV-2 PLpro protease activity by small molecules. To investigate in vitro inhibition of protease activity a PLpro enzymatic inhibition assay measuring protease activity was performed. Using a proprietary fluorogenic substrate (BPS Bioscience), fluorescence intensities at an emission wavelength of 460 nm were measured. The small molecules rutin (closed circles), and cyanidin-3-0-glucoside (open circles) were shown to result in a concentration-dependent inhibition of protease activity (A). The concentration range between 3 μM and 200 μM was investigated. Average values ± SEM from triplicate determinations are depicted. The average background (n = 3), total protease activity (n = 6), and inhibition by the noncovalent positive control GRL-0617 at 100 μM (n = 3), are highlighted (horizontal dotted lines). The % inhibition of PLpro protease activity at 100 μM concentrations of the indicated small molecule inhibitors was calculated (B). Average values ± SEM from triplicate determinations are depicted.
4. Conclusion
Overall, we have identified potential nonconvalent small molecule protease inhibitors of the SARS-CoV-2 PLpro. In particular, rutin and cyanidin-3-O-glucoside, were identified by our in silico analyses as compounds with preferential binding properties. Although not as potent as the GRL-0617 positive control, these were shown to inhibit the protease in a concentration-dependent manner with activity in the micromolar range. Hypericin, which also displayed appropriate binding characteristics in silico, exhibited activity in the in vitro enzymatic inhibition assay. Further research will be important to compare IC50 values and antiviral effects to GRL-0617. Indeed, the examination of the precise potency of hypericin, rutin and, cyanidin-3-O-glucoside requires further characterisation in antiviral model systems, and it will be interesting to determine potential deubiquitinating properties, which is also an important feature of the SARS-CoV-2 PLpro protease.
Author contributions statement
TCK and AH: conceptualization and supervision. EP: formal analysis, data curation, writing – original draft. JL: formal analysis, data curation. KV: formal analysis, data curation. TCK, AH, EP, JL, KV: writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We would like to acknowledge intellectual and financial support by McCord Research (Iowa, USA). JL are supported by an Australian Government Research Training Program Scholarship. We are indebted to Alfonso Perez Escudero and the team at Crowdfight COVID-19 for enabling access to supercomputing facilities, and to Matthew Gasperetti and the team at Hypernet Labs; Galileo, for enabling cloud computing for this project. We thank the National Computing Infrastructure (NCI), and the Pawsey Supercomputing Centre in Australia (funded by the Australian Government). Further, we thank the Spartan High Performance Computing service (University of Melbourne), and the Partnership for Advanced Computing in Europe (PRACE) for awarding the access to Piz Daint, hosted at the Swiss National Supercomputing Centre (CSCS), Switzerland.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmgm.2021.107851.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Hui D.S., IA E., Madani T.A., Ntoumi F., Kock R., Dar O., et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health - the latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020;91:264–266. doi: 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coronaviridae Study Group of the International Committee on Taxonomy of V The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4):536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. 10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26(4):450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14(8):523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen B., Tian E.K., He B., Tian L., Han R., Wang S., et al. Overview of lethal human coronaviruses. Signal Transduct Target Ther. 2020;5(1):89. doi: 10.1038/s41392-020-0190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cucinotta D., Vanelli M. WHO declares COVID-19 a pandemic. Acta Biomed. 2020;91(1):157–160. doi: 10.23750/abm.v91i1.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh A.K., Brindisi M., Shahabi D., Chapman M.E., Mesecar A.D. Drug development and medicinal chemistry efforts toward SARS-coronavirus and Covid-19 therapeutics. ChemMedChem. 2020;15(11):907–932. doi: 10.1002/cmdc.202000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280 e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pang X., Cui Y., Zhu Y. Recombinant human ACE2: potential therapeutics of SARS-CoV-2 infection and its complication. Acta Pharmacol. Sin. 2020;41:1255–1257. doi: 10.1038/s41401-020-0430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monteil V., Kwon H., Prado P., Hagelkruys A., Wimmer R.A., Stahl M., et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181(4):905–913 e7. doi: 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Callaway E. The race for coronavirus vaccines: a graphical guide. Nature. 2020;580(7805):576–577. doi: 10.1038/d41586-020-01221-y. [DOI] [PubMed] [Google Scholar]
- 13.Rego G.N.A., Nucci M.P., Alves A.H., Oliveira F.A., Marti L.C., Nucci L.P., et al. Current clinical trials protocols and the global effort for immunization against SARS-CoV-2. Vaccines. 2020;8(3) doi: 10.3390/vaccines8030474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu F.C., Guan X.H., Li Y.H., Huang J.Y., Jiang T., Hou L.H., et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2020;396:479–488. doi: 10.1016/S0140-6736(20)31605-6. 10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu F.C., Li Y.H., Guan X.H., Hou L.H., Wang W.J., Li J.X., et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. Lancet. 2020;395:1845–1854. doi: 10.1016/S0140-6736(20)31208-3. 10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folegatti P.M., Ewer K.J., Aley P.K., Angus B., Becker S., Belij-Rammerstorfer S., et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet. 2020;396:467–478. doi: 10.1016/S0140-6736(20)31604-4. 10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corbett K.S., Edwards D.K., Leist S.R., Abiona O.M., Boyoglu-Barnum S., Gillespie R.A., et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature. 2020;586:567–571. doi: 10.1038/s41586-020-2622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson L.A., Anderson E.J., Rouphael N.G., Roberts P.C., Makhene M., Coler R.N., et al. An mRNA vaccine against SARS-CoV-2 - preliminary report. N. Engl. J. Med. 2020;383:1920–1931. doi: 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antinori S., Cossu M.V., Ridolfo A.L., Rech R., Bonazzetti C., Pagani G., et al. Compassionate remdesivir treatment of severe Covid-19 pneumonia in intensive care unit (ICU) and Non-ICU patients: clinical outcome and differences in post_treatment hospitalisation status. Pharmacol. Res. 2020:104899. doi: 10.1016/j.phrs.2020.104899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pardo J., Shukla A.M., Chamarthi G., Gupte A. The journey of remdesivir: from Ebola to COVID-19. Drugs Context. 2020;9 doi: 10.7573/dic.2020-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siegel D., Hui H.C., Doerffler E., Clarke M.O., Chun K., Zhang L., et al. Discovery and synthesis of a phosphoramidate prodrug of a pyrrolo[2,1-f][triazin-4-amino] adenine C-nucleoside (GS-5734) for the treatment of Ebola and emerging viruses. J. Med. Chem. 2017;60(5):1648–1661. doi: 10.1021/acs.jmedchem.6b01594. [DOI] [PubMed] [Google Scholar]
- 22.Gordon C.J., Tchesnokov E.P., Feng J.Y., Porter D.P., Gotte M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020;295(15):4773–4779. doi: 10.1074/jbc.AC120.013056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon C.J., Tchesnokov E.P., Woolner E., Perry J.K., Feng J.Y., Porter D.P., et al. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020;295(20):6785–6797. doi: 10.1074/jbc.RA120.013679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tchesnokov E.P., Feng J.Y., Porter D.P., Gotte M. Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses. 2019;11(4) doi: 10.3390/v11040326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holshue M.L., DeBolt C., Lindquist S., Lofy K.H., Wiesman J., Bruce H., et al. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020;382(10):929–936. doi: 10.1056/NEJMoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olender S.A., Perez K.K., Go A.S., Balani B., Price-Haywood E.G., Shah N.S., et al. Remdesivir for severe COVID-19 versus a cohort receiving standard of care. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y., Zhang D., Du G., Du R., Zhao J., Jin Y., et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–1578. doi: 10.1016/S0140-6736(20)31022-9. 10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y., Zhou F., Zhang D., Zhao J., Du R., Hu Y., et al. Evaluation of the efficacy and safety of intravenous remdesivir in adult patients with severe COVID-19: study protocol for a phase 3 randomized, double-blind, placebo-controlled, multicentre trial. Trials. 2020;21(1):422. doi: 10.1186/s13063-020-04352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grein J., Ohmagari N., Shin D., Diaz G., Asperges E., Castagna A., et al. Compassionate use of remdesivir for patients with severe Covid-19. N. Engl. J. Med. 2020;382:2327–2336. doi: 10.1056/NEJMoa2007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., et al. Remdesivir for the treatment of Covid-19 - preliminary report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281(18):4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshimoto F.K. The proteins of severe acute respiratory syndrome coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J. 2020;39(3):198–216. doi: 10.1007/s10930-020-09901-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang J., Pitsillou E., Karagiannis C., Darmawan K.K., Ng K., Hung A., et al. Interaction of the prototypical alpha-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020;87:107292. doi: 10.1016/j.compbiolchem.2020.107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai W., Zhang B., Su H., Li J., Zhao Y., Xie X., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368(6497):1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jang K.-J., Jeong S., Kang D.Y., Sp N., Yang Y.M., Kim D.-E. A high ATP concentration enhances the cooperative translocation of the SARS coronavirus helicase nsP13 in the unwinding of duplex RNA. Sci. Rep. 2020;10(1):4481. doi: 10.1038/s41598-020-61432-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lei J., Kusov Y., Hilgenfeld R. Nsp3 of coronaviruses: structures and functions of a large multi-domain protein. Antivir. Res. 2018;149:58–74. doi: 10.1016/j.antiviral.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harcourt B.H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K.M., Smith C.M., et al. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004;78(24):13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clasman J.R., Everett R.K., Srinivasan K., Mesecar A.D. Decoupling deISGylating and deubiquitinating activities of the MERS virus papain-like protease. Antivir. Res. 2020;174:104661. doi: 10.1016/j.antiviral.2019.104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., et al. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U. S. A. 2006;103(15):5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83(13):6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282(44):32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bailey-Elkin B.A., Knaap R.C., Johnson G.G., Dalebout T.J., Ninaber D.K., van Kasteren P.B., et al. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014;289(50):34667–34682. doi: 10.1074/jbc.M114.609644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonvino N.P., Liang J., McCord E.D., Zafiris E., Benetti N., Ray N.B., et al. Database (Oxford); 2018. OliveNet™: a Comprehensive Library of Compounds from Olea Europaea. 2018:bay016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U. S. A. 2008;105(42):16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., et al. The protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S.J., et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. bioRxiv. 2020;6(42) doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osipiuk J., Jedrzejczak R., Tesar C., Endres M., Stols L., Babnigg G., et al. 2020. The Crystal Structure of Papain-like Protease of SARS CoV-2. [Google Scholar]
- 49.Klemm T., Ebert G., Calleja D.J., Allison C.C., Richardson L.W., Bernardini J.P., et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020;39:e106275. doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sacco M., Ma C., Wang J., Chen Y. 2020. Crystal Structure of the Native SARS-CoV-2 Papain-like Protease (PLPro) with Inhibitor GRL0617. [Google Scholar]
- 51.Báez-Santos Y.M., St John S.E., Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freitas B.T., Durie I.A., Murray J., Longo J.E., Miller H.C., Crich D., et al. Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect. Dis. 2020;6(8):2099–2109. doi: 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- 53.Agbowuro A.A., Huston W.M., Gamble A.B., Tyndall J.D.A. Proteases and protease inhibitors in infectious diseases. Med. Res. Rev. 2018;38(4):1295–1331. doi: 10.1002/med.21475. [DOI] [PubMed] [Google Scholar]
- 54.Agostini M.L., Andres E.L., Sims A.C., Graham R.L., Sheahan T.P., Lu X., et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9(2) doi: 10.1128/mBio.00221-18. e00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amici C., Di Caro A., Ciucci A., Chiappa L., Castilletti C., Martella V., et al. Indomethacin has a potent antiviral activity against SARS coronavirus. Antivir. Ther. 2006;11(8):1021–1030. [PubMed] [Google Scholar]
- 56.Chan C.N., Trinité B., Levy D.N. Potent inhibition of HIV-1 replication in resting CD4 T cells by resveratrol and pterostilbene. Antimicrob. Agents Chemother. 2017;61(9):e00408–e00417. doi: 10.1128/AAC.00408-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen H., Muhammad I., Zhang Y., Ren Y., Zhang R., Huang X., et al. vol. 10. 2019. (Antiviral Activity against Infectious Bronchitis Virus and Bioactive Components of Hypericum perforatum L). 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davies B.E. Pharmacokinetics of oseltamivir: an oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J. Antimicrob. Chemother. 2010;65 doi: 10.1093/jac/dkq015. Suppl 2(Suppl 2):ii5-ii10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gbinigie K., Frie K. Should chloroquine and hydroxychloroquine be used to treat COVID-19? A rapid review. BJGP Open. 2020;4(2) doi: 10.3399/bjgpopen20X101069. bjgpopen20X101069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huemer H.P. Possible immunosuppressive effects of drug exposure and environmental and nutritional effects on infection and vaccination. Mediat. Inflamm. 2015;2015:349176. doi: 10.1155/2015/349176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ichikawa T., Hayashi R., Suzuki K., Imanishi S., Kambara K., Okazawa S., et al. Sirtuin 1 activator SRT1720 suppresses inflammation in an ovalbumin-induced mouse model of asthma. Respirology. 2013;18(2):332–339. doi: 10.1111/j.1440-1843.2012.02284.x. [DOI] [PubMed] [Google Scholar]
- 62.Khaerunnisa S., Kurniawan H., Awaluddin R., Suhartati S., Soetjipto S. 2020. Potential Inhibitor of COVID-19 Main Protease (Mpro) from Several Medicinal Plant Compounds by Molecular Docking Study. [Google Scholar]
- 63.Kim D.E., Min J.S., Jang M.S., Lee J.Y., Shin Y.S., Song J.H., et al. Natural bis-benzylisoquinoline alkaloids-tetrandrine, fangchinoline, and cepharanthine, inhibit human coronavirus OC43 infection of MRC-5 human lung cells. Biomolecules. 2019;9(11):696. doi: 10.3390/biom9110696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim J.Y., Kim Y.I., Park S.J., Kim I.K., Choi Y.K., Kim S.-H. Safe, high-throughput screening of natural compounds of MERS-CoV entry inhibitors using a pseudovirus expressing MERS-CoV spike protein. Int. J. Antimicrob. Agents. 2018;52(5):730–732. doi: 10.1016/j.ijantimicag.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krueger J.G., Suárez-Fariñas M., Cueto I., Khacherian A., Matheson R., Parish L.C., et al. A randomized, placebo-controlled study of SRT2104, a SIRT1 activator, in patients with moderate to severe psoriasis. PloS One. 2015;10(11) doi: 10.1371/journal.pone.0142081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang J., Mantelos A., Toh Z., Tortorella S., Ververis K., Vongsvivut J., et al. Investigation of potential anti-pneumococcal effects of l-sulforaphane and metabolites: insights from synchrotron-FTIR microspectroscopy and molecular docking studies. J. Mol. Graph. Model. 2020;97:107568. doi: 10.1016/j.jmgm.2020.107568. [DOI] [PubMed] [Google Scholar]
- 67.Lin M.-H., Moses D.C., Hsieh C.-H., Cheng S.-C., Chen Y.-H., Sun C.-Y., et al. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antivir. Res. 2018;150:155–163. doi: 10.1016/j.antiviral.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu J.-W., Hsieh P.-S., Lin C.-C., Hu M.-K., Huang S.-M., Wang Y.-M., et al. Synergistic effects of combination treatment using EGCG and suramin against the chikungunya virus. Biochem. Biophys. Res. Commun. 2017;491(3):595–602. doi: 10.1016/j.bbrc.2017.07.157. [DOI] [PubMed] [Google Scholar]
- 69.Moghadamtousi S.Z., Kadir H.A., Hassandarvish P., Tajik H., Abubakar S., Zandi K. A review on antibacterial, antiviral, and antifungal activity of curcumin. BioMed Res. Int. 2014;2014:186864. doi: 10.1155/2014/186864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moghaddam E., Teoh B.-T., Sam S.-S., Lani R., Hassandarvish P., Chik Z., et al. Baicalin, a metabolite of baicalein with antiviral activity against dengue virus. Sci. Rep. 2014;4(1):5452. doi: 10.1038/srep05452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mohammadi Pour P., Fakhri S., Asgary S., Farzaei M.H., Echeverría J. The signaling pathways, and therapeutic targets of antiviral agents: focusing on the antiviral approaches and clinical perspectives of anthocyanins in the management of viral diseases. Front. Pharmacol. 2019;10(1207) doi: 10.3389/fphar.2019.01207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ren J-l, Zhang A.-H., Wang X.-J. Traditional Chinese medicine for COVID-19 treatment. Pharmacol. Res. 2020;155:104743. doi: 10.1016/j.phrs.2020.104743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Richardson P., Griffin I., Tucker C., Smith D., Oechsle O., Phelan A., et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395:e30–e31. doi: 10.1016/S0140-6736(20)30304-4. 10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rogosnitzky M., Okediji P., Koman I. Cepharanthine: a review of the antiviral potential of a Japanese-approved alopecia drug in COVID-19. Pharmacol. Rep. 2020;72(6):1509–1516. doi: 10.1007/s43440-020-00132-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vankadari N. Arbidol: a potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents. 2020;56(2):105998. doi: 10.1016/j.ijantimicag.2020.105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zakaryan H., Arabyan E., Oo A., Zandi K. Flavonoids: promising natural compounds against viral infections. Arch. Virol. 2017;162(9):2539–2551. doi: 10.1007/s00705-017-3417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery. 2020;6(1):14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47(D1):D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mendez D., Gaulton A., Bento A.P., Chambers J., De Veij M., Félix E., et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47(D1):D930–D940. doi: 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Davies M., Nowotka M., Papadatos G., Dedman N., Gaulton A., Atkinson F., et al. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015;43(W1):W612–W620. doi: 10.1093/nar/gkv352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schrödinger . Schrödinger, LLC; New York, NY: 2020. Schrödinger Release 2020-2: LigPrep; p. 2020. [Google Scholar]
- 82.Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 83.Schrödinger . Schrödinger, LLC; New York, NY: 2020. Schrödinger Release 2020-2: Glide; p. 2020. [Google Scholar]
- 84.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 85.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 86.Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J.Y., et al. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theor. Comput. 2016;12(1):281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 87.Shivakumar D., Williams J., Wu Y., Damm W., Shelley J., Sherman W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theor. Comput. 2010;6(5):1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 88.Jorgensen W.L., Maxwell D.S., Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118(45):11225–11236. [Google Scholar]
- 89.Jorgensen W.L., Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988;110(6):1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 90.Cho A.E., Guallar V., Berne B.J., Friesner R. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005;26(9):915–931. doi: 10.1002/jcc.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schrödinger . Schrödinger, LLC; New York, NY: 2020. Schrödinger Release 2020-2: QSite; p. 2020. [Google Scholar]
- 92.Murphy R.B., Philipp D.M., Friesner R.A. A mixed quantum mechanics/molecular mechanics (QM/MM) method for large-scale modeling of chemistry in protein environments. J. Comput. Chem. 2000;21(16):1442–1457. [Google Scholar]
- 93.Philipp D.M., Friesner R.A. Mixed ab initio QM/MM modeling using frozen orbitals and tests with alanine dipeptide and tetrapeptide. J. Comput. Chem. 1999;20(14):1468–1494. [Google Scholar]
- 94.Schrödinger . Schrödinger, LLC; New York, NY: 2020. Schrödinger Release 2020-2: Jaguar; p. 2020. [Google Scholar]
- 95.Bochevarov A.D., Harder E., Hughes T.F., Greenwood J.R., Braden D.A., Philipp D.M., et al. Jaguar: a high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quant. Chem. 2013;113(18):2110–2142. [Google Scholar]
- 96.Osipiuk J., Azizi S.-A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. bioRxiv. 2020 doi: 10.1038/s41467-021-21060-3. 2020.08.06.240192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jendele L., Krivak R., Skoda P., Novotny M., Hoksza D. PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019;47(W1):W345–W349. doi: 10.1093/nar/gkz424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dallakyan S., Olson A.J. In: Chemical Biology: Methods and Protocols. Hempel J.E., Williams C.H., Hong C.C., editors. Springer New York; New York, NY: 2015. Small-molecule library screening by docking with PyRx; pp. 243–250. [DOI] [PubMed] [Google Scholar]
- 99.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hypernet Labs. Galileo. https://galileoapp.io/. 2020 [Available from: https://galileoapp.io/.
- 101.D. E. Shaw Research. Molecular dynamics simulations related to SARS-CoV-2: D. E. Shaw Research Technical Data. http://www.deshawresearch.com/resources_sarscov2.html; 2020 [Available from: http://www.deshawresearch.com/resources_sarscov2.html.
- 102.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 103.Schrödinger . Schrödinger, LLC; New York, NY: 2020. Schrödinger Release 2020-2: Maestro; p. 2020. [Google Scholar]
- 104.Perola E., Walters W.P., Charifson P.S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins: Structure, Function, and Bioinformatics. 2004;56(2):235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 105.Bosken Y.K., Cholko T., Lou Y.-C., Wu K.-P., Chang C-eA. Insights into dynamics of inhibitor and ubiquitin-like protein binding in SARS-CoV-2 papain-like protease. Frontiers in Molecular Biosciences. 2020;7(174) doi: 10.3389/fmolb.2020.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. Unit. States Am. 2008;105(42):16119. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 108.Xu J., Xu Z., Zheng W. A review of the antiviral role of green tea catechins. Molecules. 2017;22(8):1337. doi: 10.3390/molecules22081337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ganeshpurkar A., Saluja A.K. The pharmacological potential of rutin. Saudi Pharmaceut. J. 2017;25(2):149–164. doi: 10.1016/j.jsps.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carmine A.A., Brogden R.N., Heel R.C., Speight T.M., Avery G.S. Cefotaxime. Drugs. 1983;25(3):223–289. doi: 10.2165/00003495-198325030-00001. [DOI] [PubMed] [Google Scholar]
- 111.Karioti A., Vincieri F.F., Bilia A.R. Rapid and efficient purification of naphthodianthrones from St. John’s wort extract by using liquid–liquid extraction and SEC. J. Separ. Sci. 2009;32(9):1374–1382. doi: 10.1002/jssc.200800700. [DOI] [PubMed] [Google Scholar]
- 112.Jacobson J.M., Feinman L., Liebes L., Ostrow N., Koslowski V., Tobia A., et al. Pharmacokinetics, safety, and antiviral effects of hypericin, a derivative of St. John’s wort plant, in patients with chronic hepatitis C virus infection. Antimicrob. Agents Chemother. 2001;45(2):517–524. doi: 10.1128/AAC.45.2.517-524.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen H., Feng R., Muhammad I., Abbas G., Zhang Y., Ren Y., et al. Protective effects of hypericin against infectious bronchitis virus induced apoptosis and reactive oxygen species in chicken embryo kidney cells. Poultry Sci. 2019;98(12):6367–6377. doi: 10.3382/ps/pez465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Romeo A., Iacovelli F., Falconi M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020;286:198068. doi: 10.1016/j.virusres.2020.198068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pitsillou E., Liang J., Karagiannis C., Ververis K., Darmawan K.K., Ng K., et al. Interaction of small molecules with the SARS-CoV-2 main protease in silico and in vitro validation of potential lead compounds using an enzyme-linked immunosorbent assay. Comput. Biol. Chem. 2020:107408. doi: 10.1016/j.compbiolchem.2020.107408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.